

Abstract
Escherichia coli dam mutants are sensitized to the cytotoxic action of base analogs, cisplatin and N-methyl-N’-nitro-N-nitrosoguanidine (MNNG), while their mismatch repair (MMR)-deficient derivatives are tolerant to these agents. We showed previously, using pulse field gel electrophoresis, that MMR-mediated double-strand breaks (DSBs) are produced by cisplatin in dam recB (Ts) cells at the non-permissive temperature. We demonstrate here that the majority of these DSBs require DNA replication for their formation, consistent with a model in which replication forks collapse at nicks or gaps formed during MMR. DSBs were also detected in dam recB(Ts) ada ogt cells exposed to MNNG in a dose- and MMR-dependent manner. In contrast to cisplatin, the formation of these DSBs was not affected by DNA replication and it is proposed that two separate mechanisms result in DSB formation. Replication-independent DSBs arise from overlapping base excision and MMR repair tracts on complementary strands and constitute the majority of detectable DSBs in dam recB(Ts) ada ogt cells exposed to MNNG. Replication-dependent DSBs result from replication fork collapse at O6-meG base pairs undergoing MMR futile cycling and are more likely to contribute to cytotoxicity. This model is consistent with the observation that fast-growing dam recB (Ts) ada ogt cells, which have more chromosome replication origins, are more sensitive to the cytotoxic effect of MNNG than the same cells growing slowly.
Keywords: DNA mismatch repair, Double-strand breaks, Recombination, Cisplatin, MNNG
1. Introduction
In Escherichia coli, almost all GATC sequences in DNA contain N6-methyladenine due to the action of Dam methyltransferase, the product of the dam gene [1-3]. The enzyme is present in limiting amounts in cells which results in a lag between DNA synthesis and methylation, thereby transiently generating hemimethylated DNA, consisting of a parental methylated strand and a newly-synthesized unmethylated strand. Short-lived hemimethylation of the DNA behind the replication fork is exploited by DNA mismatch repair (MMR) to discriminate between old and new DNA strands when repairing replication errors [4].
MMR in Escherichia coli corrects replication errors behind the replication fork and prevents recombination between similar but not identical DNA sequences (“antirecombination”) [5-7]. To correct replication errors, the MutS protein binds to mismatches in DNA and recruits MutL and MutH resulting in activation of the latent endonuclease activity of MutH to produce a nick in the newly-synthesized unmethylated DNA strand 5’ to the G in a nearby GATC sequence. The UvrD helicase loads on the nicked DNA and begins to unwind single-strand DNA either in the 5’ to 3’ direction or the 3’ to 5’ direction depending on the orientation of the mismatch to the GATC sequence. The single stranded DNA is digested either by Exo I, ExoVII or ExoX in the 3’ to 5’ direction or the RecJ or ExoVII in the 5’ to 3’ direction. The resultant gap is filled by the action of DNA polymerase III, and after the action of ligase the duplex DNA is methylated by Dam. Fully methylated DNA is not a substrate for mismatch correction.
Homeologous DNA is formed during crosses between closely related bacteria such as E. coli and Salmonella enterica serovar Typhimurium and MutS and MutL in the recipient bacteria prevent the formation of viable recombinants [8]. This antirecombinogenic effect can be demonstrated in vitro during the RecA-catalyzed strand transfer reaction, where MutS and MutL inhibit the action of the enzyme on homeologous, but not homologous, substrates [9].
E. coli dam mutants are sensitized to the cytotoxic action of base analogues [10], cisplatin [11] and methylating agents [12, 13]. Inactivation of mismatch repair, however, renders E. coli dam mutants more tolerant to these agents. For cisplatin, we have shown that MMR induces the formation of double-strand breaks (DSBs) in a mutS, mutL and mutH-dependent manner in dam cells [14] due to the recognition of platinated diguanyl intrastrand crosslinks by MutS [15, 16]. We have also shown that MutS can inhibit RecA-mediated strand exchange when one of the two homologous DNA substrates is platinated, an effect which is related to the antirecombination function of MMR [17]. These results suggest that MMR-mediated cytotoxicity might result from both the formation of DSBs and by interference of their repair.
The MutS protein binds to O6-methylguanine (O6-meG) residues paired with either C or T although the affinity for the latter is higher [16, 18]. O6-meG is a product of N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) action on DNA in E. coli and is acted upon by the Ada and Ogt methyltransferases to remove the methyl group [19, 20]. The sensitivity of dam bacteria to MNNG, but not S(N)2 methylating agents, is consistent with MMR recognition and processing of O6-meG base pairs [12]. Given the previous results with cisplatin, we wanted to test the hypothesis that MMR action at O6-meG base pairs results in the formation of DSBs. We also wanted to determine if DNA replication is necessary for MMR-mediated DSB formation in dam cells exposed to either cisplatin or MNNG.
2. Materials and methods
2.1 Media, bacterial strains, and plasmid
The rich medium used was Luria (L) broth which consists of 20 g tryptone, 10 g yeast extract, 0.5 g NaCl and 4 ml 1 M NaOH per l, supplemented with 20 μg/ml thymine and solidified when required with 16 g agar (Difco). Minimal medium contained minimal salts as described by Davis and Mingioli [21] supplemented with 0.4% glucose or succinate, 0.2 μg thiamine per ml with or without 0.2% casamino acids. Ampicillin and rifampicin were used at the concentration of 100 μg/ml. The E. coli K-12 strains used in this study were derived from AB1157 [22] and are listed in Table 1. Plasmid pBAR, a derivative of pBR322 which encodes the ada gene [23], was a gift from Dr. B. Demple (Harvard School of Public Health, Boston MA USA). Strains were constructed by P1vir transduction. The Δada∷Kan and Δogt∷Cam alleles were derived from strain KT233 [24] (Table 1). Although referred to as Δada∷Kan [24], this allele also inactivates alkB. For reasons detailed previously [25], we believe the effects reported here with this allele are due to loss of Ada and not AlkB. The phenotype of strain GM8498 (ΔmutS454∷Tet) was verified by incubating drops of different dilutions of this strain and its parent, GM56, on L plates overnight at the permissive (30°C) and non-permissive (42°C) temperatures. Strain GM8498 was temperature-resistant while strain GM56 was temperature sensitive (Ts) showing a decrease in efficiency of plating of at least 1000-fold at 42°C compared to 30°C. To monitor mutator phenotype, overnight cultures of GM56 and GM8498 were plated on L medium supplemented with 100 μg/ml rifampicin and dilutions of the saturated cultures on L plates to determine the number of viable cells.
Table 1.
Escherichia coli K-12 strains used in this study.
| Strain | Genotype | Source or reference |
|---|---|---|
| GM56 | dam-3 recB270(Ts) thr-1 araC14 leuB6(Am) Δ(gpt-proA)62 lacY1 tsx-33 supE44(AS) galK2(Oc) hisG4(Oc) rfbD1 mgl-51 rpsL31 metB1 thi-1 deoB16 (kdgK51 xylA5 mtl-1)? | [46] |
| GM8419 | As GM56 but Δada∷Kan Δogt∷Cam | This paper |
| GM8498 | As GM56 but Δada∷Kan Δogt∷Cam ΔmutS454∷Tet | This paper |
| GM8198 | As GM56 but Δada∷Kan Δogt∷Cam /pBAR (ada+) | This paper |
| KT233 | As AB1157 but Δada∷Kan Δogt∷Cam | [24] |
Abbreviations: Am, amber mutation; AS, amber suppressor; Δ, deletion; Oc, ochre mutation; Ts, temperature sensitive; Cam, chloramphenicol; Kan, kanamycin; Tet, tetracycline.
The Δada∷Kan allele also inactivates alkB.
Strains were constructed using P1vir transduction.
pBAR is referred to as pada+ throughout the manuscript.
Further information about the strains used in this study can be found at http://users.umassmed.edu/martin.marinus/dstrains.html
2.2 Cytotoxicity assay
Overnight cultures in L broth were diluted 100-fold into fresh minimal medium and grown to OD600 0.4–0.6. Cells were treated for 10 minutes with varying concentrations of MNNG. The treated cells were diluted immediately and plated on L medium to measure survival. Cells were exposed to cisplatin as described previously [14].
2.3 Amino acid starvation
Logarithmic phase cultures were grown in minimal medium for 2-3 h at 30°C, and for an additional 1-2 h at 42°C. The cultures were centrifuged and the cells resuspended in minimal salts supplemented only with 0.4% glucose and 0.2 μg thiamine /ml and without required amino acids for another 2 h at 42°C. After starvation, cells were exposed to MNNG (10 μg/ml) or cisplatin (50 μM) for 10 and 60 minutes respectively, after which the cultures were centrifuged washed, and half of each culture was resuspended in a fresh minimal medium supplemented with required amino acids and the another half in starvation medium. Cultures were then incubated for another 2 h at 42°C, harvested and prepared for pulse field gel electrophoresis (PFGE).
2.4 Preparation of plugs and PFGE migration
We have used the procedure described by Seigneur et al., [26] with modifications [14]. Cells were cultivated overnight in L medium and diluted into minimal medium containing 100 ug deoxyadenosine/ml and 5 μCi [3H]thymidine (87 Ci/mmol, Amersham Biosciences)/ml. After 2-3 h of incubation at 30°C, corresponding to OD600 0.05–0.08, the culture was shifted to 42°C. After an additional 2-3 h of incubation (OD600 0.4–0.6) the cells were centrifuged and resuspended in an equal volume of minimal salts, MNNG (usually 10 μg/ml) or cisplatin (50 μM) was added and the culture was incubated for 10 or 60 minutes respectively at 42°C. The cells were harvested by centrifugation, washed once with minimal salts, twice in SE buffer (75 mM NaCl, 25 mM EDTA, pH 7.4) and resuspended in 160 μl of distilled water. The cells were mixed with an equal volume of 2% low melt agarose (in 0.5 × TBE buffer), distributed in 60 μl portions in molds and left for a few minutes at 4°C until set. The plugs were incubated overnight at 56°C (cisplatin) or 30°C (MNNG) in 1 ml LE buffer (1% N-laurylsarcosine, 0.5 M EDTA, pH 9.6) with proteinase K (0.5%) [27]. The plugs were then washed three times with TE buffer and stored in TE buffer at 4°C. Portions of each agarose plug were loaded into the wells of 1% agarose gels (Seakem Gold Agarose) and sealed with 1.0% low melt agarose and were subjected to electrophoresis at 14°C for 24 h at 6 V/cm in 0.5 × TBE, with an initial switching time 60 s and final switching time of 120 s in a BioRad CHEF-DR II apparatus. Plugs containing the chromosomes of the yeast Saccharomyces cerevisiae were routinely used as molecular weight standards (New England Biolabs). Each lane was cut into 3 mm slices using a razor blade slicer and a total of 36 slices representing 10 cm of gel was usually made to allow the recovery of all fragments between the origin and the location corresponding to 100-200 kb fragments. To each agarose slice, 1 ml of 0.1 M HCl was added and melted at 90°C for 20 minutes. After cooling, 0.8 ml portions were added to 5 ml of scintillation fluid (EcoLume, MP Biochemicals) and the radioactivity measured in a Beckman Coulter LS6500 scintillation counter. The fraction of linear DNA was calculated by dividing the amount of tritium found in a lane (slices 4-36) by the total amount of tritium found in the lane plus the migration origin (slices 1-36). For each of the PFGE figures, the experiment was done at least three times and a representative figure is shown.
3. Results
3.1 Sensitivity of dam recB270 (Ts) and dam recB270(Ts) ada ogt strains to MNNG
RecBC protein has a high affinity for double-stranded DNA ends to initiate processive exonucleolytic degradation [28]. To preserve such ends, recBC mutations are used but since dam recBC mutants are not viable, we have used strain GM56 which contains a temperature-sensitive recB270 (Ts) mutation in a dam-3 background. The recB270 allele must retain some residual activity at 42°C because dam-3recB270 (Ts) recC271(Ts) cells grow very poorly at the permissive temperature and segregate faster growing cells with suppressor mutations at high frequency ([14] and unpublished data). This residual activity of the recB270(Ts) allele allows GM56 to grow at the permissive temperature, 30°C, while it has at least a 10,000-fold reduced efficiency of colony formation at 42°C [29]. At the non-permissive temperature, however, full viability is retained for at least the first hour after the temperature shift, if the cells are returned to the permissive temperature. Exposure of GM56, after 60 min at 42°C, to various concentrations of MNNG for 10 min indicated an increased sensitivity compared to cells grown at 30°C, consistent with reduced RecBCD activity (Fig. 1). We have shown previously that RecBCD is necessary for wildtype survival of E. coli to MNNG exposure [25].
Fig. 1.
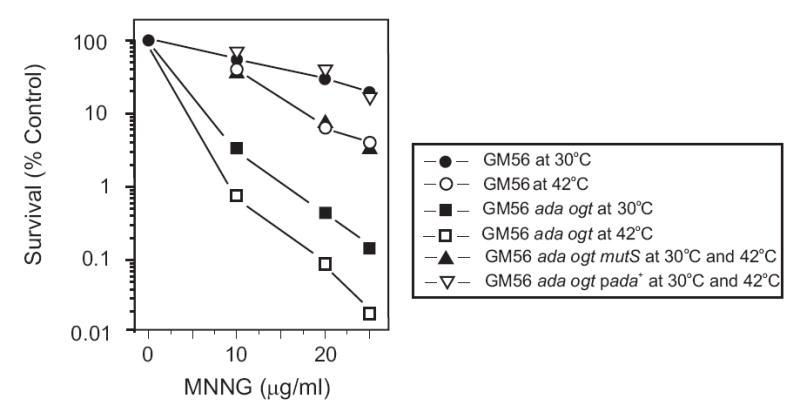
Survival of bacterial strains challenged with MNNG. Bacteria in the logarithmic phase of growth in minimal medium at 30°C were shifted to 42°C for 60 min and challenged with various doses of MNNG for 10 min. The exposure was terminated by dilution of cells into diluent for plating. GM56 at 30°C (solid circles) and 42°C (open circles); GM56 Δada Δogt at 30°C (solid squares) and 42°C (open squares); GM56 Δada Δogt mutS at 30°C and 42°C (filled triangles); GM56 Δada Δogt/pada+, at 30°C and 42°C (open triangles).
O6-meG is a substrate for the Ada and Ogt methyltransferases [19] as well as MutS [18]. To maximize the MMR effect through MutS recognition, we constructed a derivative (GM8419) of strain GM56 deleted for the ada and ogt genes which was more sensitive than GM56 to MNNG treatment at both permissive and non-permissive temperatures and the effect was greater at 42°C (Fig. 1). This result is consistent with O6-meG being processed only by MutS to produce MMR-mediated sensitization because the survival of GM56 Δada Δogt mutS at both permissive and non-permissive temperatures is increased considerably compared to GM56 Δada Δogt (Fig. 1). The survival of GM56 Δada Δogt ΔmutS at both temperatures was less than that of GM56 at 30°C (Fig. 1) indicating that Ada and Ogt are necessary to counteract MNNG damage as we have described previously [25]. Ada is also positive transcriptional regulator of the alkA and other BER genes [19, 30] and this response cannot be mounted in the Δada mutant we have used. However, at the low dose (10 μg/ml) and short exposure time (10 min) we have used in the experiments to be described here, there is no difference in survival of ada and alkA mutants compared to the wildtype [25] indicating that the contribution of AlkA is minimal. It should be noted that mutS strains have the same survival kinetics as wildtype when exposed to MNNG [12, 13].
Ada removes methyl groups from O6-meG in DNA at an exceedingly fast rate [31]. We reasoned that overproduction of Ada by the use of the multicopy plasmid pada+, should reduce the amount of O6-meG substantially in MNNG-exposed cells thereby depriving MutS of substrate. As predicted, overproduction of Ada in GM56 Δada Δogt results in a striking increase in survival after MNNG exposure, equivalent to that of GM56 at the permissive temperature (Fig. 1).
3.2 Detection of MNNG-induced double-strand breaks
We previously used pulse field gel electrophoresis (PFGE) to measure MMR-induced DSBs in chromosomal DNA of GM56 exposed to cisplatin [14]. For GM56 Δada Δogt treated with MNNG we used the same technique with one modification which was to lyse the cells at 30°C instead of 56°C. The lower temperature was used because Lundin et al [32] showed that lysis of yeast cells treated with methyl methane sulfonate (MMS) at 56°C resulted in the artifactual appearance of MMS-induced DSBs in vivo which probably resulted from hydrolysis of abasic sites after depurination in vitro at 56°C. Reducing the temperature during cell lysis to 37°C or 30°C prevented formation of these artifacts.
Circular E. coli chromosomes remain in the well during PFGE but linear chromosomal fragments migrate into the gel. In order to quantify the amount of linear DNA in each gel lane we labeled chromosomal DNA with [3H]-thymidine prior to MNNG treatment and determined the amount of radioactivity in slices of the gel after electrophoresis. Gel slices 1-3 in our procedure included the well and the radioactivity of these is not shown in the figures. Radioactivity in each slice shown in the figures was calculated as the percentage of total radioactivity which includes slices 1-3.
To test the relationship between drug dose and DSB formation, GM56 Δada Δogt was grown with tritiated thymidine at the permissive temperature, shifted to 42°C for 2-3 hr and then exposed to 0, 2.5, 5 and 10 μg MNNG/ml for 10 min at 42°C followed by harvesting and lysis at 30°C, and PFGE (Fig. 2). There was a substantial difference in the profile of radioactivity from cells exposed to MNNG compared to the unexposed control cells. In the treated cells, however, the difference was most pronounced in the region with the smallest linear fragments (slice 22-33) which corresponds to about 100 to 600 kb (Fig. 2). The total amount of linear DNA increased from about 8% in the unexposed cells to 35% in treated cells (Table 2). For subsequent experiments, 10 μg/ml MNNG was used as the standard dose.
Fig. 2.
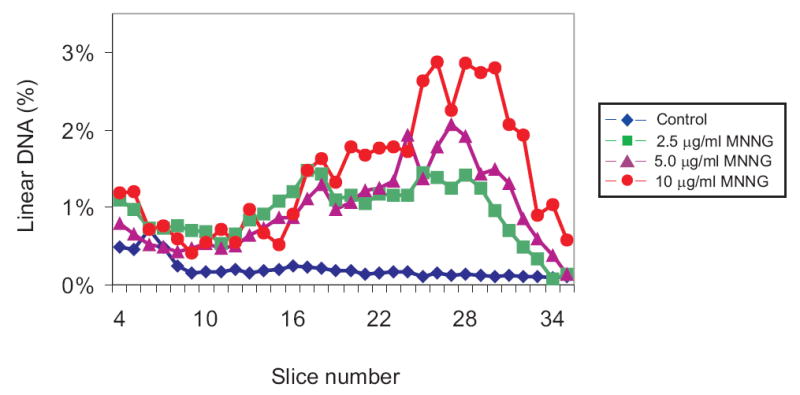
Dose-dependent distribution of linear DNA from GM56 Δada Δogt cells exposed to MNNG. GM56 Δada Δogt was grown with tritiated thymidine at 30°C, shifted to 42°C and then challenged with 0 (diamonds), 2.5 (squares), 5 (triangles) and 10 (circles) μg/ml MNNG for 10 min. The cells were harvested, lysed at 30°C, and linear DNA fragments separated by PFGE. In this and subsequent figures, the distributions of radioactivity in slices 4-35 represents linear DNA fragments that have entered the gel while radioactivity in slices 1-3 is intact DNA remaining in the well. Only fractions 4-35 are shown in the figures and the data are plotted as percent of total radioactivity in fractions 1-35.
Table 2.
Amount of linear DNA in MNNG-treated and untreated cells.
| Strain | Pertinent genotype | Per cent linear DNA | |
|---|---|---|---|
| Untreated | Treated | ||
| GM8419 | dam-3 recB270 Δada Δogt | 7.8 ± 1.4 | 34.5 ± 7.3 |
| GM8498 | dam-3 recB270Δada Δogt ΔmutS | 3.2 ± 1.0 | 3.6± 1.4 |
| GM8198 | dam-3 recB270Δada Δogt/(pada+) | 3.7 ± 1.3 | 3.9 ± 0.9 |
The numbers in the Table represent at least three separate experiments on each strain.
GM56 Δada Δogt was exposed to 10 μg MNNG/ml, and harvested immediately or incubated for a further 60 and 120 min at 42°C. After 10 min, 36% of the total DNA was linear, and at 60 min and 120 min the values were 49% and 55% respectively (Fig. 3) therefore, most of the linear DNA was formed during the 10 min of drug exposure. This result is in contrast to that obtained with GM56 and cisplatin exposure, where the amount of linear DNA increased substantially with continued incubation [14].
Fig. 3.
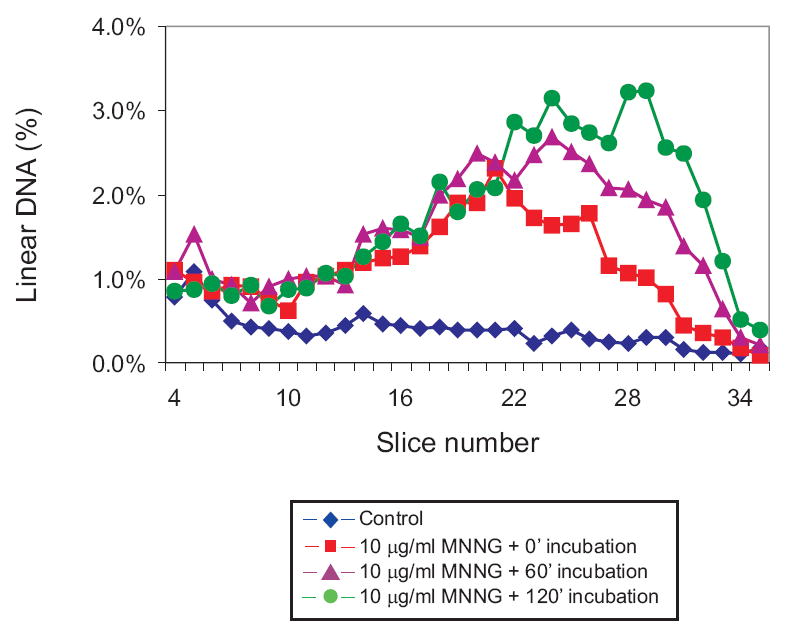
Time dependence of MNNG-induced DSBs. GM56 Δada Δogt cells growing at 42°C were exposed to 10 μg/ml MNNG for 10 min and, after removal of the MNNG, incubated for a further 0 (squares), 60 (triangles) or 120 (circles) min before harvesting, lysis and PFGE. Diamonds represent the distribution from unexposed cells. See legend to Figure 2 for further details.
The amount of linear DNA in GM56 Δada Δogt after challenge with MNNG was dependent on active MMR. There was no increase in linear DNA in a mutS deletion derivative of GM56 Δada Δogt exposed to MNNG compared to the untreated control culture (Fig. 4 and Table 2). As a second test of the contribution of MMR, overproduction of Ada protein (which removes O6-meG as a substrate for MutS) in GM56 Δada Δogt cells challenged with MNNG, produced no detectable linear DNA compared to control cells (Fig. 4 and Table 2).
Fig. 4.
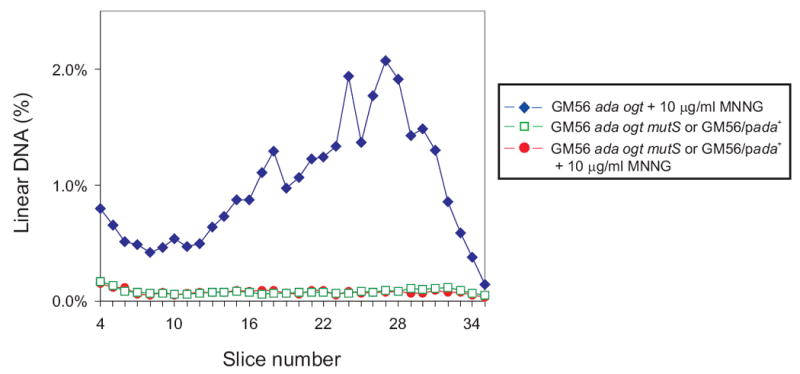
Effect of MMR inactivation and Ada overproduction on MNNG-induced DSBs. GM56 Δada Δogt (diamonds), GM56 Δada Δogt ΔmutS (squares) and GM56 Δada Δogt/pada+ (circles) cells growing at 42°C were exposed to 10 μg/ml MNNG for 10 min, harvested, lysed and subjected to PFGE. See legend to Figure 2 for further details.
3.3 DNA replication is necessary for MMR-induced DSB formation in cells exposed to cisplatin
Based on the genetic requirements for MMR-mediated DSB formation in GM56 exposed to cisplatin, we proposed [14] that such DSBs arise by replication fork collapse at gaps during MMR at cisplatin adducts (Fig. 5). A test of this prediction would be that GM56 cells not replicating their chromosomes should have fewer DSBs than those which are undergoing chromosome duplication. The method we used to stop chromosome replication was to starve growing cultures of required amino acids which allows cells to complete ongoing rounds of replication but prevents new chromosome initiations. We found that after 120 min of starvation at the non-permissive temperature, there was no significant incorporation of radioactive thymidine into DNA relative to growing cells (data not shown). Addition of required amino acids after the starvation period results in an immediate resumption of DNA synthesis, allowing a comparison between cells to which amino acids have been added and those which have not. Logarithmic phase GM56 Cultures were starved for amino acids for two hours and then exposed to 50 μM cisplatin for 60 min at 42°C. After washing the cells, amino acids were added to half the culture and the cells incubated for 120 min at 42°C before harvesting and PFGE analysis. The results in Fig. 6 show that at least 75% of DSBs are generated by chromosome replication. This result suggests that, indeed, replication fork collapse at sites of active MMR repair is the most likely explanation for DSB formation (Fig. 5).
Fig. 5.
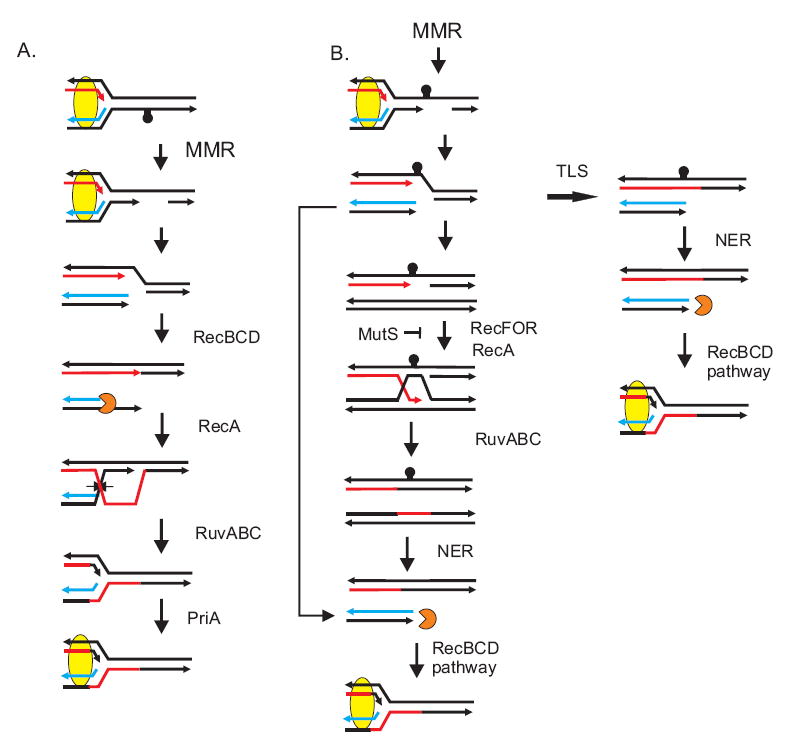
Formation of a double-stranded end following replication fork collapse. The capstan indicates a replication-blocking cisplatin adduct recognized by MutS and mismatch removal in dam strains can occur on either DNA strand. In (A), MMR acts on the strand with the adduct and in (B) on the complementary strand. In both cases, mismatch-directed excision is incomplete when the replication fork moves through the gap and collapses. This creates a double-strand end which is acted upon by RecBCD which, after encountering a Chi sequence, promotes RecA strand transfer, followed by RuvAB helicase action and RuvC cleavage to restore the replication fork. PriA indicates the replication restart pathway to reload DNA polymerase III holoenzyme. In (B), the single-strand gap remaining after fork collapse can be processed either by RecFOR recombination or by a translesion polymerase. NER indicates removal of the lesion by nucleotide excision repair. MutS indicates the step at which antirecombination prevents RecA action [39]. This figure is reproduced from reference 14 with permission from Elsevier B.V.
Fig. 6.
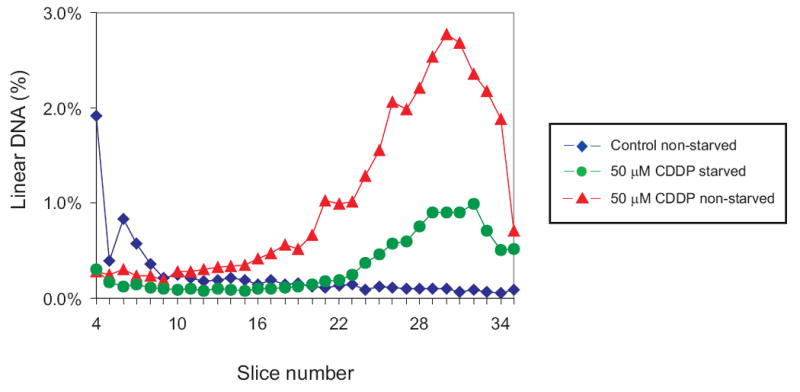
Chromosome replication is required for MMR-induced DSBs after cisplatin challenge. GM56 bacteria growing at 42°C were deprived of required amino acids for 2 hrs and then challenged with 50 μM cisplatin for 60 min. Required amino acids were added to half the culture (triangles) but not the other (circles) and both were incubated for a further 60 min before harvesting, lysis and PFGE. The diamonds indicate the distribution from untreated, non-starved cell. See legend to Figure 2 for further details.
3.4 DNA replication does not stimulate MMR-induced DSB formation in cells exposed to MNNG
Emboldened by the result with cisplatin above, we used the same starvation protocol with GM56 Δada Δogt cells exposed to 10 μg MNNG /ml. To our great surprise, there was no difference in the distribution of linear fragments in replicating versus non-replicating cultures (Fig. 7) indicating that DSBs arise in the absence of chromosome replication. This result, however, is consistent with the observation of the small increase in DSBs in cells incubated for 60 and 120 min after MNNG exposure (Fig. 3) compared with the large increase under similar conditions in cisplatin exposed cells [14].
Fig. 7.
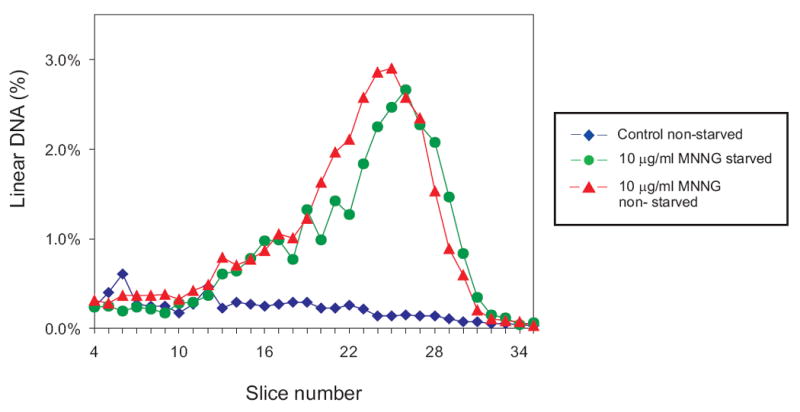
Chromosome replication is not required for MMR-induced DSBs after MNNG challenge. GM56 Δada Δogt cells growing at 42°C were deprived of required amino acids for 2 hrs, challenged with 10 ug/ml MNNG for 10 min, and required amino acids added to half the culture (triangles) but not the other (circles) and allowed to grow at 42°C for a further 120 min. before harvesting, lysis and PFGE. The diamonds indicate the distribution from untreated, non-starved cell. See legend to Figure 2 for further details.
Lundin et al [32] showed that lysis of MMS-treated yeast cells at 56°C before PFGE allowed the detection of heat labile sites which probably represent hydrolyzed abasic sites in DNA in vivo. We wanted to know if there was a difference in the frequency of these sites in GM56 Δada Δogt and its ΔmutS derivative. We treated GM56 Δada Δogt cells with 0, 2.5, 5 and 10 μg/ml MNNG for 10 min, and after harvesting and lysing the cells at 56°C, subjected them to PFGE. There was a dose-dependent increase in heat labile sites (Fig. 8) but their distribution was not significantly altered in the absence of MutS (data not shown).
Fig. 8.
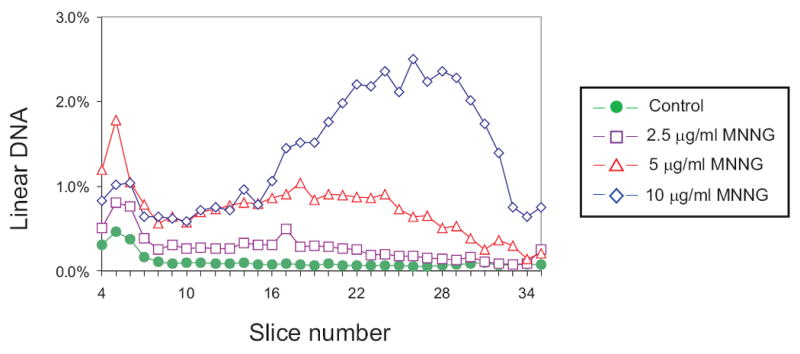
Detection of heat-labile (HL) sites after MNNG challenge. GM56 Δada Δogt cells growing at 42°C were exposed to 0 (circles), 2.5 (squares), 5 (triangles) and 10 (diamonds) μg/ml MNNG for 10 min before harvesting, lysis at 56°C and PFGE. See legend to Figure 2 for further details. Lysis of MNNG-treated cells at 30°C gives the same profile as unexposed cells.
3.5 MMR-induced DSBs in cells with a reduced number of replication forks
In E. coli the number of replication origins, and therefore the number of forks, is inversely correlated with growth rate [33]. To determine if varying the number of forks had any effect on MNNG toxicity, GM56 Δada Δogt cells were cultivated at 30°C in glucose minimal medium supplemented with casamino acids or in minimal medium with succinate as sole carbon source. Under these conditions, cell doubling times were about 45 min and 240 min respectively. Exposure to MNNG resulted in greater survival of succinate-grown cells than those cultivated with glucose as sole carbon source (Fig. 9). This result suggests that MMR-mediated toxicity is dependent principally on the number of replication forks per cell probably through replication fork collapse.
Fig. 9.
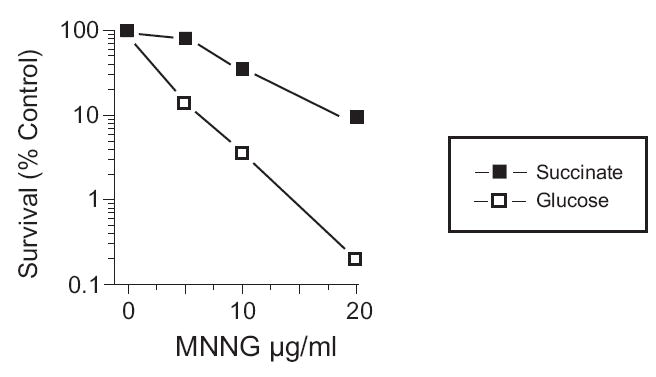
Survival after MNNG exposure of GM56 Δada Δogt cells with different doubling times. Strain GM56 Δada Δogt was grown at 30°C in either glucose minimal medium supplemented with casamino acids (open squares, 45 min doubling time) or in minimal medium with succinate as sole carbon source (filled squares, 240 min doubling time). Cultures were treated with MNNG for 10 min at the indicated concentrations.
An alternative explanation is that MMR is less efficient in succinate grown cells than glucose grown cells. This alternative is based on the findings of Feng et al [34] that cells approaching stationary phase down-regulate the levels of MutS and MutH so as to be almost negligible. To test this possibility we used two approaches. First, we found that GM56 Δada Δogt mutS454 was completely resistant to the toxic effects of MNNG under the same conditions of growth in succinate as described in Fig. 9 for GM56 Δada Δogt (data not shown). This result indicates that there is MMR activity in actively-growing GM56 Δada Δogt bacteria using succinate as carbon source. Second, we compared the survival of GM56 Δada Δogt cells growing in succinate which contained pBR322, or pET derivatives of mutS, mutL and mutH. The pET derivatives were chosen because they do not have the coding region upstream of these genes and because they complement mut deletion mutations on the chromosome in the absence of inducer [35, 36]. There was no significant difference in the survival of these strains (data not shown) indicating that alteration in the levels of MutS, MutL and MutH cannot be the explanation for the results shown in Fig. 9.
4. Discussion
MMR-induced DSBs in cisplatin-treated cells arise by a replication-dependent mechanism (Fig. 6), most likely involving replication fork collapse (Fig. 5) and there is a direct correlation between the presence of these DSBs and cytotoxicity [14]. MMR-mediated sensitization of dam cells can also be formed by base analogs such as 2-aminopurine [10, 37] which requires DNA replication for its cytotoxic action. It seems reasonable, therefore, that MMR-mediated DSB formation in MNNG exposed dam cells should also be replication-dependent. In contrast, we found no dependence on chromosome replication using cells that are actively synthesizing DNA and those which are not (Fig. 7). Even with lower or higher MNNG doses, we could not detect a replication-dependent effect (data not shown).
To explain this unexpected result (Fig. 10), we suggest that there are at least two different mechanisms for the formation and repair of MMR-induced DSBs. One of these requires chromosome replication just like base analogs or cisplatin and probably involves replication fork collapse but the number of such DSBs is masked by those arising independent of DNA replication. Our inability to detect these replication-dependent DSBs may be due to the limitations of the PFGE assay since we have shown recently that single cell microgel electrophoresis can detect DSBs that are below the limit of detection with PFGE [38]. In addition, it may be that MMR gaps induced at cisplatin lesions are more stable than those induced by MNNG thereby increasing the probability of an encounter with a replication fork.
Fig. 10.
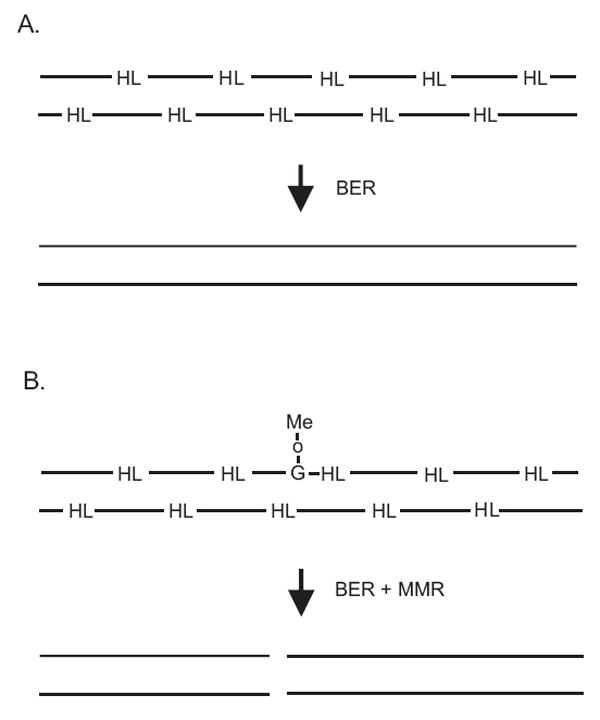
Proposed model for DSB formation in MNNG-treated dam cells. (A) MNNG damage to DNA produces heat-labile (HL) sites that are rapidly and efficiently repaired by (short-patch) BER in the absence of MMR. (B) In the presence of O6meG, MMR can produce excision tracts of hundreds or thousands of nucleotides which can overlap one or more BER repair sites to form a DSB.
Replication-independent DSBs might arise as a consequence of interference between MMR and base excision repair (BER) at abasic sites which can be formed in GM56 Δada Δogt by the spontaneous depurination of 3-methyladenine and 7-methylguanine and basal Tag glycosylase activity acting principally on 3-methyladenine residues. We have shown previously [25] that the contribution of AlkA glycosylase is minimal at (a) the low dose of MNNG (10 μg/ml) and short time of exposure (10 min) and (b) in the absence of Ada, a positive regulator for alkA gene expression [30]. These abasic sites are repaired by BER, which results in short repair patches of only a few nucleotides, on both DNA strands thereby ensuring the integrity of duplex DNA (Fig. 10A). In MMR-proficient GM56 Δada Δogt, however, some O6-meG/C base pairs are bound by MutS (such binding was demonstrated in the antirecombination assay in vitro [39]) and MMR gaps are formed which can be hundred or thousands of nucleotides in length. During this process we speculate that a BER single-strand nick or gap is encountered on the complementary strand leading to the formation of a DSB (Fig. 10B). DNA replication would not be required for DSB formation (Fig. 7) and the DSBs formed this way could be repaired efficiently by homologous recombination with a sister chromosome.
We speculate that the replication-independent DSBs are not the primary cause of cell death. We base this suggestion on the results in Fig. 9 showing that growth rate of the cells affects the degree of cytotoxicity. The level of replication-independent DSBs should be the same in cells growing fast or slow but replication fork collapse is expected to be much more frequent in fast-growing than slow-growing cells. This model would be in agreement with analogous results obtained with base analogues and cisplatin which do require DNA replication for MMR-mediated cell death of dam mutants. Using these agents as a precedent, DNA replication of O6-meG/C to form either O6-meG/C or O6-meG/T in MNNG-treated dam mutants, would provoke futile cycling where neither of these base pairs is perceived as being correct by the MMR system and leads to repeated rounds of repair [12]. During this time, a replication fork encounters this gap and collapses as shown for cisplatin in Fig. 5 and homologous recombination is required to restore the fork (Fig. 5). This scenario is supported by the data in Fig. 9, where the doubling time of the culture (and hence the number of replication forks) has a large effect on cytotoxicity. This same model can be applied to 2-aminopurine (2-AP) where 2AP/T and 2AP/C base pairs are possible. It has not been possible to test the model with 2-AP, however, due to massive chromosomal DNA breakdown presumably initiating at sites of DSBs [37]. No such DNA degradation is observed in MNNG or cisplatin-treated dam mutants.
In the models above, homologous recombination (HR) is essential to restore integrity of the chromosome after methylation damage. It has been shown recently that HR in E. coli is indeed required for resistance to methylating agents (MNNG and MMS) and its contribution is of the same magnitude as BER [25]. The genetic data indicated that both RecF (repair of single-strand gaps) and RecBCD (repair of DSBs) pathways are necessary to prevent against toxicity with the latter being more important. AP-endonuclease-induced nicks at abasic sites of methylation damage leading to the formation of DSBs were predicted to be a major substrate for repair by HR.
In mammalian cells, inactivation of MMR results in tolerance to MNNG while MMR-proficiency leads to sensitivity, a phenomenon similar to that observed with an E. coli dam mutant [7, 40, 41]. Futile cycling by MMR at O6-meG base pairs was also proposed as a model [40] and recently data were obtained in vitro that supports this concept [42]. It is not inconceivable that events similar to those in E. coli dam mutants could occur in mammalian cells by the generation of a stable MMR-induced single-strand gap as the first step and replication fork collapse as the second followed by signaling of the apoptotic response. Recent results with Saccharomyces cerevisiae exposed to MNNG indicated an MMR-dependent killing mechanism requiring HR for cell survival which is most likely mechanistically related to those in mammalian and E. coli dam cells [43]. In contrast, sensitization by MMR in mammalian cells exposed to cisplatin is now in doubt [44, 45] which may reflect the difference between the action of these agents as shown here.
Acknowledgments
We thank Sarah Leroux for technical assistance and Dr. B. Sedgwick for providing strain KT233. This work was supported by grant GM63790 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marinus MG. Methylation of DNA. In: Neidhardt FC, Curtiss R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella:Cellular and Molecular Biology. ASM Press; Washington DC: 1996. pp. 782–791. [Google Scholar]
- 2.Lobner-Olesen A, Skovgaard O, Marinus MG. Dam methylation: coordinating cellular processes. Curr Opin Microbiol. 2005;8:154–160. doi: 10.1016/j.mib.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Wion D, Casadesus J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat Rev Microbiol. 2006;4:183–192. doi: 10.1038/nrmicro1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pukkila PJ, Peterson J, Herman G, Modrich P, Meselson M. Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics. 1983;104:571–582. doi: 10.1093/genetics/104.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 6.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 7.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 8.Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- 9.Worth L, Clark SRM, Modrich P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNA. Proc Natl Acad Sci U S A. 1994;91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glickman B, van den Elsen EP, Radman M. Induced mutagenesis in dam-mutants of Escherichia coli: a role for 6-methyladenine residues in mutation avoidance. Mol Gen Genet. 1978;163:307–312. doi: 10.1007/BF00271960. [DOI] [PubMed] [Google Scholar]
- 11.Fram RJ, Cusick PS, Wilson JM, Marinus MG. Mismatch repair of cis-diamminedichloroplatinum(II)-induced DNA damage. Mol Pharmacol. 1985;28:51–55. [PubMed] [Google Scholar]
- 12.Karran P, Marinus MG. Mismatch correction at O6-methylguanine residues in E. coli DNA. Nature. 1982;296:868–869. doi: 10.1038/296868a0. [DOI] [PubMed] [Google Scholar]
- 13.Jones M, Wagner R. N-Methyl-N’-nitro-N-nitrosoguanidine sensitivity of E. coli mutants deficient in DNA methylation and mismatch repair. Mol Gen Genet. 1981;184:562–563. doi: 10.1007/BF00352542. [DOI] [PubMed] [Google Scholar]
- 14.Nowosielska A, Marinus MG. Cisplatin induces DNA double-strand break formation in Escherichia coli dam mutants. DNA Repair (Amst) 2005;4:773–781. doi: 10.1016/j.dnarep.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Fourrier L, Brooks P, Malinge JM. Binding discrimination of MutS to a set of lesions and compound lesions (base damage and mismatch) reveals its potential role as a cisplatin-damaged DNA sensing protein. J Biol Chem. 2003;278:21267–21275. doi: 10.1074/jbc.M301390200. [DOI] [PubMed] [Google Scholar]
- 16.Calmann MA, Nowosielska A, Marinus MG. Separation of mutation avoidance and antirecombination functions in an Escherichia coli mutS mutant. Nucleic Acids Res. 2005;33:1193–1200. doi: 10.1093/nar/gki263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calmann MA, Marinus MG. MutS inhibits RecA-mediated strand exchange with platinated DNA substrates. Proc Natl Acad Sci U S A. 2004 doi: 10.1073/pnas.0406104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen L, Samson L. The Escherichia coli MutS DNA mismatch binding protein specifically binds O6-methylguanine DNA lesions. Carcinogenesis. 1996;17:2085–2088. doi: 10.1093/carcin/17.9.2085. [DOI] [PubMed] [Google Scholar]
- 19.Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 20.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE. Alkylation damage in DNA and RNA--repair mechanisms and medical significance. DNA Repair (Amst) 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Davis BD, Mingioli ES. Mutants of Escherichia coli requiring methionine or vitamin B12. J Bacteriol. 1951;60:17. doi: 10.1128/jb.60.1.17-28.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewitt SK, Adelberg EA. Transduction of the attached sex factor of Escherichia coli. J Bacteriol. 1962;83:673–678. doi: 10.1128/jb.83.3.673-678.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demple B. Mutant Escherichia coli Ada proteins simultaneously defective in the repair of O6-methylguanine and in gene activation. Nucleic Acids Res. 1986;14:5575–5589. doi: 10.1093/nar/14.14.5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takano K, Nakamura T, Sekiguchi M. Roles of two types of O6-methylguanine-DNA methyltransferases in DNA repair. Mutat Res. 1991;254:37–44. doi: 10.1016/0921-8777(91)90038-q. [DOI] [PubMed] [Google Scholar]
- 25.Nowosielska A, Smith SA, Engelward BP, Marinus MG. Homologous recombination prevents methylation-induced toxicity in Escherichia coli. Nucleic Acids Res. 2006;34:2258–2268. doi: 10.1093/nar/gkl222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 27.Romling U, Grothues D, Bautsch W, Tummler B. A physical genome map of Pseudomonas aeruginosa PAO. EMBO J. 1989;8:4081–4089. doi: 10.1002/j.1460-2075.1989.tb08592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowalczykowski SC. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem Sci. 2000;25:156–165. doi: 10.1016/s0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- 29.McGraw BR, Marinus MG. Isolation and characterization of Dam+ revertants and suppressor mutations that modify secondary phenotypes of dam-3 strains of Escherichia coli K-12. Mol Gen Genet. 1980;178:309–315. doi: 10.1007/BF00270477. [DOI] [PubMed] [Google Scholar]
- 30.Landini P, Volkert MR. Regulatory responses of the adaptive response to alkylation damage: a simple regulon with complex regulatory features. J Bacteriol. 2000;182:6543–6549. doi: 10.1128/jb.182.23.6543-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindahl T, Sedgwick B, Sekiguchi M, Nakabeppu Y. Regulation and expression of the adaptive response to alkylating agents. Annu Rev Biochem. 1988;57:133–157. doi: 10.1146/annurev.bi.57.070188.001025. [DOI] [PubMed] [Google Scholar]
- 32.Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005;33:3799–3811. doi: 10.1093/nar/gki681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper S, Helmstetter CE. Chromosome replication and the division cycle of Escherichia coli B/r. J Mol Biol. 1968;31:519–540. doi: 10.1016/0022-2836(68)90425-7. [DOI] [PubMed] [Google Scholar]
- 34.Feng G, Tsui HC, Winkler ME. Depletion of the cellular amounts of the MutS and MutH methyl-directed mismatch repair proteins in stationary-phase Escherichia coli K-12 cells. J Bacteriol. 1996;178:2388–2396. doi: 10.1128/jb.178.8.2388-2396.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu TH, Marinus MG. Deletion mutation analysis of the mutS gene in Escherichia coli. J Biol Chem. 1999;274:5948–5952. doi: 10.1074/jbc.274.9.5948. [DOI] [PubMed] [Google Scholar]
- 36.Lopez de Saro FJ, O’Donnell M. Interaction of the beta sliding clamp with MutS, ligase, and DNA polymerase I. Proc Natl Acad Sci U S A. 2001;98:8376–8380. doi: 10.1073/pnas.121009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matic I, Ekiert D, Radman M, Kohiyama M. Generation of DNA-free Escherichia coli cells by 2-aminopurine requires mismatch repair and nonmethylated DNA. J Bacteriol. 2006;188:339–342. doi: 10.1128/JB.188.1.339-342.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robbins-Manke JL, Zdraveski ZZ, Marinus M, Essigmann JM. Analysis of global gene expression and double-strand-break formation in DNA adenine methyltransferase-and mismatch repair-deficient Escherichia coli. J Bacteriol. 2005;187:7027–7037. doi: 10.1128/JB.187.20.7027-7037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calmann MA, Evans JE, Marinus MG. MutS inhibits RecA-mediated strand transfer with methylated DNA substrates. Nucleic Acids Res. 2005;33:3591–3597. doi: 10.1093/nar/gki673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldmacher VS, Cuzick RA, Jr, Thilly WG. Isolation and partial characterization of human cell mutants differing in sensitivity to killing and mutation by methylnitrosourea and N-methyl-N’-nitro-N-nitrosoguanidine. J Biol Chem. 1986;261:12462–12471. [PubMed] [Google Scholar]
- 41.Bignami M, Casorelli I, Karran P. Mismatch repair and response to DNA-damaging antitumour therapies. Eur J Cancer. 2003;39:2142–2149. doi: 10.1016/s0959-8049(03)00569-0. [DOI] [PubMed] [Google Scholar]
- 42.York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cejka P, Mojas N, Gillet L, Schar P, Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Curr Biol. 2005;15:1395–1400. doi: 10.1016/j.cub.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 44.Massey A, Offman J, Macpherson P, Karran P. DNA mismatch repair and acquired cisplatin resistance in E. coli and human ovarian carcinoma cells. DNA Repair (Amst) 2003;2:73–89. doi: 10.1016/s1568-7864(02)00187-8. [DOI] [PubMed] [Google Scholar]
- 45.Claij N, Te Riele H. Msh2 deficiency does not contribute to cisplatin resistance in mouse embryonic stem cells. Oncogene. 2004;23:260–266. doi: 10.1038/sj.onc.1207015. [DOI] [PubMed] [Google Scholar]
- 46.Marinus MG, Morris NR. Biological function for 6-methyladenine residues in the DNA of Escherichia coli K12. J Mol Biol. 1974;85:309–322. doi: 10.1016/0022-2836(74)90366-0. [DOI] [PubMed] [Google Scholar]