

Abstract
The absence of dystrophin complex leads to disorganization of the force-transmitting costameric cytoskeleton and disruption of sarcolemmal membrane integrity in skeletal muscle. However, it has not been determined whether the dystrophin complex can form a mechanically strong bond with any costameric protein. We performed confocal immunofluorescence analysis of isolated sarcolemma that were mechanically peeled from skeletal fibers of mouse hindlimb muscle. A population of γ-actin filaments was stably associated with sarcolemma isolated from normal muscle and displayed a costameric pattern that precisely overlapped with dystrophin. However, costameric actin was absent from all sarcolemma isolated from dystrophin-deficient mdx mouse muscle even though it was localized to costameres in situ. Vinculin, α-actinin, β-dystroglycan and utrophin were all retained on mdx sarcolemma, indicating that the loss of costameric actin was not due to generalized membrane instability. Our data demonstrate that the dystrophin complex forms a mechanically strong link between the sarcolemma and the costameric cytoskeleton through interaction with γ-actin filaments. Destabilization of costameric actin filaments may also be an important precursor to the costamere disarray observed in dystrophin-deficient muscle. Finally, these methods will be broadly useful in assessing the mechanical integrity of the membrane cytoskeleton in dystrophic animal models lacking other costameric proteins.
Keywords: dystrophin, actin, muscular dystrophy, membrane skeleton, costameres
Introduction
In skeletal muscle, dystrophin is associated with a large, oligomeric complex of proteins that includes dystroglycans, sarcoglycans, dystrobrevins, syntrophins and sarcospan (Straub and Campbell 1997). The dystrophin complex is generally thought to link the actin-based cortical cytoskeleton and laminin-2 in the extracellular matrix (Ervasti and Campbell 1993). Genetic ablation of dystrophin or other components in the complex results in a variety of muscular dystrophies, which are often associated with defects in sarcolemmal membrane integrity (Petrof et al. 1993; Straub et al. 1997; Duclos et al. 1998; Cote et al. 1999). Thus, it is hypothesized that one important function of the dystrophin complex is to mechanically stabilize the sarcolemmal membrane from shear stresses imposed during eccentric muscle contraction (Petrof et al. 1993). A mechanically strong attachment of dystrophin with the sarcolemmal membrane has already been demonstrated (Straub et al. 1992). Regarding possible interaction with the cortical cytoskeleton, dystrophin is located adjacent to the cytoplasmic face of the sarcolemmal membrane in a discrete, rib-like lattice termed costameres (Porter et al. 1992; Straub et al. 1992), a cytoskeletal protein assembly that links the force-generating sarcomeric apparatus to the sarcolemmal membrane (Pardo et al. 1983a; Craig and Pardo 1983). Costameres transmit contractile forces laterally through the sarcolemmal membrane to the basal lamina (Danowski et al. 1992), which may be important for maintaining uniform sarcomere length between active and resting muscle fibers of different motor units. While dystrophin is not required for the assembly of several proteins comprising costamere-like structures, the absence of dystrophin in humans and mice leads to an altered costameric lattice (Minetti et al. 1992, Minetti et al. 1994; Porter et al. 1992; Ehmer et al. 1997; Williams and Bloch 1999). These data suggest that dystrophin plays an important role in organizing or stabilizing costameres, possibly via direct binding to actin filaments. Biochemical studies have demonstrated that purified dystrophin complex can bind actin filaments with an apparent dissociation constant in the sub-micromolar range (Rybakova et al. 1996). However, it has been difficult to assess whether the affinity of the dystrophin/F-actin interaction measured in vitro is sufficient to support a mechanically strong interaction in vivo.
To address this question, we isolated inside-out sarcolemmal membranes by mechanically peeling the membranes from single myofibers teased from normal mouse hindlimb muscles. We used confocal immunofluorescence microscopy to visualize the cytoskeletal proteins retained on the sarcolemma without interference from sarcomeric proteins (Straub et al. 1992). In brief, we have obtained the first evidence demonstrating that the dystrophin complex is necessary for a mechanically strong physical linkage between the sarcolemmal membrane and γ-actin of the costameric cytoskeleton. Furthermore, our data point toward destabilization of γ-actin filaments as a possible intermediate between dystrophin deficiency and global disorganization of the costameric apparatus (Minetti et al. 1992, Minetti et al. 1994; Porter et al. 1992; Ehmer et al. 1997; Williams and Bloch 1999). Finally, our approach should be broadly applicable to assessing the contributions made by other costameric proteins toward the macromolecular organization and mechanical integrity of the membrane cytoskeleton in skeletal muscle.
Materials and Methods
Antibodies and Fluorescent Probes
Rabbit 2 polyclonal antiserum against dystrophin was obtained from an animal immunized with a recombinant protein encoding the first 246 amino acids of human dystrophin (DYS246), which was expressed and purified as previously described (Rybakova et al. 1996). Rabbit 56 polyclonal antiserum against a peptide corresponding to the last 12 amino acids of utrophin (Ohlendieck et al. 1991) was the kind gift of Dr. Kevin Campbell (University of Iowa, Iowa City, IA). The pan-actin monoclonal antibody C4 was purchased from ICN. Monoclonal antibodies specific for α-actinin, α-sarcomeric actin, β-actin, and vinculin were purchased from Sigma-Aldrich. The preparation and characterization of γ-actin polyclonal antibodies was previously described (Otey and Bulinski 1988). The β-dystroglycan monoclonal antibody was purchased from Vector Laboratories. FITC- and TRITC-conjugates of anti-rabbit IgG and anti-mouse IgG were purchased from Jackson ImmunoResearch Laboratories, Inc. Alexa488- and Alexa568-conjugates of phalloidin, anti-rabbit IgG and anti-mouse IgG were purchased from Molecular Probes.
Mechanical Isolation of Single Myofibers and Sarcolemma
Age matched, or littermate control, C57BL/10ScSn-DMDmdx/J, or C57BL/6J-Lama2dy mice were obtained from Jackson ImmunoResearch Laboratories. The vastus lateralis was dissected from tendon to tendon in ice-cold Ringers solution and transferred to a 0.5-ml drop of relaxing solution (100 mM BES, 15 mM creatine phosphate, 5 mM DTT, 4.74 mM ATP, 5.43 mM MgCl2, 7 mM EGTA, and 0.02 mM CaCl2 with the ionic strength adjusted to 180 mM with potassium proprionate, pH 7.0) surrounded by silicone oil. To isolate sarcolemma membranes, one end of a single myofiber was secured with a very fine forceps, the sarcolemma was hooked near the forceps with a stainless steel micro-needle (<100 μm diameter filed to a fine point) and rolled down and off the free end of the myofiber. Isolated sarcolemma and mechanically peeled or intact single myofibers were transferred with fine-tipped glass micropipets to a 0.02-ml drop of relaxing solution stationed on a gelatin-coated glass slide. Isolated sarcolemma and mechanically peeled myofibers were fixed for 5 min with 4% paraformaldehyde in PBS. To assess the location of γ-actin in situ, intact myofibers from the vastus lateralis were fixed and permeabilized for 5 min with 4% paraformaldehyde and 0.1% Triton X-100 in PBS.
Immunofluorescence Analysis
Isolated myofibers and sarcolemmal membranes were blocked for 30 min at room temperature with 5% goat serum in PBS and incubated with primary antibodies overnight at 4°C. The samples were washed with PBS, incubated with the appropriate fluorescently tagged secondary antibodies for 30 min at 37°C, rinsed, and sealed under coverslips in an anti-fade solution. Confocal microscopy was performed with a Bio-Rad MRC1000 scan head mounted transversely to an inverted Nikon Diaphot 200 microscope in the Keck Neural Imaging Lab at the University of Wisconsin. The krypton/argon mixed gas air–cooled laser was set to allow only the 488- and 568-nm excitation lines, while the green and red emission signals were directed to separate photomultiplier tubes. Images were collected during a sequential scan to reduce the possibility of fluorescence bleed-through. To further verify that bleed-through of the green signal was not contaminating the red signal, serial optical sections (z series) were collected in some experiments using both single channel and double channel modes. Digital images were cropped using Adobe Photoshop 5.0 and figure montages prepared with Adobe Illustrator 8.0.
Miscellanea
The dystrophin/utrophin-glycoprotein complexes were amplified for immunoblot analysis from control and mdx total skeletal muscle membranes (Ohlendieck and Campbell 1991) using digitonin extraction and WGA-Sepharose chromatography (Ervasti et al. 1990). Immunofluorescence analysis of frozen cryostat sections from control and mdx muscle was performed as previously described (Ervasti and Campbell 1991).
Results and Discussion
A Population of Actin Filaments Is Tightly Associated with Costameres on Isolated Sarcolemma
Costameric proteins are typically visualized by immunofluorescence analysis of glancing longitudinal cryosections (Craig and Pardo 1983; Porter et al. 1992), or in permeabilized single myofibers from mature skeletal muscle (Straub et al. 1992; Ehmer et al. 1997). If used in combination with widely available actin probes, analysis of dystrophin/actin colocalization by either of these methods is greatly complicated by the intense and ubiquitous signal provided by sarcomeric actin (Rybakova, I.N., and J.M. Ervasti, unpublished results). Therefore, we adopted a method (Straub et al. 1992) that would enable us to visualize the costameres without interference from the sarcomeric cytoskeleton. We isolated inside-out sarcolemmal membranes by mechanical peeling of single myofibers teased from normal mouse hindlimb muscles. Sarcolemmal membranes double stained with rabbit polyclonal antibodies to dystrophin and a fluorescent conjugate of phalloidin were examined by confocal immunofluorescence microscopy, which revealed closely overlapping costameric staining patterns consisting of alternately bright and dark transverse bands (Fig. 1 a) with an average periodicity of 2.8 ± 0.3 μm (n = 7). However, only phalloidin stained the mechanically peeled myofibers (2.5 μm) while no dystrophin staining was detected (Fig. 1 b). Analysis of sarcolemma stained with other, better-characterized antibodies to dystrophin yielded similar results. However, we found that the rabbit 2 polyclonal antiserum to dystrophin raised in our laboratory yielded the greatest signal-to-noise. Since the rabbit 2 antiserum was used throughout this study and had not been previously characterized, we have included evidence documenting its specificity for dystrophin in Fig. 1 c. To confirm that phalloidin was appropriately reporting the presence of actin, we double stained sarcolemma with rabbit 2 dystrophin antibodies and a well documented pan-actin monoclonal antibody (C4) reactive with all mammalian actin isoforms (Lessard 1988). Again, dystrophin and actin staining exhibited closely overlapping staining patterns suggestive of costameres (Fig. 2, a–c). As an additional control, similar staining patterns were observed when sarcolemma were single-stained for dystrophin or actin. Finally, no staining was observed when fluorescent secondary antibodies were incubated alone with sarcolemma, nor did the secondary antibodies exhibit any inappropriate species cross-reactivity that could potentially explain the closely overlapping patterns obtained for dystrophin and actin. Thus, we conclude that a population of actin filaments and dystrophin are tightly associated with the costameric cytoskeleton of normal skeletal muscle such that both can withstand the rigors of mechanical peeling.
Figure 1.
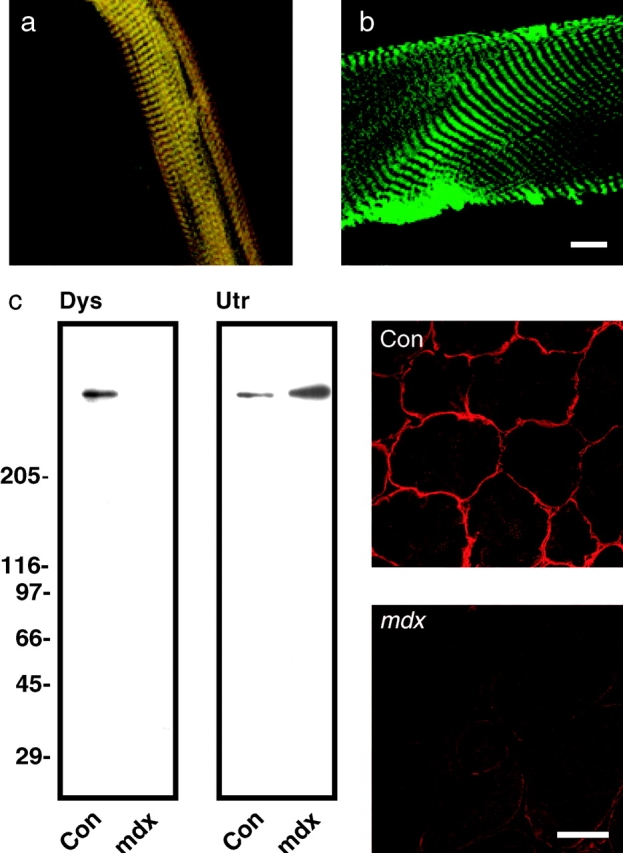
Dystrophin and F-actin colocalize on mechanically isolated sarcolemma in a costameric pattern. Shown is a mechanically isolated sarcolemma (a), or a skinned myofiber (b) both stained with Alexa488-phalloidin (green) and rabbit 2 antiserum to dystrophin (red). Red and green channels were collected simultaneously and areas of coincidence appear yellow. Shown on the left in (c) are immunoblots containing WGA-Sepharose eluates from detergent solubilized control and mdx skeletal muscle membranes stained with rabbit 2 antiserum to dystrophin (Dys), or rabbit 56 antiserum to utrophin (Utr). Shown on the right in c are transverse cryosections of control and mdx skeletal muscle stained with rabbit 2 antiserum to dystrophin. Bars: (b) 10 μm; (c) 50 μm.
Figure 2.

Costameric actin is absent from dystrophin-deficient mdx sarcolemma. Shown are representative images of sarcolemma isolated from control (a–c), dystrophin-deficient mdx (d–f), or laminin-2–deficient dy/dy (g–i) muscle stained with rabbit 2 antiserum to dystrophin (green) and monoclonal antibody C4 to actin (red). Green (a, d, and g) and red (b, e, and h) channels were collected separately, or simultaneously (c, f, and i) where areas of coincidence appear yellow. Dark ovoid shaped areas are nuclear lacunae, which mark the location of peripheral myonuclei before skinning (Straub et al. 1992). Bar, 10 μm.
Costameric Actin Is Specifically Absent from Dystrophin-deficient Sarcolemma
Because dystrophin binds actin in vitro (Rybakova et al. 1996) and the costameric cytoskeleton is altered in dystrophin-deficient muscle (Minetti et al. 1992, Minetti et al. 1994; Porter et al. 1992; Ehmer et al. 1997; Williams and Bloch 1999), we hypothesized that costameric actin should display an abnormal staining pattern on sarcolemma isolated from dystrophin-deficient mdx mice. To our surprise, we observed that costameric actin was completely absent from mdx sarcolemma as reported by monoclonal antibody C4 (Fig. 2, d–f), or phalloidin (Fig. 3, a–c). The absence of actin from mdx sarcolemma can be most directly and simply explained through an actin stabilizing function normally performed by dystrophin. Alternatively, the loss of actin from mdx sarcolemma might not be specifically due to the absence of dystrophin but rather to some other aspect of the dystrophic pathomechanism. However, we have detected costameric actin in all 24 sarcolemma isolated from 8 different control mice while costameric actin was absent from 21 different sarcolemma obtained from 7 different mdx mice. The animals used ranged from 2 to 6 months of age with the isolated sarcolemmal diameters varying from 18–60 μm for controls and 20–43 μm for mdx mice. Furthermore, we examined costameric actin distribution in sarcolemma isolated from the hindlimb muscles of severely dystrophic dy/dy mice. The dy/dy mouse is deficient in laminin-2, the major extracellular matrix ligand for α-dystroglycan in the dystrophin-glycoprotein complex (Campbell 1995). In contrast to mdx muscle, the dystrophin-glycoprotein complex is normally expressed at the sarcolemma of dy/dy muscle (Ohlendieck and Campbell 1991) and shows little evidence of sarcolemmal membrane damage (Straub et al. 1997). Sarcolemma isolated from dy/dy muscle retained both dystrophin and actin in an overlapping pattern (Fig. 2, g–i) similar to the costameric distribution observed in non-dystrophic control sarcolemma (Fig. 2, a–c). These results suggest that in the absence of dystrophin, costameric actin is not stably associated with the sarcolemma membrane.
Figure 3.

Retention of vinculin, β-dystroglycan and utrophin on mdx Sarcolemma. Shown are images of mechanically isolated sarcolemma from mdx (a–c) or control muscle (d) stained with Alexa568-phalloidin (a–d) in red, and antibodies to vinculin (a), β-dystroglycan (b), or utrophin (c and d) in green. Bar, 10 μm.
Because sarcolemmal integrity is compromised in dystrophin-deficient muscle (Straub et al. 1997), the absence of costameric actin might simply reflect a nonspecific loss of peripheral sarcolemmal proteins as a consequence of the stresses imposed during mechanical peeling. However, we found that both vinculin (Fig. 3 a) and α-actinin (Fig. 4 e) were retained on sarcolemma isolated from mdx muscle. Recently, both residual β-dystroglycan and upregulated utrophin in mdx muscle were shown to exhibit a costameric staining pattern in situ (Williams and Bloch 1999). Consistent with this study, we observed that β-dystroglycan and utrophin were also retained on mdx sarcolemma and exhibited a costameric staining pattern (Fig. 3, b–d). Importantly, these data indicate that the loss of costameric actin was not due to generalized weakness of the sarcolemma membrane but is instead a specific consequence of dystrophin deficiency. Furthermore, our data suggest that utrophin either lacks the actin filament stabilizing activity of dystrophin (Amann et al. 1999), or that the amount of costameric utrophin in mdx muscle was not sufficient to retain costameric actin on mechanically isolated sarcolemma.
Figure 4.

Costameric γ-actin is appropriately expressed and localized in mdx muscle in situ. Shown in a–c are images of sarcolemma from normal mouse muscle stained with antibodies specific to α-sarcomeric (a), β-nonmuscle (b), or γ-actin (c). Shown in d and e are images of control (d), or mdx sarcolemma (e) double stained with monoclonal antibodies to α-actinin (green) and polyclonal antibodies to γ-actin (red). Areas of coincidence between the two probes appear yellow. Shown in f and g are images of fixed and permeabilized myofibers from control (f) and mdx (g) muscle stained with antibodies specific for γ-actin. Bars, 10 μm.
Costameric Actin Is Appropriately Expressed and Localized in mdx Muscle In Situ
The γ-actin isoform was previously shown to colocalize with vinculin at costameres in chick skeletal muscle (Craig and Pardo 1983). However, the polyclonal antibody used in this study (Craig and Pardo 1983) did not significantly stain the sarcolemma of mouse diaphragm (Pardo et al. 1983b). Thus, it remained unclear which actin isoform(s) may populate costameres of mammalian muscle. Therefore, we stained normal sarcolemma with monoclonal antibodies specific for α-sarcomeric actin, or β-actin, or a polyclonal antibody raised against a peptide corresponding to the unique amino-terminus of γ-actin (Otey and Bulinski 1988). Only the γ-isoform antibody yielded a bright, costameric pattern of staining while α- and β-specific antibodies yielded only weak and diffuse background labeling (Fig. 4, a–c). As was found using monoclonal antibody C4 (Fig. 2), or phalloidin (Fig. 3), the γ-actin staining pattern evident on normal sarcolemma (Fig. 4c and Fig. d) was absent from dystrophin-deficient mdx sarcolemma (Fig. 4 e). These data indicate that costameric actin appears to be exclusively comprised of the γ-isoform.
It remained to be determined whether costameric actin was absent from mdx muscle before mechanical peeling, or was lost as a result of the peeling procedure. Therefore, we examined γ-actin staining in single fibers from control and mdx muscle that were fixed and permeabilized, but not mechanically peeled. Both control and mdx myofibers exhibited peripheral γ-actin staining patterns marked by regularly spaced transverse fluorescent bands of similar intensities (Fig. 4f and Fig. g). Finally, the γ-actin antibodies also labeled the periphery of Z-lines in mechanically peeled myofibers from both control and mdx muscle, however, the γ-actin signal appeared brighter on mdx myofibers compared with controls (not shown). Taken together, our results indicate that γ-actin is appropriately expressed and assembled into costameres in mdx muscle, but that its stable association with the sarcolemmal membrane is severely compromised.
Based on its structure, protein interactions, and membrane defects associated with its absence or abnormality in dystrophic muscle, the dystrophin complex has long been hypothesized to mechanically stabilize the sarcolemmal membrane against the stresses imposed during muscle contraction or stretch (Petrof et al. 1993; Campbell 1995). Whereas dystrophin is strongly anchored to the sarcolemmal membrane (Straub et al. 1992), no study has demonstrated a mechanically strong linkage between dystrophin and any component of the costameric cytoskeleton. Thus, ours are the first data demonstrating that the dystrophin complex is necessary for a mechanically strong physical linkage between the sarcolemmal membrane and costameric γ-actin. Furthermore, our data point toward destabilization of γ-actin filaments as a possible intermediate between dystrophin deficiency and global disorganization of the costameric apparatus (Porter et al. 1992; Minetti et al. 1992, Minetti et al. 1994; Ehmer et al. 1997; Williams and Bloch 1999). Complexes comprised of ankyrin/spectrin(Williams and Bloch 1999), vinculin/talin/α-actinin (Williams and Bloch 1999) and intermediate filament/intermediate filament associated proteins (Milner et al. 1996; Andra et al. 1997; Dalpe et al. 1999) are also localized to costameres and are capable of binding actin (Fig. 5). Therefore, it is possible that multiple, distinct costameric protein complexes act synergistically to form a strong mechanical linkage between the sarcolemmal membrane and the force-generating sarcomeric apparatus. In support of this possibility, targeted inactivation of desmin (Milner et al. 1996), plectin (Andra et al. 1997), or BPAGn1/dystonin (Dalpe et al. 1999) also cause muscular dystrophy and sarcolemmal instability. Our methods should be valuable in further assessing the macromolecular organization and mechanical integrity of the membrane cytoskeleton in dystrophic animal models lacking other components of costameres.
Figure 5.
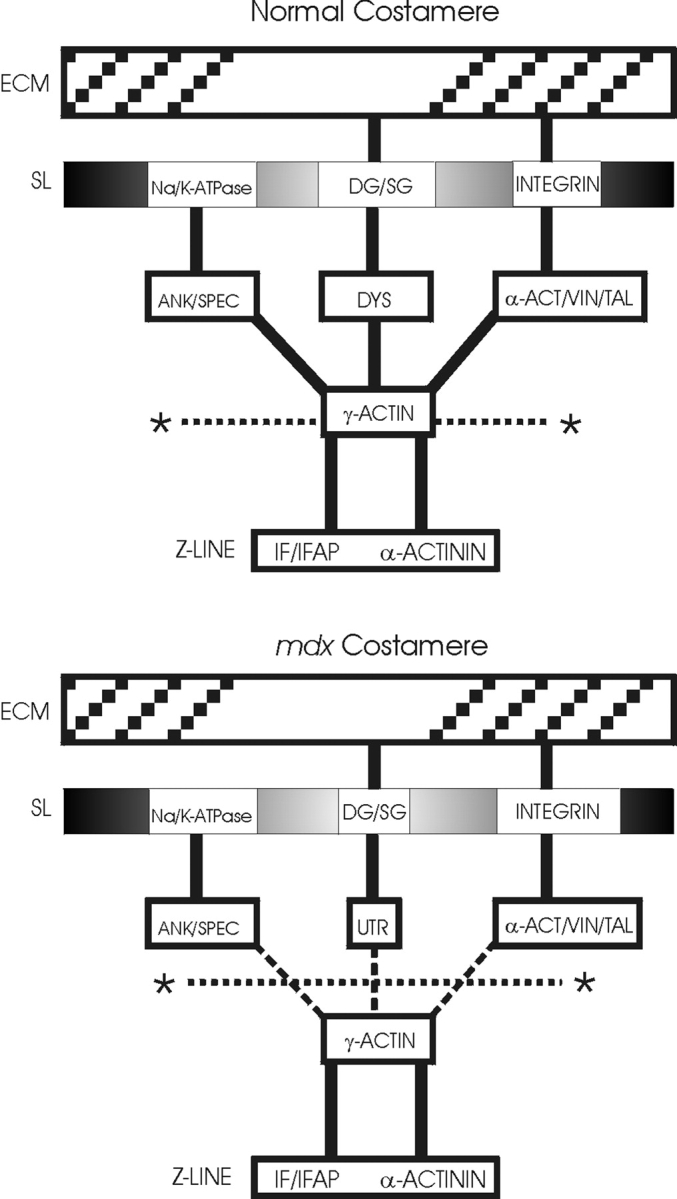
Costameric actin as a common nexus for mechanical coupling between the contractile apparatus and extracellular matrix of striated muscle. Shown is a schematic model depicting the molecular organization of costameres in normal (top) and dystrophin-deficient mdx mouse muscle (bottom). Based on evidence presented in the current report, the fracture plane effected by mechanical peeling is also indicated (*……….*).
Alternatively, dystrophin may be specifically designed to mechanically stabilize costameric actin filaments. We previously demonstrated that dystrophin contains two distinct and spatially separated actin binding sites located at the amino terminus and middle rod domains (Rybakova et al. 1996; Amann et al. 1998), which enables dystrophin to form an extended lateral association with F-actin and protect actin filaments from depolymerization in vitro (Rybakova et al. 1996; Rybakova and Ervasti 1997). Studies by others suggest that the dystrophin carboxy-terminal domain may also participate in binding actin, either directly (Howard et al. 1998), or indirectly through α-syntrophin (Iwata et al. 1998). Analysis of sarcolemma from mdx mice transgenically expressing dystrophin constructs deleted in different domains (Chamberlain et al. 1997), or mice lacking α-syntrophin (Kameya et al. 1999) will resolve which elements in the dystrophin complex are necessary for strong actin filament association with the sarcolemmal membrane. Finally, the methods used in this study will be valuable in assessing whether utrophin over-expression (Tinsley et al. 1998) can rescue the mechanically strong linkage between costameric actin and the sarcolemmal membrane observed in normal muscle.
Acknowledgments
We thank Dr. Kurt Amann for preparing the rabbit 2 antiserum to dystrophin, Dr. Kevin Campbell for providing the rabbit 56 antiserum to utrophin and Dr. J. Chloe Bulinski for providing the polyclonal antibodies specific for γ-actin. We are also grateful to Dr. Donata Oertel for providing access to her stereo dissecting microscope and Dr. Richard Moss for his expert advice and encouragement.
This work was supported by grants from the National Institutes of Health to J.M. Ervasti (AR42423 and AR01985) and a Development Grant from the Muscular Dystrophy Association to I.N. Rybakova.
References
- Amann K.J., Guo W.X.A., Ervasti J.M. Utrophin lacks the rod domain actin binding activity of dystrophin. J. Biol. Chem. 1999;274:35375–35380. doi: 10.1074/jbc.274.50.35375. [DOI] [PubMed] [Google Scholar]
- Amann K.J., Renley B.A., Ervasti J.M. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J. Biol. Chem. 1998;273:28419–28423. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- Andra K., Lassman H., Bittner R., Shorny S., Fassler R., Propst F., Wiche G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K.P. Three muscular dystrophiesLoss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Chamberlain J.S., Corrado K., Rafael J.A., Cox G.A., Hauser M., Lumeng C. Interactions between dystrophin and the sarcolemma membrane. In: Froehner S.C., Bennett V., editors. Cytoskeletal Regulation of Membrane Function. The Rockefeller University Press; New York: 1997. pp. 19–29. [PubMed] [Google Scholar]
- Cote P.D., Moukhles H., M.Lindenbaum, Carbonetto S. Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat. Genet. 1999;23:338–342. doi: 10.1038/15519. [DOI] [PubMed] [Google Scholar]
- Craig S.W., Pardo J.V. Gamma actin, spectrin, and intermediate filament proteins colocalize with vinculin at costameres, myofibril-to-sarcolemma attachment sites. Cell Motility. 1983;3:449–462. doi: 10.1002/cm.970030513. [DOI] [PubMed] [Google Scholar]
- Dalpe G., Mathieu M., Comtois A., Zhu E., Wasiak S., De Repetigny Y., Leclerc N., Kothary R. Dystonin-deficient mice exhibit an intrinsic muscle weakness and an instability of skeletal muscle cytoarchitecture. Dev. Biol. 1999;210:367–380. doi: 10.1006/dbio.1999.9263. [DOI] [PubMed] [Google Scholar]
- Danowski B.A., Imanaka-Yoshida K., Sanger J.M., Sanger J.W. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J. Cell Biol. 1992;118:1411–1420. doi: 10.1083/jcb.118.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclos F., Straub V., Moore S.A., Venzke D.P., Hrstka R.F., Crosbie R.H., Durbeej M., Lebakken C.S., Ettinger A.J., Van der Meulen J. Progressive muscular dystrophy in α-sarcoglycan-deficient mice. J. Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmer S., Herrmann R., Bittner R., Voit T. Spatial distribution of β-spectrin in normal and dystrophic human skeletal muscle. Acta Neuropathol. (Berl.) 1997;94:240–246. doi: 10.1007/s004010050699. [DOI] [PubMed] [Google Scholar]
- Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- Howard P.L., Klamut H.J., Ray P.N. Identification of a novel actin binding site within the Dp71 dystrophin isoform. Febs Letters. 1998;441:337–341. doi: 10.1016/s0014-5793(98)01566-x. [DOI] [PubMed] [Google Scholar]
- Iwata Y., Pan Y., Yoshida T., Hanada H., Shigekawa M. α1-syntrophin has distinct binding sites for actin and calmodulin. Febs Letters. 1998;423:173–177. doi: 10.1016/s0014-5793(98)00085-4. [DOI] [PubMed] [Google Scholar]
- Kameya S., Miyagoe Y., Nonaka I., Ikemoto T., Endo M., Hanaoka K., Nabeshima Y., Takeda S. α1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J. Biol. Chem. 1999;274:2193–2200. doi: 10.1074/jbc.274.4.2193. [DOI] [PubMed] [Google Scholar]
- Lessard J.L. Two monoclonal antibodies to actinOne muscle selective and one generally reactive. Cell Motil. Cytoskel. 1988;10:349–362. doi: 10.1002/cm.970100302. [DOI] [PubMed] [Google Scholar]
- Milner D.J., Weitzer G., Tran D., Bradley A., Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996;134:1255–1270. doi: 10.1083/jcb.134.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minetti C., Tanji K., Bonilla E. Immunologic study of vinculin in Duchenne muscular dystrophy. Neurology. 1992;42:1751–1754. doi: 10.1212/wnl.42.9.1751. [DOI] [PubMed] [Google Scholar]
- Minetti C., Tanji K., Rippa P.G., Morreale G., Cordone G., Bonilla E. Abnormalities in the expression of β-spectrin in Duchenne muscular dystrophy. Neurology. 1994;44:1149–1153. doi: 10.1212/wnl.44.6.1149. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J. Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K., Ervasti J.M., Matsumura K., Kahl S.D., Leveille C.J., Campbell K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- Otey C.A., Bulinski J.C. Immunolocalization of muscle and nonmuscle isoforms of actin in myogenic cells and adult skeletal muscle. Cell Motil. Cytoskel. 1988;9:337–348. doi: 10.1002/cm.970090406. [DOI] [PubMed] [Google Scholar]
- Pardo J.V., D'Angelo Siliciano J., Craig S.W. A vinculin-containing cortical lattice in skeletal muscleTransverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma Proc. Natl. Acad. Sci. USA. 80 1983. 1008 1012a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo J.V., Pittenger M.F., Craig S.W. Subcellular sorting of isoactinsselective association of γ actin with skeletal muscle mitochondria Cell. 32 1983. 1093 1103b [DOI] [PubMed] [Google Scholar]
- Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter G.A., Dmytrenko G.M., Winkelmann J.C., Bloch R.J. Dystrophin colocalizes with β-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J. Cell Biol. 1992;117:997–1005. doi: 10.1083/jcb.117.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybakova I.N., Ervasti J.M. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J. Biol. Chem. 1997;272:28771–28778. doi: 10.1074/jbc.272.45.28771. [DOI] [PubMed] [Google Scholar]
- Rybakova I.N., Amann K.J., Ervasti J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V., Campbell K.P. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr. Opin. Neurobiol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- Straub V., Bittner R.E., Léger J.J., Voit T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol. 1992;119:1183–1191. doi: 10.1083/jcb.119.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V., Rafael J.A., Chamberlain J.S., Campbell K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nature Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- Williams M.W., Bloch R.J. Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. J. Cell Biol. 1999;144:1259–1270. doi: 10.1083/jcb.144.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]