

Abstract
The sarcoglycans are a complex of four transmembrane proteins (α, β, γ, and δ) which are primarily expressed in skeletal muscle and are closely associated with dystrophin and the dystroglycans in the muscle membrane. Mutations in the sarcoglycans are responsible for four autosomal recessive forms of muscular dystrophy. The function and the organization of the sarcoglycan complex are unknown. We have used coimmunoprecipitation and in vivo cross-linking techniques to analyze the sarcoglycan complex in cultured mouse myotubes. We demonstrate that the interaction between β- and δ-sarcoglycan is resistant to high concentrations of SDS and α-sarcoglycan is less tightly associated with other members of the complex. Cross-linking experiments show that β-, γ-, and δ-sarcoglycan are in close proximity to one another and that δ-sarcoglycan can be cross-linked to the dystroglycan complex. In addition, three of the sarcoglycans (β, γ, and δ) are shown to form intramolecular disulfide bonds. These studies further our knowledge of the structure of the sarcoglycan complex. Our proposed model of their interactions helps to explain some of the emerging data on the consequences of mutations in the individual sarcoglycans, their effect on the complex, and potentially the clinical course of muscular dystrophies.
Keywords: sarcoglycans, muscular dystrophy, immunoprecipitation, cross-linking, disulfide bonds
The dystrophin–glycoprotein complex (DGC)1 consists of at least ten different proteins located at the sarcolemma and is critical for the stability of the muscle membrane (Ervasti et al., 1990; Yoshida and Ozawa, 1990; Ervasti and Campbell, 1991). The DGC can be subdivided into three subcomplexes according to their biochemical characteristics (Yoshida et al., 1994). The cytoplasmic component consists of three intracellular peripheral membrane proteins: dystrophin, syntrophin, and dystrobrevin. Dystrophin is a 427-kD rod-shaped protein that binds F-actin (Hoffman et al., 1987; Koenig et al., 1988; Hemmings et al., 1992; Ervasti and Campbell, 1993). The syntrophins are a multigene family of at least three homologous proteins of 58 kD that are directly associated with dystrophin (Ahn and Kunkel, 1995; Suzuki et al., 1995). Dystrobrevin is an 80-kD dystrophin-related protein which is also associated with dystrophin (Sadoulet-Puccio et al., 1997). Mutations in the dystrophin gene are the cause of Duchenne and Becker muscular dystrophy (Monaco et al., 1986; Koenig et al., 1987). The second subcomplex in the DGC is the dystroglycan complex, comprised of two closely associated subunits, α- and β-dystroglycan (Ibraghimov-Beskrovnaya et al., 1992). α-Dystroglycan is a 156-kD extracellular matrix protein that binds to laminin (Ervasti and Campbell, 1993). β-Dystroglycan is a 43-kD transmembrane protein that binds directly to the carboxyl terminus of dystrophin via its intracellular tail (Suzuki et al., 1994; Jung et al., 1995). Both dystroglycans are translated from a single mRNA and are expressed in many tissues (Ibraghimov-Beskrovnaya et al., 1993).
The third complex contains a group of transmembrane proteins called sarcoglycans (Yoshida et al., 1994). Unlike the dystroglycans, their expression is mostly restricted to skeletal muscle. Five sarcoglycans have been isolated and characterized to date (α, 50 kD; β, 43 kD; γ, 35 kD; δ, 35 kD; and ε, 50 kD). Four of them (α, β, γ, and δ) are known to be part of the DGC in the sarcolemma of skeletal muscle (Roberds et al., 1993; Bönnemann et al., 1995; Lim et al., 1995; Jung et al., 1996; Nigro et al., 1996b ; Ettinger et al., 1997; McNally et al., 1998). The sarcoglycans have been demonstrated to be closely associated with dystrophin (Suzuki et al., 1992; Cox et al., 1994; Greenberg et al., 1994) and the dystroglycan complex (Rafael et al., 1996).
It has been proposed that the DGC links the actin cytoskeleton with the extracellular matrix (Ohlendieck, 1996) and the proper maintenance of this connection is thought to be crucial to the mechanical stability of the sarcolemma (Ervasti and Campbell, 1993; Campbell, 1995). Although it is not clear how the sarcoglycans might be involved in this linkage, mutations in any one of the four sarcoglycans often lead to the concomitant loss of the other sarcoglycans and are responsible for four autosomal recessive forms of muscular dystrophy (Roberds et al., 1994; Bönnemann et al., 1995; Lim et al., 1995; Noguchi et al., 1995; Nigro et al., 1996a ). Even in Duchenne and Becker muscular dystrophy in which the primary defect is in the dystrophin gene, the sarcoglycans are often absent or greatly reduced at the membrane as shown by immunofluorescence (Ohlendieck and Campbell, 1991; Ohlendieck et al., 1993). Despite intensive study, the function of the sarcoglycans remains elusive. They have no homologues in the electronic databases and experiments designed to explore the structure of the sarcoglycan complex have provided only few clues to their molecular organization. The sarcoglycans can be copurified as a subcomplex from rabbit skeletal muscle but only β-, γ-, and δ-sarcoglycan can be chemically cross-linked to each other (Yoshida et al., 1994, 1997). In vitro expression of the extracellular portions of the sarcoglycans demonstrated that these domains interact with each other and as well as with the dystroglycans in coimmunoprecipitation and blot overlay experiments (Sakamoto et al., 1997).
In this report, we have investigated the molecular organization of the sarcoglycan complex in cultured mouse myotubes. Coimmunoprecipitation experiments under different stringencies suggest that α-sarcoglycan is loosely associated with the other sarcoglycans whereas the interaction between β- and δ-sarcoglycan is particularly strong. In vivo cross-linking experiments have demonstrated that β-, γ-, and δ-sarcoglycan can cross-link to form two major subcomplexes. We have also shown that δ-sarcoglycan can be cross-linked to the dystroglycan complex and of the four sarcoglycans, only β-, γ-, and δ-sarcoglycan contain intramolecular disulfide bonds. All together, these information should increase our understanding of the sarcoglycan complex and may shed light on its potential functions and pathophysiological role in muscular dystrophy.
Materials and Methods
Mouse Tissue Culture
Primary myoblasts were established from normal mouse skeletal muscle and grown on 100-mm plates in Ham's F-10 media supplemented with 20% fetal bovine serum, 2 mM l-glutamine, 50 μg ml−1 streptomycin, 50 U/ml−1 penicillin (all purchased from Sigma Chemical Co., St. Louis, MO) and 2.5 ng ml−1 basic fibroblast growth factor (Promega, Madison, WI). All plates were coated with 40 μg ml−1 extracellular matrix containing entactin, collagen IV, and laminin (Upstate Biotechnology, Lake Placid, NY) and the cultures were incubated at 37°C with 5% CO2. When the myoblasts reached confluency, the media was switched to DME (Sigma Chemical Co.) supplemented with 2% horse serum to induce differentiation. Myoblasts would normally fuse to form myotubes after 3–6 d.
Preparation of Microsomes
To minimize protein degradation, membrane microsomes were prepared at 4°C according to the method of Mitchell (Mitchell et al., 1983). In brief, ∼1 g of human skeletal muscle (or four 100-mm plates of cultured mouse myotubes) was homogenized with a Dounce homogenizer in 5 ml of ice-cold buffer A (10% sucrose, 0.5 mM EDTA, pH 7.2, 1 mM PMSF and 1× protease inhibitor cocktail purchased from Boehringer Mannheim [Mannheim, Germany]). The homogenate was centrifuged at 1,000 g for 10 min and the pellet was rehomogenized two additional times using fresh buffer A. The supernatant from each round of homogenization was subsequently combined and recentrifuged at 12,000 g for 10 min. The pellet which contained mostly lysosomes and mitochondria was discarded. Microsomes were pelleted from the postlysosomal supernatant by centrifugation at 105,000 g for 1 h using a Beckman SW 28 rotor (Palo Alto, CA). To remove proteins that were nonspecifically attached to the pellet, the microsomes were washed with 150 mM Tris and 0.5 M KCl and then recentrifuged at 105,000 g for 1 h.
Preparation of Cell Lysates and Immunoprecipitation
Cultured mouse myotubes from a 100-mm plate were washed three times with 1× PBS and lysed on ice for 15 min in 1 ml of lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS 1 mM PMSF and 1× protease inhibitor cocktail). Cell lysates were collected by centrifugation at 15,000 g for 10 min at 4°C. Protein concentration was determined by the Bio-Rad DC protein colorimetric assay using bovine serum albumin as standard (Hercules, CA). For immunoprecipitation experiments, 50–100 μg of cell lysate was first precleared with 50 μl of protein G–Sepharose beads (Sigma Chemical Co.) for 1 h at 4°C. After centrifugation at 10,000 g for 2 min, the precleared cell lysate was incubated with 2.5 μl of anti-sarcoglycan antibody on ice for 4 h and then with 10 μl of protein G–Sepharose beads on ice for 1 h. The immune complex was pelleted by centrifugation at 10,000 g for 2 min and washed three times with 1 ml of ice-cold lysis buffer. The final pellet was solubilized in 10 μl of 2× protein sample buffer (Novex, San Diego, CA) and loaded on 4–20% denaturing gradient gels (Novex). Note that the concentration of SDS in the lysis buffer was varied in some experiments.
Antibodies
Mouse monoclonal antibodies directed against α-sarcoglycan (NCL–a-sarc); β-sarcoglycan (NCL–b-sarc), γ-sarcoglycan (NCL–g-sarc), δ-sarcoglycan (NCL–d-sarc), β-dystroglycan (NCL–b-DG), and dystrophin (NCL-dys2) were all purchased from Novocastra (Newcastle-upon-Tyne, UK) and each was diluted 1:100 for Western blotting and 1:200 for immunohistochemistry. Anti–α-dystroglycan antibody (clone VIA4-1) and anti-actin antibodies (A-2066 and A-4700) were from Upstate Biotechnology and Sigma Chemical Co., respectively. Each was used in a dilution of 1:1,000 for Western blot analysis. Mouse monoclonal antibody against syntrophin was obtained from S.C. Froehner (University of North Carolina, Chapel Hill, NC) (Froehner et al., 1987) and the rabbit polyclonal antibody against δ-sarcoglycan was a generous gift from V. Nigro (Second University of Naples, Naples, Italy) (Nigro et al., 1996b ). Both antibodies were used at a dilution of 1:5,000 for Western blot analysis. The rabbit polyclonal antibody against dystrophin, 6–10, has been characterized previously (Lidov et al., 1990).
Immunoblot Analysis
Proteins were transferred from SDS-PAGE gels to nitrocellulose membranes (Schleicher & Schuell, Keene, NH) in Towbin buffer at 20 V at room temperature (Towbin et al., 1979). Membranes were blocked in western buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20, and 0.5% gelatin) for 2 h at room temperature and then incubated with primary antibodies diluted in Western buffer for another 2 h. After extensive washing in Western buffer, the membranes were incubated for 1 h with a horseradish peroxidase-conjugated donkey anti–rabbit (or anti– mouse) IgG (H + L) secondary antibody diluted at 1:10,000 in the Western buffer (Jackson ImmunoResearch Laboratories, West Grove, PA). The horseradish peroxidase-conjugated protein complex was detected by enhanced chemiluminescence according to the manufacturer's protocol (Amersham Corp., Arlington Heights, IL).
In Vivo Cross-linking of Mouse Myotubes
Cultured mouse myotubes from a 100-mm plate were washed three times with 1× PBS and cross-linked with 1 mM dithiobis (sulfosuccinimidylpropionate) (DTSSP) or 1 mM sulfo-ethylene glycolbis (sulfosuccinimidylsuccinate) (EGS) (Pierce Chemical Co., Rockford, IL) in 50 mM Hepes, pH 7.5. After a 30-min incubation at 37°C, the reaction was quenched in 5 mL of 1 M Tris, pH 7.4, for 15 min. The cross-linked myotubes were then washed three times with 1× PBS and lysed on ice as described above. DTSSP cross-linked products can be cleaved by reduction with 10 mM DTT for 30 min at 37°C. Sulfo-EGS cross-linked products can be cleaved by incubation with 1 M hydroxylamine for 4 h at 37°C, pH 8.5.
Two-dimensional Diagonal Gel Electrophoresis
Cell lysates prepared from DTSSP cross-linked mouse myotubes were separated by electrophoresis on 4–20% denaturing gradient gel (Novex) under nonreducing conditions in the first dimension. Each lane was cut into a strip and soaked in a reducing buffer of 0.1 M DTT, 0.1% SDS, 70 mM Tris, pH 6.7, and 10% glycerol at room temperature for 1 h. The reduced strip was then placed on top of a 4–20% denaturing gradient slab gel (Novex) and electrophoresed under reducing conditions in the second dimension. The identity of the cross-linked proteins was determined by Western blots using different anti-sarcoglycan antibodies. A replicate blot with known molecular weight standards was used to estimate the molecular mass of the proteins in each dimension.
Immunofluorescent Analysis of Muscle Biopsies
Muscle biopsies taken from autosomal recessive muscular dystrophy patients were frozen in isopentane cooled in liquid nitrogen and stored at −80°C until cryosectioning. Thaw-mounted sections (7 μm) were fixed in methanol for 1 min and blocked in 1× PBS with 10% fetal calf serum and 0.1% Triton X-100 for 30 min at 4°C. Incubation with primary anti-sarcoglycan and anti-dystrophin antibodies (1:200 dilution) was carried out at 4°C for overnight. The slides were washed three times for 20 min with 1× PBS, reblocked, and then incubated for 1 h at room temperature with a Cy3-conjugated donkey anti–rabbit (or anti–mouse) IgG (H + L) secondary antibody adsorbed for human with multilabel from Jackson ImmunoResearch Laboratories (diluted 1:300 in 1× PBS). After washing with 1× PBS three times for 20 min, the slides were coverslipped with Immunomount (Shandon, Pittsburgh, PA) and examined using a Leitz Orthoplan fluorescence microscope with epi-illumination (Ernst Leitz, Wetzlar, Germany).
Results
Sarcoglycans Are Localized to the Membrane Fraction of Differentiated Mouse Myotubes
Cell culture has been used widely as a model for studying the DGC. Localization of dystrophin to the cell membrane has been demonstrated by expression of recombinant dystrophin in COS cells, mouse C2C12 cells and mouse mdx myogenic cultures (Lee et al., 1991; Clemens et al., 1995; Kumar-Singh and Chamberlain, 1996). Previous studies have shown that both endogenous dystrophin and α-sarcoglycan are localized to the sarcolemma of rat L6 myotubes (Yoshida et al., 1998). In this report, we have used primary myoblasts established from skeletal muscle of normal mouse to study the organization of the sarcoglycan complex. Upon reaching confluency, mouse myoblasts in culture fuse to form multinucleated myotubes and begin to express skeletal muscle-specific proteins (Hauschka, 1994). The expression of the DGC in the differentiated myotubes was confirmed by immunoblotting of the cell lysates with antibodies against components of the DGC (data not shown).
To determine if the DGC in primary mouse myotube culture is localized to the cell membrane, membrane microsomes were prepared from myotubes culture (Fig. 1, row A). Western analysis showed that all the components of the DGC were found in the microsomal fraction (Fig. 1, lane 3) but not in the soluble protein fraction (lane 2). Similar results were obtained from mouse C2C12 myotubes (data not shown). Microsomes prepared in the same way from human skeletal muscle were shown to contain all the components of the DGC as expected (Fig. 1, row B, lane 3).
Figure 1.

Localization of the DGC. Cultured mouse myotubes (row A) and human skeletal muscle (row B) were used as starting materials to purify microsomes. Lane 1, unlysed cell pellet fraction after 1,000 g centrifugation. Lane 2, soluble protein fraction after 105,000 g centrifugation. Lane 3, microsome fraction. The composition of each fraction was determined by Western blots using antibodies (1° Ab) against the components of the DGC. Note that some α-dystroglycan from human skeletal muscle was found in the soluble protein fraction which is probably due to the presence of an extensive extracellular matrix network in human tissue but not in cell culture. Also note that actin can exist as both unpolymerized monomers in the cytoplasm (soluble protein fraction) and filaments associated with the membrane (microsome fraction).
When cell lysate from cultured mouse myotubes was immunoprecipitated with an anti-dystrophin antibody, 6–10, the immune complex was found to contain proteins recognized by antibodies against dystrophin, the syntrophins and α-, β-, γ-, and δ-sarcoglycan but not actin (Fig. 2). Neither filamin nor troponin was coimmunoprecipitated by the 6–10 antibody (data not shown). Since the DGC is defined by copurification with dystrophin, the coimmunoprecipitation of the DGC and dystrophin which is normally at the sarcolemma implied that these components form a stable complex on the plasma membrane of mouse myotubes. The assembly of the DGC at the membrane of mouse myotubes and its similar biochemical characteristics to human skeletal muscle provides an ideal cell culture system to explore the sarcoglycan complex.
Figure 2.

Coimmunoprecip-itation of the sarcoglycans with dystrophin. Cell lysate from cultured mouse myotubes was immunoprecipitated by the anti-dystrophin antibody, 6–10. The immune complex was analyzed by Western blots using antibodies (1° Ab) against the sarcoglycans, dystrophin, the syntrophins, and the actin. Lane 1, cell lysate from mouse myotubes (11% input). Lane 2, immunoprecipitated products. Note that α-sarcoglycan was the least efficiently precipitated component of the DGC.
α-, β-, γ-, and δ-Sarcoglycan Exist As a Complex in Mouse Myotubes
The sarcoglycans have been shown to copurify as a subcomplex from rabbit skeletal muscle (Yoshida et al., 1994). To determine if the sarcoglycans could also be purified as a complex from mouse myotubes, cell lysate prepared from mouse myotubes was immunoprecipitated with antibodies against different sarcoglycans followed by Western analysis. All four sarcoglycans were coprecipitated when an antibody against β-sarcoglycan (NCL–b-sarc) was used (Fig. 3), thus verifying that α-, β-, γ-, and δ-sarcoglycan form a complex in the mouse myotubes.
Figure 3.

Coimmunoprecipitation of the sarcoglycans using different anti-sarcoglycan antibodies. Cell lysate from cultured mouse myotubes were immunoprecipitated by an antibody directed against α-, β-, γ-, or δ-sarcoglycan. The immune complex was analyzed by Western blots using antibodies (1° Ab) against different sarcoglycans. Lane 1, cell lysate from mouse myotubes (13–16% input). Lane 2, immunoprecipitated products. Note that different sarcoglycans were precipitated by different anti-sarcoglycan antibodies.
When other anti-sarcoglycan antibodies were used as the primary immunoprecipitation reagents, the sarcoglycans being coimmunoprecipitated from the same cell lysate sample was different in each case (Fig. 3). A polyclonal anti–δ-sarcoglycan antibody was shown to coimmunoprecipitate only β- and δ-sarcoglycan but not α- and γ-sarcoglycan from the mouse myotubes cell lysate. An anti– α-sarcoglycan antibody (NCL–a-sarc) apparently only immunoprecipitated α-sarcoglycan while an anti–γ-sarcoglycan antibody (NCL–g-sarc) only immunoprecipitated γ-sarcoglycan. This immunoprecipitation data suggests that there is a preferential association between β- and δ-sarcoglycan.
β- and δ-Sarcoglycan Are Preferentially Associated with One Another
To examine the interaction between β- and δ-sarcoglycan more closely, the sarcoglycan complex was disrupted in the presence of different concentrations of SDS. Proteins immunoprecipitated by the anti–β-sarcoglycan antibody were immobilized on protein G–Sepharose beads and washed with buffers containing different concentrations of SDS. The final composition of the post-SDS–washed immune complex was then determined by Western blot using different anti-sarcoglycan antibodies. All four sarcoglycans were present in the immunoprecipitated complex after being washed with buffer containing no SDS (Fig. 4 a, lane 2) or 0.2% SDS (lane 3). However, increasing the SDS concentration in the washing buffer to 0.3% or above caused the complete dissociation of α-sarcoglycan from the complex (Fig. 4 a, lane 4 and 5, respectively). A slight reduction in the amount of γ-sarcoglycan in the complex was also observed at 0.3% SDS washing conditions (Fig. 4 a, lane 4). Further dissociation of γ-sarcoglycan was seen when the immune complex was washed with 0.4% SDS (lane 5). β- and δ-Sarcoglycan remained relatively well associated even after the immune complex was washed with 0.4% SDS (Fig. 4 a, lane 5).
Figure 4.

Coimmunoprecip-itation of the sarcoglycans under different stringencies. (A) Cell lysate from cultured mouse myotubes was immunoprecipitated by the anti– β-sarcoglycan antibody (NCL– b-sarc). The immune complex was washed in 1% NP-40 buffer containing no SDS (lane 2), 0.2% SDS (lane 3), 0.3% SDS (lane 4), and 0.4% SDS (lane 5). Lane 1, cell lysate from mouse myotubes (33% input). (B) Immunoprecipitation was carried out after cultured mouse myotubes were lysed in 1% NP-40 buffer containing no SDS (lane 2), 0.1% SDS (lane 3), 0.2% SDS (lane 4), 0.3% SDS (lane 5), and 0.4% SDS (lane 6). Lane 1, cell lysate from mouse myotubes (16% input). The final composition of the immune complex was determined by Western blots using antibodies (1° Ab) against different sarcoglycans. Note that α-sarcoglycan was the most sensitive to increasing SDS concentration, resulting in dissociation from the complex in both cases.
To increase the stringency of the disruption of the sarcoglycan complex, mouse myotubes were lysed in different concentrations of SDS for 1 h before immunoprecipitation was carried out. Under these conditions, the sarcoglycan complex was likely to undergo further dissociation due to the prolonged exposure to high concentrations of SDS. Similar to the results shown above, all four sarcoglycans were detected in the immunoprecipitated complex when mouse myotubes were lysed under relatively mild conditions with no SDS, 0.1% or 0.2% SDS (Fig. 4 b, lanes 2–4). α-Sarcoglycan was completely separated from the complex when myotubes were lysed at either 0.3 or 0.4% SDS (Fig. 4 b, lanes 5 and 6). However, in contrast to the previous observation that washing the immune complex with 0.3% SDS only caused a mild dissociation of γ-sarcoglycan from the complex (Fig. 4 a, lane 4), a drastic reduction of γ-sarcoglycan in the complex was observed when myotubes were lysed in 0.3% SDS (Fig. 4 b, lane 5). The amount of β- and δ-sarcoglycan found in the immunoprecipitated complex was also reduced to a greater extent in the more stringent conditions but nevertheless, a substantial amount of both sarcoglycans were still associated with one another (Fig. 4 b, lane 5). When the concentration of SDS in the lysis buffer was increased to 0.4%, all evidence of the immunoprecipitation was lost probably because such stringent conditions abolished the binding of the antibody to its antigen or the binding of the immune complex to the protein G–Sepharose beads.
The same pattern of sarcoglycan dissociation was observed when immunoprecipitation was performed on cell lysates prepared from other cell lines, such as murine C2C12 cells or a normal mouse cell line transformed with v-myc (data not shown). In addition, the experiment was repeated on cultured human myotubes using a human δ-sarcoglycan–specific antibody (NCL–d-sarc). As with the anti–β-sarcoglycan antibody, the complex immunoprecipitated by this antibody under low stringency also contained all four sarcoglycans. When the SDS concentration in the lysis buffer was increased above 0.2%, both α- and γ-sarcoglycan were found to dissociate from the complex but β- and δ-sarcoglycan remained bound together (data not shown).
Sarcoglycans in Mouse Myotubes Can Be Cross-linked In Vivo
Proteins that are tightly associated or in close proximity with one another can frequently be cross-linked with chemical reagents. To explore the spatial relationship of the sarcoglycans in the DGC, cultured mouse myotubes were cross-linked with 1 mM DTSSP (Jung and Moroi, 1983). Both uncross-linked and DTSSP cross-linked mouse myotubes were lysed in 0.3% SDS and then immunoprecipitated with the anti–β-sarcoglycan antibody. Cross-linked proteins immunoprecipitated from the DTSSP-treated myotubes were cleaved with 10 mM DTT before loading on SDS-PAGE gels. Western blots of the immunoprecipitated complex from uncross-linked myotubes showed that only β- and δ-sarcoglycan were present in the complex as one would expect under this SDS concentration (Fig. 5 a, lane 2). On the other hand, mouse myotubes cross-linked with DTSSP resulted in co-immunoprecipitation of all four sarcoglycans (Fig. 5 a, lane 3), indicating that they were in close enough proximity to be cross-linked and were located at the plasma membrane because the cross-linking reagent used in the experiments is membrane impermeable and can therefore only cross-link proteins on the extracellular surface of the cells. However, not all sarcoglycans were cross-linked to the same extent in the experiments. Comparison of the amount of sarcoglycans being immunoprecipitated (Fig. 5 b, lane 3) to the original input materials (lane 1) suggests that α-sarcoglycan was less efficiently cross-linked to the complex relative to other sarcoglycans.
Figure 5.

In vivo cross-linking of the sarcoglycans. Mouse myotubes were chemically cross-linked with 1 mM DTSSP and immunoprecipitated by anti–β-sarcoglycan antibody (A) or anti–α- or β-dystroglycan antibody (B). Lane 1, cell lysate from mouse myotubes (26–34% input). Lanes 2 and 4, immunoprecipitated products from uncross-linked myotubes. Lanes 3 and 5, immunoprecipitated products from equal amount of DTSSP cross-linked myotubes. The composition of the immune complex was determined by Western blots using antibodies (1° Ab) against different sarcoglycans and dystroglycans.
In Vivo Cross-linked Sarcoglycans Consist of Two Major Subcomplexes
The status of the DTSSP cross-linked myotubes was examined in more detail by two-dimensional (2-D) diagonal gel electrophoresis (Traut et al., 1989). The first dimension of electrophoresis was performed under nonreducing conditions and the second dimension of electrophoresis was performed under reducing conditions. Proteins that are not cross-linked will have the same electrophoretic mobility in both dimensions and will be detected on a diagonal line in the gel. On the other hand, cross-linked proteins will dissociate into their monomeric species in the second dimension under reduction and will therefore appear below the diagonal line (Fig. 6 a). By Western blot analysis, both α- and β-dystroglycan were found to cross-link to form a product of ∼200 kD in the first dimension (Fig. 6 b, X) and upon reduction with DTT, they separated from one another according to their molecular weights in the second dimension (spot 1 and 2). This is consistent with previous studies that α-dystroglycan was intimately associated with β-dystroglycan (Ibraghimov-Beskrovnaya et al., 1992; Yoshida et al., 1994) and thus validated the use of 2-D diagonal gel to detect cross-linked proteins.
Figure 6.

Analysis of the cross-linked sarcoglycans. Cell lysates from DTSSP cross-linked myotubes were examined by 2-D diagonal gel using antibodies against different sarcoglycans. (A) Schematic diagram of the principle of 2-D diagonal gel. Single circle, uncross-linked proteins; two circles joined by a line, cross-linked product. Shown in the examples are Western blots using antibodies against α-sarcoglycan, β- and α-dystroglycan (B), against β- and γ-sarcoglycan (C), and against β- and δ-sarcoglycan (D). Cross-linked proteins that appear below the diagonal line are represented by Arabic numerals: 1, α-dystroglycan; 2, β-dystroglycan; 4a and 4b, β-sarcoglycan; 5a, γ-sarcoglycan; and 6a, 6b, and 7, δ-sarcoglycan. The estimated molecular weight of the three cross-linked products X, X1, and X2 identified in this experiment are ∼200, 120, and 80 kD, respectively. Note that not all sarcoglycans and dystroglycans were cross-linked in the experiments. Uncross-linked proteins were found on the diagonal line and represented in Roman numerals: I, α-dystroglycan; II, β-dystroglycan; III, α-sarcoglycan; IV, β-sarcoglycan; V, γ-sarcoglycan; and VI, δ-sarcoglycan.
When a replicate blot was immunoblotted with the anti–β- and anti–γ-sarcoglycan antibodies, three spots were detected below the diagonal line (Fig. 6 c). Two of the spots were recognized by antibodies against β-sarcoglycan (spot 4a) and γ-sarcoglycan (spot 5a) and together they were cross-linked to form the product X1 of ∼120 kD. The third spot was also recognized by the anti–β-sarcoglycan antibody (spot 4b) but was cross-linked to form a smaller product X2 of ∼80 kD. When the sum of the molecular weight of the sarcoglycans identified in the cross-linked species X1 was obtained (43 kD + 35 kD = 78 kD), there was a missing mass of ∼40 kD in the product. Similarly, when we added up the sum of the molecular weights of the sarcoglycans identified in the cross-linked species X2 (43 kD = 43 kD), there was also a missing mass of ∼40 kD. Since δ-sarcoglycan has a molecular weight of 35 kD, it is possible that δ-sarcoglycan could be part of the cross-linked 120- and 80-kD complex. Western analysis of a replicate blot using anti–β- and anti–δ-sarcoglycan antibodies revealed at least four spots below the diagonal line (Fig. 6 d, spots 4a, 4b, 6a, and 6b). Two of the spots (4a and 6a) were recognized by the anti–β- and anti–δ-sarcoglycan antibody, respectively, and were shown to cross-link to form the same 120-kD product (Fig. 6 d, X1) identified in Fig. 6 c. The other two spots (4b and 6b) were also recognized by the anti–β- and anti–δ-sarcoglycan antibodies, respectively, and were cross-linked to form the same 80-kD product (Fig. 6 d, X2) identified in Fig. 6 c. Intriguingly, α-sarcoglycan was found to migrate as a single spot on the diagonal line and did not seem to cross-link to any proteins in these experiments (Fig. 6 b, spot III). Similar results were obtained when mouse myotubes were treated with another cross-linker, sulfo-EGS which has a longer spacer arm length (data not shown). Collectively, we have demonstrated that β- and δ-sarcoglycan could be cross-linked to form two major subcomplexes (X1 and X2) in which the 120-kD product X1 also consisted of γ-sarcoglycan. Our results suggest that both β- and δ-sarcoglycan are in close physical proximity and are consistent with our earlier observations that they are tightly associated with one another.
δ-Sarcoglycan Can Be Cross-linked to the Dystroglycan Complex
In the previous 2-D diagonal gel electrophoresis of DTSSP cross-linked mouse myotubes, a third spot recognized by the anti–δ-sarcoglycan antibody was sometimes observed below the diagonal line (Fig. 6 d, spot 7). This spot was cross-linked to form a product with an estimated molecular weight between 200–220 kD which is similar in size to the cross-linked α-/β-dystroglycan complex identified in Fig. 6 b. To determine if δ-sarcoglycan could be cross-linked to the dystroglycan complex, cell lysates from both DTSSP cross-linked and uncross-linked mouse myotubes were immunoprecipitated by antibodies against β-dystroglycan (Fig. 5 b, lanes 2 and 3) or α-dystroglycan (lane 4 and 5). Western analysis of the immune complex showed α-dystroglycan was coprecipitated with β-dystroglycan from both uncross-linked (lanes 2 and 4) and DTSSP cross-linked myotube samples (lanes 3 and 5). No sarcoglycan was found to be associated with the dystroglycans without cross-linking (lanes 2 and 4), whereas both antibodies immunoprecipitated δ-sarcoglycan from DTSSP cross-linked myotube cell lysate (lanes 3 and 5). There was no evidence that the immunoprecipitated materials contained the other three sarcoglycans as revealed by Western blot using antibodies against α-, β-, or γ-sarcoglycan (lanes 2–5 and data not shown).
Sarcoglycans in Mouse Myotubes Form Intramolecular Disulfide Bonds
Sequence analysis of the sarcoglycans revealed that there are clusters of conserved cysteine residues in their extracellular domains which have the potential to form intra- and/or intermolecular disulfide bonds (Fig. 7 a). To determine whether or not there are disulfide bonds in the sarcoglycans, their electrophoretic mobility was compared under reducing and nonreducing conditions using cell lysates prepared from mouse myotubes. Western analysis showed that β-, γ-, δ-sarcoglycan, and β-dystroglycan all had a faster electrophoretic mobility under nonreducing conditions (Fig. 7 b, lane 1, single arrowhead). This apparent shift of mobility indicated the presence of intramolecular disulfide bonds because proteins that contain such disulfide bonds will often adapt a more compact conformation and are expected to migrate faster in SDS-PAGE gel under nonreducing conditions. This approach has been used by others to demonstrate the presence of intramolecular disulfide bonds in β-dystroglycan (Deyst et al., 1995). No discernible difference in the electrophoretic mobility was observed for α-sarcoglycan or the actin negative control.
Figure 7.

Detection of intramolecular disulfide bond in the sarcoglycans. (A) Alignment of the carboxyl termini of β-, γ-, δ-, and C. elegans sarcoglycan with the consensus EGF-like repeat by MacVector Program (Oxford Molecular Group, Oxford, UK). Right, numbers correspond to the last amino acid residue in the protein; asterisk, stop codon; underline, four conserved cysteine residues. The cysteine 283 in γ-sarcoglycan (italics) is changed to tyrosine in patients with a severe form of early onset autosomal recessive muscular dystrophy (Piccolo et al., 1996). According to the consensus EGF-like repeat motif, the fifth cysteine is linked to the sixth cysteine by a disulfide bond (Abe et al., 1998). This corresponds to the two middle conserved cysteine residues in the sarcoglycans. (B) Cell lysates from cultured mouse myotubes were electrophoresed in SDS-PAGE gel under nonreducing conditions (lane 1) and reducing conditions (lane 2). After transferred to nitrocellulose membranes, blots were examined by Western using antibodies (1° Ab) against different sarcoglycans. The size of the sarcoglycans is indicated by a single arrow (nonreduced form) or a double arrow (reduced form). Note that the extra bands appearing on the nonreducing lane on the Western blot using the anti–β-sarcoglycan antibody were determined as nonspecific products unrelated to the DGC by 2-D diagonal gel electrophoresis (data not shown).
The possibility of intermolecular disulfide bonds formation in sarcoglycans was examined by 2-D diagonal gel method. The expectation would be that molecules linked by disulfide bonds will migrate in the first dimension as a large disulfide-linked complex and will dissociate into monomers upon reduction in the second dimension. All four sarcoglycans from mouse myotubes were detected on the diagonal line (Fig. 8). Since no extra spot was detected below the diagonal line, the sarcoglycans are not likely to form disulfide bridges with other proteins in mouse myotubes.
Figure 8.

Absence of intermolecular disulfide bonds between the sarcoglycans. Cell lysate from cultured mouse myotubes was electrophoresed in 2-D diagonal gel and examined by Western blots using antibodies against different sarcoglycans. Shown in the example is the Western blot using antibodies against α-, β-, and δ-sarcoglycan. Note that no extra spot was observed below the diagonal line (dashed line).
β- and γ-Sarcoglycan Mutations Have Different Effects on the Sarcoglycan Complex
Although mutations in one sarcoglycan can disrupt the sarcoglycan complex and lead to the loss or reduction of other sarcoglycans, the degree of reduction of each sarcoglycan observed in patients is often not the same. To assess the effect of different sarcoglycan mutations on the complex, we compared the immunostaining pattern of sarcoglycans in two autosomal recessive muscular dystrophy patients with homozygous mutations in different sarcoglycan genes. Both patients have a normal dystrophin pattern at the muscle membrane as revealed by immunohistochemistry (Fig. 9). Patient AL has a homozygous nonsense mutation that changes a tyrosine residue to a premature stop codon at codon 178 in the β-sarcoglycan (Bönnemann et al., 1998). This mutation is predicted to produce a truncated protein ∼25 kD. Immunofluorescence studies showed that all four sarcoglycans were completely absent from the sarcolemma (Fig. 9). Patient CR, on the other hand, carries a homozygous Δ521-T deletion in the γ-sarcoglycan gene. This patient has been previously described in another report (Bönnemann et al., 1998). The mutation has also been seen in other patients (McNally et al., 1996) and is predicted to generate a truncated γ-sarcoglycan of ∼23 kD which is similar in size to the truncated β-sarcoglycan. This patient showed a complete absence of γ-sarcoglycan staining in the muscle biopsy by immunofluorescence but the staining of the other three sarcoglycans was preserved at a significant level (Fig. 9).
Figure 9.
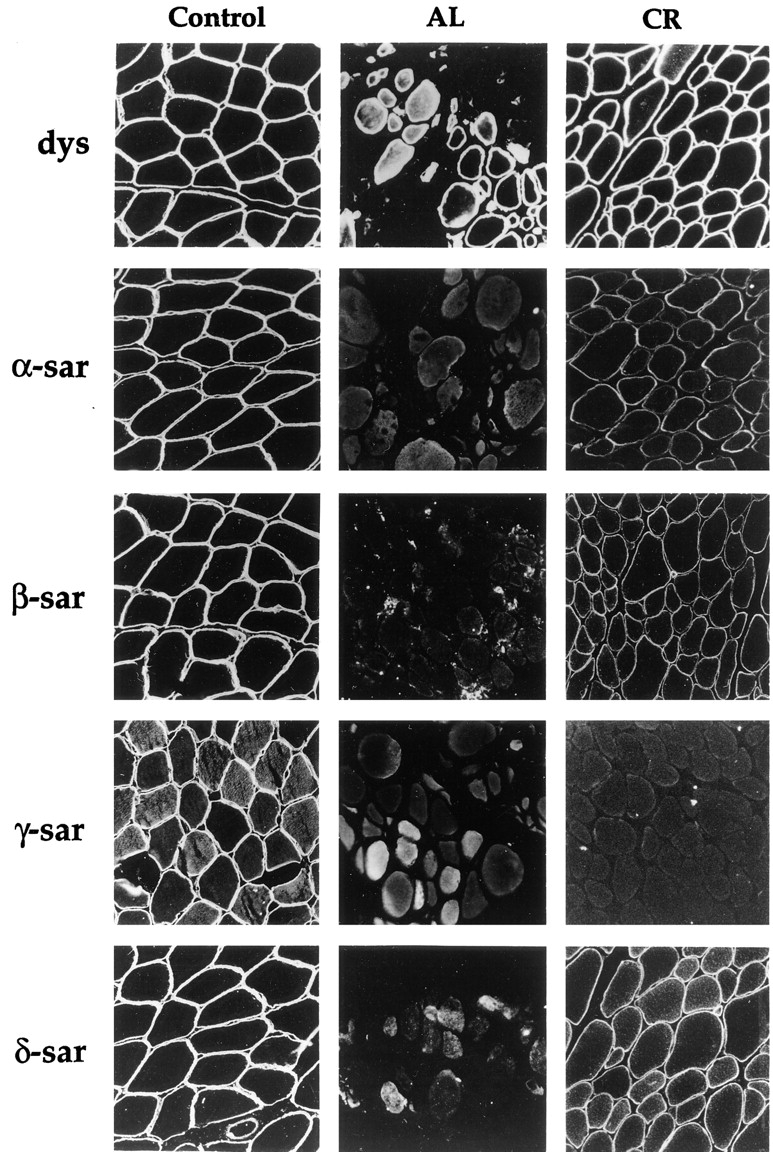
Immunofluorescence of muscle biopsies from patients with autosomal recessive muscular dystrophy. Muscle section was stained with antibodies against dystrophin (dys), α-sarcoglycan (α-sar), β-sarcoglycan (β-sar), γ-sarcoglycan (γ-sar), and δ-sarcoglycan (δ-sar). Note that patients AL and CR showed different patterns of immunostaining for each of the sarcoglycans.
Discussion
The sarcoglycans are a group of four transmembrane proteins which are part of the dystrophin-associated protein complex at the sarcolemma. Mutations in the sarcoglycans are responsible for four unique, yet clinically similar autosomal recessive forms of muscular dystrophy. Although it is thought that mutations in one sarcoglycan can lead to the disruption of the entire sarcoglycan complex, immunohistochemical studies and Western analyses have shown that the effect of mutations on the complex is often dependent on the particular sarcoglycan being mutated. This implies that the molecular organization of the sarcoglycan complex is more complicated than previously assumed. Although several attempts have been made to understand the organization of the sarcoglycans (Yoshida et al., 1994, 1997; Sakamoto et al., 1997), many of these studies have not addressed the question of how each sarcoglycan might interact differently with one another or how these interactions might be disturbed in muscular dystrophies. Coimmunoprecipitation experiments from other groups have suggested a high affinity interaction between β- and δ-sarcoglycan but the study was based on in vitro expressed sarcoglycans (Sakamoto et al., 1997). Here we have analyzed the association of sarcoglycans using tissue culture as a model system. In mouse cultured myotubes, the sarcoglycans exist as a complex of proteins (Fig. 3) and are intimately associated with dystrophin on the plasma membrane (Figs. 1 and 2). Immunoprecipitation experiments from cell lysates of these myotubes under increasingly stringent conditions showed that the interaction between β- and δ-sarcoglycan is highly resistant to SDS (Fig. 4), suggesting that they are tightly associated with one another. In contrast, α-sarcoglycan can be dissociated from the complex under relatively mild conditions (Fig. 4), implying that its association with other sarcoglycans is relatively weaker.
Previous cross-linking experiments by other groups using sarcoglycans purified from rabbit skeletal muscle have identified two cross-linked products of 69 and 110 kD but their exact compositions were not determined (Yoshida et al., 1994). It has also been shown that α-sarcoglycan is not readily cross-linked to other sarcoglycans (Yoshida et al., 1994, 1997). In this report, when cultured mouse myotubes were in vivo cross-linked with 1 mM DTSSP, two major sarcoglycan subcomplexes of similar size (80 and 120 kD) were identified using 2-D diagonal gel electrophoresis (Fig. 6). Using a panel of sarcoglycan-specific antibodies, we have demonstrated that the 120-kD product consists of β-, γ-, and δ-sarcoglycan and the 80-kD product contains β- and δ-sarcoglycan. This preferential cross-linking between β- and δ-sarcoglycan provides additional support that they are the most tightly associated sarcoglycans. There was no evidence of cross-linking between α-sarcoglycan and other proteins as detected by the 2-D diagonal gel method, although coimmunoprecipitation experiments have shown that a small amount of α-sarcoglycan can be cross-linked to other sarcoglycans (Fig. 5 a). This discordance is likely due to the low sensitivity of 2-D diagonal gel electrophoresis. All together, these results suggest that α-sarcoglycan may be spatially separated from other sarcoglycans.
Our results also showed that δ-sarcoglycan was able to cross-link to the dystroglycan complex. However, it is not possible to distinguish to which of the two dystroglycans δ-sarcoglycan was cross-linked because the anti–α- or β-dystroglycan antibody used in this study resulted in coimmunoprecipitation of both α- and β-dystroglycan and δ-sarcoglycan (Fig. 5 b). In the study of the BIO 14.6 hamster model which has a deletion in the δ-sarcoglycan gene, there is not only a complete absence of all sarcoglycans but also a dramatic reduction of α-dystroglycan (Holt et al., 1998). Our data thus support a possible interaction between δ-sarcoglycan and the dystroglycan complex.
In our coimmunoprecipitation experiments, there was a difference in the immunoprecipitation pattern of the sarcoglycans with different anti-sarcoglycan antibodies (Fig. 3). Although β-sarcoglycan was always coimmunoprecipitated with δ-sarcoglycan, α- and γ-sarcoglycan were often immunoprecipitated without the other sarcoglycans. Such a pattern is consistent with our findings that β-sarcoglycan is preferentially associated with δ-sarcoglycan and α-sarcoglycan is less strongly associated with other sarcoglycans. However, the antibodies used in the experiments might also play a role. Depending on the epitopes recognized by these antibodies, the binding of the antibody to the sarcoglycan complex might be sterically hindered or might disrupt existing interactions among the sarcoglycans. It is worthwhile to note that both anti-β-sarcoglycan and anti-human δ-sarcoglycan antibodies were raised against their intracellular domains and were able to precipitate all four sarcoglycans from cultured myotubes. The other three anti-sarcoglycan antibodies used in this study were all directed against the extracellular portion of the proteins and were only able to immunoprecipitate one or two sarcoglycans. Thus, our results suggest that the extracellular domain of the sarcoglycans might be important for their association. In support of this view, other studies have shown that all four sarcoglycans interact with one another via the extracellular but not intracellular domains in an in vitro system (Sakamoto et al., 1997). Moreover, almost all mutations identified in the sarcoglycans are in the extracellular domains of the respective proteins.
The Sarcoglycans As Two Distinct Subunits
Based on the above results, a structural model of the sarcoglycan complex is proposed (Fig. 10). In this model, the sarcoglycans are separated into two subunits: one consisting of α-sarcoglycan and the other consisting of β-, γ-, and δ-sarcoglycan in which the association between β- and δ-sarcoglycan is particularly strong. Structural analysis of the sarcoglycans predicts that β-, γ-, and δ-sarcoglycan are all type II transmembrane proteins (amino terminus on the intracellular side) while α-sarcoglycan belongs to the type I family (amino terminus on the extracellular side). Sequence comparison between β-, γ-, and δ-sarcoglycan also reveals they share significant homology with each other and have the same number of amino acid residues on the extracellular side (231 residues). Recently, an α-sarcoglycan homologue, called ε-sarcoglycan, has been identified (Ettinger et al., 1997; McNally et al., 1998). However, the level and the localization of the ε-sarcoglycan was shown to be normal in transgenic mice lacking either α-sarcoglycan or γ-sarcoglycan (Duclos et al., 1998b ; Hack et al., 1998). Our preliminary data also showed that ε-sarcoglycan did not coimmunoprecipitate with other sarcoglycans, suggesting that it might not be an integral part of the sarcoglycan complex (data not shown).
Figure 10.

Structural model of the sarcoglycan complex and the dystroglycan complex. The four sarcoglycans (left) are represented by α, β, γ, and δ. α-DG and β-DG denote α- and β-dystroglycan (right), respectively. Branch structure corresponds to N-glycoside sugar chain. SH, disulfide linkage. Double-headed arrow, potential interaction between δ-sarcoglycan and the dystroglycan complex. In the model, β-sarcoglycan is tightly associated with δ-sarcoglycan. α-Sarcoglycan is placed apart from other sarcoglycans and is viewed as a separate subunit within the sarcoglycan complex.
We have also identified intramolecular disulfide bonds in β-, γ-, and δ-sarcoglycan (Fig. 7). Although α-sarcoglycan did not show a shift of electrophoretic mobility under nonreducing conditions, it has been suggested that it might also have disulfide bonds based on the observation that the binding of a monoclonal antibody specific to α-sarcoglycan on immunoblots required nonreducing conditions (Roberds et al., 1993). It is possible that the change in the electrophoretic mobility might be too small to be detected by our assay due to the different position of the cysteine residues in α-sarcoglycan as opposed to the conserved cysteines found at the carboxyl terminus of the other sarcoglycans.
The exact cysteine residues that are involved in these disulfide bonds were not determined here but sequence analysis reveals that the last four cysteine residues located at the carboxyl terminus of β-, γ-, and δ-sarcoglycan are highly conserved (Fig. 7 a). The importance of these cysteine residues is demonstrated in a report that a missense mutation in one of the conserved cysteines (C283Y) in γ-sarcoglycan is sufficient to cause the limb-girdle muscular dystrophy type 2C (Piccolo et al., 1996). Comparison to other cysteine-rich motifs that are known to form intramolecular disulfide bonds has shown that they belong to the family of laminin-type EGF-like repeats commonly found in many proteins such as laminin or the EGF receptor (McNally et al., 1996). Since these proteins often exist as oligomers and serve as receptors, it is possible that the β-, γ-, and δ-sarcoglycan subunit also functions as a receptor for an yet unidentified ligand. In this regard, they may actually resemble other receptor molecules on the cell surface and potentially be part of a ligand-induced receptor signaling pathway. α-Sarcoglycan, as a separate subunit, could be the downstream effector. If this is the case, the sarcoglycan complex could also be viewed as two distinct but interrelated functional subunits. In support of this hypothesis, the DGC has been linked with other focal adhesion assembly proteins such as integrins (Kramarcy and Sealock, 1990; Lakonishok et al., 1992; Yoshida et al., 1998) and suggested to function analogously to the integrins as a mechanochemical transducer in skeletal muscle (Brown and Lucy, 1993). Recently, an ecto-ATPase activity has been associated with α-sarcoglycan (Salviati et al., 1997).
The Integrity of the Sarcoglycan Complex is Dependent on β- and δ-Sarcoglycan
The tight association between β- and δ-sarcoglycan raises the possibility that they may form a functional core for the assembly of the sarcoglycan complex as suggested earlier by Bönnemann et al. (1996). According to our model, mutations in either β- or δ-sarcoglycan are expected to have a profound effect on the sarcoglycan complex, whereas the overall effect of α-sarcoglycan mutations on the complex would be less disruptive since it is proposed to loosely associate with the other sarcoglycans and is possibly spatially separated from others as well. The available immunofluorescence and Western blot analyses of muscle biopsy samples taken from autosomal recessive muscular dystrophy patients support this hypothesis. Of the small number reported cases of patients with primary β- or δ-sarcoglycan mutations, they uniformly showed a complete absence or a drastic reduction of all sarcoglycans at the sarcolemma (Bönnemann et al., 1995, 1996, 1998; Lim et al., 1995; Vainzof et al., 1996; Nigro et al., 1996a ; Duclos et al., 1998a ). In contrast, patients with mutations in α- and γ-sarcoglycan often showed a reduced but, nevertheless, detectable level of the other sarcoglycans by immunofluorescence (McNally et al., 1996; Sewry et al., 1996; Carrie et al., 1997; Barresi et al., 1997; Jones et al., 1998; Higuchi et al., 1998). We were able to confirm this pattern of sarcoglycan staining in two patients with similar homozygous mutations and show that the effect of β- and γ-sarcoglycan mutations on the complex is different (Fig. 9). Consistent with the model is that patients showing a complete absence of just α- or γ-sarcoglycan in their muscle biopsies have been reported but patients missing only β- or δ-sarcoglycan have never been documented.
In conclusion, the combined use of immunoprecipitation and cross-linking techniques provides a useful tool to dissect the organization of the sarcoglycan complex. Such an approach may also be useful in identifying new proteins that associate with the DGC. Our structural model for the sarcoglycan complex may enable us to predict the overall effect of specific sarcoglycan mutations on the complex and perhaps explain the correlation between the genotype and the phenotype of patients, including the immunohistochemical findings. The molecular characterization of the sarcoglycans may provide important information on the possible functions of the sarcoglycans and thus the pathogenesis of muscular dystrophy.
Acknowledgments
The authors would like to thank M. Brosius, J. Chu, E. Gussoni, S. Lacy, K. O'Brien, J. Scharf, and T. Thompson (all from Children's Hospital, Boston, MA) for their helpful advice and comments. The authors would also like to thank K. Walrich (Children's Hospital) for her expert technical assistance on immunocytochemistry and M. MacCollin (Massachusetts General Hospital, Boston, MA) for providing clinical data on the β-sarcoglycan patient.
This work has been supported by a grant from the National Institutes of Health to H.G.W. Lidov (KO 8NS 01739). L.M. Kunkel is an investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper
- α-dys
α-dystroglycan
- β-dys
β-dystroglycan
- 2-D
two-dimensional
- DGC
dystrophin–glycoprotein complex
- DSP
dithiobis (succinimidylpropionate)
- DTSSP
dithiobis (sulfosuccinimidylpropionate)
- EGS
ethylene glycolbis (sulfosuccinimidylsuccinate)
- sar
sarcoglycan
Footnotes
C.G. Bönnemann's present address is Department of Neuropediatrics, University Children's Hospital, Göttingen 37075, Germany.
References
- Abe Y, Odaka M, Inagaki F, Lax I, Schlessinger J, Kohda D. Disulfide bond structure of human epidermal growth factor receptor. J BiolChem. 1998;273:11150–11157. doi: 10.1074/jbc.273.18.11150. [DOI] [PubMed] [Google Scholar]
- Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi R, Confalonieri V, Lanfossi M, Di Blasi C, Torchiana E, Mantegazza R, Jarre L, Nardocci N, Boffi P, Tezzon F, et al. Concomitant deficiency of β- and γ-sarcoglycans in 20 α-sarcoglycan (adhalin)-deficient patients: immunohistochemical analysis and clinical aspects. Acta Neuropathologica. 1997;94:28–35. doi: 10.1007/s004010050668. [DOI] [PubMed] [Google Scholar]
- Bönnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP, et al. β-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nature Genetics. 1995;11:266–272. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- Bönnemann CG, Passos-Bueno MR, McNally EM, Vainzof M, Moreira ES, Marie SK, Pavanello RCM, Noguchi S, Ozawa E, Zatz M, Kunkel LM. Genomic screening for β-sarcoglycan gene mutations: missense mutations may cause severe limb-girdle muscular dystrophy type 2E (LGMD 2E) Hum Mol Genet. 1996;5:1953–1961. doi: 10.1093/hmg/5.12.1953. [DOI] [PubMed] [Google Scholar]
- Bönnemann CG, Wong J, Hamida CB, Hamida MB, Hentati F, Kunkel LM. LGMD 2E in Tunisia is caused by a homozygous missense mutation in β-sarcoglycan exon 3. Neuromuscular Disorders. 1998;8:193–197. doi: 10.1016/s0960-8966(98)00014-5. [DOI] [PubMed] [Google Scholar]
- Brown SC, Lucy JA. Dystrophin as a mechanochemical transducer in skeletal muscle. Bioessays. 1993;15:413–419. doi: 10.1002/bies.950150608. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Carrie A, Piccolo F, Leturcq F, de Toma C, Azibi K, Beldjord C, Vallat J-M, Merlini L, Voit T, Sewry C, et al. Mutational diversity and hot spots in the α-sarcoglycan gene in autosomal recessive muscular dystrophy (LGMD2D) J Med Genet. 1997;34:470–475. doi: 10.1136/jmg.34.6.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens PR, Krause TL, Chan S, Korb KE, Graham FL, Caskey CT. Recombinant truncated dystrophin minigenes: construction, expression, and adenoviral delivery. Hum Gene Therapy. 1995;6:1477–1485. doi: 10.1089/hum.1995.6.11-1477. [DOI] [PubMed] [Google Scholar]
- Cox GA, Sunada Y, Campbell KP, Chamberlain JS. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nature Genetics. 1994;8:333–839. doi: 10.1038/ng1294-333. [DOI] [PubMed] [Google Scholar]
- Deyst KA, Bowe MA, Leszyk JD, Fallon JR. The α-dystroglycan-β-dystroglycan complex. J Biol Chem. 1995;270:25956–25959. doi: 10.1074/jbc.270.43.25956. [DOI] [PubMed] [Google Scholar]
- Duclos F, Broux O, Bourg N, Straub V, Feldman GL, Sunada Y, Lim LE, Piccolo F, Cutshall S, Gary F, Quetier F, Kaplan J-C, Jackson CE, Beckman JS, Campbell KP. β-sarcoglycan: genomic analysis and identification of a novel mutation in the LGMD2E Amish isolate. Neuromuscular Disorders. 1998a;8:30–38. doi: 10.1016/s0960-8966(97)00135-1. [DOI] [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, Venzke DP, Hrstka RF, Crosbie RH, Durbeej M, Lebakken CS, Ettinger AJ, van der Meulen J, et al. Progressive muscular dystrophy in α-sarcoglycan-deficient mice. J Cell Biol. 1998b;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti JM, Ohlenidieck K, Kahl SD, Graver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- Ettinger AJ, Feng G, Sanes JR. ε-sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem. 1997;272:32534–32538. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- Froehner SC, Murnane AA, Tobler M, Peng HB, Sealock R. A postsynaptic M r58,000 (58K) protein concentrated at acetylcholine receptor-rich sites in Torpedo electroplaques and skeletal muscle. J Cell Biol. 1987;104:1633–1646. doi: 10.1083/jcb.104.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DS, Sunada Y, Campbell KP, Yaffe D, Nudel U. Exogenous Dp71 restores the levels of dystrophin associated proteins but does not alleviate muscle damage in mdx mice [see Comments] Nature Genetics. 1994;8:340–834. doi: 10.1038/ng1294-340. [DOI] [PubMed] [Google Scholar]
- Hack AA, Ly CT, Jiang F, Clendenin CJ, Sigrist KS, Wollmann RL, McNally EM. γ-Sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschka, S.D. 1994. The Embryonic Origin of Muscle. In Myology. A.G. Engel and C. Franzini-Armstrong, editors. McGraw-Hill Press, New York, NY. 3–16.
- Hemmings L, Kuhlman PA, Critchley DR. Analysis of the actin-binding domain of alpha-actinin by mutagenesis and demonstration that dystrophin contains a functionally homologous domain. J Cell Biol. 1992;116:1369–1380. doi: 10.1083/jcb.116.6.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi I, Kawai H, Umaki Y, Kawajiri M, Adachi K, Fukunaga H, Nakagawa M, Arimura K, Osame M. Different manners of sarcoglycan expression in genetically proven α-sarcoglycan deficiency and gamma-sarcoglycan deficiency. Acta Neuropathol. 1998;96:202–206. doi: 10.1007/s004010050882. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Holt KH, Lim LE, Straub V, Venzke DP, Duclos F, Anderson RD, Davidson BL, Campbell KP. Functional rescue of the sarcoglycan complex in the bio 14.6 hamster using δ-sarcoglycan gene transfer. Mol Cell. 1998;1:841–848. doi: 10.1016/s1097-2765(00)80083-0. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Milatovich A, Ozcelik T, Yang B, Koepnick K, Francke U, Campbell KP. Human dystroglycan: skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum Mol Genet. 1993;2:1651–1657. doi: 10.1093/hmg/2.10.1651. [DOI] [PubMed] [Google Scholar]
- Jones KJ, Kim SS, North KN. Abnormalities of dystrophin, the sarcoglycans, and laminin α2 in the muscular dystrophies. J Med Genet. 1998;35:379–386. doi: 10.1136/jmg.35.5.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SM, Moroi M. Cross-linking of platelet glycoprotein 1b by N-succinimidyl (4-azidophenyldithio) propionate and 3,3-dithiobis (sulfo-succinimdyl propionate) Biochem Biophys Acta. 1983;761:152–162. doi: 10.1016/0304-4165(83)90224-6. [DOI] [PubMed] [Google Scholar]
- Jung D, Yang B, Meyer J, Chamberlain J, Campbell KP. Identification and characterization of the dystrophin anchoring site of beta-dystroglycan. J Biol Chem. 1995;45:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- Jung D, Leturcq F, Sunada Y, Duclos F, Tome FMS, Moomaw C, Merlini L, Azibi K, Chaouch M, Slaughter C, et al. Absence of γ-sarcoglycan (35 DAG) in autosomal recessive muscular dystrophy linked to chromosome 13q12. FEBS (Fed Eur Biochem Soc)Lett. 1996;381:15–20. doi: 10.1016/0014-5793(96)00056-7. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffmann EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–226. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- Kramarcy NR, Sealock R. Dystrophin as a focal adhesion protein. FEBS (Fed Eur Biochem Soc) Lett. 1990;274:171–174. doi: 10.1016/0014-5793(90)81356-s. [DOI] [PubMed] [Google Scholar]
- Kumar-Singh R, Chamberlain JS. Encapsidated adenovirus minichromosomes allow delivery and expression of a 14 kb dystrophin cDNA to muscle cells. Hum Mol Genet. 1996;5:913–921. doi: 10.1093/hmg/5.7.913. [DOI] [PubMed] [Google Scholar]
- Lakonishok M, Muschler J, Horwitz AF. The α5β1integrin associates with a dystrophin-containing lattice during muscle development. Dev Biol. 1992;152:209–220. doi: 10.1016/0012-1606(92)90129-5. [DOI] [PubMed] [Google Scholar]
- Lee CC, Pearlman JA, Chamberlain JS, Caskey CT. Expression of recombinant dystrophin and its localization to the cell membrane. Nature. 1991;349:334–336. doi: 10.1038/349334a0. [DOI] [PubMed] [Google Scholar]
- Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature. 1990;348:725–728. doi: 10.1038/348725a0. [DOI] [PubMed] [Google Scholar]
- Lim, L.E., F. Duclos, O. Broux, N. Bourg, Y. Sunada, V. Allamand, J. Meyer, I. Richard, C. Moomaw, C. Slaughter, et al. β-sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nature Genetics. 11:257–265. [DOI] [PubMed]
- McNally EM, Duggan D, Gorospe JR, Bönnemann CG, Fanin M, Pegoraro E, Lidov HGW, Noguchi S, Ozawa E, Finkel RS, et al. Mutations that disrupt the carboxyl-terminus of γ-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5:1841–1847. doi: 10.1093/hmg/5.11.1841. [DOI] [PubMed] [Google Scholar]
- McNally EM, Ly CT, Kunkel LM. Human ε-sarcoglycan is highly related to α-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene. FEBS (Fed Eur Biochem Soc) Lett. 1998;422:27–32. doi: 10.1016/s0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- Mitchell RD, Palade P, Fleischer S. Purification of morphologically intact triad structures from skeletal muscle. J Cell Biol. 1983;96:1008–1016. doi: 10.1083/jcb.96.4.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- Nigro V, Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passo-Bueno MR, Zatz M. Autosomal recessive limb-girdle muscular dystrophy, LGMD 2F, is caused by a mutation in the δ-sarcoglycan gene. Nature Genetics. 1996a;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- Nigro V, Piluso G, Belsito A, Politano L, Puca AA, Papparella S, Rossi E, Viglietto G, Esposito MG, Abbondanza C, Medici N, Molinari AM, Nigro G, Puca GA. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Hum Mol Genet. 1996b;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- Noguchi S, McNally EM, Othmane KB, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bönnemann CG, Gussoni E, Denton PH, et al. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–821. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur J Cell Biol. 1996;69:1–10. [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Matsumura K, Ionasescu VV, Towbin JA, Bosch EP, Weinstein SL, Sernett SW, Campbell KP. Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology. 1993;43:795–800. doi: 10.1212/wnl.43.4.795. [DOI] [PubMed] [Google Scholar]
- Piccolo, F., M. Jeanpierre, F. Leturcq, C. Dode, K. Azibi, A. Toutain, L. Merlini, L. Jarre, C. Navarro, R. Krishnamoorthy, et al. A founder mutation in the γ-sarcoglycan gene of Gypsies possibly predating their migration out of India. Hum. Mol. Genet. 5:2019–2022. [DOI] [PubMed]
- Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberds SL, Anderson RD, Ibraghimov-Beskrovnaya O, Campbell KP. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin) J Biol Chem. 1993;268:23739–23742. [PubMed] [Google Scholar]
- Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tome FM, Romero NB, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Sadoulet-Puccio H, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. Proc Natl Acad Sci USA. 1997;94:12413–12418. doi: 10.1073/pnas.94.23.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, Masaki T, Toyo-Oka T, Hanaoka F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, δ-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci USA. 1997;94:13873–13878. doi: 10.1073/pnas.94.25.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salviati, G., L. Senter, L. Agnolucci, S. Ceoldo, and R. Betto. 1997. Role of dystrophin and dystrophin associated proteins in the pathogenesis of duchenne muscular dystrophy. Scientific Convention of Telethon, Bologna.
- Sewry CA, Taylor J, Anderson LVB, Ozawa E, Pogue R, Piccolo F, Bushby K, Dubowitz V, Munton F. Abnormalities in α-, β- and γ-sarcoglycan in patients with limb-girdle muscular dystrophy. Neuromuscular Disorders. 1996;6:467–474. doi: 10.1016/s0960-8966(96)00389-6. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Yamamoto H, Ozawa E. Glycoprotein-binding site of dystrophin is confined to the cysteine-rich domain and the first half of the carboxy-terminal domain. FEBS (Fed Eur Biochem Soc) Lett. 1992;308:154–160. doi: 10.1016/0014-5793(92)81265-n. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E. Molecular organization at the glycoprotein-complex-binding site of dystrophin: three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. Eur J Biochem. 1994;220:283–292. doi: 10.1111/j.1432-1033.1994.tb18624.x. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Yoshida M, Ozawa E. Mammalian alpha 1- and beta 1-syntrophin bind to the alternative splice-prone region of the dystrophin COOH terminus. J Cell Biol. 1995;128:373–381. doi: 10.1083/jcb.128.3.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin J, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut, R.R., C. Casiano, and N. Zecherie. 1989. Crosslinking of protein subunits and ligands by the introduction of disulphide bonds. In Proteins Function: A Practical Approach. T.E. Creighton, editor. IRL Press, Oxford, UK. 101–133.
- Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES, Pavanello RCM, Marie SK, Anderson LVB, Bönnemann CG, McNally EM, Nigro V, et al. The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet. 1996;5:1963–1969. doi: 10.1093/hmg/5.12.1963. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Suzuki A, Yamamoto H, Noguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-d-glucoside. Eur J Biochem. 1994;222:1055–1061. doi: 10.1111/j.1432-1033.1994.tb18958.x. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Noguchi S, Wakabayashi E, Piluso G, Belsito A, Nigro V, Ozawa E. The fourth component of the sarcoglycan complex. FEBS (Fed Eur Biochem Soc) Lett. 1997;403:143–148. doi: 10.1016/s0014-5793(97)00040-9. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Pan Y, Hanada H, Iwata Y, Shigekawa M. Bidirectional signaling between sarcoglycans and the integrin adhesion system in cultured L6 myocytes. J Biol Chem. 1998;273:1583–1590. doi: 10.1074/jbc.273.3.1583. [DOI] [PubMed] [Google Scholar]