

Abstract
Several recent studies suggest the isolation of stem cells in skeletal muscle, but the functional properties of these muscle-derived stem cells is still unclear. In the present study, we report the purification of muscle-derived stem cells from the mdx mouse, an animal model for Duchenne muscular dystrophy. We show that enrichment of desmin+ cells using the preplate technique from mouse primary muscle cell culture also enriches a cell population expressing CD34 and Bcl-2. The CD34+ cells and Bcl-2+ cells were found to reside within the basal lamina, where satellite cells are normally found. Clonal isolation and characterization from this CD34+Bcl-2+ enriched population yielded a putative muscle-derived stem cell, mc13, that is capable of differentiating into both myogenic and osteogenic lineage in vitro and in vivo. The mc13 cells are c-kit and CD45 negative and express: desmin, c-met and MNF, three markers expressed in early myogenic progenitors; Flk-1, a mouse homologue of KDR recently identified in humans as a key marker in hematopoietic cells with stem cell-like characteristics; and Sca-1, a marker for both skeletal muscle and hematopoietic stem cells. Intramuscular, and more importantly, intravenous injection of mc13 cells result in muscle regeneration and partial restoration of dystrophin in mdx mice. Transplantation of mc13 cells engineered to secrete osteogenic protein differentiate in osteogenic lineage and accelerate healing of a skull defect in SCID mice. Taken together, these results suggest the isolation of a population of muscle-derived stem cells capable of improving both muscle regeneration and bone healing.
Keywords: dystrophin, gene transfer, BMP-2, stem cells, bone formation
Introduction
The development of muscle stem cells for transplantation or gene transfer (ex vivo approach) as a new method for treatment of patients with muscle disorders has become more attractive and challenging in the past few years (Barroffio et al., 1996; Cornelison and Wold 1997; Gussoni et al. 1999; Miller et al. 1999). Duchenne muscular dystrophy (DMD) is a progressive muscle weakness characterized by a lack of dystrophin expression at the sarcolemma of muscle fibers (Hoffman et al. 1987; Bonilla et al. 1988; Watkins et al. 1988; Zubryzcka-Gaarn et al. 1988; Arahata et al. 1989). Dystrophin is associated with a large oligomeric complex of glycoprotein called dystrophin-associated protein (DAPs), which provide linkage to the extracellular matrix (Ervasti and Campbell 1991). Although the transplantation of myoblasts has been found capable of transiently delivering dystrophin and improving the strength of injected dystrophic muscle, this approach has been hindered by various limitations: immune rejection, poor cellular survival, and the limited spread of the injected cells (Morgan et al. 1988, Morgan et al. 1990, Morgan et al. 1993; Karpati et al. 1989, Karpati et al. 1992; Partridge et al. 1989; Partridge 1991; Gussoni et al. 1992, Gussoni et al. 1997; Huard et al. 1992a,Huard et al. 1992b, Huard et al. 1994a,Huard et al. 1994b,Huard et al. 1994c; Tremblay et al. 1993; Beauchamps et al. 1994, Beauchamps et al. 1999; Kinoshita et al. 1994; Mendell et al. 1995; Vilquin et al. 1995; Fan et al. 1996; Guerette et al. 1997; Qu et al. 1998; Qu and Huard 2000a,Qu and Huard 2000b).
The efficiency of cell transfer might be improved by using muscle stem cells, which potentially display unique features, including: self-renewing cells that produce progeny; arise early in development and persist throughout life; and long-term proliferation and multipotency. In fact, muscle satellite cells have long been considered the myogenic cells responsible for postnatal muscle growth, regeneration and repair for the maintenance of skeletal muscle (Bichoff 1994). However, the recent isolation of cells from bone marrow (Ferrari et al. 1998; Bittner et al. 1999; Gussoni et al. 1999) and the embryonic vasculature (De Angelis et al. 1999) that are capable of differentiating in the myogenic lineage led us to question whether the satellite cells are the only cells capable of supporting muscle growth and regeneration in postnatal life (see review by Seale and Rudnicki 2000).
It has been established that mesenchymal stem cells derived from bone marrow (Caplan 1991; Pittenger et al. 1999) and other connective tissues (Young et al. 1993, Young et al. 1995; Lucas, et al. 1995) have the potential to differentiate into different lineage upon changes in external stimuli. Skeletal muscle tissue has been extensively investigated as a potential source for isolation of pluripotent stem cells (Katagiri et al. 1995; Cornelison and Wold 1997; Gussoni et al. 1999; Jackson et al. 1999; Bosch et al. 2000). In particular, stem cell populations from muscle and nonstromal fraction of bone marrow were recently shown to partially restore the expression of dystrophin in the mdx mouse, an animal model of DMD (Gussoni et al. 1999). Also, a myogenic cell line from mouse skeletal muscle has been shown to differentiate into osteoblastic lineage in vitro upon stimulation with bone morphogenetic protein, a class of proteins intimately involved in growth and differentiation of mesenchymal cells (Katagiri et al. 1995). Thus, at least one subpopulation of cells within skeletal muscle can differentiate into multiple lineages.
The satellite cells, a subpopulation of muscle-derived cells, may have stem cell-like characteristics (Baroffio et al. 1996). When cultured in a monolayer system, subpopulations of satellite cells remain mononucleated and desmin positive, dividing to produce fusing and nonfusing myoblasts (Baroffio et al. 1996). When transplanted into a host muscle, myoblasts can either fuse with host myofibers or fuse together into myotubes that will consequently differentiate into muscle fibers (Morgan et al. 1988, Morgan et al. 1990, Morgan et al. 1993; Karpati et al. 1989, Karpati et al. 1992; Partridge 1991; Gussoni et al. 1992, Gussoni et al. 1997; Huard et al. 1992a,Huard et al. 1992b, Huard et al. 1994a,Huard et al. 1994b,Huard et al. 1994c; Tremblay et al. 1993; Beauchamps et al. 1994, Beauchamps et al. 1999; Kinoshita et al. 1994; Mendell et al. 1995; Vilquin et al. 1995; Fan et al. 1996; Guerette et al. 1997; Qu et al. 1998; Qu and Huard 2000a,Qu and Huard 2000b). However, the vast majority of transplanted myoblasts die rapidly after the injection (Beauchamps et al. 1994, Beauchamps et al. 1999; Huard et al. 1994c; Fan et al. 1996; Qu et al. 1998), which may be partly related to inflammatory reactions (Guerette et al. 1997). Although an improvement of cell survival was achieved by blocking inflammation, a dramatic loss of injected cells was still observed (Guerette et al. 1997; Qu et al. 1998).
A recent report has suggested that only a discrete minority of myoblasts can survive after implantation and thus may represent a population of myogenic stem cells (Beauchamps et al. 1999). Indeed, we have recently identified a specific population of highly purified muscle-derived cells by the preplate technique that significantly improved cell survival after transplantation when injected intramuscularly (Qu et al. 1998). Although the mechanism by which these specific muscle-derived cells display a high cell survival is unclear, their ability to rapidly fuse with various types of host myofibers may help to improve their survivability in skeletal muscle (Qu et al. 1998). In fact, we have recently observed that the fusion of myoblasts with host myofibers after transplantation is muscle fiber-dependent (Qu and Huard 2000a,Qu and Huard 2000b). Our results suggest that myoblasts fuse with myofibers expressing the same type of myosin heavy chains (MyHCs); consequently, matching host muscle and donor myoblasts for MyHCs improves myoblast transfer therapy (Qu and Huard 2000a,Qu and Huard 2000b). Alternatively, the use of these specific muscle-derived cells that can fuse with both types of myofibers can also be used to improve the efficiency of myoblast transplantation (Qu and Huard 2000a,Qu and Huard 2000b).
Taken together, these results suggest that satellite cells are highly heterogeneous in nature. This prompted our attempt to investigate whether our highly purified muscle-derived cells (preplate technique) will express markers of stem cells and differentiate into osteogenic lineage. In this report, we show that the highly purified myogenic cells derived by the preplate technique express markers indicative of stem cells. Isolation and characterization of a clonal population from these highly purified myogenic cells revealed that a clonal cell population (mc13) express both stem cell and satellite cell-specific markers. More importantly, the mc13 cells have the capacity to differentiate into both myogenic and osteogenic lineage in vitro and in vivo. Thus, our results suggest that a subpopulation of muscle-derived cells possess stem cell-like characteristics and can differentiate into multiple lineages.
Materials and Methods
Isolation of Muscle-derived Cells
Primary muscle cells were isolated from 3-wk-old mdx mice (C57BL/10ScSn mdx/mdx, The Jackson Laboratory) using a technique previously described (Rando and Blau 1994; Qu et al. 1998). The isolated cells were then suspended in the growth medium (DME supplemented with 10% FBS, 10% horse serum, 0.5% chick embryo extract, and 2% penicillin/streptomycin). The cells were then preplated in collagen-coated flasks (Rando and Blau 1994; Qu et al. 1998). After ∼1 h, the supernatant was withdrawn from the flask and replated in a fresh collagen-coated flask. The cells that adhered rapidly within this 1-h incubation were mostly fibroblasts (Rando and Blau 1994; Qu et al. 1998; Qu and Huard 2000a,Qu and Huard 2000b). The serial replating of the supernatant was repeated when 30–40% of the cells had adhered to each flask. After ∼5–6 serial platings, the culture was enriched with small, round cells (pp6; Qu et al. 1998; Qu and Huard 2000a,Qu and Huard 2000b).
Clonal Isolation of Purified Muscle-derived Cells
To isolate clones from pp6, the slow adhering primary muscle cells were transfected with a plasmid encoding for the β-galactosidase, minidystrophin (Yuasa et al. 1998; gift from Dr. Takeda, Kumamoto University School of Medicine, Kumamoto, Japan) and the neomycin resistance gene. The highly purified muscle-derived cells were transfected with 10 μg of the linear plasmid containing minidystrophin, lacZ, and neomycin resistance gene using the lipofectamine reagent (GIBCO BRL) according to the manufacturer's instructions. At 72 h after transfection, cells were selected with 3,000 μg/ml of G418 (GIBCO BRL) for 10 d until colonies appeared. Colonies were then picked up after transfection, expanded to obtain large quantity of the transfected cells, and tested for expression of lacZ and dystrophin genes. One of these clones, mc13, was chosen for further study.
We have recently investigated two additional clones of muscle-derived cells from the highly purified muscle-derived cells (pp6). These clones of muscle-derived cells (MD1 and MD2) were not transfected by the plasmid that encodes for the minidystrophin, lacZ, and neomycin resistance genes. These cells were cloned using a limiting dilution derived technique by which the cells from the preplate #6 (pp6) population were: seeded at a low density in culture flasks; cultivated for 1 wk until colonies appeared; and single colonies were then trypsinized and the detached cells from the individual colonies picked up and seeded in a culture flask. The expansion of these new clonal cells in large quantity was performed using a similar protocol as described for mc13.
Immunohistochemistry on Muscle Cells In Vitro
Cells were plated in a 6-well culture dish and fixed with cold methanol for 1 min. After rinsing with PBS, cells were blocked with 5% horse serum at room temperature for 1 h. The primary antibodies were diluted in PBS as follows: antidesmin (1:100; Sigma-Aldrich), biotinylated anti-mouse CD34 (1:200; BD PharMingen), rabbit anti–mouse Bcl-2 (1:1,000; BD PharMingen); rabbit anti–mouse m-cadherin (1:50); mouse anti–mouse MyoD (1:100; BD PharMingen); mouse anti–rat myogenin (1:100; BD PharMingen); rabbit anti–mouse Flk-1 (1:100; Research Diagnostics); and biotinylated anti-Sca-1 (1:200; BD PharMingen). The primary antibodies were applied overnight at room temperature. Appropriate biotinylated secondary antibodies for nonbiotinylated primary antibodies were applied for 1 h at room temperature. The cells were rinsed with PBS then incubated at room temperature with 1/300 streptavidine conjugated with Cy3 fluorochrome for 1 h. The cells were visualized by fluoroscopy, and the percentage of positive cells was calculated by counting positively stained cells in 10 randomly chosen 20× fields.
RT-PCR Analysis of Muscle-derived Cells
Total RNA was isolated using TRIzol reagent (Life Technologies). Reverse transcription (RT) was carried out using SuperScript™ preamplification system for first strand cDNA synthesis (Life Technologies) according to the instructions of the manufacturer. PCR amplification of the targets was performed in 50 μl reaction mixture containing 2 μl of RT material, 5 U/100 μl Taq DNA polymerase (Life Technologies), and 1.5 mM MgCl2. CD34 primers were designed by using Oligo software. The sequences of the other primers are from references as follows: myogenin (Rohwedel et al. 1995); c-met (Cornelison and Wold 1997); and MNF (Yang et al. 1997). The following parameters were used: 94°C 45 s, 50°C (for CD34) 60 s; 60°C (for myogenin and c-met) 60 s; and 58°C (for MNF) 60 s, 72°C 90 s for 40 cycles. PCR products were checked by 1% agarose-TBE-ethidium bromide gels. The expected products sizes are: CD34, 147 bp; myogenin, 86 bp; c-met, 370 bp; and MNF, 305 bp. To exclude genomic DNA contamination, two controls were used: parallel RT without reverse transcriptase, and amplification of β-actin using a primer set that spans an intron (CLONTECH Laboratories, Inc.).
Cell Characterization by Flow-cytometry Analysis
Cultures of muscle-derived cells were harvested before analysis with a 1:2 dilution of trypsin/EDTA solution diluted in HBSS (0.5% trypsin/5.3 mM EDTA initial concentration; Life Technologies). Cells were then spun, washed, counted, and divided into two groups (experimental and control). A 1:10 mouse serum (Sigma-Aldrich) in PBS solution (0.5% BSA, 0.1% sodium azide) and Fc Block (rat anti–mouse CD16/CD32; BD PharMingen) were added to each cell pellet for 10 min on ice. Optimal amounts of rat anti–mouse mAbs were predetermined and added directly to each tube for 30 min. Each experimental tube received FITC-conjugated CD45, R-PE–conjugated CD117(c-kit) and biotin-conjugated Sca-1 (all from BD PharMingen). A control tube for each cell type received equivalent amounts of FITC-conjugated, biotin-conjugated, and R-PE-conjugated isotype standards (BD PharMingen). After several rinses, streptavidin allophycocyanin conjugate (APC; BD PharMingen) was added to each tube, including controls, and incubated on ice for 20 min. Just before analysis, 7-amino-actinomycin D (7-AAD, Via-Probe, BD PharMingen) was added to each tube for dead cell exclusion. A minimum of 10,000 live cell events were analyzed on a FACSCalibur (Becton Dickinson) flow cytometer.
Immunohistochemistry on Muscle Tissue In Vivo
The cryosections of muscle samples from a 4-wk-old normal mouse (C-57 BL/6J, The Jackson Laboratory) was fixed with cold acetone for 2 min and preincubated in 5% horse serum diluted in PBS for 1 h. For CD34, Bcl-2, and laminin/collagen type IV, the following primary antibodies were used: biotin anti-mouse CD34 (1:200 in PBS; BD PharMingen); rabbit anti–mouse Bcl-2 (1:1,000; BD PharMingen); and rabbit antilaminin (1:100 in PBS; Sigma-Aldrich) or anti–mouse collagen type IV (1:100 in PBS; Chemicon). The CD34 and Bcl-2 was also colocalized with Hoechst 33258 (bis-Benzimide, 1/100 in PBS; Sigma-Aldrich) to stain the nuclei. For dystrophin staining, sheep anti–human D-10 antibody (1:250 dilution in PBS) was used as the primary antibody. After several rinses in PBS, a biotin-conjugated anti-sheep was subsequently used (1:250 dilution in PBS). Streptavidin-FITC (bone) and streptavidin-Cy3 (muscle) at a dilution of 1:250 dilution in PBS were used; immunoreaction was observed by fluorescence microscopy (Nikon, Eclipse E-800). Finally, the colocalization of β-galactosidase, osteocalcin, and 4′,6-diaminido-2-phenylindole (DAPI; Sigma-Aldrich) stain nuclei was performed using the following protocol. The muscle sections were incubated with DAPI at a dilution of 1/100 in PBS to stain the nuclei; a biotinylated anti–β-galactosidase antibody (1/100 in PBS; Sigma-Aldrich), followed by streptavidin conjugated to fluorescein (Gal-13, 1/300 in PBS; Sigma-Aldrich) to stain the β-galactosidase expressing nuclei; and a goat anti–mouse osteocalcin (1:100 in PBS; Chemicon Co), followed by an incubation with a Cy3-conjugated anti-goat antibody (1/100 in PBS; Sigma-Aldrich) to label the osteocalcin expressing cells. The colocalization of the cells expressing β-galactosidase, osteocalcin with the nuclear labeling was visualized by fluorescence microscopy using an E-800 Nikon microscope.
Stimulation with rhBMP-2, Osteocalcin Staining, and Alkaline Phosphatase Assay
The mc13 and nonpurified muscle-derived cells (npmc) were plated in triplicate at a density of 1–2 × 104 per well in 12-well collagen-coated flasks. The cells were stimulated by addition of 200 ng/ml recombinant human BMP-2 (rhBMP-2) to the media. The media was changed on days 1, 3, and 5 after initial plating. The control group also had media changed on these days, without rhBMP-2. After 6 d of rhBMP-2 stimulation, cells were counted using a hemacytometer, and cell lysates were harvested by repeated freeze-thaw cycles. The alkaline phosphatase activity in the cell lysate was measured using a commercially available kit (Sigma-Aldrich) that utilizes color change in the reagent due to the hydrolysis of inorganic phosphate from p-nitrophenyl phosphate. The color change was analyzed on a spectrophotometer, and the data was expressed in international units: ALP activity per liter normalized to 106 cells (U/L/mil cells). Statistical difference among the different groups was analyzed using t test (*P < 0.05). The mc13 cells with or without rhBMP-2 stimulation were also analyzed on day 6 for expression of desmin. The desmin immunoreactivity was determined with a mouse antidesmin (1:100; Sigma-Aldrich), followed by a biotinylated anti-mouse (1/100; Sigma-Aldrich), and finally a streptavidin-conjugated Cy3 (1/300; Sigma-Aldrich). The immunofluorescence was visualized by an inverted miscropscope (Diaphot, Nikon); the number of desmin expressing cells were monitored and compared between the rhBMP-2 stimulated and nonstimulated cells.
In Vivo Differentiation of Muscle-derived Cells in Myogenic and Osteogenic Lineages
Myogenic.
The mc13 cells were injected (5 × 105 cells) intramuscularly in the hind limb muscle of mdx mice. The mdx mice were killed at 7 d after injection, and the injected muscles were frozen, cryostat sectioned, and assayed for dystrophin (see above) and LacZ expression (Qu et al. 1998). The mc13 cells (5 × 105 cells) were also injected intravenously in the tail vein of mdx mice. The mice were killed at 7 d after injection and various tissues, including the hind limb muscle, lung, liver, spleen, kidney, and brain, were isolated and assayed for dystrophin and LacZ using the same protocol described above.
Osteogenic.
The mc13 and npmc were transduced with an adenovirus encoding for rhBMP-2 and injected intramuscularly in the hind limb muscles of SCID mice. Genetics Institute, Cambridge, MA, generously provided the BMP-2-125 plasmid that contains the rhBMP-2 cDNA. A replication defective, E1 and E3 gene-deleted adenoviral vector was engineered to encode rhBMP-2 under the human cytomegalovirus promoter (Bosch et al. 2000; Musgrave et al. 2000).
The mc13 and the npmc were transduced with the adenoviral vector (MOI = 50). After 4 h of incubation with the adenovirus at 37°C, equal volume of serum containing media was added to the cell culture for 24 h. The transduced cells were then trypsinized, centrifuged, washed twice with HBSS, and 0.5–1.0 × 106 cells were injected into exposed triceps surae of SCID mice (The Jackson Laboratory) using a 30-gauge needle on a gas-tight syringe. At 14–15 d, animals were killed with cervical dislocation. The injected hind limbs were analyzed by radiography, and triceps surae were isolated and flash frozen in 2-methylbutane buffered in PBS, precooled in liquid nitrogen. The frozen samples were cut into 5–10-μm sections using a cryostat (Microm, HM 505 E, Fisher Scientific) and stored at −20°C until further study.
Skull Defect Assay
3 6–8-wk-old female SCID mice (The Jackson Laboratory) were used in both the control and experimental groups. The mice were anesthetized with methoxyflurane and placed prone on the operating table. Using a number 10 blade, the scalp was dissected to the skull, and the periosteum was stripped. A 5-mm full-thickness circular skull defect was created using a dental burr, with minimal penetration of the dura. A collagen sponge matrix (Helistat™, Colla-Tec, Inc) seeded with 0.5–1.0 × 106 mc13 cells, either with or without adBMP-2 transduction, was placed to fill the skull defect. The scalp was closed using a 4-0 nylon suture, and the animals were allowed to ad lib food and activity. After 14 d, the mice were killed, and the skull specimens were analyzed both grossly and microscopically. For von Kossa staining, slides were fixed in 4% formaldehyde and were then soaked in 0.1 M AgNo3 solution for 15 min. After exposure to light for at least 15 min, the slides were washed with PBS and stained with hematoxylin and eosin for histological evaluation. The identification of the mc13 within the newly formed bone in the skull defect was performed using the LacZ staining. The number of LacZ positive mc13 cells within and outside the osteoid was consequently monitored.
Fluorescent In Situ Hybridization (FISH) Using Y-probes
The FISH technique was used to follow the fate of the injected male npmc genetically engineered to express BMP-2 into the skeletal muscle of female mice (Bosch et al. 2000). The Y chromosome-specific probe (Fan et al. 1996) was biotinylated using a BioNick kit (GIBCO BRL) according to the manufacturer's instructions. The hybridized probe was detected with fluorescein-labeled avidin (ONCOR, Inc). The nuclei were counterstained with 10 ng/ml ethidium bromide in Vectashield mounting medium (Vector, Inc).
Standard Cytogenetic Method for Metaphase Preparations of mc13
The standard cytogenetic method for metaphase preparation of mc13 was performed using a previously described protocol (Barch 1991). In brief, the cells were grown to near confluency in a T75 collagen-coated flask in DME supplemented with 10% horse serum, 10% FBS, 0.5% chick embryo extract, and 1% Pen-Strep solution (all from GIBCO BRL). Cells had 15 ml of fresh medium added along with 0.2 μg/ml Colcemid solution (GIBCO BRL) and were allowed to incubate an additional 2 h at 37°C. Cells were then harvested by adding 0.1% Trypsin-EDTA to the cells until they lifted. Cells were pelleted and resuspended in 5 ml 0.75 M KCl and incubated 8 min at 37°C. 1 ml Carnoy's fixative (3:1 methanol to glacial acetic acid) was next added to solution and the cells pelleted. Fresh fixative was added and cells were again pelleted. This rinsing and pelleting was performed three times. Cells were then dropped on glass slides, heated at 60°C for 30 min and GTG banded. 32 metaphase cells were then counted for modal number using at 630×. Two images were captured on a Cytoscan cytogenetics analyzer.
Soft Agar Technique
The technique of cell growth on soft agar was performed as previously described (Tremblay et al. 1991). A 1.275% bacto-Agar (Difco) solution was first prepared in distilled water and sterilized in an autoclave. The agar medium containing 20 ml of culture medium at twice the concentration used was mixed with 20 ml of 1.275% bacto-Agar at 45°C and 10 ml of FBS. Petri dishes (60 mm) were filled with 5 ml of agar medium and the agar was allowed to solidify during 1 h at room temperature. The cells to be tested were trypsinized and resuspended in the culture medium with 15% FBS to obtain a concentration of 500,000 cells per ml. This suspension was passed twice through a 20 gauge needle to dissociate any aggregate. 1,000,000 cells (2 ml) were then mixed with 5 ml of agar medium. A sample of 1.5 ml of this agar cell suspension was finally plated over the solidified agar in the Petri dishes. When the new agar layer containing the cells solidified, 1 ml of culture medium was added to the Petri dishes. The culture medium overlaying the agar was changed once a week for 2 wk. The cell behavior was monitored every 2 d and picture was taken. Different cell populations were tested: mc13 late passages (>20), mc13 early passage (<5), HEK293 cells (adenovirus permissive cells), and a freshly isolated primary myoblast cell culture.
Results
Characterization of the pp6 Cells In Vitro
Cells isolated from primary muscle tissue contain a mixture of fibroblasts, myoblasts, adipocytes, and hematopoietic cells. However, the muscle-derived cells can be enriched using the preplate technique based on their differential adherence characteristics of primary muscle cells to collagen-coated flasks (Rando and Blau 1994; Qu et al. 1998; Qu and Huard 2000a,Qu and Huard 2000b). Cells that are slow to adhere tend to be morphologically round, express high levels of desmin, and have the ability to fuse and differentiate into multinucleated myotubes (Rando and Blau 1994; Qu et al. 1998; Qu and Huard 2000a,Qu and Huard 2000b). To further analyze the possibility of the existence of stem cells among the slow adhering cells, the fractions of slowly adhering, round cells (pp6) were isolated from mdx mice and tested for the expression of various markers using RT-PCR, immunohistochemistry, and flow cytometry (see Table ).
Table 1.
Immunohistochemical, RT-PCR, and Flow Cytometry Analysis of pp6, mc13, MD1, MD2, and Fibroblasts
| pp6 | mc13 | MD1 | MD2 | Fibroblasts | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immuno | RT-PCR | Flow | Immuno | RT-PCR | Flow | Immuno | RT-PCR | Flow | Immuno | RT-PCR | Flow | Immuno | RT-PCR | Flow | |
| Desmin | + | nd | nd | + | nd | nd | + | nd | nd | + | nd | nd | − | nd | nd |
| CD34 | + | + | nd | − | − | nd | + | + | nd | + | + | nd | − | − | nd |
| Bcl-2 | + | nd | nd | +/− | nd | nd | nd | − | nd | nd | − | nd | − | nd | nd |
| Flk-1 | + | nd | nd | + | nd | nd | + | nd | nd | + | nd | nd | − | nd | nd |
| Sca-1 | + | nd | + | + | nd | + | + | nd | + | + | nd | + | − | nd | − |
| M-cadherin | −/+ | nd | nd | +/− | nd | nd | − | nd | nd | − | nd | nd | − | nd | nd |
| Myogenin | −/+ | + | nd | +/− | + | nd | nd | + | nd | nd | + | nd | − | − | nd |
| C-met | nd | + | nd | nd | + | nd | nd | + | nd | nd | + | nd | nd | − | nd |
| MNF | nd | + | nd | nd | + | nd | nd | + | nd | nd | + | nd | nd | − | nd |
| MyoD | +/− | + | nd | nd | + | nd | nd | + | nd | nd | + | nd | nd | − | nd |
| C-kit | nd | nd | − | nd | nd | − | nd | nd | − | nd | nd | − | nd | nd | − |
| CD45 | nd | nd | − | nd | nd | − | nd | nd | − | nd | nd | − | nd | nd | − |
+, >95%; −, <2%; +/−, 40–80% of cells in the culture expressed the antigen; −/+, 5–30% of cells expressed the antigen; nd, not determined.
As shown in Table , cells in pp6 fractions were expressing myogenic markers, including desmin+, MyoD+/−, myogenin−/+. The pp6 cells were also c-met and MNF positive (RT-PCR), two genes which are expressed at an early stage of myogenesis (Miller et al. 1999). The pp6 showed a lower percentage of cells expressing m-cadherin (−/+), a satellite cell-specific marker (Irintchev et al. 1994), but a higher percentage of cells expressing Bcl-2, a marker limited to cells in the early stages of myogenesis (Dominov et al. 1998), and CD34, a marker identified in human hematopoietic progenitor cells, as well as stromal cell precursors in bone marrow (Civin et al. 1984; Andrews et al. 1986; Fina et al. 1990; Simmons and Torok-Storb 1991). The pp6 cells were also highly positive for the expression of Flk-1, a mouse homologue of human KDR gene that was recently identified as a marker of hematopoietic cells with stem cell-like characteristics (Ziegler et al. 1999). Similarly, the pp6 cells were also found positive for Sca-1, a marker present in subpopulations of both skeletal muscle and hematopoietic cells with stem cell-like characteristics (Gussoni et al. 1999). Finally, the pp6 cells were also found CD45 and c-kit negative (see Table ).
Marker Analysis of the Clonal Muscle-derived Cells Isolated from pp6
The biochemical markers expressed by mc13 cells were analyzed using RT-PCR, immunohistochemistry, and flow cytometry. The markers expressed by mc13 clone were compared with those of pp6 and fibroblasts. As summarized in Table , mc13 cells were positive for the expression of desmin, c-met, MNF, myogenin (+/−), and MyoD (RT-PCR). These results suggest that this clonal population of mc13 contained cells at different stages of differentiation. The mc13 cells were positive for m-cadherin (+/−) and Bcl-2 (+/−), but negative for CD34 expression. They were highly positive for the expression of Flk-1 and Sca-1. Similar to that observed with the pp6 cells, the mc13 cells were negative for CD45 and c-kit (see Table ).
Two additional clones (MD1/MD2) were also investigated. These clonal cells have been isolated from the pp6, but were not transfected with the plasmid to express β-galactosidase, minidystrophin, and the neomycin resistance gene. These two clones share similarities with mc13 and pp6 since they express desmin, MyoD+, myogenin+, c-met+, MNF+, and Flk-1+ (see Table ). The MD1 and MD2 cells are also CD45− (see Table ); in contrast to the mc13, they are positive for CD34 and negative for m-cadherin and Bcl-2 (see Table ). The MD1 and MD2 cells were also compared with the pp6 and mc13 for their expression of Sca-1 and c-kit. Similar to that observed with pp6 and mc13, the MD1 and MD2 cells are Sca-1+ and c-kit− (see Table ).
In Vivo Localization of CD34+ and Bcl-2+ Cells
To identify the location of CD34+ and Bcl-2+ cells in vivo, muscle tissue sections from gastrocnemius of normal mice were stained using anti-CD34 and anti–Bcl-2 antibodies. The CD34 positive cells constituted a small population of muscle-derived cells (Fig. 1 A). Colocalization of CD34 expressing cells (Fig. 1 A) with laminin, which stained the basal lamina (Fig. 1 B), revealed the location of these CD34+ cells within the basal lamina (Fig. 1C and Fig. D). The colocalization of the CD34+ cells with a nuclear staining (Hoescht 33258) indicated the presence of cells instead of small blood vessels that are also positive for CD34 (Fig. 1C and Fig. D). In fact, the expression of CD34 by vascular endothelial cells has been shown in previous studies (Fina et al. 1990). The Bcl-2 expressing cells (Fig. 1 E) were colocalized with collagen type IV (Fig. 1 F) and identified within the basal lamina (Fig. 1 G). The Bcl-2 expressing cells were also colocalized with a nuclear staining (Fig. 1 G; Hoechst 33258). The sections were stained for m-cadherin to identify the location of satellite cells (Fig. 1 H). The satellite cells were identified at similar locations as CD34+ and Bcl-2+ cells (Fig. 1 H, arrow). The expression of BCL-2 (Fig. 1 E), as well as m-cadherin (Fig. 1 H) was not found uniformly distributed in the cell membrane. Multiple attempts to colocalize CD34 or Bcl-2 with m-cadherin were unsuccessful, suggesting that m-cadherin expressing cells do not express either Bcl-2 or CD34. This is consistent with our result that pp6, which express CD34 and Bcl-2, but express minimal levels of m-cadherin−/+ (see Table ). Similarly, mc13 are expressing m-cadherin (+/−) and are negative for CD34 (see Table ).
Figure 1.
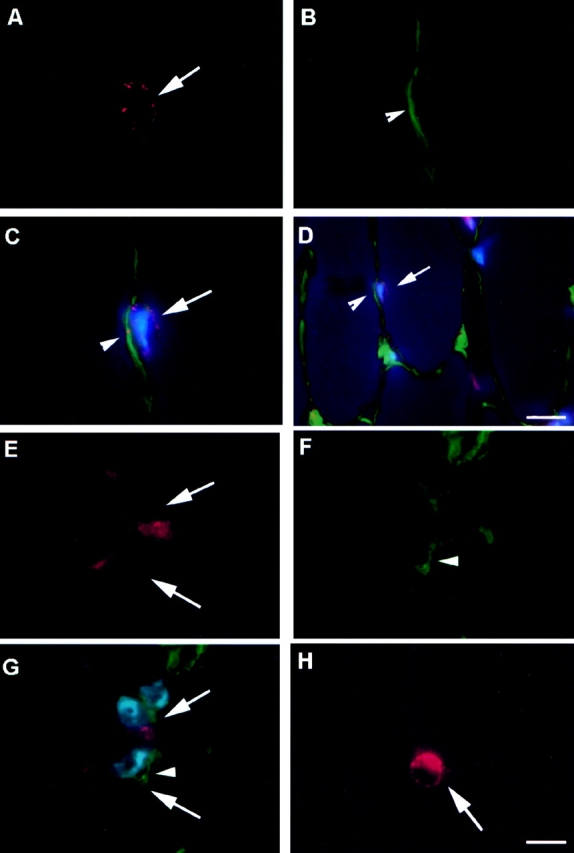
Colocalization of CD34 and Bcl-2 positive cells with laminin/collagen type IV. Muscle sections from normal mice were stained with anti-CD34 antibody and visualized with fluorescence (A, arrow). The sections were costained with antilaminin antibody to outline the basal lamina (B, arrowhead), and Hoechst to demonstrate nuclei (C and D, blue fluorescence). The CD34 positive cells were located within the basal lamina (C and D). The muscle sections were also stained with Bcl-2, collagen type IV, and Hoechst in a similar manner. The Bcl-2 positive cells (E, arrow), which colocalized with nuclei staining (G), were found located within the basal lamina (F and G). M-cadherin staining showed that satellite cells are found beneath the basal lamina at similar locations as CD34+ or Bcl-2+ cells (H). Bar: (A–C, E–H) 10 μm; (D) 25 μm.
In Vitro Differentiation of Clonal Muscle Stem Cells into Osteogenic Lineage
To further characterize the subpopulation of muscle-derived cells that may have stem cell-like capabilities, the mc13 clone isolated from the pp6 population was further subjected to in-depth analysis. We investigated whether mc13 cells have the potential to differentiate into different lineages by examining their response to rhBMP-2 stimulation. The cells were plated on a 6-well culture dish in identical density and allowed to become confluent with and without exposure to 200 ng/ml rhBMP-2. Within 3–4 d, there was a striking morphologic difference between mc13 cells exposed to rhBMP-2 and control cells. Without stimulation of rhBMP-2, mc13 cells started to fuse into multinucleated myotubes (Fig. 2 A). When exposed to 200 ng/ml rhBMP-2, however, cells remained mononucleated and did not fuse (Fig. 2 B). When cell density reached >90% confluency, the untreated culture fused to form multiple myotubes (Fig. 2 C), whereas the treated cells became circular and hypertrophic (Fig. 2 D, see arrows). Using immunohistochemistry, these hypertrophic cells were analyzed for the expression of osteocalcin. Osteocalcin is a matrix protein that is deposited on bone, specifically expressed by osteoblasts. These hypertrophic cells in the rhBMP-2–treated mc13 were found highly positive for expression of osteocalcin (Fig. 2 E, see arrows). This suggests that the rhBMP-2–stimulated mc13 cells can no longer fuse into myotubes and differentiate into osteoblasts.
Figure 2.
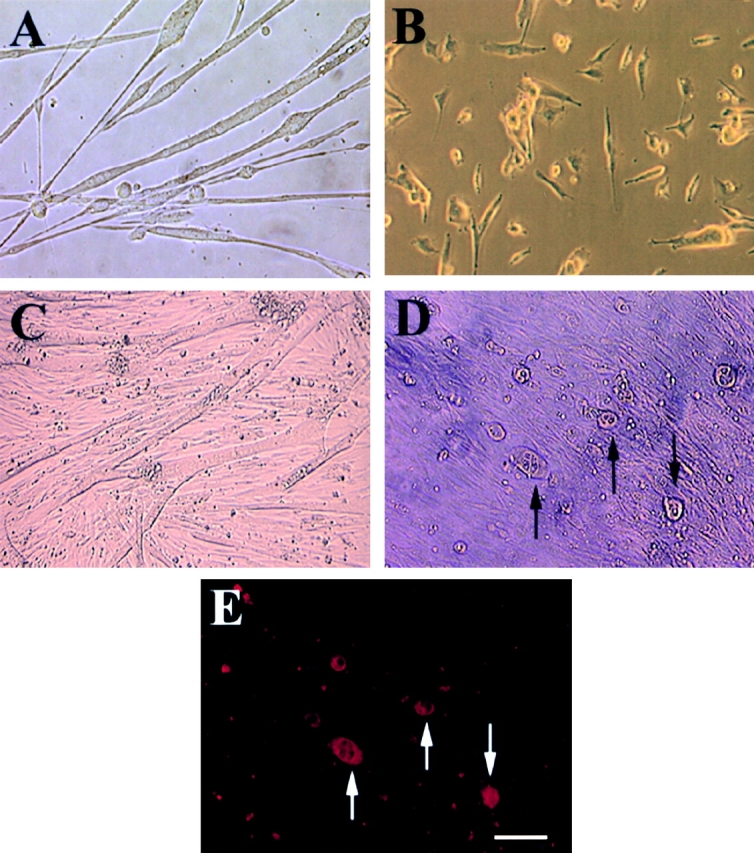
Morphologic change and expression of osteocalcin by mc13 cells with exposure to rhBMP-2. Mc13 cells were incubated in growth media without rhBMP-2 for 6 d. When cells became >50% confluent, they began to fuse and form multinucleated myotubes (A). When mc13 cells were incubated in growth media containing 200 ng/ml rhBMP-2, cells remained mononucleated and did not fuse (B). When cells reached >90% confluency without rhBMP-2, almost all the cells fused to form myotubes (C). With rhBMP-2, when cells reached >90% confluency, round, hypertrophic cells began to appear in the culture (D, arrows). These round, hypertrophic cells were highly positive for osteocalcin expression (E, arrows). Bar, 50 μm.
We monitored the level of desmin as mc13 cells undergo morphologic differentiation with rhBMP-2 stimulation as described above. Freshly isolated mc13 cells were uniformly positive for desmin (Fig. 3A and Fig. B). However, within six days of exposure to rhBMP-2, the percentage of desmin positive cells significantly decreased to 30–40% (*P < 0.05), whereas the control cells not exposed to rhBMP-2 remained 90–100% desmin positive (Fig. 3 C). This result indicates that a large number of mc13 cells lose their desmin expression upon stimulation with rhBMP-2 (Fig. 3 C). It is unclear whether this decrease in percentage of desmin positive cells was due to increased proliferation of a small number of cells that lose desmin expression or due to a large number of cells responding to rhBMP-2. However, in light of the complete absence of multinucleated myotubes in flasks containing rhBMP-2, it seems more likely the decreased percentage of desmin positive cells is due to the loss of myogenic characteristics of mc13 cells.
Figure 3.



The effect of rhBMP-2 on the expression of desmin and alkaline phosphatase by the mc13 cells. Desmin staining of freshly isolated mc13 clones (A) showed that all of these cells express desmin. The phase-contrast view (B) is shown to demonstrate that a high percentage of the cells were desmin positive. Mc13 cells were incubated in growth media containing 200 ng/ml rhBMP-2 for 6 d. Cells were then stained for desmin expression, and percent desmin positive cells calculated by visualization with immunofluorescence. As a control, mc13 cells were grown in parallel without addition of rhBMP-2. When grown without rhBMP-2, mc13 cells remained uniformly (90–100%) desmin positive (C). With exposure to rhBMP-2, there is a significant decrease (*P < 0.05) in relative number of desmin positive cells (30–40%) within 6 d (C). The npmc and mc13 cells were also analyzed for expression of alkaline phosphatase after 6 d of growth with and without exposure to 200 ng/ml rhBMP-2. With rhBMP-2 stimulation, mc13 cells show a >600-fold increase in alkaline phosphatase activity than nonstimulated cells (D). The npmc show only minimal alkaline phosphatase activity with or without rhBMP-2 (D). *, Indicates a significant difference using the t test (P < 0.05). Bar, 100 μm.
We also tested for alkaline phosphatase activity in rhBMP-2–stimulated mc13 cells. The alkaline phosphatase activity has been used as a biochemical marker for cells differentiating in osteoblastic lineage (Katagiri et al. 1995). As shown in Fig. 3 D, alkaline phosphatase expression of mc13 cells, in contrast, increased >600-fold in response to rhBMP-2. The npmc did not give rise to increased alkaline phosphatase activity in response to rhBMP-2 (Fig. 3 D). Taken together, these data demonstrate that mc13 cells lose the expression of desmin and do not fuse into myotubes, but differentiate into an osteogenic lineage in response to rhBMP-2 in vitro.
In Vivo Differentiation of mc13 Cells into Myogenic and Osteogenic Lineages
To assess the in vivo characteristics of mc13 cells, the cells were intramuscularly injected into hind limb musculature of mdx mice to determine whether these clones were capable of differentiating through the myogenic lineage in vivo. The β-galactosidase and dystrophin expression were followed to confirm the ability of mc13 cells to enhance muscle regeneration and partially restore dystrophin. Before injection, LacZ staining of the stably transfected mc13 revealed that 90–100% of cells were expressing β-galactosidase (data not shown). 5 × 105 mc13 cells were injected intramuscularly in the hind limb muscles of mdx mice; the animals were killed at 7 d after transplantation. The hind limbs of the injected animals were harvested for histology and immunohistochemical analysis. Multiple LacZ (Fig. 4 A) and dystrophin positive myofibers (Fig. 4 B) were readily identified at the injection site (Fig. 4A and Fig. B). We have monitored the number of myofibers that coexpress LacZ/dystrophin and found 379 ± 256 positive myofibers at 7 d after injection (Fig. 4 E). This demonstrates that mc13 cells, when injected into the dystrophin deficient mdx mice, can differentiate through the myogenic lineage in vivo and consequently enhance muscle regeneration and partially restore dystrophin expression in the dystrophic muscle.
Figure 4.


In vivo differentiation of mc13 cells into myogenic lineage after i.m. and i.v. injection. The mc13 cells were stably transfected with a plasmid DNA construct encoding LacZ, dystrophin, and neomycin resistance genes and injected intramuscularly into hind limbs of mdx mice. After 7 d, mice were killed and hind limb musculature was isolated for histology. Many LacZ positive myofibers (A) were found at the injected site that colocalized with dystrophin positive myofibers (B). Some LacZ (C,*) and dystrophin positive myofibers (D,*) were also found in the hind limb muscle of mdx mice after i.v. injection of mc13. The number of myofiber that coexpressed LacZ and dystrophin was counted (E) and compared between the i.m. (IM) and i.v. (IV) groups. Bar: (A and B) 100 μm; (C and D) 50 μm.
More importantly, we have tested whether mc13 cells can be systemically delivered to dystrophic muscles. The mc13 cells (5 × 105) were intravenously injected in the tail vein of mdx mice, and the animals were killed at 7 d after injection. We observed a small number of LacZ positive myofibers (Fig. 4 C) coexpressing dystrophin in the hind limb of the injected animals (Fig. 4C and Fig. D, asterisk). A lower number of LacZ and dystrophin positive myofibers were observed after the systemic delivery of the mc13 cells when compared with the i.m. injection (Fig. 4 E). This result suggests that mc13 cells can be delivered systemically to the target tissue for partial restoration of dystrophin expression. We have also investigated whether the mc13 cells were also disseminated in other tissues by testing the presence of dystrophin positive cells in nonmuscle tissues of the injected animals. We have been unable to detect any dystrophin positive cells in the lung, spleen, liver, kidney, and brain of the injected animals at 7 d after injection (data not shown).
The colocalization of dystrophin and β-galactosidase expression in the membrane of various myofibers after i.m. and i.v. injection of mc13 is unclear. In fact, some myofibers display a different level of β-galactosidase expression in the membrane versus the cytoplasm, whereas other myofibers are β-galactosidase negative in the cytoplasm and positive in the membrane. The potential fusion of β-galactosidase with dystrophin and their transport to the membrane may explain this predominance of β-galactosidase expression in the membrane.
To test the pluripotent characteristics of mc13 in vivo, the cells were transduced with an adenoviral vector encoding rhBMP-2 (adBMP-2). The mc13 cells were then injected into hind limbs of SCID mice, and bone formation was monitored radiographically and histologically at 14–15 d after injection. The LacZ and dystrophin were assessed in vivo to follow the fate of the injected cells. Previous experiments have shown that 70–90% of these cells are typically successfully transduced with our adenoviral vectors (our unpublished data). ELISA of mc13 cells transduced with adBMP-2 showed that infected cells are capable of producing a significantly higher amount of rhBMP-2 (*P < 0.05) when compared with control cells that were not transduced by the vector (Fig. 5 A). The BMP-2 detected in the nontreated cells with the ELISA technique is attributed to nonspecific background detection. Radiographic analysis of hind limbs of injected SCID mice revealed robust ectopic bone formation within 14 d of injection (Fig. 5 B, see arrow). Histologic analysis using LacZ staining of the ectopic bone showed that LacZ positive mc13 cells were uniformly located within the mineralized matrix or lacunae, a typical location where osteoblasts and osteocytes are found (Fig. 5 C). To further confirm the role of mc13 in formation of the ectopic bone, the muscle sections were also stained for the expression of dystrophin. As shown in Fig. 5 D, the ectopic bone contained cells highly positive for dystrophin, further implicating that mc13 cells are intimately participating in bone formation.
Figure 5.

In vivo differentiation of mc13 cells into osteogenic lineage after genetic engineering to express BMP-2. The amount of BMP-2 secreted by the mc13 cells that were transduced with adBMP-2 was found significantly higher (*P < 0.05) than the nontransduced mc13 cells (A). 0.5–1.0 × 106 cells genetically engineered to express BMP-2 were injected into hind limbs of SCID mice. After 14 d, mice were killed, and the hind limb muscle tissues were analyzed radiographically for evidence of bone formation. There was a robust ectopic bone formation (seen radiographically) within skeletal muscle in all mice injected with mc13 cells transduced with adBMP-2 (B, arrow). The injected muscle containing the ectopic bone was then harvested and stained for β-galactosidase activity to locate injected cells. The LacZ positive cells were uniformly found within the lacunae, a location where osteoblasts and osteocytes are normally found (C). The ectopic bone was also stained for presence of dystrophin. As indicated by green fluorescence, the ectopic bone contained abundant cells expressing dystrophin, confirming that mc13 cells were active participants in formation of bone (D). To determine whether the genetically engineered mc13 expressing BMP-2 can express bone protein, we colocalized β-galactosidase expressing nuclei, osteocalcin expression, and nuclei staining (DAPI) by immunohistochemistry (E–H). We identified nuclei expressing β-galactosidase (see Fig. 6 F, arrow, FITC/green) that expressed osteocalcin (see Fig. 6 G, arrow, cy3/red) and colocalized with nuclei staining (see Fig. 6 E, arrow, DAPI/blue). The triple colocalization of DAPI/osteocalcin and β-galactosidase (Fig. 6 H, arrows) suggests that the genetically engineered mc13 can express bone protein (osteocalcin). We have also observed β-galactosidase expressing nuclei (Fig. 6 F, arrowhead) that were not colocalized with osteocalcin expressing cells (Fig. 6 G, arrowhead), suggesting that some of the engineered mc13 were not expressing osteocalcin (Fig. 6, E–H, arrowheads). Bar: (C and D) 50 μm; (E–H) 25 μm.
To determine whether the genetically engineered mc13 expressing BMP-2 can express bone protein, we have colocalized β-galactosidase expressing nuclei, osteocalcin expression, and nuclei staining (DAPI) by immunohistochemistry (Fig. 5, E–H). We have identified nuclei expressing β-galactosidase (Fig. 5 F, FITC/green, see arrow), which express osteocalcin (Fig. 5 G, cy3/red, see arrow), and colocalized with nuclei staining (Fig. 5 E, DAPI/blue, see arrow). The triple colocalization of DAPI/osteocalcin and β-galactosidase (Fig. 5 H, see arrow) suggest that the genetically engineered mc13 can differentiate in bone lineage and consequently express bone protein (osteocalcin). We were also capable of detecting β-galactosidase expressing cells (Fig. 5 F, arrowhead) that were not colocalized with osteocalcin positive cells (Fig. 5 G, arrowhead), suggesting that some of the injected mc13 were not expressing osteocalcin (Fig. 5, E–H, arrowheads).
As a control, similar experiments were carried out with male npmc, which are highly fibroblastic in nature. We have observed that npmc genetically engineered to express BMP-2 also supported robust ectopic bone formation in skeletal muscle (Fig. 6 A). The FISH technique was used to identify the Y chromosome positive cells and revealed that the injected npmc cells were located, in contrast to mc13, outside of the osteoid (Fig. 6 B, arrow); a complete absence of Y chromosome positive cells was found within the newly formed osteoid (Fig. 6 C, arrowheads). This suggests, as previously described by our group (Bosch et al. 2000; Musgrave et al. 2000), that the npmc are capable of delivering rhBMP-2 to form ectopic bone, but are unable to differentiate into osteoblasts. In this case, the cells participating in mineralization of the ectopic bone are most likely from the host tissue. This confirms that the capacity to differentiate into osteoblasts, both in vivo and in vitro, is inherent to highly purified muscle-derived cells such as mc13.
Figure 6.

The inability of BMP-2 expressing npmc to differentiate into osteogenic lineage. The npmc were isolated from a male mdx mouse and injected in the hind limb muscle of a female SCID mice. We have observed that npmc genetically engineered to express BMP-2 also leads to ectopic bone formation when injected in skeletal muscle. A, The FISH technique was used to identify the Y chromosome positive cells (donor cells) and revealed that the injected cells were located outside of the osteoid (B, arrow); a complete absence of Y chromosome positive cells was found within the newly formed osteoid (C, arrows). These cells were therefore incapable of differentiating into osteogenic lineage in vivo. Bar: (A) 100 μm; (B and C) 50 μm.
Enhancement of Bone Healing by Genetically Engineered mc13 Cells
To test the clinical applicability of our findings and to further support the functional properties of mc13, we investigated whether the mc13 cells engineered to express BMP-2 can be used to enhance healing of a bone defect. We created 5-mm skull defects in skeletally mature (6–8-wk-old) female SCID mice using a dental burr. A 5-mm size skull defect has been shown to be a nonhealing defect in previous mouse models (Krebsbach et al. 1998). This skull defect was filled with a collagen sponge matrix that was seeded with mc13 cells with or without adBMP-2 transduction. The SCID mice were killed at 14 d, and the healing of the skull defect was analyzed both grossly and microscopically. As shown in Fig. 7 A, the control group treated with mc13 cells that were not transduced to express rhBMP-2 shows no evidence of healing of the defect (see arrows showing the edge of the bone defect). The experimental group treated with mc13 transduced to express rhBMP-2 shows a full closure of the skull defect at two weeks (Fig. 7 B). The von Kossa staining, which highlights mineralized bone, shows robust new bone formation in the group treated with mc13 transduced to express rhBMP-2 (Fig. 7 D), but little evidence of new bone formation by the control group (Fig. 7 C, see arrow to indicate the defect site). The area of new bone in the experimental group injected with mc13 engineered to express BMP-2 (Fig. 7 D, square E), was also analyzed by LacZ staining to determine whether the transplanted cells were participating in the bone formation. As shown in Fig. 7 E, LacZ positive nuclei were identified within the newly formed bone (arrows), indicating active participation of transplanted cells in bone formation under the influence of rhBMP-2. Greater than 95% of the LacZ positive mc13 cells were found integrated within the newly formed bone. LacZ negative cells were also identified within the newly formed skull indicating participation of host-derived cells. This experiment shows that mc13 can be induced to differentiate into osteoblasts and consequently, be used to heal a nonhealing bone defect under the influence of rhBMP-2.
Figure 7.
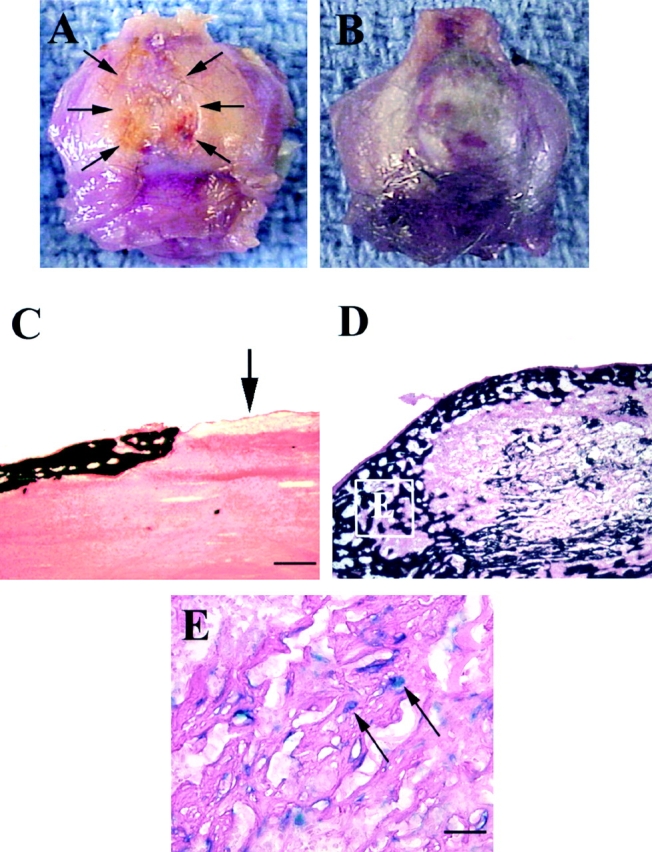
Enhancement of bone healing by rhBMP-2 producing mc13 cells. A 5-mm skull defect was created in a SCID mice using a dental burr, and the defect was filled with a collagen sponge seeded with mc13, with or without adBMP-2 transduction. The mice were killed at 14 d and analyzed grossly and microscopically for healing of the defect. A gross specimen from the control group was treated with collagen sponge seeded with mc13 without adBMP-2. At 14 d, there was no apparent healing of the skull defect (A, arrows). A representative specimen from mice treated with collagen sponge seeded with mc13 transduced with adBMP-2 (B). There was almost a complete closure of the defect within 14 d (B). A von Kossa staining of the histological specimen from the control group showed no evidence of new bone formation (C, arrow shows defect site). von Kossa stain of the adBMP-2–treated group showed robust bone formation at 14 d (D). The mc13 cells transduced with adBMP-2 were followed using the LacZ staining (E). The vast majority of the β-galactosidase expressing nuclei (>95%) was found within the newly formed bone (E, arrows). Bar: (C and D) 100 μm; (E) 50 μm.
Standard Cytogenetic and Tumorigenicity Assay on mc13
We have also investigated the karyotype of these cells and tested whether they can be grown in soft agar as an indicator of their tumorigenicity (not illustrated). The mc13 cells analyzed had an unknown passage history, but likely over 20 passages. The normal diploid number of chromosomes (2n) seen in mice (Mus musulus) is 40 (Francke and Nesbitt 1971). The majority of cells counted in our clone was 40 with few cells missing 1–4 chromosomes (known as hypodiploidy). Also observed were tetraploid (4n) cells (metaphase with 80 chromosomes). The hypodiploid and tetraploid cells are probably due to normal tissue culture artifact (Barch 1991). In fact, tetraploidy is commonly seen in cultured cells and the hypodiploidy is often the result of mechanical overspreading of the metaphase cells when making the slides, resulting in a loss of a chromosome(s) (Barch 1991).
The soft agar test (Tremblay et al. 1991) was additionally performed to characterize whether the mc13 can grow on soft agar as an indicator of malignant processes within the cell population. The mc13 used for this experiment were totally incapable of growing on soft agar. In fact, we compared these data with a very early passage of the mc13 and, similarly, they were not capable of growing in soft agar. As a positive control, we used the HEK293 cells (adenovirus permissive cells), which grew vigorously and presented large cellular clumps. Our negative control was a freshly isolated primary myoblast cell isolation that displayed no growth and consequently no evidence of tumorigenicity. To further substantiate that these cells are not tumorigenic, we have never histologically observed any adverse effect(s), such as tumor development, after their injection in SCID mice skeletal muscle or other tissues, including the bone defect model.
Discussion
We have observed that the preplate technique enriches for a population of muscle-derived cells that express both early myogenic markers, including desmin, c-met, MNF, and Bcl-2, and stem cell markers (Sca-1, Flk-1, CD34). In vivo staining showed that these cells were localized within the basal lamina, a location normally occupied by satellite cells. Clonal analysis from these highly purified muscle-derived cells (pp6) showed that, as observed with the pp6 cells (Bosch et al. 2000), the mc13 clonal cells are able to differentiate into myogenic, as well as osteogenic lineage, in vitro and in vivo. The marker analysis of the clone showed expression of both stem cell and satellite cell markers, including m-cadherin, Flk-1, and Sca-1. The i.m. as well as i.v. injection of mc13 can enhance muscle regeneration and partially restore dystrophin in mdx mice. Finally, the genetic engineering of the mc13 to express osteogenic protein (BMP-2) was capable of enhancing the healing of a skull defect in SCID mice.
An immortalized clonal cell line from mouse myoblast, C2C12, has been shown to decrease expression of myogenin and MyoD mRNA, and increase expression of alkaline phosphatase, osteocalcin, and parathyroid-dependent 3′, 5′-cAMP in response to rhBMP-2 in vitro (Katagiri et al. 1995). However, no in vivo data with C2C12 cells are available. Our marker analysis with RT-PCR, as well as immunohistochemistry, shows that C2C12 cells are also Flk-1−, m-cadherin−, and c-met− (not shown). This suggests that mc13 cells are different from C2C12 cells. In addition, morphologically, C2C12 cells are polygonal and fibroblast-like, whereas mc13 cells are small and round when cultured in a monolayer system. Whether mc13 cells and C2C12 cells represent a completely distinct subgroup of cells or a different maturation stage of the same population is still unknown.
A recent in vivo study has demonstrated that in human hematopoietic cells, CD34+KDR+ population had the highest pluripotent characteristics (Ziegler et al. 1999). The mouse homologue of KDR is Flk-1. The mc13 population is Flk-1+, similar to the human hematopoietic pluripotent cells. However, mc13 cells express myogenic markers such as desmin, MyoD, myogenin, c-met, and MNF, and they are CD34−, indicating that these cells are distinct from the hematopoietic stem cells. The expression of myogenic markers indicative of both early (c-met, desmin, MNF, and Bcl-2) and late stage of myogenesis (myogenin, MyoD) is probably related to the myogenic differentiation of some muscle-derived cells within the mc13 population with the cell culturing.
Interestingly, the pp6 cells are CD34+, whereas mc13 cells are CD34−. However, we cannot rule out that mc13 initially expressed CD34, but the extensive selection with G418 may have differentiated the mc13 clone, resulting consequently in their loss of CD34 and the gain of m-cadherin expression. In fact, we have recently investigated two new clones (MD1 and MD2) from the pp6 to determine whether the marker expression by the mc13 were unique or also expressed by other clonal cell populations originated from the same pp6 population. These clonal cells have been isolated from the pp6, but were not genetically engineered with the plasmid to express β-galactosidase and dystrophin. These two clones were similar to mc13 for most of the myogenic and stem cell marker expression in vitro, but in contrast to mc13, they are positive for CD34 and negative for m-cadherin, as well as Bcl-2. These results suggest that mc13 might be isolated from a small population of CD34− cells within the pp6 population or the mc13 were originally CD34+, but during the selection, they differentiated into CD34− cells.
Although the expression of CD34 can account for a difference between the mc13 and pp6/MD1/MD2, recently it has been published (Goodell 1999; Sato et al. 1999) that the expression of CD34 is reversible on hematopoietic stem cells. In fact, these papers suggest that CD34 is probably a marker of activated stem cells, but it is not necessarily expressed in all stem cells. Although the reversible expression of CD34 remains to be determined in muscle-derived stem cells, the use of CD34 as a marker of muscle-derived stem cells should at least be used with caution.
The relationship between mc13 and other populations of muscle-derived stem cells, such as the side population (SP) of muscle-derived cells described by Gussoni et al. 1999, was also investigated. The SP population of cells from Gussoni et al. 1999 derived from muscle was Sca-1+, cKit−, CD43−, CD45−, Lin−, CD34−. The mc13 are CD34−, Bcl-2+/−, Flk-1+, Sca-1+, m-cadherin+/−, myogenin+/−, c-met+, MNF+, c-kit−, and CD45−. Although mc13 is highly similar to the muscle-derived SP cells, more markers (especially myogenic markers for the SP cells) should be characterized to determine their relationship. Similarly, a recent paper by Jackson et al. 1999 also described a new population of muscle-derived stem cells that express stem cell antigens (Sca-1 and cKit) and lack CD45. However, based on their methodology, the cells used for the experiments were derived from the pp2. Since the mc13 was isolated from the pp6 and the mc13 are c-Kit−, we believe that mc13 are different than the muscle-derived cells described by Jackson et al. 1999. In fact, the cells isolated at pp2 in our experiments are highly different than the cells isolated at pp6 in term of marker expression in vitro as well as their functional properties in vivo (Qu et al. 1998). Clearly, more studies are required to accurately assess the origin and, more importantly, the functional properties of these various population of muscle-derived stem cells.
The relationship of the clonal mc13 cells with satellite cells is still unclear, but various features of these cells suggest a close relationship with satellite cells. These features include: the expression of myogenic markers including m-cadherin, which is known as a specific marker for satellite cells by the mc13 cells (Irintchev et al. 1994); the myogenic ability of the mc13 to differentiate into myotubes in vitro and regenerate skeletal muscle in vivo; and the location of Bcl-2 expressing cells within the basal lamina, where the satellite cells are normally located, also suggest a close relationship of mc13 with the satellite cells, especially knowing that mc13 are Bcl-2+/−. Additional studies are required to determine the origin of mc13 and consequently elucidate their relationship with satellite cells.
Although mc13 can efficiently regenerate skeletal muscle after i.m. injection, their ability to migrate to skeletal muscle after i.v. injection remains an important feature of these cells. Whereas a lower number of LacZ and dystrophin positive myofibers were found after i.v. injection of mc13 when compared with the i.m. injection, the mechanism by which these cells can be disseminated by the bloodstream and return to the skeletal muscle is important and requires further investigation. The development of approaches to improve the systemic delivery of these cells will be investigated. The absence of dystrophin positive cells at 7 d after injection within the lung, liver, spleen, kidney, and brain after i.v. injection of mc13 is surprising. These results suggest that the injected cells get specifically disseminated in the skeletal muscle, although we cannot rule out that the injected cells were present on these nonmuscle tissues at an early time point after injection, but died in the nonmuscle tissue.
The number of positive myofibers found in the injected animals after i.m. and i.v. injection of mc13 was also compared with that reported by Beauchamps et al. 1999 and Gussoni et al. 1999. In fact, the number of dystrophin positive myofibers found in the injected muscle after i.m. injection of 500,000 mc13 cells led to 379 ± 256 dystrophin/LacZ positive myofibers; only 16 ± 7 dystrophin/LacZ positive myofibers were found after i.v. injection of the same number of mc13 cells. The number of dystrophin positive myofibers in our experiment after the i.v. injection was slightly lower to the i.v. injection of muscle-derived SP cells as described by Gussoni et al. 1999. Various factors may have accounted for the slight difference of cell transfer between the two cell populations including: different populations of muscle-derived cells or a different stage of maturation of a similar population of cells; the times after injection; and the irradiation treatment that may improve the success of myoblast transfer (Morgan et al. 1990, Morgan et al. 1993).
We have also compared our results with a recent publication from Beauchamps et al. 1999. They reported that only 1% of the injected cells persisted at 24 h, but an extensive proliferation of the surviving population equivalent to 23.5% of the initial population of cells were observed at four days after injection. The extensive proliferation of this small fraction of cells led to the formation of a large number of dystrophin positive myofibers at three weeks after injection. Although the number of dystrophin positive myofibers is not available, these results are also similar to our current data, as well as to data previously reported by our group (Qu et al. 1998). In fact, by counting the number of m-cadherin and CD34 positive cells, we have approximated that the purified cells (pp6) represent about one percent of the satellite cell population. The i.m. injection of these highly purified cells also leads to a significant enhancement of myoblast transfer when compared with regular myoblasts (Qu et al. 1998).
The presence of stem cells in skeletal muscle makes this tissue an attractive source of cells for cell-transplantation therapy to enhance the healing of various tissues of the musculoskeletal system. We have in fact investigated in this study whether the genetic engineering of mc13 cells to express rhBMP-2 can be used to enhance closure of a nonhealing skull defect. Our results suggest that the mc13 was capable of inducing and, more importantly, participating in ectopic and orthopic bone formation when genetically engineered to express BMP-2. It is likely that the transplanted muscle cells are acting as a delivery vehicle for rhBMP-2, as well as a source of cells that differentiate into osteoblasts. The observation that 95% of the transplanted mc13 genetically engineered to express BMP-2 are located within the newly formed bone suggests that the vast majority of these cells are differentiating into osteogenic lineage and participating in bone formation. Based on these results, the stimulation with BMP-2 is required to differentiate these mc13 cells in osteogenic lineage to consequently improve bone healing. Our data suggest that the mc13 will differentiate into skeletal muscle, but the addition of an extra stimuli, such as the BMP-2 used in this experiment, will push them to differentiate in osteogenic lineage. These results suggest that muscle tissue is a valuable resource for osteoprogenitor cells to be used in clinical setting to improve bone healing.
Finally, we have investigated the karyotype of these cells and tested whether they can be grown in soft agar as an indicator of their tumorigenicity. Our results suggest that the number of chromosomes within the mc13 cells are normal and more importantly, they are totally incapable of growing on soft agar. This result, in addition to the lack of any adverse effect(s), at least histologically, such as tumor development, after their injection (mc13) in either skeletal muscle or other tissues in immunodeficient SCID mice, suggest that the mc13 are not tumorigenic.
In summary, the isolation and purification of muscle-derived stem cells offers an opportunity to elucidate cellular and molecular mechanisms of organogenesis. Taken together, our results suggest the isolation of a clonal population of muscle-derived stem cells capable of improving both muscle regeneration and bone healing. These new results will shed more light on the functional properties of the muscle-derived stem cells and further support that muscle tissue may become a valuable resource for the isolation of osteoprogenitor cells capable of improving bone healing. Further characterization of these muscle-derived stem cells will open an array of possibilities for advancement of tissue engineering and tissue transplantation techniques.
Acknowledgments
The authors wish to thank Marcelle Pellerin and Ryan Pruchnic for their technical assistance, and Dr. Lilian Hsu and Dana Och for their assistance with the manuscript. The authors also wish to thank the Genetics Institute (Cambridge, MA) for the human recombinant BMP-2 and the antibody against BMP-2, Stephen Hardy for graciously providing us with the Cre-lox adenoviral system, and Dr. Miranda Grounds for providing us with the Y-probes.
This work was supported in part by grants to Dr. Johnny Huard from the National Institutes of Health (1 P60 AR44811-01, 1PO1 AR45925-01), the Pittsburgh Tissue Engineering Initiative (PTEI), and the William F. and Jean W. Donaldson Chair at Children's Hospital of Pittsburgh.
Footnotes
Joon Yung Lee and Zhuqing Qu-Petersen contributed equally to the work.
Abbreviations used in this paper: adBMP-2, adenovirus bone morphogenetic protein-2 construct; ALP, alkaline phosphatase; DAPI, 4′,6-diaminido-2-phenylindole; DMD, Duchenne muscular dystrophy; MNF, myocyte nuclear factor; npmc, nonpurified muscle-derived cells; pp, preplate; rhBMP-2, recombinant human BMP-2; RT, reverse transcription.
References
- Andrews R.G., Singer J.W., Bernstein I.D. Monoclonal antibody 12-8 recognizes a 115-kd molecule present on both unipotent and multipotent hematopoietic colony-forming cells and their precursors. Blood. 1986;67:842–845. [PubMed] [Google Scholar]
- Arahata K., Ishiura S., Ishiguro T., Tsukahara T., Suhara Y., Egicjo C., Ishihara T., Nonak I., Ozawa E., Sugita H. Immunostaining of skeletal and cardiac muscle surface membrane with antibody against Duchenne muscular dystrophy peptide. Nature. 1989;333:861–863. doi: 10.1038/333861a0. [DOI] [PubMed] [Google Scholar]
- Barch, M.J. 1991. Chromosome analysis. In The ACT Cytogenetics Laboratory Manual. 2nd edition. Raven Press, Ltd., NY, NY. 349.
- Baroffio A., Hamann M., Bernheim L., Bochaton-Piallat M.L., Gabbiani G., Bader C.R. Identification of self-renewing myoblasts in the progeny of single human muscle satellite cells. Differentiation. 1996;60:47–57. doi: 10.1046/j.1432-0436.1996.6010047.x. [DOI] [PubMed] [Google Scholar]
- Beauchamps J.R., Morgan J.E., Pagel C.N., Partridge T.A. Quantitative studies of the efficacy of myoblast transplantation. Muscle Nerve. 1994;18:S261. [Google Scholar]
- Beauchamps J.R., Morgan J.E., Pagel C.N., Partridge T.A. Dynamics of myoblast transplantation reveal a discrete minority of precursors with stem cell-like properties as the myogenic source. J. Cell Biol. 1999;144:1113–1122. doi: 10.1083/jcb.144.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichoff, R. 1994. The satellite cells and muscle regeneration. In Myology, Basic and Clinical. 2nd edition. McGraw-Hill, Inc., NY. A.G. Engel and C. Franzini-Armstrong, editors. 97–118.
- Bittner R.E., Schofer C., Weipoltshammer K., Ivanova S., Streubel B., Hauser E., Freilinger M., Hoger H., Elbe-Burger A., Wachtler F. Recruitment of bone marrow derived cells by skeletal and cardiac muscle in adult dystrophic mdx mice. Anat. Embryol. 1999;199:391–396. doi: 10.1007/s004290050237. [DOI] [PubMed] [Google Scholar]
- Bonilla E.C.E., Samitt A.F., Miranda A.P., Hays G., Salviati S., Dimauro S., Kunkel L.M., Hoffman E.P., Rowland L.P. Duchenne muscular dystrophydeficiency of dystrophin at the muscle cell surface. Cell. 1988;54:447–452. doi: 10.1016/0092-8674(88)90065-7. [DOI] [PubMed] [Google Scholar]
- Bosch P., Musgrave D.S., Lee J.Y., Cummins J., Shuler F., Ghivizzani S.C., Evans C.H., Robbins P.D., Huard J. Osteoprogenitor cells within skeletal muscle. J. Orthop. Res. 2000;In press doi: 10.1002/jor.1100180613. [DOI] [PubMed] [Google Scholar]
- Caplan A.I. Mesenchymal stem cells. J. Orthop. Res. 1991;9:641–650. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- Civin C.I., Strauss L.C., Brovall C., Fackler M.J., Schwartz J.F., Shaper J.H. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG- 1a cells. J. Immunol. 1984;133:157–165. [PubMed] [Google Scholar]
- Cornelison D.D., Wold B.J. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol. 1997;191:270–283. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- DeAngelis L.D., Berghella L., Coletta M., DeAngelis M.G.C., Lattanzi L., Ponzetto C., Cossu G. Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to post-natal muscle growth and regeneration. J. Cell Biol. 1999;147:869–878. doi: 10.1083/jcb.147.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominov J.A., Dunn J.J., Miller J.B. Bcl-2 expression identifies an early stage of myogenesis and promotes clonal expansion of muscle cells. J. Cell Biol. 1998;142:537–544. doi: 10.1083/jcb.142.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin–glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Fan Y., Maley M., Beilharz M., Grounds M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve. 1996;19:853–860. doi: 10.1002/(SICI)1097-4598(199607)19:7<853::AID-MUS7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Ferrari G., Cusella-De Angelis G., Coletta M., Paolucci E., Stornaiuolo A., Cossu G., Mavilio F. Muscle regeneration by bone marrow derived myogenic progenitors. Science. 1998;279:1528–1530. doi: 10.1126/science.279.5356.1528. [DOI] [PubMed] [Google Scholar]
- Fina L., Molgaard H.V., Robertson D., Bradley N.J., Monaghan P., Delia D., Sutherland D.R., Baker M.A., Greaves M.F. Expression of the CD34 gene in vascular endothelial cells. Blood. 1990;75:2417–2426. [PubMed] [Google Scholar]
- Francke U., Nesbitt M. Identification of the mouse chromosomes by quinacrine mustard staining. Cytogenetics. 1971;10:356–366. doi: 10.1159/000130154. [DOI] [PubMed] [Google Scholar]
- Goodell MA. CD34+ or CD34−does it really matter? Blood. 1999;94:2545–2547. [PubMed] [Google Scholar]
- Guerette B., Asselin I., Skuk D., Entman M., Tremblay J.P. Control of inflammatory damage by anti-LFA-1increase success of myoblast transplantation. Cell Transplant. 1997;6:101–107. doi: 10.1177/096368979700600203. [DOI] [PubMed] [Google Scholar]
- Gussoni E., Pavlath P.K., Lancot A.M., Sharma K., Miller R.G., Steinman L., Blau R.M. Normal dystrophin transcripts detected in DMD patients after myoblast transplantation. Nature. 1992;356:435–438. doi: 10.1038/356435a0. [DOI] [PubMed] [Google Scholar]
- Gussoni E., Blau H.M., Kunkel L.M. The fate of individual myoblasts after transplantation into muscles of DMD patients. Nat. Med. 1997;3:970–977. doi: 10.1038/nm0997-970. [DOI] [PubMed] [Google Scholar]
- Gussoni E., Soneoka Y., Strickland C.D., Buzney E.A., Khan M.K., Flint A.F., Kunkel L.M., Mulligan R.C. Dystrophin expression in the mdx mouse restored by stem cell transplantation. Nature. 1999;401:390–394. doi: 10.1038/43919. [DOI] [PubMed] [Google Scholar]
- Hoffman E.P., Brown J., Kunkel L.M. Dystrophinthe protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Huard J., Bouchard J.P., Roy R., Malouin F., Dansereau G., Labrecque C., Albert N., Richards C.L., Lemieux B., Tremblay J.P. Human myoblast transplantationpreliminary results of 4 cases Muscle Nerve. 15 1992. 550 560a [DOI] [PubMed] [Google Scholar]
- Huard J., Roy R., Bouchard J.P., Malouin F., Richards C.L., Tremblay J.P. Human myoblast transplantation between immunohistocompatible donors and recipients produces immune reactions Transplant. Proc. 24 1992. 3049 3051b [PubMed] [Google Scholar]
- Huard J., Guerette B., Verreault S., Tremblay G., Roy R., Lille S., Tremblay J.P. Human myoblast transplantation in immunodeficient and immunosuppressed miceevidence of rejection Muscle Nerve. 17 1994. 224 234a [DOI] [PubMed] [Google Scholar]
- Huard J., Verreault A., Roy R., Tremblay M., Tremblay J.P. High efficiency of muscle regeneration following human myoblast clone transplantation in SCID mice J. Clin. Invest. 93 1994. 586 599b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard J., Acsadi G., Massie B., Karpati G. Gene transfer to skeletal muscles by isogenic myoblasts Hum. Gene Ther 5 1994. 949 958c [DOI] [PubMed] [Google Scholar]
- Irintchev A., Zeschnigk M., Starzinski-Powitz A., Wernig A. Expression pattern of m-cadherin in normal, denervated, and regenerating mouse muscle. Dev. Dyn. 1994;199:326–337. doi: 10.1002/aja.1001990407. [DOI] [PubMed] [Google Scholar]
- Jackson K.A., Mi T., Goodell M.A. Hematopoietic potential of stem cells isolated from murine skeletal muscle. Proc. Natl. Acad. Sci. 1999;USA. 96:14482–14486. doi: 10.1073/pnas.96.25.14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpati G., Pouliot Y., Zubrzycka-Gaarn E., Carpenter S., Ray P.N., Worton R.G., Holland P. Dystrophin is expressed in mdx skeletal muscle fibers after normal myoblast implantation. Am. J. Pathol. 1989;135:27–32. [PMC free article] [PubMed] [Google Scholar]
- Karpati G., Holland P., Worton R.G. Myoblast transfer in DMDproblems and interpretation of efficiency. Muscle Nerve. 1992;15:1209–1210. doi: 10.1002/mus.880151016. [DOI] [PubMed] [Google Scholar]
- Katagiri T., Yamaguchi A., Komaki M., Abe E., Takahashi N., Ikeda T., Rosen V., Wozney J.M., Fujisawa-Sehara A., Suda T. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 1995;127:1755–1766. doi: 10.1083/jcb.127.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita I., Vilquin J.T., Guerette B., Asselin I., Roy R., Tremblay J.P. Very efficient myoblast allotransplantation in mice under FK506 immunosuppression. Muscle Nerve. 1994;17:1407–1415. doi: 10.1002/mus.880171210. [DOI] [PubMed] [Google Scholar]
- Krebsbach P.H., Mankani M.H., Satomura K., Kuznetsov S.A., Robey P.G. Repair of craniotomy defects using bone marrow stromal cells. Transplantation. 1998;66:1272–1278. doi: 10.1097/00007890-199811270-00002. [DOI] [PubMed] [Google Scholar]
- Lucas P.A., Calcutt A.F., Southerland S.S., Warejcka D., Young H.E. A population of cells resident within embryonic and newborn rat skeletal muscle is capable of differentiating into multiple mesordermal phenotypes. Wound Rep. Reg. 1995;3:457–468. doi: 10.1046/j.1524-475X.1995.30409.x. [DOI] [PubMed] [Google Scholar]
- Mendell J.R., Kissel J.T., Amato A.A., King W., Signore L., Prior T.W., Sahenk Z., Benson S., McAndrew P.E., Rice R. Myoblast transfer in the treatment of Duchenne's muscular dystrophy. N. Engl. J. Med. 1995;333:832–838. doi: 10.1056/NEJM199509283331303. [DOI] [PubMed] [Google Scholar]
- Miller J.B., Schaefer L., Dominov J.A. Seeking muscle stem cells. Curr. Topics Dev. Biol. 1999;43:191–219. doi: 10.1016/s0070-2153(08)60382-8. [DOI] [PubMed] [Google Scholar]
- Morgan J.E., Watt D.J., Slopper J.C., Partridge T.A. Partial correction of an inherited defect of skeletal muscle by graft of normal muscle precursor cells. J. Neurol. Sci. 1988;86:137–147. doi: 10.1016/0022-510x(88)90093-7. [DOI] [PubMed] [Google Scholar]
- Morgan J.E., Hoffman E.P., Partridge T.A. Normal myogenic cells from newborn mice restore normal histology to degenerating muscle of the mdx mouse. J. Cell Biol. 1990;111:2437–2449. doi: 10.1083/jcb.111.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan J.E., Pagel C.N., Sherrat T., Partridge T.A. Long-term persistence and migration of myogenic cells injected into pre-irradiated muscles of mdx mice. J. Neurol. Sci. 1993;115:191–200. doi: 10.1016/0022-510x(93)90224-m. [DOI] [PubMed] [Google Scholar]
- Musgrave D.S., Bosch P., Lee J.Y., Pelinkovich D., Ghivizzani S.C., Whalen J., Niyibizi C., Huard J. Ex vivo gene therapy to produce bone using different cell types. Clin. Orthop. Related Res. 2000;377:1–16. doi: 10.1097/00003086-200009000-00040. [DOI] [PubMed] [Google Scholar]
- Partridge T.A. Myoblast transfera possible therapy for inherited myopathies. Muscle Nerve. 1991;14:197–212. doi: 10.1002/mus.880140302. [DOI] [PubMed] [Google Scholar]
- Partridge T.A., Morgan J.E., Coulton G.R., Hoffman E.P., Kunkel L.M. Conversion of mdx myofibers from dystrophin negative to positive by injection of normal myoblast. Nature. 1989;337:176–179. doi: 10.1038/337176a0. [DOI] [PubMed] [Google Scholar]
- Pittenger M.F., MacKay A.M., Beck S.C., Jaiswal R.K., Douglas R., Mosca J.D., Moorman M.A., Simonetti D.W., Craig S., Marshak D.R. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Qu Z., Huard J. Matching host muscle and donor myoblasts for myosin heavy chain improves myoblast transfer therapy Gene Therapy. 7 2000. 428 437a [DOI] [PubMed] [Google Scholar]
- Qu Z., Huard J. The influence of muscle fiber type in myoblast-mediated gene transfer to skeletal muscle Cell Transplan t.In press2000. b [DOI] [PubMed] [Google Scholar]
- Qu Z., Balkir L., van Deutekom J.C., Robbins P.D., Pruchnic R., Huard J. Development of approaches to improve cell survival in myoblast transfer therapy. J. Cell Biol. 1998;142:1257–1267. doi: 10.1083/jcb.142.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando T.A., Blau H.M. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwedel J., Horak V., Hebrok M., Fuchtbauer E.M., Wobus A.M. M-twist expression inhibits mouse embryonic stem cell-derived myogenic differentiation in vitro. Exp. Cell. Res. 1995;220:92–100. doi: 10.1006/excr.1995.1295. [DOI] [PubMed] [Google Scholar]
- Sato T., Laver J.H., Ogawa M. Reversible expression of CD34 by murine hematopoietic stem cells. Blood. 1999;94:2548–2554. [PubMed] [Google Scholar]
- Seale P., Rudnicki M.A. Reviewa new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev. Biol. 2000;218:115–124. doi: 10.1006/dbio.1999.9565. [DOI] [PubMed] [Google Scholar]
- Simmons P.J., Torok-Storb B. CD34 expression by stromal precursors in normal human adult bone marrow. Blood. 1991;78:2848–2853. [PubMed] [Google Scholar]
- Tremblay J.P., Roy B., Goulet M. Human myoblast transplantationa simple assay for tumorogenicity. Neuromuscular Disorders. 1991;1:341–343. doi: 10.1016/0960-8966(91)90120-h. [DOI] [PubMed] [Google Scholar]
- Tremblay J.P., Malouin F., Huard J., Bouchard J.P., Satoh A., Richards C.L. Results of a blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell. Trans. 1993;2:99–112. doi: 10.1177/096368979300200203. [DOI] [PubMed] [Google Scholar]
- Vilquin J.T., Wagner E., Kinoshita I., Roy R., Tremblay J.P. Successful histocompatible myoblast transplantation in dystrophin-deficient mdx dystrophin. J. Cell Biol. 1995;131:975–988. doi: 10.1083/jcb.131.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins S.C., Hoffman E.P., Slayter H.S., Kunkel L.M. Immunoelectron microscopic localization of dystrophin in myofibers. Nature. 1988;333:863–866. doi: 10.1038/333863a0. [DOI] [PubMed] [Google Scholar]
- Yang Q., Bassel-Duby R., Williams R.S. Transient expression of a winged-helix protein, MNF-beta, during myogenesis. Mol. Cell. Biol. 1997;17:5236–5243. doi: 10.1128/mcb.17.9.5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young H.E., Ceballos E.M., Smith J.C., Mancini M.L., Wright R.P., Ragan B.L., Bushell I., Lucas P.A. Pluripotent mesenchymal stem cells reside within avian connective tissue matrices. In Vitro Cell Dev. Biol. 1993;29A:723–736. doi: 10.1007/BF02631429. [DOI] [PubMed] [Google Scholar]
- Young H.E., Mancini M.L., Wright R.P., Smith J.C., Black A.C., Jr., Reagan C.R., Lucas P.A. Mesenchymal stem cells reside within the connective tissues of many organs. Dev. Dynam. 1995;202:137–144. doi: 10.1002/aja.1002020205. [DOI] [PubMed] [Google Scholar]
- Yuasa K., Miyagoe Y., Yamamoto K., Nabeshima Y., Dickson G., Takeda S. Effective restoration of dystrophin associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett. 1998;425:329–336. doi: 10.1016/s0014-5793(98)00251-8. [DOI] [PubMed] [Google Scholar]
- Ziegler B.L., Valtieri M., Porada G.A., De Maria R., Muller R., Masella B., Gabbianelli M., Casella I., Pelosi E., Bock T. KDR receptora key marker defining hematopoietic stem cells. Science. 1999;285:1553–1558. doi: 10.1126/science.285.5433.1553. [DOI] [PubMed] [Google Scholar]
- Zubryzcka-Gaarn E.E., Bulman D.E., Karpati B., Burghes Bellfall B., Klamut H.J., Talbot J., Hodges R.S., Ray P.N., Worton R.G. The Duchenne muscular dystrophy gene is localized in the sarcolemma of human skeletal muscle. Nature. 1988;333:466–469. doi: 10.1038/333466a0. [DOI] [PubMed] [Google Scholar]