

Abstract
Calpains and caspases are two cysteine protease families that play important roles in regulating pathological cell death. Here, we report that m-calpain may be responsible for cleaving procaspase-12, a caspase localized in the ER, to generate active caspase-12. In addition, calpain may be responsible for cleaving the loop region in Bcl-xL and, therefore, turning an antiapoptotic molecule into a proapoptotic molecule. We propose that disturbance to intracellular calcium storage as a result of ischemic injury or amyloid β peptide cytotoxicity may induce apoptosis through calpain- mediated caspase-12 activation and Bcl-xL inactivation. These data suggest a novel apoptotic pathway involving calcium-mediated calpain activation and cross-talks between calpain and caspase families.
Keywords: calcium, Alzheimer's disease, Bcl-xL, endoplasmic reticulum, ER stress
Introduction
Calpains and caspases are two families of cysteine proteases that may be involved in mediating both acute and chronic neuronal cell deaths (Chan and Mattson 1999). Calpains are cytosolic calcium-activated neutral cysteine endopeptidases, and are ubiquitously distributed in all animal cells (Croall and Demartino 1991; Saido et al. 1994). The calpain family has at least six members, which can be divided into two groups based upon their tissue distribution: ubiquitous and tissue specific. The best characterized calpains are two ubiquitously expressed isozymes, μ- and m-calpain, which can be distinguished by their in vitro requirement for different levels of calcium for activation. μ-Calpain is activated in the presence of micromolar concentrations of calcium, whereas m-calpain requires millimolar concentrations of calcium (Croall and Demartino 1991; Saido et al. 1994). Calpain-mediated proteolysis proceeds in a limited manner but does not require a specific amino acid residue like that of caspases. Calpain has been proposed to play a role in regulating normal signal transduction processes by cleaving cytoskeletal proteins such as talin and α-actinin; membrane proteins such as the EGF receptor, integrin, N-CAM, and cadherin; and enzymes such as protein kinase C and calmodulin-dependent kinase (Saido et al. 1994). Calpain may play an important role in pathogenesis of diseases. Calpain activation has been proposed to contribute to neuronal cell death in ischemic brain injury (Saido et al. 1994), Alzheimer's disease (Saito et al. 1993; Tsuji et al. 1998), and myelin protein degradation in multiple sclerosis (Shields et al. 1998). Calpain inhibitors have been shown to have neuroprotective effects in models of focal (Markgraf et al. 1998) or global (Li et al. 1998) cerebral ischemia, delayed neuronal death after transient ischemia (Yokota et al. 1999), and myocardial infarction (Toda et al. 1989).
Caspases are mammalian homologues of Caenorhabditis elegans cell death gene product Ced-3, and they play an important role in regulating apoptosis (Zheng et al. 1999). 14 members of the mammalian caspase family have been identified that are widely expressed in a variety of tissues and cell types. Like calpain, caspases normally exist in cells as proenzymes, which may be activated through recruitment into activating complexes or direct proteolytic cleavage by another caspase (Cryns and Yuan 1998). Caspase activation has been shown to contribute to cell death in the ischemic brain (Namura et al. 1998), ischemic heart injury (Bialik et al. 1999) and neuronal loss in chronic neurodegenerative diseases such as Alzheimer's disease (Gervais et al. 1999) and Huntington's disease–related polyglutamine repeat expansion diseases (Sanchez et al. 1999; Sawa et al. 1999). Although calpain and caspase have both been proposed to play important roles in regulating pathological neuronal cell death, the interactions of these two families of proteases under pathological conditions are not clear.
In contrast to calpains, which mainly exist in the cytosol when inactive, procaspases may be present in different subcellular locations. For example, caspase-1 and caspase-3 are predominantly cytosolic (Nakagawa et al. 2000) while at least a portion of caspase-9 is mitochondrial (Li et al. 1997). Recently, we demonstrated a novel ER-specific apoptosis pathway mediated by caspase-12 (Nakagawa et al. 2000). We showed that procaspase-12 is predominantly localized at the ER, and is specifically activated by disturbances to ER homeostasis such as ER stress and mobilization of intracellular calcium ion store (Nakagawa et al. 2000). Caspase-12−/− mice are resistant to tunicamycin-induced renal epithelial cell death and lethality. Caspase-12−/− cortical neurons are resistant to amyloid-β protein–mediated neurotoxicity (Nakagawa et al. 2000). Since caspase-12 may play a critical role in regulating pathological cell death, it is important to understand its mechanism of activation.
ER plays a central role in protein biosynthesis and is also the major intracellular organelle involved in calcium storage. Intracellular calcium concentration is regulated by a combination of actions of calcium channels, calcium-binding proteins, and sequestration to the ER and other intracellular spaces. The calcium ion is involved in regulating many physiological events, such as fertilization, proliferation, and learning and memory in the central nervous system (Berridge 1993). Physiological release of calcium from ER storage is mediated through binding of inositol 1,4,5-triphosphate to inositol 1,4,5-trisphosphate receptor (IP3R) at the ER (Furuichi et al. 1989). Disruption of intracellular calcium homeostasis can often be lethal to cells (Berridge et al. 1998). Inhibition of N-glycosylation by tunicamycin and alterations of intracellular calcium homeostasis result in accumulation of unfolded proteins in ER and lead to induction of chaperone proteins and elevation of intracellular calcium, a phenomenon termed unfolded protein response or ER stress (Buckley and Whorton 1997; Sidrauski et al. 1998). Treatment of thapsigargin, an inhibitor of the intracellular calcium-ATP transporter and calcium ionophore (A23187), induces the elevation of calcium concentration in the cytosol. Disruption of intracellular calcium homeostasis also has been shown to contribute to neuronal degenerative diseases. Mutations in presenilin-1, a protein predominantly localized in ER, have been linked to the majority of inherited early onset Alzheimer's disease (Alzheimer Disease Collaborative Group 1995). Expression of mutant presenilin-1 disturbs the intracellular calcium homeostasis (Leissring et al. 1999). Synaptosomes from transgenic mice harboring presenilin-1 mutations exhibit elevated cytoplasmic calcium (Begley et al. 1999). These data suggest that the disturbance in the intracellular calcium homeostasis may contribute to the pathogenesis of Alzheimer's disease involving expression of mutant presenilin-1. Recently, calpain has been shown to cleave p35 to generate a toxic p25 fragment under neurotoxic conditions including ischemia and amyloid-β treatment (Patrick et al. 1999; Lee et al. 2000). These data indicated that disturbance in calcium homeostasis and calpain activation may contribute to ischemic neuronal injury and amyloid-β cytotoxicity.
Since both calpain and caspase-12 are activated by elevated intracellular calcium and both may play important roles in mediating neuronal degeneration, we examined the possible interaction between these two cysteine proteases. We demonstrate here that calpain is required for caspase-12 activation. Furthermore, we show that ischemia-induced calpain activation results in the Bcl-xL cleavage and most likely transformation of this antiapoptotic molecule into a proapoptotic form. We propose a novel pathway of calpain-mediated caspase-12 activation induced by disturbance of intracellular calcium homeostasis.
Materials and Methods
Materials
We used the following antibodies for Western blotting and chemicals for cell culture. Bcl-xL/S polyclonal antibody and Bcl-2 monoclonal antibody were purchased from Santa Cruz Biotechnology, Inc. Tubulin antibody was purchased from Sigma Chemical Co. IP3R antibody was purchased from Calbiochem. Grp78 and 94, calreticulin antibodies were purchased from Stressgen. α-Spectrin antibody was a gift from M.S. Lee and L.H. Tsai (Harvard Medical School, Boston, MA). Calpain inhibitor I (N-acetyl-Leu-Leu-Nle-CHO), II (N-acetyl-Leu-Leu-Met-CHO), and E-64d were purchased from Sigma Chemical Co. E-64 was purchased from Boehringer Mannheim. MDL28170 was a gift from Hoechst Marion Roussel, zVAD and amyloid β were purchased from Bachem.
Limited Digestion of Microsomal Proteins
Kidney microsome was prepared as described previously (Nakagawa et al. 2000). Microsomal proteins were digested by 0.05% trypsin-EDTA (GIBCO BRL) or HBSS (−) at 37°C for 5 or 10 min, and the reaction was stopped by adding SDS sample buffer. Proteins were analyzed by Western blotting using anti–caspase-12, anti-IP3R, anti-grp78 (BiP) and grp94, and anticalreticulin antibodies.
Cell Culture and Induction of Cell Death
Mixed glial cells were prepared from C57BL/6J mice around embryonic day 18. In brief, mouse brain cortex, free of meninges, was dissected and passed through a mesh (200–300 μm). Cells were collected by centrifugation (160 g, 5 min) and resuspended in culture medium containing MEM with 10% FBS, 5 mg/ml insulin, and 2% glucose. Cells were washed three times with DME without glucose (oxygen and glucose deprivation [OGD]), or with complete medium (oxygen deprivation [OD]), and then transferred to anaerobic chamber for hypoxia.
Primary cortical neurons were prepared as described previously (Nakagawa et al. 2000). 5 × 105/ml of cortical neurons were suspended in medium containing B27 in DME/F-12, and 4 d later 40 μM fibrillar Aβ (25-35) or 40 μM of fibrillar Aβ (25-35) plus 28 μM E-64 or 10 μM MDL28170 were added to cells. Calpain inhibitors were added every 24 h. Cells were collected after 48-h treatment.
2 × 105/ml of Rat1 cells were suspended in medium containing 10% FBS in DME. Full-length (1.6 μg) or truncated caspase-12 (1.6 μg) or pcDNA3 vector (1.6 μg) was transfected into Rat1 cells by the calcium phosphate method. The percentages of cell death were determined by morphology 24 h after transfection.
Cerebral Cortical S-100 and Cleavage Assay
Cortical S-100 was prepared in buffer B (20 mM Tris-HCl, pH 7.6, 2 mM EDTA, 10 mM EGTA, 0.25 M sucrose). A total of 300 μg S-100 was used for the cleavage assay in buffer C (12 mM Tris-HCl, pH 8.0, 5 mM β-mercaptoethanol, 0.2 mM EDTA, 1 mM EGTA, 25 mM sucrose plus calcium) at 30°C. 2 mM EDTA and 5 mM EGTA were added for calcium chelating.
In Vitro Cleavage Reaction by m-Calpain
pcDNA3.1 (Invitrogen)-Bcl-xL and pcDNA3/caspase-12 were transcribed and translated in vitro using a TNT kit (Promega) in the presence of [35S]methionine. In vitro cleavage reactions were performed in a buffer containing 150 mM NaCl, 20 mM Tris-Cl, pH 7.6, 1 mM DTT, and 100 mg/ml BSA with 5 mM calcium and m-calpain (1 U/ml; Sigma Chemical Co.) at 25°C. Reactions were terminated by the addition of SDS buffer and boiling. Samples were analyzed by SDS-PAGE and autoradiography.
Amino Acid Sequencing
pRSET (Invitrogen)-truncated mutant caspase-12 or pGEX-6P (Pharmacia Biotech) full-length Bcl-xL fusion proteins were cleaved in vitro by m-calpain (2 U) for 15 min at room temperature. Cleaved products were transferred to PVDF membrane (BioRad) and NH2-terminal peptides were sequenced by PE/ABD Procise 494 HT protein sequencing system (Harvard MicroChem).
Active Caspase-12 Bacteria Lysate and In Vitro Reaction
Escherichia coli HMS (DE3) pLysS carrying either pGEX-2T (Pharmacia Biotech) or expression plasmids, pRSET-truncated (T159-N419) caspase-12, was cultured in LB medium at 18°C for 12 h with 0.4 mM isopropyl-β-d-thiogalactopyranoside for induction of the recombinant protein. E. coli was harvested, washed in PBS, and broken by lysozyme and sonication in 25 mM Hepes, pH 7.5, 10% sucrose, 0.1% CHAPS, and 0.1 mM PMSF. The supernatant was used as active lysates after centrifugation at 16,000 g for 30 min. 35S-truncated caspase-12 was incubated with bacterial lysates in cleavage buffer (100 mM Hepes, pH 7.5, 10% sucrose, 0.1% CHAPS, 10 mM DTT, and 0.1 mM PMSF) at 37°C for 3 h, and analyzed by SDS-PAGE and autoradiography.
Results
Expression Pattern of Mouse Caspase-12
To determine the tissue expression pattern of caspase-12, we stained Western blots of mouse tissue lysates using an anti–caspase-12 antibody. We found that caspase-12 is ubiquitously expressed in postnatal day 4 mouse tissues (Fig. 1 A). Caspase-1 is also ubiquitously expressed, but the expression levels of caspase-1 are different from that of caspase-12 (Fig. 1 A). Caspase-12 expression was detected in the lysates of purified cortical neurons and glial cells (data not shown). We concluded that caspase-12 is widely expressed in a variety of cells.
Figure 1.

The expression of caspase-12. (A) A Western blot of caspase-12. Anti–caspase-1 and antitubulin antibodies were used as controls. Caspase-12 is constitutively expressed in postnatal day 4 mouse tissues. (B) Caspase-12 localizes on the outer membrane of the ER. IP3R, a transmembrane protein, was readily digested by trypsin as that of caspase-12, whereas ER lumenal proteins, grp78, grp94, and calreticulin were protected against trypsin digestion. ut, untreated.
Caspase-12 Is Localized on the Cytoplasmic Side of the ER
We have shown that procaspase-12 is localized in the ER (Nakagawa et al. 2000). To understand the mechanism of caspase-12 activation, we determined whether caspase-12 is localized on the lumenal or cytoplasmic side of the ER. We prepared the mouse kidney microsomal fraction and analyzed the orientation of caspase-12 by limited trypsin digestion (Fig. 1 B). We found that caspase-12, like the IP3 receptor, an ER resident protein facing the cytoplasmic side (Furuichi et al. 1989) was quickly digested by trypsin treatment (Fig. 1 B). In contrast, grp78 (BiP), grp94, and calreticulin, which are localized on the lumenal side of ER, were protected from trypsin digestion. These data indicate that caspase-12 is localized on the cytoplasmic side of the ER.
Cleavage of Caspase-12 and Bcl-xL after Oxygen and Glucose Deprivation
To determine the mechanism of caspase-12 activation, we examined caspase-12 processing in apoptotic glial cells. We have previously shown that caspase-12 is specifically activated by apoptotic stimuli with an ER stress component (Nakagawa et al. 2000). We found that oxygen and glucose deprivation (OGD) in cultured glial cells, which mimics ischemic condition in vivo, induced an ER stress response efficiently, as indicated by the induction of grp78 (BiP) and grp 94 using Western blot analysis (Fig. 2 A). Caspase-12 was cleaved into a number of protein fragments ∼35 kD in glial cells after treatment of OGD (Fig. 2 A), thapsigargin (2 μM), or A23187 (2 μM; Fig. 2 B). Consistent with the requirement of ER stress response for caspase-12 activation (Nakagawa et al. 2000), hypoxia (OD) alone did not induce BiP expression nor did it induce caspase-12 cleavage (Fig. 2 A). Interestingly, Bcl-xL, but not Bcl-2, was cleaved into a 25-kD proteolytic fragment in oxygen- and glucose-deprived glial cells (Fig. 2 A). We ruled out the possibility that this 25-kD protein was Bcl-xS, an alternative spliced form of Bcl-xL (Boise et al. 1993). Although Bcl-xL mRNA was detected, Bcl-xS mRNA was not detected in glial cells after OGD treatment by reverse transcriptase–PCR (data not shown). Since an antibody generated against the COOH-terminal end of Bcl-xL detects this 25-kD protein, the cleavage site is most likely in the loop domain of Bcl-xL (see below).
Figure 2.




The requirement of calpain for caspase-12 activation in vivo. (A) Caspase-12 and Bcl-xL are cleaved in glial cells under oxygen and glucose deprivation (OGD, 9-h treatment), but not under hypoxia condition (oxygen deprivation, OD). ER stress is induced by OGD but not by OD, as indicated by the induction of BiP in OGD but not OD-treated cells. Antitubulin was used as a control (bottom). UT; untreated. (B) Caspase-12 is cleaved in glial cells treated by ER stress–inducing agents (thapsigargin, 2 μM; A23187, 2 μM), which induces ER stress as indicated by BiP induction. (C and D) The effects of pan-caspase inhibitor (zVAD), and calpain inhibitor (calpain inhibitors I, II, and E64d) in glial cells treated by OGD. Calpain inhibitors (calpain inhibitors I and II; CI-I and -II, respectively), but not z-VAD, inhibited caspase-12 and Bcl-xL cleavages (C and D). zVAD and calpain inhibitor partially protected glial cell death induced by OGD (D). Calpain activation correlates with caspase-12 cleavage activity. Endogenous calpain substrate, α-spectrin, was cleaved to 145- and 150-kD fragments after OGD treatment (D), which indicates calpain activation. (E) Fibrillar Aβ peptide induced caspase-12 cleavage, and calpain inhibitors prevented caspase-12 cleavage in primary cortical neurons. The cleavage product of caspase-12 was detected by Western blotting after 48 h of treatment with 40 μM fibrillar Aβ (25-35). Arrows indicate cleaved caspase-12 fragment. no suppl: no supplement.
Inhibition of Caspase-12 and Bcl-xL Cleavage by Calpain Inhibitors
Since ER stress agents, tunicamycin, thapsigargin, and A23187, are all capable of inducing increases in cytosolic calcium concentration and calpain has been shown to play a role in mediating ischemic brain injury (Saido et al. 1994) and kidney necrosis (Edelstein et al. 1995), we examined the possible involvement of calpain in caspase-12 activation (Fig. 2c and Fig. d). Glial cells were subjected to oxygen and glucose deprivation in the presence of calpain inhibitors. Treatment of calpain inhibitors (calpain inhibitor I and II, E64d) resulted in inhibition of caspase-12 cleavage induced by OGD; on the contrary, the pan-caspase inhibitor zVAD.fmk (200 μM) had no effect on the cleavage of caspase-12 (Fig. 2 D). Caspase-12 cleavage correlated with calpain activation as indicated by calpain-specific cleavage of nonerythroid α-spectrin cleavage (Nath et al. 1996; Fig. 2 D). OGD-induced Bcl-xL cleavage was also inhibited by calpain inhibitors but not by zVAD (Fig. 2c and Fig. d). Proteasome inhibitors also had no effect on the cleavage of caspase-12 or Bcl-xL (data not shown). Furthermore, the cleavage product of Bcl-xL was detected in caspase-12−/− glial cells (data not shown). These data indicate that OGD-induced cleavage of both caspase-12 and Bcl-xL is mediated by calpain but not by caspases.
Although zVAD did not inhibit OGD-induced caspase-12 and Bcl-xL cleavage, zVAD did rescue a significant portion of cell death (Fig. 2 D). Although zVAD cannot inhibit caspase-12 cleavage, zVAD is effective in inhibiting caspase-12 activity (see Fig. 4 B). It is also possible that zVAD is inhibiting caspases in addition to caspase-12, which may be activated in parallel or downstream from caspase-12. E64d and calpain inhibitor I also rescued a significant portion of oxygen- and glucose-deprived glial cells (viability: 47.2% and 45.5% by MTT assay, respectively; Fig. 2 D), although E64d and calpain inhibitor I, like other inhibitors of calpain, are toxic to cells when incubated for an extended period of time. This could reflect the involvement of calpain in normal cell homeostasis (Wang et al. 1989; Shumway et al. 1999).
Figure 4.
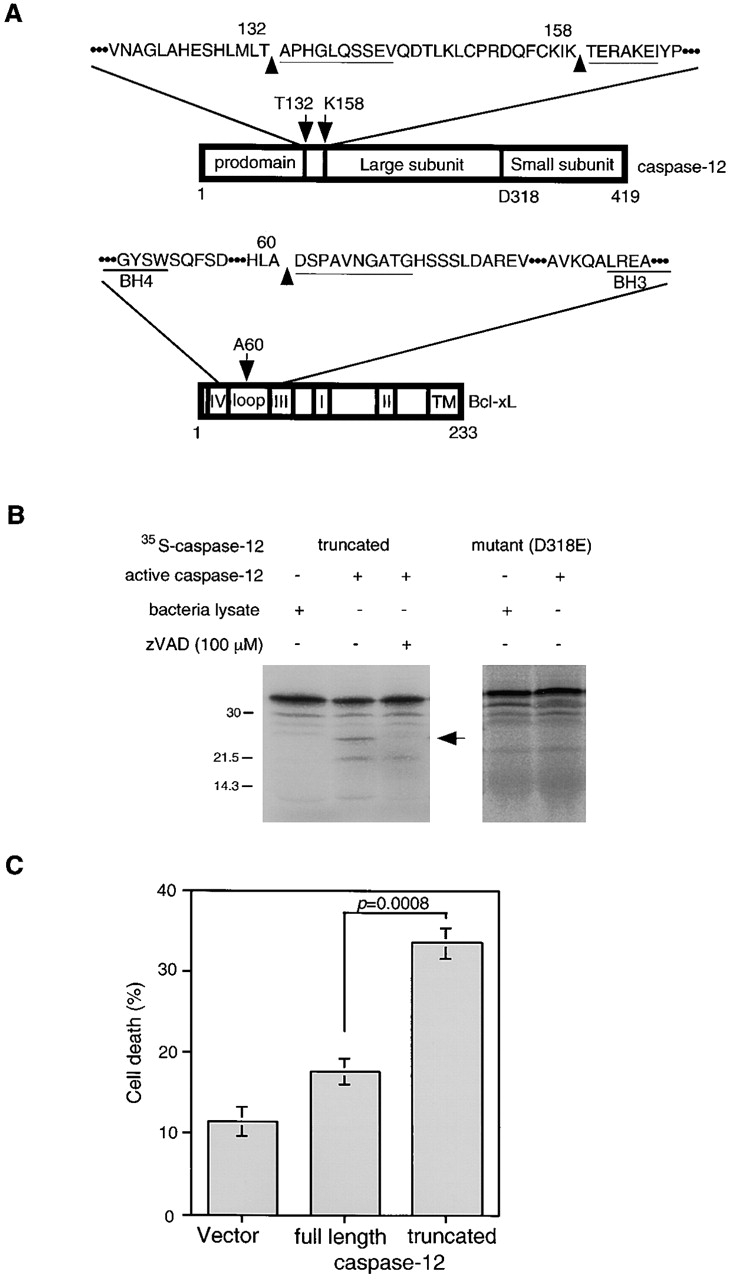
The cleavage sites in caspase-12 and Bcl-xL by m-calpain. (A) Caspase-12 is cleaved in at least two sites at T132/A133 and K158/T159. Bcl-xL is cleaved at one site, A60/D61. The cleavage site in Bcl-xL is close to that of caspase-1 or -3, (D61/S62 by caspase-1 and -3, D76/A77 by caspase-3; Clem et al. 1998; Fujita et al. 1998). The cleavage site of Bcl-xL is not conserved in Bcl-2. Underlines show sequence results. (B) Truncated (T159-N419) caspase-12 (t-caspase-12) expressed in bacteria is active in cleaving the wild-type caspase-12 but not D318E mutant. Caspase-12 activity is inhibited by zVAD. Arrow indicates the cleaved product of caspase-12. (C) t-Caspase-12 induced cell death in Rat1 cells. The percentage of cell death was determined by morphology 24 h after transfection. Data are averages (± SEM).
Caspase-12 Activation Is Induced by Fibrillar Aβ and Inhibited by Calpain Inhibitor in Primary Cortical Neurons
We have shown that caspase-12−/− cortical neurons are significantly more resistant to cell death induced by amyloid β peptide (1-40) than that of wild-type neurons, suggesting a critical role for caspase-12 in mediating neuronal cell death induced by fibrillar Aβ peptide (Nakagawa et al. 2000). To test if calpain plays a role in mediating neuronal cell death induced by amyloid β peptide, we incubated mouse primary cortical neurons with fibrillar Aβ in the presence or absence of calpain inhibitors (E64 and MDL28170) for 48 h. Western blotting with anti–caspase-12 antibody showed in cortical neurons after treatment of fibrillar Aβ, caspase-12 was cleaved into a fragment identical to that of the caspase-12 fragment observed after OGD treatment (Fig. 2 E). Since there were only 20–30% of cortical neurons sensitive to fibrillar Aβ neurotoxicity, the majority of caspase-12 stayed intact in these cultures. The appearance of this caspase-12 fragment was inhibited by the addition of calpain inhibitors E64 or MDL28170 (Fig. 2 E). From these results, we concluded that calpain is also required for fibrillar Aβ peptide–induced caspase-12 activation.
The Involvement of m-Calpain in Calcium-dependent Caspase-12 Cleavage
Since calcium is essential for activation of calpain, we reasoned that calcium should be essential for caspase-12 activation as well. To examine the requirement of calcium for caspase-12 cleavage, we used the S-100 fraction of mouse cerebral cortex as a source of calpain to cleave in vitro–translated caspase-12. We found that the cortical S-100 cleaved the full-length caspase-12 into three fragments each ∼35 kD in the presence of millimolar but not micromolar calcium (Fig. 3 A, lane 3). This cleavage was inhibited by the addition of calcium chelators EGTA and EDTA (Fig. 3 A, lane 4). zVAD was totally ineffective in inhibiting caspase-12 cleavage similar to that of in vivo cleavage of caspase-12 induced by OGD (data not shown). This result suggests that caspase-12 is cleaved by m-calpain, rather than μ-calpain in cortical S-100.
Figure 3.


Cleavage of caspase-12 by calpain in vitro. (A) Cleavage of caspase-12 by cerebral cortex–soluble (S-100) proteins in a calcium-dependent manner. Caspase-12 is effectively cleaved in the presence of millimolar, but not micromolar, calcium. Activation of caspase-12 is inhibited by calcium chelators (EGTA and EDTA). (B) Caspase-12 and Bcl-xL are cleaved by purified m-calpain in vitro. (a) Caspase-12 is cleaved by m-calpain to generate three major fragments each ∼35 kD, which may autoactivate themselves to generate fragments of 20 and 10 kD (arrows), which may be the large and small subunits of caspase-12, respectively. The 20-kD fragment is detected by anti–caspase-12 p20 antibody after a longer incubation with calpain (b). (c) Bcl-xL is cleaved by m-calpain in vitro. The arrow indicates the cleaved product that was sequenced. (C) The cleavage products of caspase-12 in vivo are similar in sizes to those generated by in vitro cleavage assays (S100 and purified m-calpain). The arrows point to the procaspase-12 (arrows 1 and 2) and the cleaved products of caspase-12 (arrows 3–5). Cleaved products (arrows 3 and 5) were sequenced. Cleavage sites of 3 and 5 are T132/A133 and K158/T159, respectively. The large subunit of caspase-12 (p20) is produced in a time-dependent manner.
To confirm the involvement of m-calpain in caspase-12 cleavage, we examined in vitro cleavage of 35S-caspase-12 and 35S-Bcl-xL by purified m-calpain (Fig. 3 B, a and c). Incubating 35S-caspase-12 with purified m-calpain resulted in the formation of three fragments of caspase-12 ∼35 kD and a 20-kD fragment, all of which were recognized by the anti–caspase-12 p20 large subunit antibody (Fig. 3 B, b) and very similar in their respective sizes to that observed in vivo. Cleavage of Bcl-xL by calpain generated two fragments of 25 and 13 kD; the 25-kD fragment was recognized by the anti–COOH-terminal antibody of Bcl-xL.
To further confirm that the cleavage products of caspase-12 by calpain in vitro are indeed the same as those observed in vivo, we compared the sizes of endogenous caspase-12 fragments in apoptotic glial cells induced by OGD and those caspase-12 cleavage products generated by incubating with S-100 or purified m-calpain side by side on the same Western blot (Fig. 3 C). OGD induced expression of a protein (asterisk) that is slightly bigger than the full-length caspase-12 (1). This protein is expressed at a very low level under control conditions and may be related to caspase-12, as it is recognized by the anti–caspase-12 antibody. Caspase-12 is expressed in two forms, the larger one (1) in vivo (Fig. 3 C, first four lanes) exhibit slightly faster mobility on SDS-PAGE than the larger one (1) in vitro (Fig. 3 C, middle four lanes). The smaller form (2) is identical in sizes in vivo and in vitro (Fig. 3 C). Three caspase-12 fragments ∼35 kD were detected in OGD-treated glial cells; three caspase-12 fragments (3–5) of very similar sizes were also detected in S-100 and calpain-cleaved caspase-12 in vitro (Fig. 3 C). Since m-calpain and S-100 in the presence of calcium produced three fragments very similar to that in vivo, we concluded that calpain may be involved in caspase-12 and bcl-xL cleavage in OGD-treated cells.
Activation of Caspase-12 and Inactivation of Bcl-xL by m-Calpain
To identify the cleavage sites in caspase-12 and Bcl-xL by m-calpain, we incubated recombinant caspase-12 and Bcl-xL with m-calpain and the cleavage products were isolated and sequenced. The two major calpain cleavage sites in caspase-12 were T132/A133 and K158/T159, corresponding to the fragments 3 and 5 (Fig. 3 C). Both cleavage sites are between the prodomain and the large subunit (Fig. 4 A). Bcl-xL was cleaved at one site in the loop domain, A60/D61 (Fig. 4 A), which corresponds to the 25-kD Bcl-xL fragment. Interestingly, this cleavage site in Bcl-xL by calpain is very close to the reported cleavage sites in Bcl-xL by caspase-1 (D61/S62; Clem et al. 1998) and caspase-3 (D61/S62 and D76/A77; Fujita et al. 1998). Since the cleaved products of Bcl-xL by caspase-1 or -3 have proapoptotic activity (Clem et al. 1998; Fujita et al. 1998), the cleaved product of Bcl-xL by m-calpain most likely has a similar function. These data suggest that active calpain may convert procaspase-12 into active caspase-12 and Bcl-xL from an antiapoptotic protein into a proapoptotic form.
To confirm that cleavage of caspase-12 at K158 indeed activates caspase-12, we next examined whether bacterially expressed T159-N419 caspase-12 (t-caspase-12), mimicking calpain cleavage product of caspase-12, is active. Although bacterially expressed t-caspase-12 is largely present in the inclusion body and poor in solubility, the bacterial lysate expressing t-caspase-12, but not control lysate, was active in cleaving itself, and its cleavage activity was inhibited by zVAD (Fig. 4 B). In contrast, the full-length caspase-12 expressed in E. coli was not active (data not shown). Procaspases are usually cleaved between the large and the small subunits during activation (Thornberry et al. 1992). Caspase-12 has a predicted cleavage site, D318, which is located between the large and the small subunits. To determine the cleavage site of caspase-12, we generated a mutant caspase-12, D318E. Active caspase-12 cleaved the wild-type but not the D318E caspase-12 (Fig. 4 B). These data suggest that calpain-cleaved 35-kD caspase-12 fragments are active, which may further cleave themselves between the large and small subunits to generate mature caspase-12. To further compare the activity of full-length and truncated caspase-12, we transfected the expression vectors of full-length versus the 35-kD fragment into Rat1 cells, and compared cell death after 24 h. The truncated caspase-12 was significantly more active in inducing cell death (33.6 ± 2.0%) than that of full-length caspase-12 (17.7 ± 1.6%). From these results, we concluded that cleavage of caspase-12 before T159 likely results in the activation of caspase-12.
Discussion
In this paper, we described a new mode of caspase activation. Previously, two modes of caspase activation have been described. The long prodomain caspases may be activated through recruitment into caspase activating complexes and activated through the induced proximity model (Budihardjo et al. 1999). For example, caspase-8 is activated through direct recruitment into Fas and TNF death signaling complex, whereas caspase-9 is activated through recruitment into Apaf-1/cytochrome c complex. On the other hand, the short prodomain-containing caspases such as caspase-3 and caspase-7 are activated through direct proteolytic cleavage by another active caspase. In this paper, we described that caspase-12 may be activated by direct proteolytic cleavage by a noncaspase protease, namely calpain. Procaspase-12 is localized on the cytoplasmic side of ER, whereas inactive calpain is mainly cytosolic. On the other hand, calpain, upon activation by elevated intracellular calcium, is known to translocate from the cytosol to the membrane (Suzuki 1987), where it may cleave procaspase-12. It is possible that membrane association of caspase-12 is a part of its inhibitory mechanism to prevent its activation under normal conditions.
It is interesting to note that although caspase-12 is downstream from calpain in mediating OGD-induced cell death, zVAD-fmk is considerably more effective in inhibiting OGD-induced cell death than that of calpain inhibitors. Calpain inhibitors consistently exhibit significant toxicity toward normal living cells; this is likely because of the role of calpain in regulating cytoskeleton, signal transduction, and metabolic pathways of normal cells. On the other hand, caspase inhibitors have exhibited significantly lower toxicity than that of calpain towards living cells. As caspase-12 is downstream from calpain, the effectiveness of the caspase inhibitor also suggests that calpain activation is not sufficient to induce apoptosis. In general, caspase inhibitors may be more suitable as therapeutical agents than that of calpain inhibitors. Recently, however, it was shown that caspase-8 may play a role in regulating erythropoiesis by cleavage of GATA-1, a transcription factor required for the terminal differentiation of erythroid precursor cells (De Maria et al. 1999). It remains to be seen whether other caspases may have a role in processes other than apoptosis. Thus, it may be critical to develop specific caspase inhibitors that can discriminate difference between caspases. Since caspase-12−/− mice develop and behave normally, caspase-12 and its human homologue may be better suited as a disease target for treatment of chronic neurodegenerative diseases.
Active caspase-12 is not the only cytotoxic agent activated by calpain when neurons and glias are subjected to ischemic injury in vivo or OGD in culture since caspase-12−/− cells are not resistant to OGD. In contrast, amyloid β peptide–induced neurotoxicity appears to be mediated predominantly through caspase-12 as caspase-12−/− cortical neurons are more resistant to Aβ than that of wild type (Nakagawa et al. 2000). The basis of such dichotomy may be due to the scale of calpain activation as well as the kinetics of cleavage. Although we can detect Bcl-xL cleavage readily in OGD-treated cells, we cannot detect the cleavage of Bcl-xL in Aβ peptide–treated neurons (data not shown). OGD may not target the ER specifically; it has impact on mitochondria as well (Perez-Pinzon et al. 1999). Thus, OGD may have caused calcium release from mitochondria as well as from the ER, which may have resulted in higher increases in intracellular calcium concentration and more generalized activation of calpain than cells treated by Aβ peptide. In addition, we noticed that in vitro, caspase-12 is cleaved much more readily than that of Bcl-xL by calpain (Fig. 3 B). Thus, OGD may have created at least two killer proteins, active caspase-12 and truncated Bcl-xL (Clem et al. 1998; Fujita et al. 1998), which may explain why the loss of caspase-12 alone is not sufficient for inhibiting apoptosis induced by OGD, whereas treatment of Aβ peptide activates caspase-12 specifically and inhibition of caspase-12 is sufficient to block Aβ peptide–induced cell death. We conclude that caspase-12 may be an attractive therapeutic target for Alzheimer's disease.
Acknowledgments
We thank Dr. Yong-Keun Jung (Kwangju Institute of Science and Technology, Korea) for helpful discussion and Drs. Honglin Li, Or Gozani, and Alexei Degterev (Yuan's lab) for critical reading of this manuscript. We thank M.S. Lee and Dr. L.H. Tsai for α-spectrin antibody.
This work was supported in part by grants from Hoechst-Marion-Roussle and Harvard Medical School project (to J. Yuan) and from the Human Frontier Science Program Organization and TOYOBO Biotechnology Foundation (to T. Nakagawa).
Footnotes
Abbreviations used in this paper: Aβ, amyloid β; IP3R, inositol 1,4,5-trisphosphate receptor; OD, oxygen deprivation; OGD, oxygen and glucose deprivation; t-caspase-12, T159-N419 caspase-12.
References
- Alzheimer's Disease Collaborative Group The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat. Genet. 1995;11:219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- Begley J.G., Duan W., Chan S., Duff K., Mattson M.P. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J. Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- Berridge M.J. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berridge M.J., Bootman M.D., Lipp P. Calciuma life and death signal. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- Bialik S., Cryns V.L., Drincic A., Miyata S., Wollowick A.L., Srinivasan A., Kitsis R.N. The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes. Circ. Res. 1999;85:403–414. doi: 10.1161/01.res.85.5.403. [DOI] [PubMed] [Google Scholar]
- Boise L.H., Gonzalez-Garcia M., Postema C.E., Ding L., Lindsten T., Turka L.A., Mao X., Nunez G., Thompson C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- Buckley B.J., Whorton A.R. Tunicamycin increases intracellular calcium levels in bovine aortic endothelial cells. Am. J. Physiol. 1997;273:C1298–C1305. doi: 10.1152/ajpcell.1997.273.4.C1298. [DOI] [PubMed] [Google Scholar]
- Budihardjo I., Oliver H., Lutter M., Luo X., Wang X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Chan S.L., Mattson M.P. Caspase and calpain substratesroles in synaptic plasticity and cell death. J. Neurosci. Res. 1999;58:167–190. [PubMed] [Google Scholar]
- Clem R.J., Cheng E.H., Karp C.L., Kirsch D.G., Ueno K., Takahashi A., Kastan M.B., Griffin D.E., Earnshaw W.C., Veliuona M.A., Hardwick J.M. Modulation of cell death by Bcl-XL through caspase interaction. Proc. Natl. Acad. Sci. USA. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croall D.E., Demartino G.N. Calcium-activated neutral protease (calpain) systemstructure, function, and regulation. Physiol. Rev. 1991;71:813–847. doi: 10.1152/physrev.1991.71.3.813. [DOI] [PubMed] [Google Scholar]
- Cryns V., Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- De Maria R., Zeuner A., Eramo A., Domenichelli C., Bonci D., Grignani F., Srinivasula S.M., Alnemri E.S., Testa U., Peschle C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. 1999;401:489–493. doi: 10.1038/46809. [DOI] [PubMed] [Google Scholar]
- Edelstein C.L., Wieder E.D., Yaqoob M.M., Gengaro P.E., Burke T.J., Nemenoff R.A., Schrier R.W. The role of cysteine proteases in hypoxia-induced rat renal proximal tubular injury. Proc. Natl. Acad. Sci. USA. 1995;92:7662–7666. doi: 10.1073/pnas.92.17.7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N., Nagahashi A., Nagashima K., Rokudai S., Tsuruo T. Acceleration of apoptotic cell death after the cleavage of Bcl-XL protein by caspase-3-like proteases. Oncogene. 1998;17:1295–1304. doi: 10.1038/sj.onc.1202065. [DOI] [PubMed] [Google Scholar]
- Furuichi T., Yoshikawa S., Miyawaki A., Wada K., Maeda N., Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–38. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- Gervais F.G., Xu D., Robertson G.S., Vaillancourt J.P., Zhu Y., Huang J., LeBlanc A., Smith D., Rigby M., Shearman M.S. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Lee M.S., Kwon Y.T., Li M., Peng J., Friedlander R.M., Tsai L.H. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Leissring M.A., Paul B.A., Parker I., Cotman C.W., LaFerla F.M. Alzheimer's presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J. Neurochem. 1999;72:1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li P.A., Howlett W., He Q.P., Miyashita H., Siddiqui M., Shuaib A. Postischemic treatment with calpain inhibitor MDL 28170 ameliorates brain damage in a gerbil model of global ischemia. Neurosci. Lett. 1998;247:17–20. doi: 10.1016/s0304-3940(98)00266-3. [DOI] [PubMed] [Google Scholar]
- Markgraf C.G., Velayo N.L., Johnson M.P., McCarty D.R., Medhi S., Koehl J.R., Chmielewski P.A., Linnik M.D. Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke. 1998;29:152–158. doi: 10.1161/01.str.29.1.152. [DOI] [PubMed] [Google Scholar]
- Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A., Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Namura S., Zhu J., Fink K., Endres M., Srinivasan A., Tomaselli K.J., Yuan J., Moskowitz M.A. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath R., Raser K.J., Stafford D., Hajimohammadreza I., Posner A., Allen H., Talanian R.V., Yuen P., Gilbertsen R.B., Wang K.K. Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1beta-converting-enzyme-like protease(s) in apoptotic cellscontributory roles of both protease families in neuronal apoptosis. Biochem. J. 1996;319:683–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick G.N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., Tsai L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon M.A., Xu G.P., Born J., Lorenzo J., Busto R., Rosenthal M., Sick T.J. Cytochrome c is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J. Cereb. Blood Flow Metab. 1999;19:39–43. doi: 10.1097/00004647-199901000-00004. [DOI] [PubMed] [Google Scholar]
- Saido T.C., Sorimachi H., Suzuki K. Calpainnew perspectives in molecular diversity and physiological-pathological involvement. FASEB (Fed. Am. Soc. Exp. Biol.) J. 1994;8:814–822. [PubMed] [Google Scholar]
- Saito K., Elce J.S., Hamos J.E., Nixon R.A. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer's diseasea potential molecular basis for neuronal degeneration. Proc. Natl. Acad. Sci. USA. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I., Xu C.J., Juo P., Kakizaka A., Blenis J., Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- Sawa A., Wiegand G.W., Cooper J., Margolis R.L., Sharp A.H., Lawler J.F., Jr., Greenamyre J.T., Snyder S.H., Ross C.A. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- Shields D.C., Tyor W.R., Deibler G.E., Hogan E.L., Banik N.L. Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc. Natl. Acad. Sci. USA. 1998;95:5768–5772. doi: 10.1073/pnas.95.10.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumway S.D., Maki M., Miyamoto S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. J. Biol. Chem. 1999;274:30874–30881. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- Sidrauski C., Chapman R., Walter P. The unfolded protein responsean intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998;8:245–249. doi: 10.1016/s0962-8924(98)01267-7. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Calcium-activated neutral protease and its endogenous inhibitor. Activation at the cell membrane and biological function. FEBS (Fed. Eur. Biochem Soc.) Lett. 1987;220:271–277. doi: 10.1016/0014-5793(87)80828-1. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Bull H.G., Calaycay J.R., Chapman K.T., Howard A.D., Kostura M.J., Miller D.K., Molineaux S.M., Weidner J.R., Aunins J. A novel heterodimeric cysteine protease is required for interleukin-1beta processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Toda G., Matsushita S., Kuramoto K., Oda S., Ezaki H., Hattori A., Kawashima S. Calcium-activated neutral protease inhibitor (E-64c) and reperfusion for experimental myocardial infarction. Jpn. Heart. J. 1989;30:375–386. doi: 10.1536/ihj.30.375. [DOI] [PubMed] [Google Scholar]
- Tsuji T., Shimohama S., Kimura J., Shimizu K. m-Calpain (calcium-activated neutral proteinase) in Alzheimer's disease brains. Neurosci. Lett. 1998;248:109–112. doi: 10.1016/s0304-3940(98)00348-6. [DOI] [PubMed] [Google Scholar]
- Wang K.K., Villalobo A., Roufogalis B.D. Calmodulin-binding proteins as calpain substrates. Biochem. J. 1989;262:693–706. doi: 10.1042/bj2620693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota M., Tani E., Tsubuki S., Yamaura I., Nakagaki I., Hori S., Saido T.C. Calpain inhibitor entrapped in liposome rescues ischemic neuronal damage. Brain Res. 1999;819:8–14. doi: 10.1016/s0006-8993(98)01334-1. [DOI] [PubMed] [Google Scholar]
- Zheng T.S., Hunot S., Kuida K., Flavell R.A. Caspase knockoutsmatters of life and death. Cell Death Differ. 1999;6:1043–1053. doi: 10.1038/sj.cdd.4400593. [DOI] [PubMed] [Google Scholar]