

Abstract
The biogenesis of secretory granules embodies several morphological and biochemical changes. In particular, in neuroendocrine cells maturation of secretory granules is characterized by an increase in size which has been proposed to reflect homotypic fusion of immature secretory granules (ISGs). Here we describe an assay that provides the first biochemical evidence for such a fusion event and allows us to analyze its regulation. The assay reconstitutes homotypic fusion between one population of ISGs containing a [35S]sulfate-labeled substrate, secretogranin II (SgII), and a second population containing the prohormone convertase PC2. Both substrate and enzyme are targeted exclusively to ISGs. Fusion is measured by quantification of a cleavage product of SgII produced by PC2. With this assay we show that fusion only occurs between ISGs and not between ISGs and MSGs, is temperature dependent, and requires ATP and GTP and cytosolic proteins. NSF (N-ethylmaleimide–sensitive fusion protein) is amongst the cytosolic proteins required, whereas we could not detect a requirement for p97. The ability to reconstitute ISG fusion in a cell-free assay is an important advance towards the identification of molecules involved in the maturation of secretory granules and will increase our understanding of this process.
Keywords: fusion, immature secretory granule, prohormone convertase 2, secretogranin, NSF
Secretory granules are specialized exocytic vesicles responsible for the controlled release of bioactive molecules, such as hormones. During the lifetime of the secretory granule, from formation to consumption, a series of regulatory events control its composition and size. Control of these events is crucial for the fidelity of regulated secretion. Secretory granules originate from the TGN with a selected subset of lumenal and membrane proteins: this protein selection during formation is presumably controlled by proteins both within the lumen of the TGN and in the cytoplasm (Tooze, 1998). Cargo selection may be via receptor–ligand type interactions. For example, carboxypeptidase E recognizes a motif present in several prohormones and has been implicated as a general cargo receptor (Cool et al., 1997) although this is currently the subject of intense debate (Irminger et al., 1997; Thiele et al., 1997). To date, no cytoplasmic coat proteins have been implicated in the selection of cargo receptors or secretory granule membrane proteins. Patches of clathrin have been detected on the TGN membrane in the region of the budding immature secretory granule (ISG)1 although there is no evidence that clathrin is involved in formation of secretory granules (for review see Urbé et al., 1997b ). Formation of ISGs is regulated by heterotrimeric G proteins (Barr et al., 1991) and proteins required for lipid metabolism (Chen et al., 1997; Ohashi et al., 1995) although how these are involved in protein sorting during ISG formation is not known.
Additional proteins and machinery will undoubtedly be identified and found to be involved in secretory granule biogenesis at the TGN. However, it is already well established that the selection mechanisms operative in the TGN alone are not sufficient to produce a mature secretory granule (MSG). Isolation of ISGs (Tooze et al., 1991) confirmed the previous morphological data (Salpeter and Farquhar, 1981) showing that ISGs are smaller than MSGs. Subsequently, proteins were found in ISGs that are not present in MSGs, such as furin (Dittié et al., 1997), lysosomal enzymes (Kuliawat et al., 1997) and the mannose-6-phosphate receptor (Klumperman, 1998). In addition, the lumenal pH of the ISG is ∼6.3 whereas the lumenal pH of the MSG is ∼pH 5.5 (Orci et al., 1994; Urbé et al., 1997a ).
The differences in size and content between ISGs and MSGs have led to the proposal that the nascent ISG, which buds from the TGN is an intermediate stage in secretory granule biogenesis (Tooze et al., 1991). Maturation of the intermediate ISG to an MSG would require both fusion and budding events. Fusion events between ISGs would result in an increase in the size of ISGs to that of MSGs, whereas budding of vesicles from the ISG would allow removal of nonessential proteins and excess membrane. The coordinated action of budding and fusion would exert a tight control on both the composition and quantal size of the released secretory granule content upon stimulation. This hypothesis is supported by morphological data demonstrating that MSGs in endocrine cells have a defined size depending on the cell type. For example, within the anterior pituitary gland the mammotrophs have MSGs 350 μm in diameter, whereas the gonadotrophs have MSGs 200 μm in diameter (Smith and Farquhar, 1966). The size differences may be related to the biophysical properties of the major content protein and the structure of the dense core, although the mechanisms that underlie this phenomenon are not known (Burgess and Kelly, 1987).
We are interested in understanding the mechanisms that control secretory granule composition and size, and determining if these might involve fusion and budding from the ISG. In one approach we have developed a cell-free assay that reconstitutes fusion between ISGs. The assay is based on the interaction of an endopeptidase with its substrate, both of which are specifically sorted into secretory granules. We have designed the assay so that the substrate, which is labeled by radioactive sulfate on tyrosine residues, is present in one ISG population, whereas the enzyme is present in another population of ISGs. Upon fusion of the two ISG populations the enzyme will cleave the labeled substrate into discrete fragments. Fusion is measured by immunoprecipitation with a specific antibody which only recognizes one of these fragments and not the intact substrate.
With this assay we have investigated the requirements for cytosolic components involved in ISG fusion. In particular, we have asked if either one of the two well-characterized N-ethylmaleimide (NEM)–sensitive proteins, NEM sensitive fusion protein (NSF), or p97 are involved in ISG fusion. NSF, first identified by Rothman and colleagues (Block et al., 1988) is involved in vesicular transport along the secretory pathway, fusion events in the endocytic pathway, and regulated exocytosis (for review see Whiteheart and Kubalek, 1995; Woodman, 1997). p97, or its yeast homologue cdc48p, was shown to be necessary for homotypic fusion events involved in organelle maintenance (Acharya et al., 1995a ; Latterich et al., 1995; Rabouille et al., 1995a ). Both are ATPases which bind to membranes via attachment proteins, soluble NSF attachment proteins (SNAPs) (Whiteheart et al., 1993) in the case of NSF (Clary et al., 1990; Weidman et al., 1989) and p47 for p97 (Kondo et al., 1997; Rabouille, 1998). NSF and SNAPs interact with detergent-solubilized soluble NSF attachment protein receptors (SNAREs) to form a 20 S particle (Sollner et al., 1993). Over 20 SNAREs have been identified (for review see Advani et al., 1998; Bock and Scheller, 1997) which are classified into two families, v-SNAREs found on the vesicle and t-SNAREs found on the target membrane. The structural similarity of the SNAREs, combined with their restricted localization is the basis of the current model for vesicular transport through the secretory pathway (Rothman, 1994; for review see Götte and von Mollard, 1998). Recently, interaction of a v-SNARE with a t-SNARE has been shown to be the minimal requirement for fusion of liposomes (Weber et al., 1998).
The interaction of p97 or cdc48p with membranes also requires a SNARE protein, the t-SNARE syntaxin 5 (Rabouille, 1998), and Ufe1p (Patel, 1998), respectively. It has recently been proposed that whereas heterotypic fusion requires the pairing of cognate v- and t-SNAREs, homotypic fusion can also proceed via the pairing of t-SNAREs alone (Nichols et al., 1997) and may be in some cases mediated by cdc48p or p97 (Patel, 1998; Rabouille, 1998, respectively).
Using the in vitro system we describe here we report that fusion of ISGs can be reconstituted in a cell-free assay, and provide the first biochemical evidence that ISG– ISG fusion can occur. This fusion requires ATP, GTP, and elevated temperature plus cytosolic components. We have found that NSF, but not p97, is required for homotypic ISG fusion. Further understanding of the role of ISG–ISG fusion during maturation, and how the generation of MSGs may be controlled by ISG fusion will be greatly facilitated with this assay.
Materials and Methods
Materials and Reagents
Carrier-free [35S]sulfate as a disodium salt was supplied by Amersham Pharmacia Biotech (Piscataway, NJ). Fine chemicals were supplied by Sigma Chemical Co. (St. Louis, MO) or Boehringer Mannheim (Mannheim, Germany). PC12 cells (clone 251) were originally obtained from H. Thoenen (Max Planck Institute, Martinsried, Germany) (Heumann et al., 1983). PC12/PC2 cell lines stably expressing the endoprotease PC2 have been previously characterized (Dittié and Tooze, 1995). PC12 and PC12/PC2 cells were grown in DME supplemented with 10% horse serum and 5% fetal calf serum at 10% CO2.
Antibodies and Recombinant Proteins
Horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgGs were supplied by Amersham (Little Chalfont, UK). Antibodies raised against SgII (175 and 100) have been previously described (Dittié et al., 1996). Anti-NSF antibody was provided by M. Tagaya (Tokyo University, Tokyo, Japan), anti–α-SNAP by T. Söllner (Sloan-Kettering, New York, NY), purified p97 and his-tagged p47, anti-p97, and p47 by H. Kondo (Imperial Cancer Research Fund [ICRF], London, UK). His-tagged NSF and His-tagged α-SNAP were a kind gift of G. Schiavo (ICRF).
Antibodies specific for the free COOH terminus of p18 were raised against the peptide (KEENSRENPF) from rat SgII (Gerdes et al., 1988). Polyclonal antisera were obtained from rabbits (ab69), whereas the monoclonal antibody was developed from hybridomas using a standard procedure (6B1). Both the polyclonal and monoclonal antibodies are specific for the free COOH terminus of p18 as assessed by standard Western blotting, immunoprecipitation, and immunofluorescence techniques using lysates obtained from either PC12 cells or PC12/PC2 cells. Experiments have been performed with either or both polyclonal antibody (ab69) and monoclonal antibody (6B1).
Preparation of Postnuclear Supernatant, ISGs, and MSGs
A postnuclear supernatant (PNS) was prepared in HB-PMSF (250 mM sucrose, 10 mM Hepes-KOH, pH 7.2, 1 mM Mg[OAc]2, 1 mM EDTA, pH 7.2, with 0.5 mM PMSF) as described (Tooze and Huttner, 1990) and contained ∼10 mg/ml of protein as determined by Bio-Rad protein microassay (using bovine IgG as a standard; Hercules, CA). PC12 cells were labeled with [35S]sulfate and chased as previously described (Tooze and Huttner, 1990) for the times specified in the figure legends.
ISGs and MSGs were prepared as described in Dittié et al. (1996). In brief, 1.3 ml of PC12 or PC12/PC2 PNS was loaded on linear sucrose gradients of 0.3–1.2 M sucrose in 10 mM Hepes-KOH, pH 7.2, and subjected to velocity controlled centrifugation. Fractions from the top of the velocity gradient containing ISGs and MSGs were pooled and subjected to equilibrium sucrose gradient centrifugation. Fractions of 1 ml were collected from the top and fractions seven to nine were pooled for the ISGs whereas fractions 10–12 were pooled for the MSGs. Aliquots were stored in liquid nitrogen and used within 6 mo. All gradient preparations were routinely checked using [35S]sulfate labeling to ensure that the preparations were consistent and correct sorting of the soluble proteins was achieved (Tooze and Huttner, 1990).
Preparation of PC12 Cytosol
PC12 cells (∼107–108 cells) were homogenized in HB-PMSF and a PNS was prepared as described above. The PNS was then subjected to centrifugation for 30 min at 30,000 g av, followed by a second centrifugation of the supernatant at 100,000 g av for 90 min at 4°C. Cytosol was stored in liquid nitrogen and used within 3 mo. The protein concentration of the PC12 cytosol was determined using the Bio-Rad microassay and was typically between 8 and 13 mg/ml.
Preparation of Rat Liver Cytosol
Rat livers either from Wistar or Sprague-Dawley male or female rats were used. The livers were washed, minced, and then homogenized either in FB (20 mM Hepes-KOH, pH 7.4, 50 mM KOAc, 3 mM MgCl2, 1 mM DTT), containing 250 mM sucrose or protease inhibitors (0.5 mM PMSF, 10.5 μM leupeptin) and then centrifuged as described for the PC12 cell cytosol above. Alternatively, cytosol was prepared as described (Rabouille et al., 1995b ) in 0.5 M sucrose, 0.1 M KPO4, pH 6.7, 5 mM MgCl2 and protease inhibitors. After centrifugation both cytosol preparations were precipitated with 40% ammonium sulfate, resuspended in FB containing 250 mM sucrose and protease inhibitors, and either dialyzed overnight against FB containing 250 mM sucrose or desalted into FB containing 250 mM sucrose with a PD10 column (Bio-Rad). Aliquots were stored in liquid nitrogen and used within 3 mo. The protein concentration of these cytosols was typically between 20 and 40 mg/ml.
Cell-free Assays for ISG–ISG Fusion and Processing
Reconstitution of ISG Fusion with a PNS.
PC12 cells were pulse-labeled with [35S]sulfate or pulse-labeled and chased, before being washed, harvested, and homogenized to prepare a PNS as described above. PC2-ISGs were thawed rapidly at 37°C, diluted at 4°C with 2 vol of 10 mM Hepes-KOH, pH 7.2, pelleted by centrifugation at 100,000 g av for 1 h. The pellets were resuspended in HB-PMSF at 50-fold the concentration of the original ISG pool. If not indicated otherwise, standard conditions were as follows: 100 μl of the PC12 PNS per condition were supplemented with 10× FB, ATP-regenerating system (see below), and 10 μl PC2-ISGs in a final volume of 120–160 μl.
After incubation at 4° or 37°C the samples were transferred to ice and diluted with low pH buffer (50 mM MES, pH 5.5, in 0.3 M sucrose) to a final volume of 800 μl. The samples were preincubated for 30 min on ice to equilibrate the pH in the ISGs with the buffer and then transferred to 37°C to allow processing of SgII by PC2 to take place. After this processing incubation, the samples were placed on ice and 200 μl of 5× lysis buffer (500 mM Tris-HCl, pH 7.5, 750 mM NaCl, 1.5% Triton X-100, 25 mM EDTA for ab69, or 500 mM Tris-HCl, pH 7.5, 750 mM NaCl, 5% Triton X-100, 0.5% SDS, 5% Na deoxycholate for 6B3) was added. The lysed samples were precleared by a 5-min spin in an Eppendorf centrifuge (Eindhoven, The Netherlands) and subjected to immunoprecipitation as described below.
ATP-regenerating system (Davey et al., 1985) stock solutions contained 100 mM ATP, pH 7, 800 mM creatine phosphate, and 3,200 U/ml creatine phosphokinase that were mixed before use in equal volumes to give a 30× stock. ATP depletion was performed with 3.3 mM d-glucose and 25 U/ml hexokinase. AMP-PNP (final concentration 100 μM), GTP (final concentration 300 μM), and GTPγS (final concentration 100 μM) were made up as a 100× stock solutions in H2O.
Immunoprecipitation and Quantitation of [35S]sulfate-labeled p18.
For standard immunoprecipitations from cell-free fusion assay incubations polyclonal ab69 and protein A–Sepharose (Amersham Pharmacia Biotech) or monoclonal 6B1 and protein G–Sepharose (Sigma Chemical Co.) were used. The immunoprecipitates were washed with lysis buffer, eluted from the beads by boiling in Laemmli sample buffer, and then analyzed by SDS-PAGE using a modified Laemmli system (Lee and Huttner, 1983), followed by fluorography with 1 M sodium salicylate for 30 min and exposure to a preflashed Kodak XAR5 film (Eastman-Kodak, Rochester, NY). The fluorographs were usually exposed to film for 3–5 d, except for those obtained with material labeled as in Fig. 3 a where exposures longer than 2 wk were required. Quantitation of [35S]sulfate-labeled p18 was by densitometry using the NIH Image package version 68k/1.58 (Bethesda, MD). If not indicated otherwise, the PC2-independent signal was subtracted from the signal obtained in the presence of PC2-ISGs.
Figure 3.
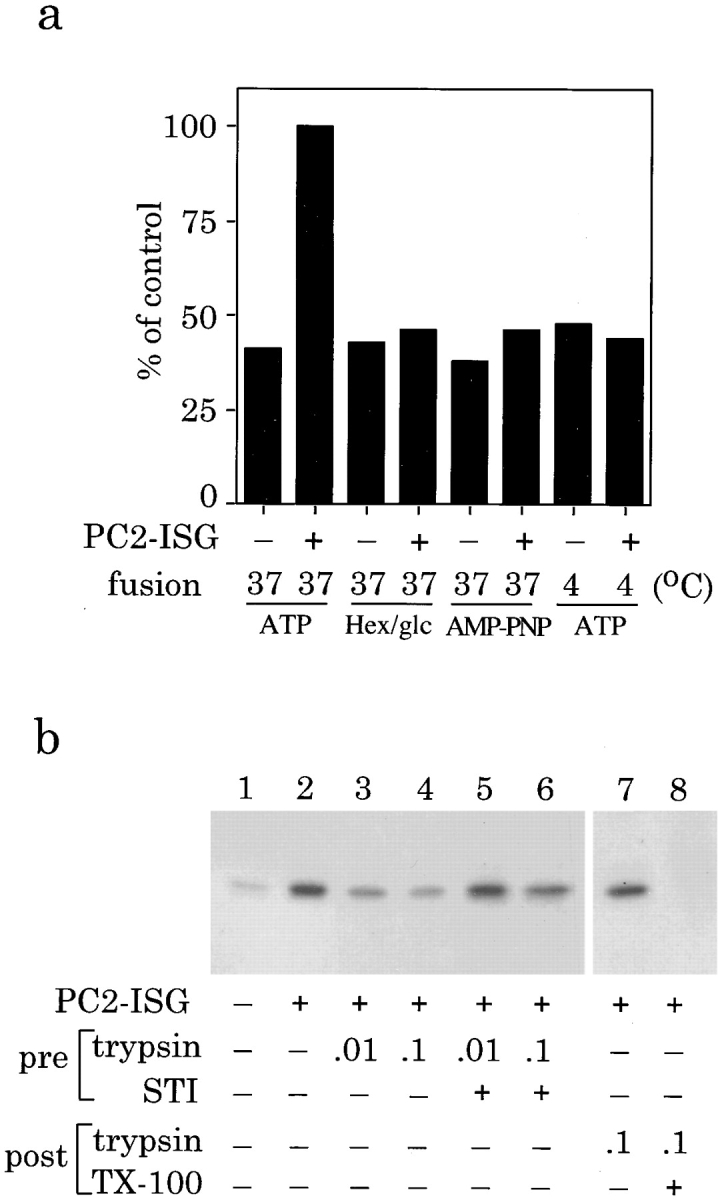
Requirements for ISG fusion. (a) Nucleotides (1 mM ATP and ATP-regenerating system, or 100 mM AMP-PNP) or ATP-depletion mixture (25 U hexokinase in 3 mM glucose) were added to the fusion reaction containing 100 μl of PNS from PC12 cells labeled for 30 min with [35S]sulfate with or without PC2-ISGs. The reactions were incubated at 37° or 4°C for 30 min and then incubated for processing for 90 min. (b) Fusion requires trypsin sensitive proteins and p18 is generated in the lumen of membrane enclosed organelles. PC2-ISGs were pretreated with trypsin (0.01 and 0.1 mg/ml trypsin [final concentration] which corresponds to 20 and 200 ng trypsin/μg ISG protein) for 15 min at 37°C in the absence (lanes 3 and 4) or presence of STI (lanes 5 and 6). STI was added to the reactions shown in lanes 3 and 4, and fusion was assayed in a standard reaction by incubation for 30 min at 37°C, followed by processing for 45 min at 37°C. Alternatively, the reaction mixture was treated with the highest concentration of trypsin (0.1 mg/ml) for 15 min at 37°C after the processing reaction in the absence (lane 7) or presence (lane 8) of 0.3% Triton X-100. p18 was immunoprecipitated and then subjected to SDS-PAGE and autoradiography. Quantitation in a was performed by densitometry. The data shown are from a representative experiment done in duplicates.
Treatment of PC2-ISGs with Trypsin Before and After Fusion.
PC2-ISGs were incubated with trypsin (20 ng trypsin/μg ISG protein or 200 ng trypsin/μg ISG protein) either with or without 25 μg of soybean trypsin inhibitor (STI) for 15 min at room temperature, placed on ice, and then PMSF and STI were added. The ISGs were then pelleted for 1 h, 5 min at 100,000 g av. Postfusion and postprocessing treatment with trypsin was performed with a final concentration of 0.1 mg/ml trypsin (200 ng trypsin/μg ISG protein) in the absence or presence of 0.3% Triton X-100 for 15 min at 37°C. After digestion STI was added and the samples were solubilized in Laemmli sample buffer.
Cell-free Assay Using a Membrane Pellet.
To obtain a membrane pellet, the PNS was layered on top of a 1 ml sucrose cushion (0.5 M sucrose in 10 mM Hepes-KOH, pH 7.2). The membranes were pelleted by centrifugation for 1 h, 5 min at 100,000 g av at 4°C. The supernatant was removed and the membranes were resuspended in HB or cytosol to their original volume. An ATP-regenerating system, 10× FB, and PC2-ISGs were added as described for the standard cell-free assay using a PNS. GTP was included when rat liver cytosol was used.
Cell-free Assay Using Sucrose Gradient Fractions.
For the type of experiment shown in Fig. 8, the PNS was first subjected to velocity controlled sucrose centrifugation to separate the TGN and post-TGN vesicles. The fractions from the top of the velocity gradient (fractions 2–4) were pooled, diluted with 10 mM Hepes-KOH, pH 7.2, and subjected to centrifugation as above. The membranes were then resuspended in cytosol or HB in a volume corresponding to the volume of the PNS loaded and used as described.
Figure 8.

Fusion occurs with enriched acceptor ISGs. A membrane pellet (MP) or velocity gradient (VG) fraction (pool of fractions 2–4) were isolated from a PNS obtained from PC12 cells labeled with [35S]sulfate for 30 min. A standard reaction was performed with or without 10 μl of PC2-ISGs and with or without rat liver cytosol. After a 30-min incubation for fusion, processing was performed for 90 min at 37°C. p18 was immunoprecipitated and quantitated as above. Data shown is of representative experiment performed in triplicate (error bars are ±SD of the mean) expressed as percent of the control. For comparison, the PC2-dependent signal is shown relative to the PC2-independent signal as an inset to emphasize that ISG–ISG fusion is comparable in the MP and VG fractions.
NEM Treatment of Cytosol and Membranes.
NEM treatment of PNS or cytosol was performed using freshly made 50 mM NEM stock solution in dH2O added to a final concentration of 3 mM. The samples were mixed immediately, incubated for 10 min on ice, and then quenched by addition of 3.6 mM glutathione for 15 min on ice. The membranes were then pelleted through a sucrose cushion as described above while cytosol was used without further treatment. Control samples were treated with glutathione only.
Results
Experimental System to Reconstitute Fusion of ISGs
To study the molecular requirements for fusion of ISGs we have developed an assay that allows reconstitution of fusion in a cell-free extract. The assay is based on content mixing after fusion of ISGs which allows an enzyme– substrate interaction to occur. Prohormone convertases cleave prohormones, and other regulated secretory proteins containing a dibasic amino acid site, in a pH and Ca2+-dependent fashion (Steiner, 1991). PC2, a prohormone convertase that preferentially cleaves dibasic amino acid sites composed of lysine and arginine at a pH optimum of 5.5 and at micromolar Ca2+ concentrations (Bennett et al., 1992) was used in this assay. As the substrate for PC2, we have used secretogranin II (SgII), an abundant resident secretory granule protein present in all neuroendocrine cells that contains nine potential dibasic amino acid cleavage sites. SgII is sulfated on tyrosine by tyrosylprotein sulfotransferase (TPST), an enzyme that is localized to the TGN (Niehrs et al., 1992). Tyrosine sulfation has been frequently exploited to label proteins in the TGN (for example see Tooze and Huttner, 1990; Carnell and Moore, 1994; Itin, 1997). The processing of SgII by PC2 has been studied in PC12 cells either stably transfected with the cDNA encoding PC2 (PC12/PC2 cells; Dittié and Tooze, 1995) or after infection with recombinant vaccinia virus encoding PC2 (Laslop et al., 1998). In PC12/ PC2 cells, sulfated SgII exits from the TGN in secretory granules and is cleaved by PC2 within the secretory granule into several fragments (Urbé et al., 1997a ). The sulfated fragments are derived from the NH2 terminus and include p18, the end product of the PC2-catalyzed processing reaction (Fig. 1 a).
Figure 1.

Experimental design of the ISG fusion assay. (a) Processing of SgII by PC2. The major sulfated processing products of SgII (38, 24, and 18 kD) are illustrated with the full-length protein (86 kD). Solid boxes, sequences recognized by the antibodies used in this study; arrowheads, the di-basic amino acid sites used by PC2; solid dots, the tyrosine residue which is sulfated by TPST. (b) Scheme of ISG– ISG fusion assay. *, [35S]sulfate-labeled proteins. See Materials and Methods for details. (c) Immunoprecipitation with ab100, ab175, and ab69 using lysates obtained from [35S]sulfate-labeled PC12/PC2 cells (lanes 1–3) or PC12 cells (lane 4). Arrows, position of SgII (p86), and the processing products p38, p24, and p18.
A scheme of the assay is shown in Fig. 1 b. The acceptor ISG fraction containing [35S] SgII is present in a PNS derived from wild-type PC12 cells labeled with [35S]sulfate. Since wild-type PC12 cells have no PC1 or PC2 enzymes, SgII is present in an unprocessed form of 86 kD. The donor is an enriched population of ISGs containing PC2 (PC2-ISGs) isolated by subcellular fractionation from unlabeled PC12/PC2 cells (Dittié and Tooze, 1995). After preparation of the fusion partners, the donor PC2 ISGs are added to the acceptor PNS and incubated at either 4° or 37°C in the presence or absence of ATP. During this reaction, referred to as the fusion reaction, the wild-type ISGs fuse with the PC2-ISGs and the granule contents mix. Next, the samples are diluted with a pH 5.5 buffer, equilibrated at 4°C for 30 min, and then warmed to 37°C for 45–90 min for the processing reaction. During this second incubation the PC2 enzyme recognizes the dibasic amino acid sites in the [35S]sulfate-labeled SgII and at pH 5.5, processes it into several fragments. The sulfated fragments, including the processing end product p18, will then accumulate in the hybrid ISGs. The processing reaction ensures that the readout of this assay (i.e., processing of SgII) is independent of the conditions used during the fusion reaction. Lysis buffer is then added to the sample and the end product of processing p18 is immunoprecipitated with p18-specific antibodies.
Characterization of the Antibody Specific for the Fusion Product p18
To detect the [35S]sulfate-labeled SgII fragment (p18) generated by PC2 as a result of fusion we developed antibodies that only recognize the free COOH terminus of p18. Polyclonal antibody ab69 and monoclonal antibody 6B3 were characterized using immunoblotting and immunoprecipitation techniques with extracts obtained from both PC12/PC2 cells and PC12 cells. As previously shown, [35S]sulfate-labeled SgII in PC12/PC2 cells is processed by PC2 into several smaller molecular weight fragments including p18 (Dittié and Tooze, 1995). All the sulfated processing products as well as the full length SgII can be visualized by immunoprecipitation with ab100 (Fig. 1 c, lane 1) whereas ab175 recognizes only the intact SgII (Fig. 1 c, lane 2). In addition to the full-length protein (p86), the sulfated fragments of SgII immunoprecipitated are 38, 24, and 18 kD. Of these fragments only the 18-kD protein was immunoprecipitated with ab69 or 6B3 from [35S]sulfate-labeled extracts from PC12/PC2 cells (Fig. 1 c, lane 3). Immunoprecipitation with ab69 and 6B3 of extracts from unlabeled or [35S]sulfate-labeled wild-type PC12 cells confirmed that there was no detectable p18 (Fig. 1 c, lane 4) as expected since there is no PC2 in PC12 cells.
Optimization of the Processing Reaction
As detection of fusion between ISGs depends on the catalytic activity of the PC2 enzyme it is important that the conditions within the fused ISGs support optimal PC2 activity to ensure that the processing reaction is not rate limiting and will proceed to completion. In particular, the lumenal pH within the ISGs after fusion must be acidic to achieve optimal enzymatic activity. The ISG preparations from both the PC12 cells and the PC12/PC2 cells have a lumenal pH equivalent to that of the buffer used during their isolation, i.e., pH 7.2. We have shown that when these isolated ISGs are incubated at 4°C in a low pH buffer they will equilibrate to the pH of the buffer. Using this equilibration step, performed at 4°C for 30 min, we can manipulate the lumenal pH of the ISG to promote efficient processing of SgII by PC2 in isolated ISGs (Urbé et al., 1997a ).
We have previously demonstrated that p18 can be produced from [35S]sulfate-labeled SgII in ISGs isolated from PC12/PC2 cells after equilibration at pH 5.5 followed by incubation at 37°C (Urbé et al., 1997a ). To obtain evidence that the conditions required for fusion do not interfere with the processing of SgII in vitro by PC2 we performed a mock fusion assay with a PNS obtained from PC12/PC2 cells pulse labeled with [35S]sulfate for 10 min and chased for 15 min. After this pulse labeling condition the [35S]sulfate-labeled SgII in this PNS is unprocessed (Urbé et al., 1997a ). If this PNS, which contains [35S]sulfate-labeled SgII and PC2 in the same ISG, is kept at 4°C, equilibrated to pH 5.5, and then incubated at 37°C, SgII processing can be detected by immunoprecipitation of p18 (Fig. 2 a, lane 1). Incubation at 37°C before the equilibration to pH 5.5 in the presence of ATP (Fig. 2 a, lane 2) under conditions which mimic the fusion step in our assay does not interfere with the generation of p18. In a parallel processing reaction preceded by a mock fusion incubation using a PNS from PC12 cells labeled and isolated by an identical procedure, no p18 is detected (Fig. 2 a, lanes 3 and 4) as expected in the absence of PC2.
Figure 2.
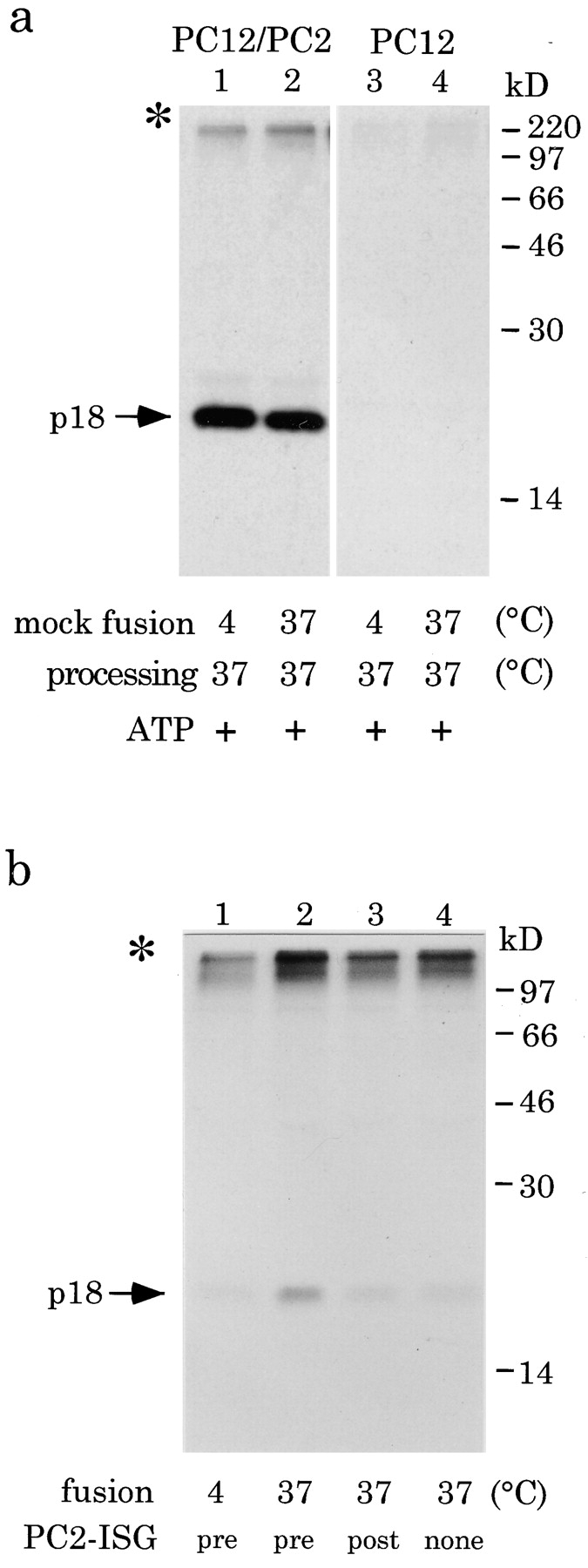
Generation of p18 in a cell-free fusion assay. (a) Processing of SgII in PC12/PC2 ISGs is not affected by previous incubation for fusion. A PNS was prepared from PC12/PC2 cells (lanes 1 and 2) or PC12 cells (lanes 3 and 4) labeled with a 10-min pulse followed by a 15-min chase. Each PNS was then incubated either at 4° or 37°C under conditions which allow fusion in the presence of ATP, then for processing at 37°C for 45 min. (b) Reconstitution of ISG fusion. A PNS was prepared from PC12 cells labeled as above and incubated in the presence of 7 μl of PC2-ISGs and ATP at 4° (lane 1) or 37°C (lane 2) for 45 min. Alternatively, the PC2-ISGs were added after the fusion incubation (lane 3) or omitted entirely (lane 4). After equilibration for 60 min on ice, processing reactions were incubated at 37°C for 45 min. [35S]sulfate-labeled p18 was immunoprecipitated and subjected to SDS-PAGE and autoradiography. Note the autoradiograph shown in b was exposed for 2 mo. *, a high molecular weight band which is brought down during the immunoprecipitation step by binding nonspecifically to both protein A– and G–Sepharose.
Reconstitution of ISG–ISG Fusion In Vitro
Fusion of ISGs, as detected by the appearance of p18, can be observed when the donor PC2-ISGs are added to an acceptor PNS derived from [35S]sulfate-labeled PC12 cells and the reaction mixture is incubated at 37°C for fusion and then processing (Fig. 2 b, lane 2). A low amount of p18 can be detected if the fusion reaction is kept at 4°C (Fig. 2 b, lane 1). Addition of the PC2-ISGs after the fusion reaction does not result in a significant signal over background demonstrating that the processing reaction does not support fusion (Fig. 2 b, lane 3). In the absence of the donor PC2-ISGs a low amount of p18 is detected at this exposure, and we define this as the PC2-independent background. We attribute this background p18 signal (seen in Fig. 2 b, lane 4) to a minor protease activity present in PC12-ISGs. To maximize the p18 signal a 30-min labeling protocol of the PC12 cells was thereafter adopted and routinely used. All of the controls performed in Fig. 2 were done with the extended labeling period and gave the same result (data not shown). The high molecular weight material present in all conditions in Fig. 2 is an unidentified [35S]sulfate-labeled molecule which binds nonspecifically to the protein A– and protein G–Sepharose independently of added antibody.
The fusion reaction is both ATP and temperature dependent (Fig. 3 a). Incubation at 4°C or addition of hexokinase in the presence of glucose resulted in no increase in the production of p18 over the background level. Nonhydrolyzable analogues of ATP were unable to substitute for ATP (Fig. 3 a) suggesting that ATP hydrolysis is necessary for fusion to occur. The PC2-dependent p18 signal routinely obtained in the presence of ATP after a 37°C incubation in the assay was two- to five-fold greater than the PC2-independent signal.
Characterization of ISG–ISG Fusion
ISG–ISG fusion required the presence of proteins on the cytoplasmic surface of the donor ISGs. If the donor PC2-ISGs were pretreated with trypsin before addition to the assay there was a significant decrease in p18 production (Fig. 3 b, lanes 3 and 4) compared with untreated (Fig. 3 b, lane 2) and control samples (Fig. 3 b, lanes 5 and 6). To ensure that the p18 produced in the assay is not a consequence of lysis we added trypsin to the reaction mixture after the processing incubation. The trypsin was then quenched by the addition of soybean trypsin inhibitor, lysis buffer was added, and then the p18 was immunoprecipitated (Fig. 3 b, lane 7). Even after the low pH incubation, p18 was protected from the action of trypsin and was thus generated from SgII by PC2 within the fused ISGs and not by external proteases. As expected, no p18 was detected if trypsin was added in combination with Triton X-100 to allow the trypsin to gain access to the [35S]sulfate-labeled p18 (Fig. 3 b, lane 8).
Optimization of the Assay
We estimate, based on the cell number in the original starting material, that the amount of ISGs contained in 100 μl of acceptor PNS is equivalent to 3 μl of donor ISGs. Therefore, addition of 10 μl of donor ISGs containing PC2 (PC2-ISGs) to 100 μl of acceptor PNS would give approximately a 3:1 ratio between donor ISGs containing the enzyme and acceptor ISGs containing the substrate. To optimize the PC2-dependent fusion signal, donor ISGs were titrated into the assay at ratios from 1:1 to 10:1. Addition of increasing amounts of donor PC2-ISGs, from 2.5–30 μl, to the acceptor PNS stimulated the production of p18. Production of p18 reached a plateau after addition of 20–30 μl of PC2-ISGs (Fig. 4 a). A second round of immunoprecipitation with anti-p18 antibodies demonstrated the first immunoprecipitation was quantitative and sufficient even when using 30 μl of PC2-ISGs with 100 μl of acceptor PNS (data not shown).
Figure 4.


Optimization of the fusion assay conditions. (a) Increasing amounts of PC2-ISGs (from 0 to 30 μl) were added to 100 μl of PNS from PC12 cells labeled for 30 min with [35S]sulfate and then assayed for fusion by incubation for 30 min at 37°C followed by the processing reaction for 90 min at 37°C. Top and bottom panel are from separate representative experiments. (b) Time course of fusion. 20 μl of PC2-ISGs were added to the assay mixture and incubated for fusion. At the time points shown the samples were transferred to ice and diluted with pH 5.5 buffer and subjected to the processing at 37°C for 45 min. (c) Time course of processing. Fusion assays were performed with either 0 or 20 μl of PC2-ISGs for 45 min at 37°C. The reactions were then subjected to processing reactions at 37°C for times ranging from 0 to 150 min. p18 was immunoprecipitated and quantified as described in Fig. 3 and the PC2-independent background was subtracted. Data shown are from representative experiments done in duplicate.
To optimize the efficiency of the assay, and to ensure that neither incubation was limiting, the length of both the fusion incubation and the processing incubation was varied. Fusion between ISGs, assayed with a 45-min processing reaction, occurred with a relatively linear increase over time until ∼30 min when the production of p18 reached a plateau (Fig. 4 b). Processing of SgII by PC2, assayed after a 45-min fusion incubation, also increased with time (Fig. 4 c) and was significantly greater than the PC2-independent signal at all time points. The PC2-independent signal remained constant over time during the fusion reaction whereas it increased with time during the processing incubation until 90 min, suggesting that the background activity is dependent upon acidic pH. For the remainder of the experiments described, fusion was performed for 30 min and the processing reaction for 90 min ensuring that neither fusion nor processing were limiting.
Characterization of Fusion Product
Incubation of ISGs containing [35S]sulfate-labeled SgII with PC2-ISGs results in the appearance of [35S]sulfate-labeled p18. To ensure that the vesicles which contained the p18 were derived from the fusion of two populations of ISGs, we studied the sedimentation behavior of the fused ISGs by subcellular fractionation on sequential velocity and equilibrium sucrose gradients. A fusion incubation was performed as above and chilled to 4°C while an equal portion of the [35S]sulfate-labeled PC12-PNS was kept on ice (starting material). Both the fusion reaction and the starting material were subjected to velocity controlled centrifugation. ISG containing fractions (fractions 2–4) were pooled and either loaded on equilibrium gradients or a portion (one-fifth) of each pool was incubated for processing. The latter was analyzed after processing for the content of [35S]sulfate-labeled SgII or p18 (Fig. 5, a and b). In the pool obtained after fusion (postfusion) [35S]sulfate-labeled p18 (Fig. 5 b) can be detected but not in the pool kept at 4°C (prefusion). As expected, a decrease in the total [35S]sulfate-labeled SgII is detectable after fusion (Fig. 5 a), reflecting the processing of SgII to fragments including p18 (Fig. 1). To verify that the vesicles containing p18 were ISGs, the remaining four-fifths of the velocity gradient pools were subjected to equilibrium gradient centrifugation. After equilibrium gradient centrifugation and fractionation, the fractions were analyzed for [35S]sulfate-labeled SgII (Fig. 5 c) or [35S]sulfate-labeled p18 (Fig. 5 d). The p18 containing vesicles produced by fusion (Fig. 5 d) sedimented to the same position on the equilibrium gradient as the ISGs containing [35S]sulfate-labeled SgII in the starting material (Fig. 5 c). This confirms that the fusion of two ISGs results in a “hybrid” ISG with similar properties as the starting ISG. Interestingly, the hybrid ISG (Fig. 5 d) displayed a slight increase in density, suggesting they were larger than the original population of ISGs. This experiment was also used to calculate the percentage of SgII processed after fusion by comparing the amount of p86 in the starting material with p86 after fusion and processing (Fig. 5 a, Table I, and see below).
Figure 5.


Characterization of the membranes containing p18 after fusion. A PNS was prepared from PC12 cells labeled with [35S]sulfate for 30 min. One-half of this PNS was kept on ice while the remainder was incubated under standard conditions for fusion with PC2-ISGs. Both the starting material and the reaction mixture were loaded onto velocity gradients and subjected to fractionation. Fractions 2–4 from the velocity gradients were pooled, and one-fifths were analyzed for p18 using the standard incubation for processing while the remaining four-fifths were subjected to equilibrium gradient centrifugation and fractionation. (a) Samples of the velocity gradient pools analyzed for p86, or (b) subjected to processing and then immunoprecipitation with anti-p18 antibodies from either the starting material (pre fusion) or after fusion (post fusion). (c) Samples of equilibrium gradient fractions 5–11 from the gradient loaded with the starting material were analyzed for p86, or (d) equilibrium gradient fractions 5–11 from the gradient loaded with the fusion reaction were subjected to processing followed by immunoprecipitation to detect p18 as above. Analysis of p86 was performed by SDS-PAGE followed by fluorography.
Table I.
Determination of the Fusion Efficiency
| PNS | PNS | VG | ||||
|---|---|---|---|---|---|---|
| PC2-ISGs fusion NR* + processing ip 175 | hybrid ISGs + fusion + processing ip 175 | hybrid ISGs + fusion + processing total p86 | ||||
| p86 in starting material (st) | 76.0 | 62.73 | 34.2 | |||
| p86 after incubations (fin) | 23.6 | 46.76 | 20.8 | |||
| Percent processing efficiency | 69.0% | 25.5% | 39.2% | |||
| (p86st − p86fin) × 100 | ||||||
| (p86st) | ||||||
| Percent fusion efficiency | 100% | 37% | — |
NR, not required. A PNS was prepared from PC12 or PC12/PC2 cells labelled for 30 min with [35S]sulphate. Fusion assays were performed as described using a PNS or enriched fractions (VG). The amount of p86 before (starting material = st) and after the fusion reaction (final = fin) was determined by immunoprecipitation with Ab 175 (ip 175) or by direct analysis of the total p86 from enriched fractions containing ISGs (Fig. 5) and is expressed as arbitrary units. Processing efficiency is calculated from the values shown which are from representative experiments. Fusion efficiency in hybrid ISGs was expressed as a percentage of SGII processing in intact PC2-ISGs using the values obtained with the PNS samples.
Comparison of SgII Processing in Fused ISGs with PC2-ISGs
Direct comparison of the extent of processing in the hybrid ISGs with the PC2-ISGs where the enzyme and substrate are copackaged when ISGs are formed in the cell also allowed us to estimate the fusion efficiency under the standard conditions of the assay (Table I). A PNS from PC12/PC2 cells labeled with [35S]sulfate was isolated and incubated in a fusion assay under standard conditions except that no unlabeled PC2-ISGs were added. The extent of processing obtained was then compared with that observed for the hybrid ISGs formed in a typical fusion assay. The relative processing index in hybrid ISGs was found to be ∼25% compared with 69% for PC2-ISGs. Thus, the efficiency of fusion between ISGs in the assay was estimated to be ∼37%.
Comparison of the time course of SgII processing in the hybrid ISG (Fig. 4 c) and in ISGs isolated from PC12/PC2 cells in a previous study (Urbé et al., 1997a ) reveals that SgII processing in hybrid ISGs occurs with different kinetics. In the PC2-ISGs 50% of the maximum processing signal is reached after 15 min of incubation (Fig. 5 in Urbé et al., 1997a ) whereas the processing signal in the hybrid ISGs is less than 10% of the maximum signal at that time. In addition, the maximum p18 signal in the PC2-ISGs is reached at 60 min compared with 90 min for the hybrid ISGs. These differences may be due to the fact that in the hybrid ISGs the PC2 enzyme will process the PC12-ISG derived [35S]sulfate-labeled SgII as well as the unlabeled SgII contained in the PC2-ISGs. If one assumes that one PC2-ISG fuses with one PC12-ISG containing [35S]sulfate-labeled SgII to generate a hybrid ISG, at most only half of the total SgII processed at any one time will be [35S]sulfate-labeled, thus processing of [35S]sulfate-labeled SgII will take about twice as long in the hybrid ISG.
Fusion Only Occurs between ISGs and ISGs
The ISG is defined as an early stage intermediate that can undergo changes such as acidification, membrane remodeling, and homotypic fusion (Tooze, 1991) whereas the MSG is a late stage stable organelle which has a constant size and content. One prediction of these definitions is that fusion should be specific to the early stage and will not occur at the later mature granule stage. Two approaches were used to investigate the stage specificity of fusion: first we asked whether MSGs were able to substitute for ISGs in the fusion assay and second whether ISGs or MSGs could compete for generation of labeled p18.
MSGs, isolated from PC12/PC2 cells, were substituted for the PC2-ISGs in a standard assay (Fig. 6 a). MSGs from PC12/PC2 cells contain large amounts of the mature PC2 enzyme (Dittié and Tooze, 1995; data not shown) and thus the processing of the [35S]sulfate-labeled SgII should be readily detected if fusion occurs. Substitution of the PC2-ISGs with either one or two times the amount of PC2-MSGs did not result in the appearance of appreciable amounts of p18 over the PC2-independent background.
Figure 6.

MSGs cannot fuse with ISGs. (a) PC2-ISGs, or one or two times the volume of MSGs prepared from PC12/PC2 cells were added to the fusion reactions containing 100 μl of PNS from PC12 cells labeled with 30 min [35S]sulfate. (b) Increasing amounts of ISGs (open squares) or 30 μl of MSGs (solid diamond) prepared from PC12 cells, were added to fusion reactions containing 10 μl of PC2-ISGs and 100 μl of PNS from labeled PC12 cells. Fusion reactions, carried out for 30 min at 37°C were followed by processing reactions for 90 min at 37°C. p18 was immunoprecipitated from the reactions and quantitated as above. Results shown are after subtraction of the PC2-independent background and are representative experiments done in duplicate.
The specificity of fusion was next tested by competition for fusion using ISGs isolated from PC12 cells (PC12-IGSs) that contain cold substrate but no enzyme. Addition of increasing amounts of cold PC12-IGSs to the complete reaction mixture (containing 100 μl of [35S]sulfate-labeled PNS from PC12 cells, 10 μl PC2-ISGs, ATP, and an ATP-regenerating system) resulted in a decrease in the amount of p18 produced (Fig. 6 b, open squares). When the ratio of PC12-ISGs to PC2-ISGs added to the assay was ∼3:1, a 50% reduction in the p18 signal was detected. Importantly, the addition of an equivalent amount of MSGs, prepared from PC12 cells (PC12-MSGs), to the assay (i.e., 3 MSGs:1 ISG) resulted in no competition (Fig. 6 b, closed diamond) confirming the observation that MSGs do not fuse with ISGs.
Competition leading to a decrease of the total p18 signal could result from either competition for fusion or enzyme. If the ISGs have a limited capacity to fuse they will become refractory to fusion after a defined number of fusion events. Thus, addition of excess ISGs will result in competition and a decrease in the p18 produced. However, it is equally possible that competition is the result of unlabeled substrate competing with labeled substrate for enzyme in an unrestricted fusion reaction where PC2 is limiting. Further experiments need to be carried out to confirm the mode of competition, however, whether it is a result of competition for fusion or competition for substrate, both depend on an initial fusion event between ISGs.
Cytosol Dependence of ISG–ISG Fusion
To determine if ISG–ISG fusion required cytosolic components we modified the assay by isolating the membranes and membrane bound organelles from the acceptor PNS derived from the [35S]sulfate-labeled PC12 cells. The isolated membranes were resuspended in buffer or cytosol prepared from PC12 cells, and tested for activity in the fusion assay. Comparison of the results obtained with the PNS assay showed that the efficiency of the fusion reaction with the membrane pellet was similar and required cytosol (Fig. 7 a). To obtain a more abundant source of cytosol we explored the feasibility of substituting cytosol from tissues, such as rat liver, for the PC12 cell cytosol. Rat liver cytosol was demonstrated to be active in the fusion assay and the maximum fusion signal was obtained with ∼7 mg/ml cytosol which was comparable to PC12 cell cytosol.
Figure 7.


Cytosol-dependent reconstitution of ISG fusion. (a) A membrane pellet (MP) was prepared from 100 μl of PNS obtained from PC12 cells labeled for 30 min with [35S]sulfate. An equal volume of PNS was used as control. PC2-ISGs (10 μl), and PC12 cell cytosol (final concentration 7 mg/ml) was added to the reaction mixtures as indicated. Fusion and processing was carried out under standard conditions. p18 was immunoprecipitated and quantitated as described above. (b) A standard fusion reaction was performed using a MP prepared from the PNS of PC12 cells labeled as above, in the presence or absence of 10 μl of PC2-ISGs, rat brain cytosol (final amounts 7 mg/ml), and nucleotides as indicated. p18 was immunoprecipitated and quantitated as described above. Data shown are representative experiments performed in duplicate.
We next investigated the requirements for the nucleotides ATP and GTP in the cytosol-dependent assay. Rat liver cytosol was prepared and added to the assay with either ATP, GTP, or GTPγS. Both ATP and GTP together were required for maximum efficiency, and interestingly GTPγS could substitute for GTP but not ATP (Fig. 7 b). Although the requirement for ATP may be attributed to a requirement for NSF (see below) we do not yet understand the GTP requirement. It may be that a GTP-binding protein is required in its GTP bound form for fusion to occur.
Fusion of ISGs Can Occur with an Enriched Preparation of Acceptor ISGs
To ensure that the fusion we measure was homotypic, that is occurring between identical populations of ISGs, we attempted to reconstitute fusion using an acceptor ISG fraction of comparable purity to the donor PC2-ISGs in the presence of cytosol. Several attempts to do so failed (data not shown) and we speculate that centrifugation through two consecutive sucrose gradients removes essential components that are required on at least one ISG population. To overcome this problem we used a partially purified pool enriched in post-Golgi vesicles, in particular ISGs, which does not contain detectable levels of TGN membranes. Isolation of acceptor ISGs from PC12 cells labeled with [35S]sulfate for 30 min was performed using the sucrose velocity gradients normally used to prepare donor PC2-ISGs. The pool of fractions 2–4 which was then used in a standard assay supplemented with rat liver cytosol with or without PC2 ISGs. Rat liver cytosol was added because the velocity gradient fractions are largely depleted of cytosolic proteins which remain in the load. Fusion was detected in the reaction mixtures containing the velocity gradient fractions and PC2-ISGs and cytosol (Fig. 8). Although the total signal was reduced compared with that obtained with a membrane pellet derived from a PNS of [35S]sulfate-labeled PC12 cells, when normalized to background the PC2-dependent fusion signal obtained with the enriched donor pool (1.9-fold over background) was comparable to that obtained with a membrane pellet (1.7-fold over background, Fig. 8).
Role of NEM-sensitive Proteins in ISG Fusion
Previously identified proteins involved in membrane fusion reactions include the ATPases NSF and p97 both of which can be inactivated by NEM treatment. Analysis of the distribution of these NEM-sensitive proteins in the membrane fractions and rat liver cytosol used in the assay is shown in Fig. 9 a. The majority of the NSF contained in the PNS (∼3 μg NSF per mg of total protein) remains associated with the membranes after preparation of the membrane pellet (MP). Most of the α-SNAP present in the PNS was recovered on the membrane pellet, but very little was detected on PC2-ISGs. The distribution of p97 on the PNS and membrane pellet is similar compared with NSF. However, there was very little p47 detected on the membrane pellet or the PC2-ISG fraction.
Figure 9.
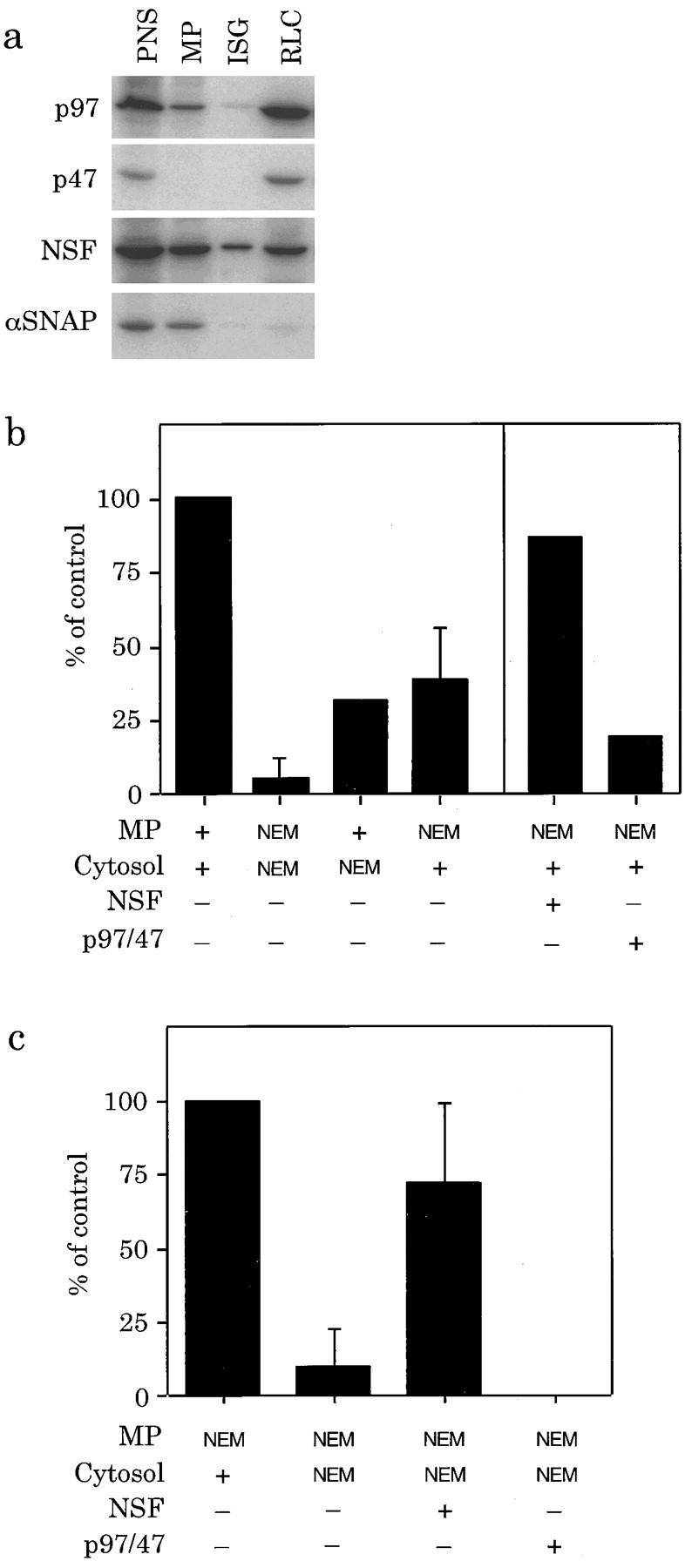
NSF can rescue the inhibition of ISG fusion after NEM treatment. (a) Immunoblot of a PNS, membrane pellet (MP) obtained from the PNS, PC2-ISGs, and rat liver cytosol with antibodies directed against p97, p47, NSF, and α-SNAP. The blot was prepared from material loaded in amounts which are proportional to that in the assay, i.e., 110 μg of a PNS, 38.5 μg of MP, 4.1 μg of ISGs, 49 μg of rat liver cytosol. Bound antibodies were detected using 125I-protein A. (b) NEM treatment was performed on the membrane pellet obtained from a PNS of PC12 cells labeled with [35S]sulfate for 30 min or rat liver cytosol or both. A fusion assay was performed with the treated or untreated components using 10 μl of PC2-ISGs. NSF (19 μg/ml; 230 nM) or p97/ p47 (28 μg/ml p97; 290 nM p97) was added to NEM-treated membranes and cytosol before incubation for fusion. (c) NSF can rescue NEM inhibition of fusion. NEM-treated membrane pellet was incubated with either rat liver cytosol or NEM-treated rat liver cytosol in the presence or absence of NSF, p94/p47 in a standard fusion assay with 10 μl of PC2-ISGs. Values shown are after subtraction of the PC2-independent background and are representative of three independent experiments for NSF and two independent experiments for p97/47 performed in duplicates or triplicates (error bars are ±SD of the mean).
To explore the possibility that one, or both of these complexes, are required for fusion we treated either the PC12 cell membrane pellet, containing [35S]sulfate-labeled SgII, rat liver cytosol, or both with NEM (Fig. 9 b, left). Treatment of both membranes and cytosol inhibited fusion more than 90%. In the presence of NEM treated rat liver cytosol the efficiency of fusion of untreated membranes was only ∼30% of the control value. The fusion efficiency of NEM-treated membranes could be recovered to ∼40% of the original fusion efficiency by the addition of untreated rat liver cytosol to the NEM treated membranes.
To determine if we could attribute the decrease in fusion efficiency after NEM treatment to inactivation of NSF or p97 we added recombinant NSF and α-SNAP, or purified p97 and recombinant p47 to the fusion assay after various NEM treatments. Using NEM-treated membranes, addition of NSF to NEM treated rat liver cytosol could restore fusion nearly to the level observed after addition of untreated rat liver cytosol (Fig. 9 c). Addition of α-SNAP did not increase the rescue by NSF, in fact was slightly inhibitory (data not shown). Interestingly, supplementing untreated cytosol with similar amounts of NSF stimulated the recovery of NEM-treated membranes to almost 90% of the control value (Fig. 9 b, right). Addition of a complex of purified p97 and his-tagged p47 to NEM-treated membranes and NEM-treated cytosol did not restore fusion (Fig. 9 c). Neither did the addition of p97/47 to untreated cytosol restore the ability of NEM-treated membranes to undergo fusion (Fig. 8 b, right). Experiments were also performed using purified p97 alone, or a crude p97–p47 fraction (purified by 40% ammonium sulfate precipitation of rat liver cytosol, then sucrose gradient centrifugation using the method of Kondo et al. [1997]), both of which were unable to rescue NEM-treated membranes and cytosol.
Discussion
Fusion of ISGs has been reconstituted in a cell-free assay based on the proteolytic processing of a substrate prohormone (SgII) by a prohormone convertase (PC2). An important feature of the assay is the fact that proteolytic processing of substrates by PC2 only occurs in the regulated secretory pathway after the substrates and enzymes are packaged into secretory granules (Urbé et al., 1997a ). Using this assay we have determined that ISG fusion is dependent upon cytosolic and membrane associated proteins, ATP, GTP, and physiological temperature. Fusion is specific for ISGs in that MSGs cannot fuse with ISGs, and competition is observed after addition of excess ISGs, but not MSGs, derived from unlabeled PC12 cells.
In the absence of PC2-ISGs we can detect a low amount of endopeptidase activity which processes the labeled SgII in the PC12-ISGs. This background signal is produced during the processing reaction by an endogenous protease present in PC12 cell ISGs. This endopeptidase activity is not detected in intact PC12 cells after prolonged [35S]sulfate-labeling during which time the MSGs accumulate unprocessed [35S]sulfate-labeled SgII. Furthermore, subcellular fractionation data suggest that the activity is found in ISGs but not MSGs (data not shown). We speculate that this unknown endopeptidase activity may have a pH optimum closer to pH 5.5 than 6.3, and therefore is normally inactive in ISGs. Furthermore, this enzyme might be removed from ISGs during maturation and thus never reach the more acidic milieu of the MSG where it would be active. Additional information about the nature and function of this enzyme requires further study.
The assay described reconstitutes ISG–ISG fusion, and was developed with the aim of understanding the role of fusion in MSG biogenesis. Homotypic fusion, defined as the fusion of two like compartments, is thought to be involved in the steady-state maintenance of organelles, unlike heterotypic fusion which results in the transfer of molecules and lipids from one compartment to another. Examples of homotypic fusion include the reassembly of the ER (Latterich et al., 1995; Turner et al., 1997), the Golgi complex (Acharya et al., 1995b ; Rabouille et al., 1995b ), endosome-endosome fusion (Gruenberg and Howell, 1986; Diaz et al., 1988; Woodman and Warren, 1988), lysosome–lysosome fusion (Ward et al., 1997) and vacuole–vacuole fusion (Conradt et al., 1992).
In the case of assays which rely on internalized markers to define the endocytotic compartments (for example see Gruenberg and Howell, 1986; Diaz et al., 1988), fusion is assumed to be homotypic if the compartments undergoing fusion contain markers internalized for the same length of time. In the ISG–ISG fusion assay described here fusion can potentially occur amongst heterologous populations of ISGs defined by the [35S]sulfate-labeling protocol. ISGs form de novo from the TGN then mature into MSGs. Maturation of ISGs takes place over a relatively long period of time (t 1/2 = ∼45 min), compared with the time it takes an ISG to bud (budding t 1/2 = ∼5 min; Tooze and Huttner, 1990). The possibility therefore exists that the labeled ISGs in the assay are at different stages of maturation, and consequently might display variable fusionogenicity. Although there might be subpopulations of ISGs between which we can not discriminate, we define this fusion as homotypic in contrast with heterotypic fusion of ISGs with the plasma membrane.
Key components required for either or both heterotypic and homotypic membrane fusion are the related ATPases, NSF and p97 (for review see Mellman, 1995; Whiteheart and Kubalek, 1995; Woodman, 1997). To begin to understand the exact nature of ISG–ISG fusion we asked if either or both of these molecules were required. We found that NSF, but not p97, is required for fusion of ISGs. NEM treatment of cytosol and membranes resulted in inhibition of fusion, which could be rescued by the addition of NSF. We found that α-SNAP was not required for NSF activity, and the addition of α-SNAP alone had no effect. In light of the results from the reconstitution of homotypic fusion between Golgi membranes (Acharya et al., 1995a ; Rabouille et al., 1995a ) one might have anticipated a role for p97 in ISG fusion. We found that addition of p97, or a complex of p97 and p47, to NEM-treated membranes and cytosol was not sufficient to rescue fusion.
The distinction between the function of NSF and p97 in heterotypic fusion versus homotypic fusion is somewhat blurred by the ability of NSF to rescue NEM-treated Golgi membranes (Acharya et al., 1995a ; Rabouille et al., 1995a ) in the presence of α-SNAP, γ-SNAP, and p115, and the demonstration that both p47 and α-SNAP bind the t-SNARE syntaxin 5 (Rabouille, 1998). In addition, homotypic fusion between yeast vacuoles, mammalian endosomes, and lysosomes has been shown to require NSF (Diaz et al., 1989; Rodriguez, et al., 1994; Robinson, et al., 1997) and fusion between yeast vacuoles and mammalian endosomes has been shown not to require cdc48p (Mayer et al., 1996) or p97 (Robinson et al., 1997), respectively. Thus, until we obtain more data on the exact role of either NSF or p97 in membrane fusion, a requirement for one or the other does not seem to be a primary factor in classifying fusion events as either homotypic or heterotypic.
What is the purpose of ISG–ISG fusion? We propose that fusion of ISGs with each other is an integral part of the maturation process and may be a prerequisite for post-TGN sorting of proteins from the maturing ISG. Fusion of ISGs may be necessary to create enough excess membrane to form a clathrin-coated vesicle (CCV) from the ISG. Formation of one CCVs from an ISG (80-nm-diam) would result in the removal of nearly one-half the surface area of the ISG. If three to five ISGs fuse to form a MSG (120-nm-diam) (Tooze et al., 1991) enough excess membrane would become available to allow two to seven CCVs to form. In addition, fusion of ISGs might be a prerequisite for formation of CCVs by increasing for example the number of AP-1–binding sites present in the ISG. Furin, a TGN-resident endopeptidase and both mannose-6-phosphate receptors (MPRs) associate with AP-1 and are removed in CCVs which bud from the ISGs (Dittié et al., 1996, 1997; Klumperman, 1998; Kuliawat, et al., 1997). Other molecules which are not found in MSGs such as certain isoforms of peptidylglycine-α-amidating-mono-oxygenase (Milgram et al., 1994), as well as soluble molecules such as C-peptide (Kuliawat and Arvan, 1994) may also be present in these CCVs although how they are selected is not clear.
If ISG fusion is part of the maturation process it follows that after maturation is completed, homotypic fusion would no longer be required. In agreement with this we have shown here that MSGs can no longer fuse with ISGs. It is possible that the ISG–ISG fusion machinery maybe gradually inactivated during maturation. We have previously shown that ISGs have a less acidic luminal pH than MSGs (pH 6.3 versus pH 5.5) and proposed that the decrease in pH during maturation may be a direct consequence of the membrane remodelling events during maturation (Urbé et al., 1997a ). Increasing lumenal acidity may influence the properties of both the membrane and proteins within the membrane and might thus modify the fusion competence of the secretory granule. An alternative but not exclusive explanation maybe that factors involved in homotypic fusion of ISGs, for instance a t-SNARE, are removed during maturation by budding of CCVs or constitutive-like secretory vesicles such that the MSG is left with only the molecule(s), for instance a v-SNARE, required to mediate heterotypic fusion with the plasma membrane during regulated exocytosis. Control of the final size of the MSG would then be dictated by the number of ISG fusion events needed to generate enough vesicles to remove all the SNAREs involved in ISG fusion.
There are to date two possible candidate t-SNAREs that may be involved in ISG fusion, syntaxin 3 and 6. Syntaxin 3 has been localized to zymogen granules (Gaisano et al., 1996) whereas syntaxin 6 has been localized to ISGs in PC12 cells (Klumperman, 1998). Syntaxin 6 has been shown to be present in coated buds emanating from the ISGs as well as in CCVs, endosomes, (Klumperman, 1998), and in TGN membranes (Bock, et al., 1997; Klumperman, 1998). It has been proposed that syntaxin 6 is involved in targeting the CCVs to an endosomal compartment (Klumperman, 1998). It is equally plausible that syntaxin 6 is involved in ISG fusion and only subsequently removed by CCVs together with furin and MPR and targeted to the endosome and/or returned to the TGN.
Selection and removal of putative sorting receptors, membrane proteins, or missorted cargo before regulated exocytosis is the most important goal of maturation and is critical for the homeostasis of the organism. Although sorting and fusion events during secretory granule biogenesis are likely to be linked, more experiments are required to understand when and why ISGs lose their ability to fuse with each other and how this is controlled to generate a MSG. The assay described here represents an ideal experimental system to investigate secretory granule biogenesis.
Acknowledgments
We thank all the past and present members of the Tooze and Warren labs for helpful discussions. We are grateful to G. Warren, G. Schiavo, and J. Tooze (all from ICRF, London, UK) for critically reading the manuscript as well as for discussions, advice, and encouragement. We thank H. Kondo for generously supplying purified p97, p97–p47 complex, and his-tagged p47 as well as for his time and advice.
Abbreviations used in this paper
- CCV
clathrin-coated vesicle
- ISG
immature secretory granules
- MP
membrane pellet
- MPR
mannose-6-phosphate receptor
- MSG
mature secretory granule
- NEM
N-ethylmaleimide
- NSF
N-ethylmalemide–sensitive fusion protein
- PC2
prohormone convertase 2
- PNS
postnuclear supernatant
- SgII
secretogranin II
- SNAP
soluble NSF attachment protein
- SNARE
SNAP receptor
- STI
soybean trypsin inhibitor
- t-SNARE
target SNARE
- v-SNARE
vesicle SNARE
Footnotes
S. Urbé's present address is Physiological Laboratory, University of Liverpool, Crown Street, Liverpool L34BU, UK.
References
- Acharya U, Jacobs R, Peters JM, Watson N, Farquhar MG, Malhotra V. The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell. 1995a;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Acharya U, McCaffery JM, Jacobs R, Malhotra V. Reconstitution of vesiculated Golgi membranes into stacks of cisternae: requirement of NSF in stack formation. J Cell Biol. 1995b;129:577–589. doi: 10.1083/jcb.129.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Advani RJ, Bae H-R, Bock JB, Chao DS, Doung Y-C, Prekeris R, Yoo J-S, Scheller RH. Seven novel mammalian SNARE proteins localize to distinct membrane compartments. J Biol Chem. 1998;273:10317–10324. doi: 10.1074/jbc.273.17.10317. [DOI] [PubMed] [Google Scholar]
- Barr FA, Leyte A, Mollner S, Pfeuffer T, Tooze SA, Huttner WB. Trimeric G-proteins of the trans-Golgi network are involved in the formation of constitutive secretory vesicles and immature secretory granules. FEBS (Fed Eur Biochem Soc) Lett. 1991;294:239–243. doi: 10.1016/0014-5793(91)81438-e. [DOI] [PubMed] [Google Scholar]
- Bennett DL, Bailyes EM, Nielsen E, Guest PC, Rutherford NG, Arden SD, Hutton JC. Identification of the type 2 proinsulin processing endopeptidase as PC2, a member of the eukaryote subtilisin family. J Biol Chem. 1992;267:15229–15236. [PubMed] [Google Scholar]
- Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc Natl Acad Sci USA. 1988;85:7852–7856. doi: 10.1073/pnas.85.21.7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock JB, Scheller RH. Protein transport A fusion of new ideas. Nature. 1997;36:133–135. doi: 10.1038/387133a0. [DOI] [PubMed] [Google Scholar]
- Bock JB, Klumperman J, Davanger S, Scheller RH. Syntaxin 6 functions in trans Golgi network vesicle trafficking. Mol Biol Cell. 1997;8:1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess TL, Kelly RB. Constitutive and regulated secretion of proteins. Annu Rev Cell Biol. 1987;3:243–293. doi: 10.1146/annurev.cb.03.110187.001331. [DOI] [PubMed] [Google Scholar]
- Carnell L, Moore HP. Transport via the regulated secretory pathway in semi-intact PC12 cells: role of intra-cisternal calcium and pH in the transport and sorting of secretogranin II. J Cell Biol. 1994;127:693–705. doi: 10.1083/jcb.127.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Siddhanta A, Austin CD, Hammond SM, Sung TC, Frohman MA, Morris AJ, Shields D. Phospholipase D stimulates release of nascent secretory vesicles from the trans-Golgi network. J Cell Biol. 1997;138:495–504. doi: 10.1083/jcb.138.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary D, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- Conradt B, Shaw J, Vida T, Emr S, Wickner W. In vitro reactions of vacuole inheritance in Saccharomyces cerevisiae. . J Cell Biol. 1992;119:1469–1479. doi: 10.1083/jcb.119.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool DR, Normant E, Shen F-s, Chen H-C, Pannell L, Zhang Y, Loh YP. Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpefatmice. Cell. 1997;88:73–83. doi: 10.1016/s0092-8674(00)81860-7. [DOI] [PubMed] [Google Scholar]
- Davey J, Hurtley SM, Warren G. Reconstitution of an endocytic fusion event in a cell-free system. Cell. 1985;43:643–652. doi: 10.1016/0092-8674(85)90236-3. [DOI] [PubMed] [Google Scholar]
- Diaz R, Mayorga L, Stahl P. In vitro fusion of endosomes following receptor-mediated endocytosis. J Biol Chem. 1988;263:6093–6100. [PubMed] [Google Scholar]
- Diaz R, Mayorga L, Weidman PJ, Rothman JE, Stahl PD. Vesicle fusion following receptor-mediated endocytosis requires a protein active in Golgi transport. Nature. 1989;339:398–400. doi: 10.1038/339398a0. [DOI] [PubMed] [Google Scholar]
- Dittié A, Tooze S. Characterization of the endopeptidase PC2 activity towards SgII in stably transfected PC12 cells. Biochem J. 1995;310:777–787. doi: 10.1042/bj3100777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittié AS, Hajibagheri N, Tooze SA. The AP-1 adaptor complex binds to immature secretory granules from PC12 cells, and is regulated by ADP- ribosylation factor. J Cell Biol. 1996;132:523–536. doi: 10.1083/jcb.132.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittié AS, Thomas L, Thomas G, Tooze SA. Interaction of furin in immature secretory granules from neuroendocrine cells with the AP-1 adaptor complex is modulated by casein kinase II phosphorylation. EMBO (Eur Mol Biol Organ) J. 1997;16:4859–4870. doi: 10.1093/emboj/16.16.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisano HY, Ghai M, Malkus PN, Sheu L, Bouquillion A, Bennett MK, Trimble WS. Distinct cellular locations of the syntaxin family of proteins in rat pancreatic acinar cells. Mol Biol Cell. 1996;7:2019–2027. doi: 10.1091/mbc.7.12.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes H-H, Phillips E, Huttner WB. The primary structure of rat secretogranin II deduces from a cDNA sequence. Nucl Acids Res. 1988;16:11811. doi: 10.1093/nar/16.24.11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götte M, von Mollard GF. A new beat for the SNARE drum. Trends Cell Biol. 1998;8:215–218. doi: 10.1016/s0962-8924(98)01272-0. [DOI] [PubMed] [Google Scholar]
- Gruenberg JE, Howell KE. Reconstitution of vesicle fusions occurring in endocytosis with a cell-free system. EMBO (Eur Mol Biol Organ) J. 1986;5:3091–3101. doi: 10.1002/j.1460-2075.1986.tb04615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumann R, Kachel V, Thoenen H. Relationship between NGF-mediated volume increase and “priming effect” in fast and slow reacting clones of PC12 pheochromocytoma cells. Exp Cell Res. 1983;145:179–190. doi: 10.1016/s0014-4827(83)80019-6. [DOI] [PubMed] [Google Scholar]
- Irminger J-C, Verchere B, Meyer K, Halban PA. Proinsulin targeting to the regulated pathway is not impaired in carboxypeptidase E-deficient Cpefat/Cpefatmice. J Biol Chem. 1997;272:27532–27534. doi: 10.1074/jbc.272.44.27532. [DOI] [PubMed] [Google Scholar]
- Itin C, Rancanos C, Nakajima Y, Pfeffer SR. A novel assay reveals a role for soluble N-ethylmaleimide-sensitive fusion attachment protein in mannose 6-phosphate receptor transport from endosomes to the trans-Golgi network. J Biol Chem. 1997;272:27737–27744. doi: 10.1074/jbc.272.44.27737. [DOI] [PubMed] [Google Scholar]
- Klumperman J, Kuliawat R, Griffith JM, Geuze HJ, Arvan P. Mannose 6-phosphate receptors are sorted from immature secretory granules via adaptor protein AP-1, clathrin, and syntaxin 6-positive vesicles. J Cell Biol. 1998;141:359–371. doi: 10.1083/jcb.141.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. p47 is a cofactor for p97-mediated membrane fusion. Nature. 1997;388:75–78. doi: 10.1038/40411. [DOI] [PubMed] [Google Scholar]
- Kuliawat R, Arvan P. Distinct molecular mechanisms for protein sorting within immature secretory granules of pancreatic β-cells. J Cell Biol. 1994;126:77–86. doi: 10.1083/jcb.126.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliawat R, Klumperman J, Ludwig T, Arvan P. Differential sorting of lysosomal enzymes out of the regulated secretory pathway in pancreatic β-cells. J Cell Biol. 1997;137:595–608. doi: 10.1083/jcb.137.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslop A, Weiss C, Savaria D, Eiter C, Tooze SA, Seidah NG, Winkler H. Proteolytic processing of chromogranin B and secretogranin II by prohormone convertases. J Neurochem. 1998;70:374–383. doi: 10.1046/j.1471-4159.1998.70010374.x. [DOI] [PubMed] [Google Scholar]
- Latterich M, Frohlich KU, Schekman R. Membrane fusion and the cell cycle: Cdc48p participates in the fusion of ER membranes. Cell. 1995;82:885–893. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- Lee RWH, Huttner WB. Tyrosine-O-sulfated proteins of PC12 pheochromocytoma cells and their sulfation by a tyrosylprotein sulfotransferase. J Biol Chem. 1983;258:11326–11334. [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Mellman I. Enigma variations: protein mediators of membrane fusion. Cell. 1995;82:869–872. doi: 10.1016/0092-8674(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Milgram SL, Eipper BA, Mains DE. Differential trafficking of soluble and integral membrane secretory granule-associated proteins. J Cell Biol. 1994;124:33–41. doi: 10.1083/jcb.124.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols JB, Ungermann C, Pelham HRB, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t-and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Niehrs C, Stinchcombe J, Huttner WB. Two membrane-bound forms of tyrosylprotein sulfotransferase as revealed by phase partitioning in Triton X-114. Eur J Cell Biol. 1992;58:35–43. [PubMed] [Google Scholar]
- Ohashi M, Jan de Vries K, Frank R, Snoek G, Bankaitis V, Wirtz K, Huttner WB. A role for phosphatidylinositol transfer protein in secretory vesicle formation. Nature. 1995;377:544–547. doi: 10.1038/377544a0. [DOI] [PubMed] [Google Scholar]
- Orci L, Halban P, Perrelet A, Amherdt M, Ravazzola M, Anderson RGW. pH-independent and -dependent cleavage of proinsulin in the same secretory vesicle. J Cell Biol. 1994;126:1149–1156. doi: 10.1083/jcb.126.5.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SK, Indig FE, Olivieri N, Levine ND, Latterich M. Organelle membrane fusion: a novel function for the syntaxin homolog Ufe1p in ER membrane fusion. Cell. 1998;92:611–620. doi: 10.1016/s0092-8674(00)81129-0. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Levine TP, Peters JM, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995a;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Misteli T, Watson R, Warren G. Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J Cell Biol. 1995b;129:605–618. doi: 10.1083/jcb.129.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 1998;92:603–610. doi: 10.1016/s0092-8674(00)81128-9. [DOI] [PubMed] [Google Scholar]
- Robinson LJ, Aniento F, Gruenberg J. NSF is required for transport from early to late endosomes. J Cell Sci. 1997;110:2079–2087. doi: 10.1242/jcs.110.17.2079. [DOI] [PubMed] [Google Scholar]
- Rodriguez L, Stirling CJ, Woodman PG. Multiple N-ethylmaleimide-sensitive components are required for endosomal vesicle fusion. Mol Biol Cell. 1994;5:773–783. doi: 10.1091/mbc.5.7.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Salpeter MM, Farquhar MG. High resolution analysis of the secretory pathway in mammotrophs of the rat anterior pituitary. J Cell Biol. 1981;91:240–246. doi: 10.1083/jcb.91.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RE, Farquhar MG. Lysosome function in the regulation of the secretory process in cells of the anterior pituitary gland. J Cell Biol. 1966;31:319–347. doi: 10.1083/jcb.31.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Bennet MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Steiner DF. Prohormone convertases revealed at last. Curr Biol. 1991;1:375–377. doi: 10.1016/0960-9822(91)90198-6. [DOI] [PubMed] [Google Scholar]
- Thiele C, Gerdes HH, Huttner WB. Protein secretion: puzzling receptors. Curr Biol. 1997;7:496–500. doi: 10.1016/s0960-9822(06)00247-8. [DOI] [PubMed] [Google Scholar]
- Tooze SA. Biogenesis of secretory granules Implications arising from the immature secretory granule in the regulated pathway of secretion. FEBS (Fed Eur Biochem Soc) Lett. 1991;285:220–224. doi: 10.1016/0014-5793(91)80805-d. [DOI] [PubMed] [Google Scholar]
- Tooze SA. Biogenesis of secretory granules in the trans-Golgi network of neuroendocrine and endocrine cells. Biochim Biophys Acta. 1998;1404:231–244. doi: 10.1016/S0167-4889(98)00059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze SA, Huttner WB. Cell-free protein sorting to the regulated and constitutive secretory pathways. Cell. 1990;60:837–847. doi: 10.1016/0092-8674(90)90097-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze SA, Flatmark T, Tooze J, Huttner WB. Characterization of the immature secretory granule, an intermediate in granule biogenesis. J Cell Biol. 1991;115:1491–1503. doi: 10.1083/jcb.115.6.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MD, Plutner H, Balch WE. A Rab GTPase is required for homotypic assembly of the endoplasmic reticulum. J Biol Chem. 1997;272:13479–13483. doi: 10.1074/jbc.272.21.13479. [DOI] [PubMed] [Google Scholar]
- Urbé S, Dittié S, Tooze SA. pH-dependent processing of secretogranin II by the endopeptidase PC2 in isolated immature secretory granules. Biochem J. 1997a;321:65–74. doi: 10.1042/bj3210065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbé S, Tooze SA, Barr FA. Formation of secretory vesicles in the biosynthetic pathway. Biochim Biophys Acta. 1997b;1358:6–22. doi: 10.1016/s0167-4889(97)00050-5. [DOI] [PubMed] [Google Scholar]
- Ward DM, Leslie JD, Kaplan J. Homotypic lysosome fusion in macrophages: analysis using an in vitro assay. J Cell Biol. 1997;139:665–673. doi: 10.1083/jcb.139.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westerman B, Gmachi M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Weidman PJ, Melancon P, Block MR, Rothman JE. Binding of an N-ethylmaleimide–sensitive fusion protein to golgi membranes requires both a soluble protein(s) and an integral membrane protein. J Cell Biol. 1989;108:1589–1596. doi: 10.1083/jcb.108.5.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrov SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain- specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- Whiteheart SW, Kubalek EW. SNAPs and NSF: general members of the fusion apparatus. Trends Cell Biol. 1995;5:64–68. doi: 10.1016/s0962-8924(00)88948-5. [DOI] [PubMed] [Google Scholar]
- Woodman P. The roles of NSF, SNAPs and SNAREs during membrane fusion. Biochim Biophys Acta. 1997;1357:155–172. doi: 10.1016/s0167-4889(97)00039-6. [DOI] [PubMed] [Google Scholar]
- Woodman PG, Warren G. Fusion between vesicles from the pathway of receptor-mediated endocytosis in a cell-free system. Eur J Biochem. 1988;173:101–108. doi: 10.1111/j.1432-1033.1988.tb13972.x. [DOI] [PubMed] [Google Scholar]