

Abstract
Axotomized neurons have several characteristics that are different from intact neurons. Here we show that, unlike established cultures, the axotomized sympathetic neurons deprived of NGF become committed to die before caspase activation, since the same proportion of NGF-deprived neurons are rescued by NGF regardless of whether caspases are inhibited by the pan-caspase inhibitor Boc-Asp(O-methyl)-CH2F (BAF). Despite prolonged Akt and ERK signaling induced by NGF after BAF treatment has prevented death, the neurons fail to increase protein synthesis, recover ATP levels, or grow. Within 3 d, all the mitochondria disappear without apparent removal of any other organelles or loss of membrane integrity. Although NGF does rescue intact BAF-treated 6-d cultures after NGF deprivation, rescue by NGF fails when these neurons are axotomized before NGF deprivation and BAF treatment. Moreover, cytosolic cytochrome c rapidly kills axotomized neurons. We propose that axotomy induces signals that make sympathetic neurons competent to die prematurely. NGF cannot repair these NGF-deprived, BAF-treated neurons because receptor signaling (which is normal) is uncoupled from protein renewal, and the mitochondria (which are damaged) go on to be eliminated. Hence, the order of steps underlying neuronal death commitment is mutable and open to regulation.
Keywords: apoptosis, caspases, mitochondria, NGF, survival signaling
Introduction
The concept of death commitment is used to describe the time after which cells exposed to death-inducing stimuli can no longer be rescued by survival factors. Determining the mechanism of death commitment is important for prevention of cell degeneration; yet, the molecular basis for death commitment is still poorly understood. In the nervous system, caspase inhibitors such as z-Val-Ala-Asp-(O-methyl)-CH2F (zVAD.fmk) or Boc-Asp(O-methyl)-CH2F (BAF) have been reported to prevent neuronal apoptosis after a wide variety of insults (Hara et al. 1997; Yakovlev et al. 1997; Chen et al. 1998; Cheng et al. 1998; Fink et al. 1998; Braun et al. 1999; Chaudhary et al. 1999; Kermer et al. 1999; Schierle et al. 1999; Schulz et al. 1999) and, therefore, it has been proposed that caspase inhibition could be a useful therapeutic strategy in pathologies involving neuronal loss (Kumar 1999; Schulz et al. 1999). However, in many cases, it is not yet clear whether caspase inhibition prevents death upstream of death commitment and, hence, is sufficient to maintain the long-term survival of cells.
Evidence suggesting that death commitment is commensurate with caspase activation in established sympathetic (superior cervical ganglia [SCG]) neuron cultures was provided by Deshmukh et al. 1996, who showed that inhibition of apoptosis by BAF was sufficient to support cells that could be rescued by NGF even after several days of NGF deprivation. Although neurons had decreased markedly in size, they resumed normal growth upon readdition of NGF. In support, Neame et al. 1998 showed that cytochrome c (cyt c), a cofactor for caspase activation (Liu et al. 1996), was necessary for apoptosis in SCG neurons and was released from the mitochondria after NGF deprivation, but was not sufficient for death since injection of cyt c into the cytosol of NGF-maintained neurons did not trigger apoptosis. Deshmukh and Johnson 1998 invoked the development of a second event also required for the activation of caspases and death which they termed “competence-to-die.” Further evidence that death commitment of established SCG neuron cultures is caspase-dependent was provided by Martinou et al. 1999, who showed that the addition of NGF to NGF-deprived/BAF-treated neurons that had progressed to the point where cyt c had been degraded promoted resynthesis of cyt c, refilling of the mitochondria and normal growth.
However, we have reported that NGF addition fails to rescue newly isolated SCG neurons after death by apoptosis is prevented by BAF (Xue et al. 1999). Thus, in these neurons, death commitment appears to be caspase-independent, which is similar to several other cells that cannot be rescued from survival factor deprivation, an insult that is easily reversible (Miller et al. 1997; Villa et al. 1998; Gregoli and Bondurant 1999; Johnson et al. 1999; Stefanis et al. 1999). Furthermore, there is now evidence that treatment with caspase inhibitors in vivo may also provide only temporary protection against death (Kermer et al. 1999). Several explanations have been proposed for caspase-independent death commitment. Some cells in which mitochondria are damaged during an apoptotic stimulus may die by necrosis if ATP falls below a critical threshold (Hirsch et al. 1997; Leist et al. 1997). However, many cells in which apoptosis is prevented by caspase inhibitors do not die immediately by necrosis (McCarthy et al. 1997; Xiang et al. 1998; Xue et al. 1999). Activation of alternative death mechanisms that override caspase inhibition have been suggested, such as one involving autophagy (Xue et al. 1999).
The above results show that in SCG neurons, death commitment may be either caspase-dependent or caspase-independent, suggesting that the steps that control death commitment can vary in the same cell type. Several differences between established cultures and newly isolated neurons may explain the alteration in the steps leading to death commitment. For example, newly isolated neurons are removed at a stage when programmed cell death is about to begin and are highly vulnerable to NGF deprivation, whereas established cultures have been exposed to high amounts of NGF and have developed resistance to NGF deprivation since their intrinsic rate of death is slower than that of younger neurons (Edwards and Tolkovsky 1994; Easton et al. 1997). Alternatively, newly isolated neurons may have partially initiated death signaling events since they have undergone the recent trauma of axotomy that induces stress proteins such as c-Jun (Virdee et al. 1997), which has been associated with NGF deprivation–induced death (Estus et al. 1994; Ham et al. 1995), whereas established cultures have regenerated neurites and no longer express c-Jun protein.
In this study, we explore the reasons why death commitment in NGF-deprived newly isolated neurons cannot be delayed by inhibition of caspases, although caspase inhibition blocks apoptosis. We show that axotomy reverts the behavior of established neurons to that of newly isolated neurons by delivering signals that both advance the death commitment point and makes neurons competent-to-die in response to cytosolic cyt c. We further show that death commitment is due to an underlying failure in mitochondrial function, which cannot be repaired by NGF despite normal signaling through Trk. Damaged mitochondria are selectively eliminated and neurons eventually die by a slow process of starvation, which we propose to name “limoktonia.” (Part of these results were presented at the Mitochondrial and Cell Death Workshop. 666th Biochemical Society meeting. Sheffield University, UK, 1998).
Materials and Methods
Antibodies
Antiactive mitogen-associated protein kinase was from Promega, anti–phospho-Akt from New England Biolabs, anti-ERK (MK12) from Transduction Laboratories, and anti–cyt c antibodies were from PharMingen.
Cell Culture
SCGs were dissected from 1-d-old Wistar rat pups, and sympathetic neurons were extracted, purified, and cultured as described previously (Virdee and Tolkovsky 1995). To purify the neurons, cells were preplated twice for 0.5–1 h each on collagen-coated culture dishes in L15-CO2 medium containing 5% rat serum under an atmosphere of 5% CO2 at 37°C, and nonadherent neurons were collected by centrifugation. Purified neurons were grown on a poly-l-lysine/laminin substrate in L15-CO2 medium containing 0.6% glucose, 20 μM uridine/fluorodeoxyuridine, 3% rat serum, with or without 50 ng/ml 2.5S NGF and 100 μM BAF (Enzyme System Products) for varying periods of time as indicated. Some cultures were grown for 6 d (6 DIV) with 50 ng/ml NGF before experimentation. The half-life of biologically active BAF (100 μM) in medium bathing SCG neurons was determined by reusing it to inhibit apoptosis of HeLa cells treated with TNFα and cycloheximide, which die by apoptosis within 4–8 h. About 50% of the original BAF activity remained in the medium after 24 h (a loss which also occurred in the absence of the neurons). However, 50 μM BAF was still fully capable of blocking neuronal apoptosis and caspase activation (Fletcher, 1999). BAF was replenished every 24 h when NGF deprivation exceeded 1 d.
Neuritotomy
Small aggregates (∼1,000 neurons) of purified 0-DIV neurons were created by growing the neurons overnight in suspension in growth medium containing 50 ng/ml NGF. Aggregates were plated as individual islets on laminin and grown for 6 d. Neurites surrounding each islet were cut with a small needle, and aggregates were collected, washed free of serum, trypsinized, and triturated according to the procedure used for isolating the neurons from ganglia. Neurons were plated as described above in various media as indicated.
Apoptosis, Survival, Cell Size, and MTT Assay Measurements
Apoptosis was scored after staining nuclei with 5 μg/ml Hoechst 33342 by counting the percentage of neurons with fragmented nuclei (at least 200 neurons per well) that we previously showed corresponds to the percentage of nuclei labeled by in situ end labeling (Edwards and Tolkovsky 1994). To score for survival, all of the phase-bright neurons in a prescribed field were counted at the beginning of the experiment, and counting was repeated at various times thereafter. At least 200 neurons were counted per sample. To measure the size of the cell bodies, images of several random fields of neurons were captured using a Panasonic CCD video camera, digitized using Apple Video Player, and imported into NIH Imager 1.62. The circumference of each cell body was traced with the freehand tool, and the area enclosed was determined. Cell body volume was estimated assuming spherical geometry. To relate cell size to metabolic activity, in some experiments, neurons were preincubated in the presence of 0.5 mg/ml 3-[4,5 dimethylthiazol-2-phenyl]-2,5-diphenyl-tetrazolium (MTT) for 30 min, and then imaged immediately as described above. The intensity of formazan staining in each cell was determined at the same time as cell size measurements.
Cyt c Trituration
Whole ganglia or aggregates of 6-DIV–neuritotomized neurons were trypsinized, washed, and triturated by uptake and expulsion 20 times through a 200-μl Gilson pipette tip in groups of 10 ganglia per point (Nobes and Tolkovsky 1995; Nobes et al. 1996). Trituration solutions (25 mg/ml of bovine cyt c or yeast cyt c [Sigma Chemical Co.] without or with 100 μM BAF) were prepared in L15 medium containing 5% rat serum. After trituration, cells were immediately preplated on collagen in the presence of NGF with or without BAF and cultured on laminin. Neurons were either scored for apoptosis after 4–12 h, or left to grow for several days. To calculate the amount of exogenous cyt c triturated into the neurons, cohort samples of neurons were collected and cyt c content was analyzed by Western blotting and quantified against known amounts of rat heart cyt c on the same blot using the Leica Q500 Quantimet image analysis system. To measure the number of cells loaded with cyt c, biotinylated cyt c (25 mg/ml; Sigma Chemical Co.), which does not induce apoptosis, was triturated into SCG neurons. After washing and preplating, neurons were cultured on poly-l-lysine/laminin–coated coverslips for 4 h in the presence of 50 ng/ml NGF, fixed with 3% paraformaldehyde, and stained using an antibiotin HRP-conjugated antibody with diaminobenzidine as the substrate. At least 80% of the neurons contained strong, diffuse biotin staining (data not shown).
ATP Measurement
Neurons (16,000 per well) were collected into an Eppendorf tube, washed once gently in ice-cold PBS by centrifugation, and rapidly lysed by boiling for 40 s in 30 μl of preheated glycine buffer (200 mM glycine-potassium hydroxide, pH 7.5). Samples were cooled, spun at 15,000 rpm for 10 min at 4°C, and the supernatants were stored at −20°C. Luciferase/luciferin reagent (freeze-dried in glycine buffer; Sigma Chemical Co.) was used to measure the amount of ATP luminometrically (Jade luminometer; Labtech International) according to manufacturer's instructions with the following modifications: the reagent was dissolved in water to a final concentration of 10 mg/ml and 100-μl aliquots were dispensed into a series of LP3 tubes. For each sample, background reading was set to zero, 10 μl of the sample was added, the tube was spun briefly, and readings were taken every 10 s over 1 min, yielding an average value. A standard curve was similarly constructed using known amounts of ATP. The assay was log linear between 0.1–100 fmol ATP/tube. Samples that fell outside the range of the standard curve were diluted and reread.
Protein Synthesis
Protein synthesis was measured as previously described (Buckmaster et al. 1991). In brief, neurons were incubated in the appropriate growth medium with [35S]methionine (Translabel™; ICN Biomedicals) for ∼18 h. Cultures were washed once with unlabeled medium and incubated three times with 5% ice-cold TCA to precipitate the protein in the wells. Protein precipitate was dissolved in 200 μl 15% SDS and a 100-μl aliquot was counted in a scintillation counter.
Preparation of Cell Extracts and Western Blot Analysis
After treatment, SCG neurons were washed three times with ice-cold PBS, collected by centrifugation, and solubilized in lysis buffer (20 mM Tris-HCl, pH 7.4, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 10 mM sodium-glycerophosphate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 0.1% 2-mercaptoethanol, 1 mM benzamidine, 5 mg/ml leupeptin, 0.2 mM PMSF fluoride, and 1% Triton X-100). Cell lysates were fractionated by SDS-PAGE, electrophoretically transferred to nitrocellulose, and the membrane was probed with antibodies as indicated. Immunocomplexes were detected using enhanced chemiluminescence, and the intensity of bands was quantified using the Leica Q500 Quantimet image analysis system. Images of blots were imported into Microsoft PowerPoint 4.0 without any change.
Immunocytochemistry
SCG neurons grown on poly-l-lysine/laminin–coated coverslips were fixed with 3% paraformaldehyde in PBS, blocked with 1% BSA and 0.1% saponin in PBS, and probed with a mouse mAb against cyt c, followed by anti-mouse IgG antibody conjugated with Cy3. The nuclei were stained with 2.5 μg/ml Hoechst 33342 during the final wash. Images of the neurons were captured using a Leica TCS-NT confocal system. Aperture settings were fixed throughout the experiment to compare intensities between different treatments without distortion. Images were imported into PowerPoint 4.0 without change.
Electron Microscopy
SCG neurons grown on poly-l-lysine/laminin–coated dishes were fixed with 3% glutaraldehyde in 0.1 M Pipes buffer, pH 7.2, containing 2 mM CaCl2 at 4°C for 4 h. The cells were collected by scraping and centrifugation at 1,000 g for 5 min. After four washes with the same buffer, the pellets were postfixed with 1% osmium ferricyanide in the above buffer for 1 h at 4°C, followed by three washes of H2O and 1-h block staining with 2% uranyl acetate in 0.5 M maleate buffer, pH 5.5. The samples were dehydrated and embedded in Spurr's epoxy resins. Ultrathin sections were stained with uranyl acetate and lead citrate, and observed in a Philips CM100 electron microscope. The total numbers of mitochondria (and in some cases, autophagic particles) in several individual cell sections were counted under the EM viewer. Data were sorted into 12 groups (sections containing 0–10 or >10 mitochondria) and plotted as a frequency of distribution.
Results
BAF Prevents Apoptotic Death of Newly Isolated (0-DIV) SCG Neurons but These Cells Cannot Be Rescued by NGF
Caspase inhibition using BAF has been shown to inhibit apoptotic death after NGF deprivation in both 0-DIV (Xue et al. 1999) and 6-DIV neurons, and it has been shown that 6-DIV neurons can recover and grow after readdition of NGF (Deshmukh et al. 1996). To investigate whether BAF also prevents newly isolated neurons from becoming committed to die, neurons were pretreated for 1 d in the absence or presence of 50 ng/ml NGF and/or 100 μM BAF, after which 100 ng/ml NGF was added to all the cultures and survival was followed for the next 25 d (Fig. 1). It can be seen that after a 1-d pretreatment, only 9 ± 5% (mean ± SEM, four independent experiments) of the NGF-deprived neurons survived but >95% of the BAF-treated/NGF-deprived neurons survived, which is similar to the survival of neurons cultured in the presence of NGF (in the absence or presence of BAF). After NGF was added, the BAF-treated/NGF-deprived neurons continued to survive during the next day but by 3 d ∼15% of the neurons had died despite the presence of NGF. From this time, more neurons died each day, 50% death being measured by ∼6 d after the beginning of the culture period (Xue et al. 1999). No toxic or metabolic effects of BAF treatment were apparent since ∼90% of the neurons cultured continuously in the presence of NGF and BAF survived up to 25 d and neurite outgrowth was not diminished (see Fig. 4).
Figure 1.

0-DIV SCG neurons deprived of NGF in the presence of BAF cannot be rescued by NGF, in contrast to established neuronal cultures. (A) 0-DIV SCG neurons were cultured for 24 h in the presence or absence of 50 ng/ml NGF and/or 100 μM BAF (treatment). After 24 h, half the medium was replaced with medium containing 200 ng/ml NGF (addition indicated by arrow), and NGF was replenished every 3 d. Between 150–300 neurons were counted per well, initially after 4–5 h of plating and in the same areas every 2–3 d as indicated, up to 25 d. (B) Purified SCG neurons were cultured for 6 d in medium containing 50 ng/ml NGF, after which some cultures were deprived of NGF by removal of medium and addition of anti-NGF antibody (treatment). NGF-maintained or NGF-deprived neurons were cultured in the absence or presence of 100 μM BAF for 3 d, after which NGF was added to all the wells as described above (addition indicated by right-most arrow). Neurons in a defined area were counted once immediately after medium replacement, and surviving cells were counted over the next 25 d. Percent survival was calculated by normalizing the counts to the counts of neurons maintained continuously in the presence of NGF (55–95% survival between different experiments). Data represent mean ± SD derived from four independent experiments in which deviations between triplicate wells were <5%.
Figure 4.
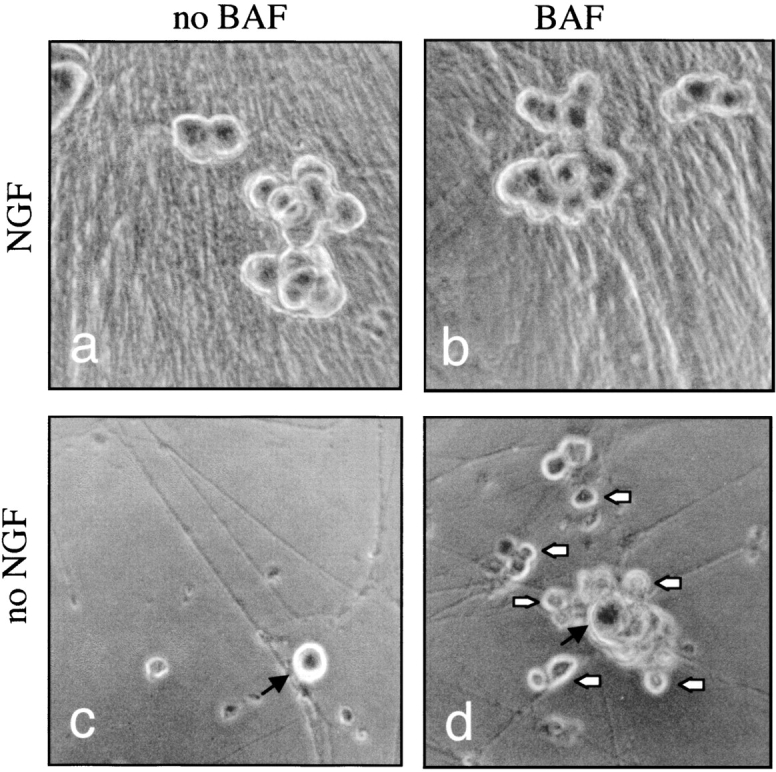
0-DIV SCG neurons fail to respond trophically to NGF stimulation after NGF deprivation and BAF treatment. 0-DIV neurons were initially cultured in the absence or presence of 50 ng/ml NGF and/or 100 μM BAF for 24 h, and then treated with 100 ng/ml NGF and photographed 9 d later. Initial conditions: (a) medium containing NGF; (b) medium containing NGF and BAF; (c) medium without NGF or BAF; (d) medium containing BAF but no NGF. Note small, live neurons in d and large neurons, which were rescued by NGF, in c and d.
Since these results were different to those reported by Deshmukh et al. 1996 for established SCG neurons, we treated 6-DIV cultures of SCG neurons with 100 μM BAF during 3 d of NGF deprivation (when >95% of the NGF-deprived neurons had died), and followed their survival after readdition of NGF. As shown in Fig. 1 B, NGF rescued ∼85% of the 6-DIV neurons after 22 d, in contrast to the lack of rescue of 0-DIV neurons. Survival of 6-DIV BAF-treated/NGF-deprived neurons rescued with NGF was maintained for at least 6 wk. Thus, BAF prevents the attainment of death commitment induced by NGF deprivation in 6-DIV but not newly isolated neurons.
The mode of death of BAF-treated/NGF-deprived 0-DIV neurons was nonapoptotic since there was no sign of DNA degradation, condensation, or fragmentation (see Fig. 4 and Fig. 6). Further, caspase activity was completely blocked since there was also no increase in the cleavage of the caspase substrates DEVD.AMC or VDVAD.AMC (VDVAD.AMC is a preferred caspase-2 substrate; Talanian et al. 1997; Mesner et al. 1999) in extracts from BAF-treated/NGF-deprived neurons, whereas cleavage of both substrates increased between 6–13-fold in extracts from NGF-deprived neurons (Xue et al. 1999) (no change in cleavage of other caspase substrates was detectable; Fletcher, 1999). Death was also not typically necrotic since cells did not become permeable to propidium iodide until they became phase-dark, and nuclei did not form the typical tight pyknotic knot associated with necrotic SCG neurons (induced, for example, by profound oxygen and glucose deprivation). These data show that caspase activity and execution of apoptosis was blocked by BAF in both 0-DIV and 6-DIV cultures of neurons (see Fig. 10). Thus, it appears that death commitment is caspase-independent in 0-DIV cells, but is caspase-dependent in 6-DIV SCG neurons.
Figure 6.

Mitochondria of NGF-deprived/BAF-treated 0-DIV neurons, but not 6-DIV neurons, fail to refill mitochondria with cyt c after NGF stimulation. (A) 0-DIV neurons were either grown with NGF for 4 d, or NGF-deprived and BAF-treated for 1 d, and then stimulated with 100 ng/ml NGF for 0 or 7 d. At each time, some cultures were fixed with 3% paraformaldehyde and stained with anti–cyt c antibody (cyt c), and with Hoechst 33342 to visualize nuclei (DNA), or proteins were extracted and analyzed by Western blotting with anti–cyt c followed by anti-ERK1/2 to normalize for loading. (B) 6-DIV neurons were either maintained with NGF or deprived of NGF for 3 d in the presence of BAF, after which the BAF-treated neurons were restimulated with NGF for 0 or 1 d and analyzed for cyt c as described in A.
Figure 10.


0-DIV and neuritotomized 6-DIV neurons are competent-to-die. (A) Neurons were triturated in the presence of 25 mg/ml bovine cyt c (a, c, d, and f) or yeast cyt c (b and e) in the absence or presence of 100 μM BAF as indicated. After purification, neurons were cultured in the presence of NGF (100 ng/ml) for 12 h and stained for DNA (a–c), or left to grow for 13 d (b–f). (Apoptosis after 12 h: cyt c, 74 ± 3%; cyt c + BAF, 5.3 ± 2%; and yeast cyt c, 1.7 ± 0.8%.) (B) 6-DIV neurons were neuritotomized as described in Fig. 8 and triturated under the same conditions described above alongside 0-DIV. After 4 h, neurons were stained with Hoechst 33342 and scored for apoptosis. Results are mean ± range of duplicate samples.
Death Commitment of 0-DIV NGF-deprived Neurons Is Unaltered by Caspase Inhibition
In the experiments above, NGF was added to 0-DIV or 6-DIV BAF-treated/NGF-deprived neurons when ∼90% of the control neurons had died (after 1 and 3 d, respectively). Since 0-DIV neurons become committed to die faster than 6-DIV neurons when deprived of NGF (Edwards and Tolkovsky 1994), it was possible that NGF failed to rescue BAF-treated 0-DIV neurons because of some secondary changes that occur during the period of NGF deprivation itself. Therefore, we tested whether NGF might rescue more 0-DIV neurons if the period of BAF treatment and NGF deprivation was reduced. Therefore, 0-DIV neurons were NGF-deprived in the presence of BAF for 8, 15, and 24 h before NGF was added, and survival over the next 15 d was compared with that of neurons that were deprived of NGF in the absence of BAF. Fig. 2 shows that at each time (8, 15, or 24 h) after NGF deprivation, NGF rescued the same proportion of neurons irrespective of BAF treatment (survival after 8 h of deprivation, 85 ± 3% or 81 ± 6% with or without BAF; after 15 h of deprivation, 50 ± 8% or 51 ± 1% with or without BAF; after 24 h of deprivation, 11 ± 2% or 13 ± 3% with or without BAF, respectively). Although NGF-deprived neurons died much faster in the absence of BAF (with 42 and 85% of the neurons dying by apoptosis within 15 and 24 h of NGF deprivation), there was no period of time during which treatment with BAF increased the potential of 0-DIV neurons to be rescued by NGF. These data strengthen the conclusion that 0-DIV neurons undergo a caspase-independent mechanism of death commitment.
Figure 2.
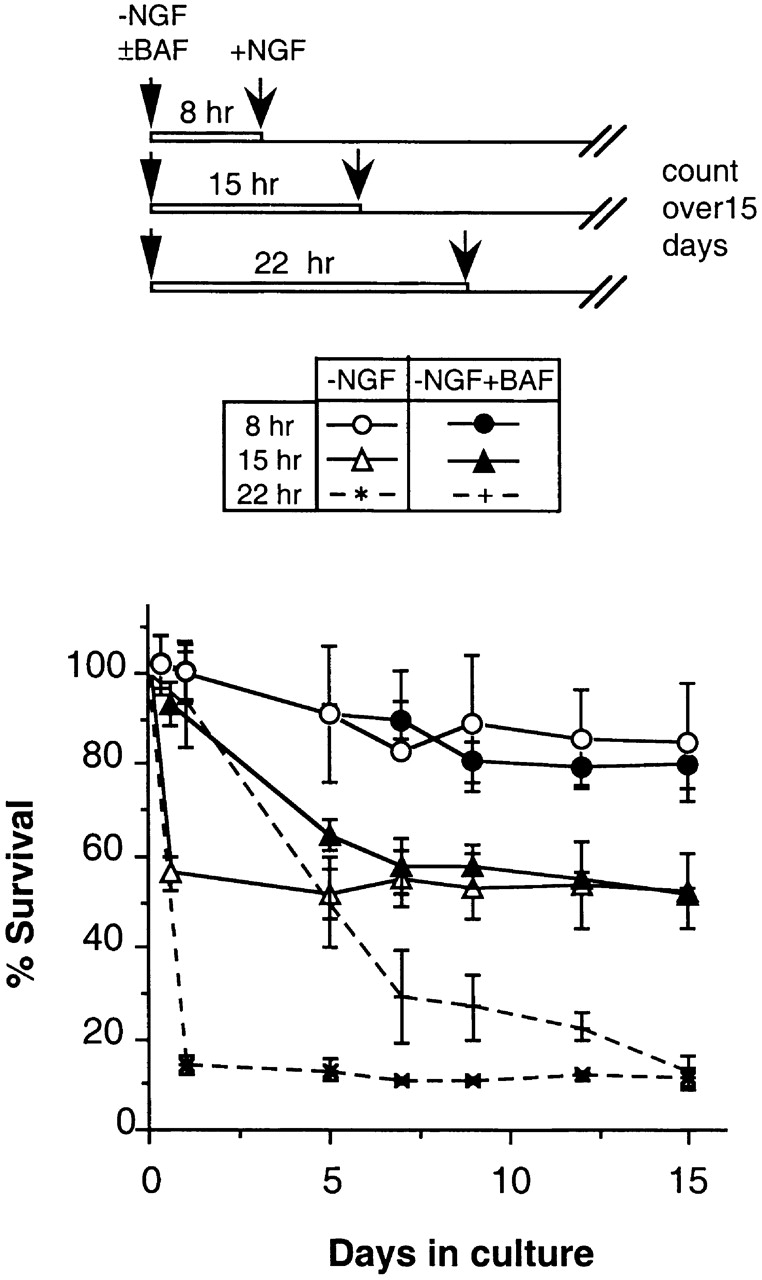
Death commitment of 0-DIV neurons is defined by the period of NGF deprivation and is unaffected by BAF. Three sets of 0-DIV neurons were plated in the absence of NGF, and in the absence (○, ▵, and *) or presence (•, ▴, and +) of 100 μM BAF. One set of neurons was transferred to 100 ng/ml NGF after 8 h (○ and •), another set after 15 h (▵ and ▴) and the final set after 22 h (* and +), and their survival was followed up to 15 d. Data represent mean ± range of two independent experiments in which deviations between triplicate wells were <5%.
BAF-treated/NGF-deprived Neurons Transduce Prolonged Akt/ERK Signals in Response to NGF
One explanation for why BAF-treated/NGF-deprived 0-DIV neurons were not rescued by NGF was that the neurons were not capable of responding to NGF. The kinase Akt is crucial for NGF-mediated survival in both 0-DIV and 6-DIV SCG neurons (Philpott et al. 1997; Crowder and Freeman 1998; Virdee et al. 1999; Xue et al. 2000), whereas ERK MAP kinases inhibit a p53-dependent mechanism of apoptosis induced by treatment with cytosine arabinoside (Anderson and Tolkovsky 1999). To determine if 0-DIV BAF-treated/NGF-deprived neurons could respond to NGF, we examined the activation of these two kinase systems in 0-DIV neurons that were pretreated for 1 d with BAF in the absence of NGF before NGF addition. As shown in Fig. 3, addition of NGF induced a rapid, robust, and sustained increase in phosphorylation of both ERK1/2 and Akt, which was maintained for at least 22 h (the longest time tested). The response was similar to that induced by NGF in freshly isolated neurons or in neurons maintained for 1 d in the presence of CPTcAMP, a survival-promoting agent which neither activates Akt in 6-DIV (Crowder and Freeman 1999) or 0-DIV SCG neurons (Virdee, K., unpublished data) nor activates ERK1/2 in either culture system (Virdee and Tolkovsky 1995). Thus, death of NGF-deprived, BAF-treated neurons after NGF addition was not because of an inability to transduce Trk-mediated signals required for survival. Hence, survival signaling must be interrupted downstream of Akt/ERK activation.
Figure 3.
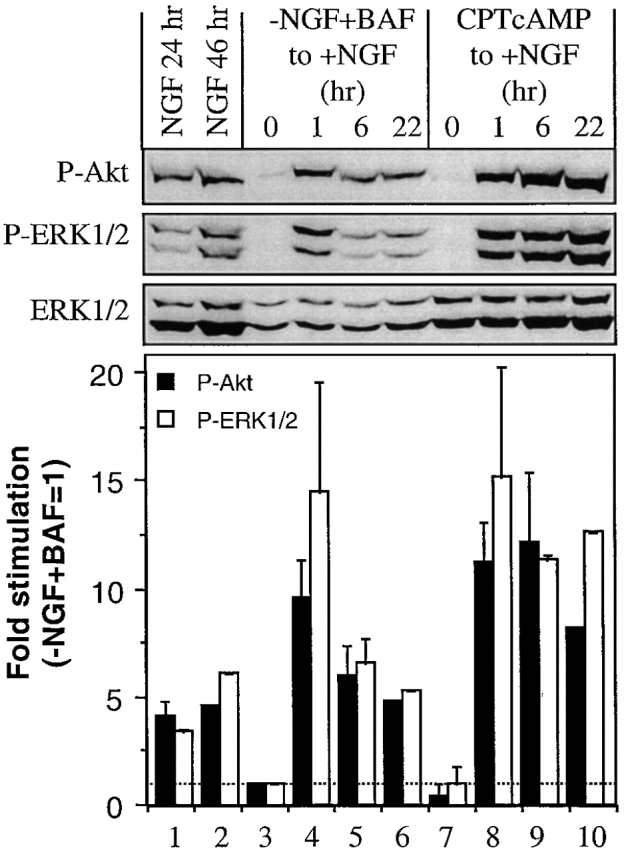
NGF induces prolonged phosphorylation of Akt and ERK1/2 in NGF-deprived/BAF-treated neurons. 0-DIV SCG neurons were deprived of NGF in the presence of 100 μM BAF for 24 h, or maintained for 24 h in the presence of 50 ng/ml NGF or 0.5 mM 8-(4-chlorophenyl thio) cAMP (CPTcAMP), after which one set of neurons was lysed immediately, whereas the other three sets were stimulated for 1, 6, or 22 h with 100 ng/ml NGF. Neuronal extracts were analyzed by Western blotting with anti–phospho(ser-473)-Akt (P-Akt, top panel) and anti–phospho-ERK (P-ERK1/2, middle panel). After development by ECL, the blot was stripped and reprobed with anti-ERK (ERK1/2, bottom panel) to normalize for protein loading. Bottom panel shows quantitative analysis of fold stimulation normalized to the value obtained from NGF-deprived/BAF-treated neurons (lane 3). Experiments were performed independently two to three times, and show mean ± range (n = 2) or ± SD (n = 3).
BAF-treated/NGF-deprived 0-DIV Neurons Fail to Grow Neurites or Mount a Hypertrophic Response after Stimulation with NGF
Despite the presence of protracted NGF signaling, neurons produced little neurite outgrowth in response to NGF (a response which normally occurs within minutes of plating neurons on laminin; Virdee et al. 1999), and failed to mount a hypertrophic cell body response. On the contrary, a defining feature of these neurons was that their cell bodies shrunk day by day until they became very small and ultimately phase-dark. Fig. 4 shows micrographs of 0-DIV SCG neurons treated with or without NGF and/or BAF for 1 d, and then treated for 9 d with NGF. It can be seen that most of the BAF-treated/NGF-deprived neurons are small compared with the size of the control neurons (Fig. 4, a and b). The large neurons are rescued neurons whose size is similar to the size of neurons rescued from NGF-deprived conditions (Fig. 4c and Fig. d). The nuclei of the small neurons stained evenly with Hoechst dye until the neurons died, and remained the same intensity as the nuclei of rescued neurons (see Fig. 6 A, 7-d time point), further suggesting that the death of these neurons is not apoptotic.
To investigate more closely whether NGF addition could reverse any of the metabolic effects of NGF deprivation (Deckwerth and Johnson 1993; Franklin and Johnson 1998), we measured four parameters: cell size, reduction of MTT into formazan (Fig. 5 A), ATP content, and net protein synthesis (Fig. 5 B), which was measured after 2.5 d of continuous incorporation of [35S]methionine into proteins. We chose to measure protein and ATP content because it has been suggested that NGF signaling controls neuron size by coupling the rates of protein synthesis and degradation (Franklin and Johnson 1998), while Buttgreit and Brand 1995 have demonstrated that protein synthesis in cells is limited by the amount of available ATP. Fig. 5 A (left) shows that after 1 d, there was no difference in the size or the extent of formazan production between BAF-treated/NGF-deprived neurons, and those neurons maintained in the presence of NGF. However, 3 d later (right), the set of neurons maintained continuously with NGF grew significantly and increased formazan production (P < 0.01, two-tailed t test), but there was no increase in the size of BAF-treated/NGF-deprived neurons that had been treated with NGF for 3 d. Formazan production was visibly diminished, although the numbers did not achieve statistical significance.
Figure 5.


NGF-deprived/BAF-treated 0-DIV neurons fail to recover metabolic activity in response to NGF. (A) Neurons were maintained in the presence of 50 ng/ml NGF, or deprived of NGF in the presence of 100 μM BAF for 24 h (left), at which time similarly treated neurons were stimulated with 100 ng/ml NGF for 3 d (right). Exactly 30 min before analysis, 5 mg/ml MTT was diluted 10-fold into the medium. Random fields of neurons from triplicate wells were captured by videomicroscopy, and the intensity of formazan staining and the area of cell bodies were quantified as described in Materials and Methods. The scatter plot shows the results of both measurements for 100–200 cells per treatment. (B) Neurons, which were treated as described in A, were analyzed for cell volume, ATP content, and net incorporation of [35S]methionine into proteins as described in Materials and Methods. Values were normalized to those obtained from cultures treated continuously with NGF, and represent means ± range of two independent experiments, where deviation between triplicate wells was ≤ 10%.
Fig. 5 B shows that ATP content and the net incorporation of [35S]methionine into proteins were reduced to 21 ± 2% and 19 ± 3% in neurons that were BAF-treated/NGF-deprived for 1 d, followed by 3 d of NGF addition, compared with the values measured in neurons maintained continuously in the presence of NGF (mean ± range, two independent experiments) while cell volume was reduced to 36 ± 9% of that of NGF-maintained neurons. In keeping with its lack of effect on cell growth (Fig. 4), the addition of BAF to NGF-maintained cultures did not alter any of the above parameters (data not shown). The concomitant loss of ATP content and protein synthesis, together with the observation that the reduction in ATP and protein content was larger than that of cell volume, suggest that the observed cell shrinkage (Fig. 4) is driven by the inability of NGF to couple signaling to the renewal of net protein synthesis, which is impaired, in part, by the loss of ATP. Although the neurons were metabolically impaired, it is clear that glycolysis provided sufficient ATP to preserve plasma membrane integrity until the neurons died, since the removal of glucose from these cultures at any stage caused necrosis within a few hours.
BAF-treated/NGF-deprived 0-DIV Neurons Do Not Refill Their Mitochondria with cyt c after NGF Addition
To further pinpoint which process might cause the metabolic impairment and uncouple NGF signaling from growth, we examined cellular cyt c levels. BAF-treated/NGF-deprived 0-DIV neurons lose cyt c from their mitochondria, releasing it into the cytoplasm where it is degraded (Xue et al. 1999), which is similar to 6-DIV neurons treated with BAF or zVAD.fmk (Deshmukh and Johnson 1998; Neame et al. 1998; Martinou et al. 1999). In Fig. 6, the pattern of immunostaining of cyt c in 0-DIV and 6-DIV neurons and the amount of cyt c determined by Western blotting are compared. Both sets of neurons lost >95% of the cyt c from their mitochondria during the period of NGF deprivation in the presence of BAF (0 time point). However, although 6-DIV neurons were deprived of NGF for 3 d whereas 0-DIV neurons were only deprived for 1 d, cyt c packaging into the mitochondria of 6-DIV neurons resumed within 1 d after NGF readdition (confirming the data of Martinou et al. 1999). But no filling of mitochondria with cyt c was observed in 0-DIV neurons after NGF addition even after 7 d, during which time the nuclei of surviving cells were still normal.
Neurons that Cannot Be Rescued by NGF after BAF Treatment Lose Their Mitochondria
The data above show that NGF does not induce net synthesis of cyt c. One reason may be that NGF signaling fails to engage the mechanism that couples protein synthesis and degradation rates (Franklin and Johnson 1998). Alternatively, the mitochondria themselves could be irreversibly altered so that they cannot be refilled with cyt c. Since NGF deprivation of 0-DIV neurons for 16 h induces autophagy of mitochondria, which is not inhibited significantly by BAF (Xue et al. 1999), we wished to examine whether we could detect any structural changes in mitochondria of 0-DIV neurons that had been BAF-treated/NGF-deprived for 1 d and then stimulated with NGF for 3 d. Fig. 7 shows that most of the neurons that had been NGF-deprived and BAF-treated for 1 d still contained several normal looking mitochondria, although autophagic activity was also very prominent (Fig. 7 B; >5 autophagic particles detected in 93/99 sections). Surprisingly, however, the majority of neurons that had been subsequently treated with NGF for 3 d contained no detectable mitochondria whatsoever, although the plasma membrane, Golgi apparatus, ER, nuclei, centrioles, microtubules, and cytoplasm all appeared to be normal (Fig. 7 C). Fig. 7 D shows a frequency plot of the proportion of cell sections contained in categories of 0–10 or >10 mitochondria. Over 50% of the cell sections contained no detectable mitochondrial structures, whereas analysis of cohort cultures showed that 20% of neurons contained >10 mitochondria and were also rescued by NGF. Thus, NGF addition failed to induce refilling of mitochondria with cyt c, in part, because the mitochondria were dysfunctional and eliminated. To investigate whether NGF addition to BAF-treated/NGF-deprived neurons prompted the loss of mitochondria, or whether the damage occurred during the period of NGF deprivation, we also cultured BAF-treated/NGF-deprived neurons in BAF alone, without adding NGF. We not only found that these neurons died at similar rates to those neurons to which NGF was added (Xue et al. 1999), but also that mitochondria disappeared from these neurons with the same kinetics (Xue, L., unpublished data), implying that the signals that prompt the loss of mitochondria are induced during the period of NGF deprivation and not by the subsequent addition of NGF.
Figure 7.
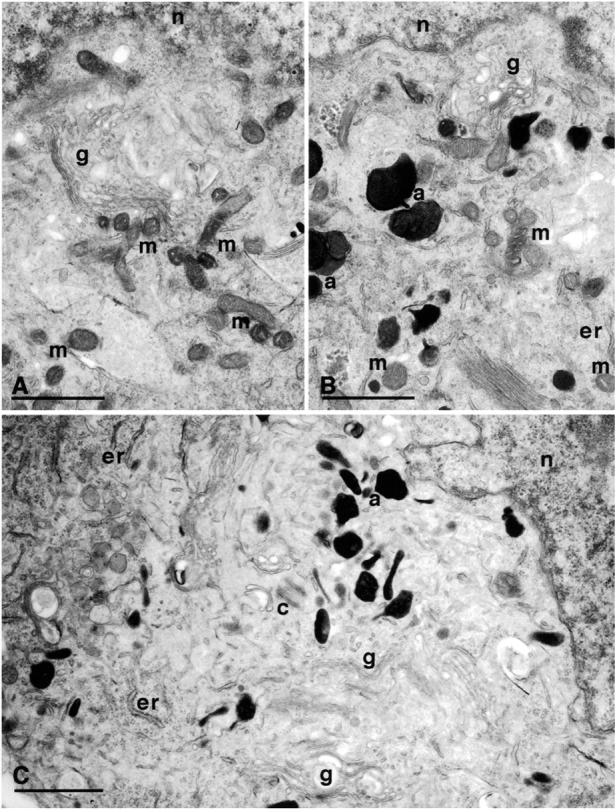

NGF-deprived, BAF-treated 0-DIV neurons lose their mitochondria within 3 d of NGF addition. 0-DIV SCG neurons were fixed for EM after being: (A) treated with NGF for 1 d; (B) NGF-deprived and BAF-treated for 1 d; and (C) NGF-deprived and BAF-treated for 1 d, followed by stimulation with NGF for 3 d. m, mitochondria; g, Golgi; n, nucleus; a, autophagic bodies; c, centriole. Note the complete absence of mitochondria in C. (D) Up to 202 individual cell sections of neurons from each of the three treatments described above were visualized under the EM viewer, and the number of mitochondria per section was counted. Data were sorted into 12 groups (sections containing 0–10 or >10 mitochondria) and plotted as a percentage of neurons containing 0–10 or >10 mitochondria per cell section. Cells maintained with NGF contained 59.2 ± 10.3 mitochondria per section (mean ± SD) and <2 autophagic particles per section, whereas 93/99 sections shown in B contained at least 6 autophagic particles.
Rescue by NGF Is a Function of Maturation and Is Abrogated by Injury
Therefore, it appears that there is a fundamental difference in the responses of 0-DIV and established cultures of SCG neurons to NGF after NGF deprivation and BAF treatment. Since 6-DIV neurons result from a culture of 0-DIV neurons, we wondered when the shift between the two cell phenotypes occurred. Therefore, we examined the potential of 0-, 2-, 4- and 6-DIV BAF–treated/NGF-deprived neuronal cultures to be rescued by NGF. For each culture set, neurons were BAF-treated and NGF-deprived until ∼90% of the neurons that had been NGF-deprived in the absence of BAF had died (43 h for 2-DIV neurons, 48 h for 4-DIV neurons, and 60 h for 6-DIV neurons). Then, NGF was added and survival was scored after 14 d. Fig. 8 A shows that there was a progressive increase in the capacity of NGF to rescue the BAF-treated/NGF-deprived neurons as the time in culture increased before the treatment commenced, with NGF rescuing ∼50% of 2-DIV BAF–treated/NGF-deprived cultures. By 4-DIV, neurons behaved the same as 6-DIV neurons. These data show that there is a progressive change in the mechanisms that underlie death commitment as neurons age in culture.
Figure 8.
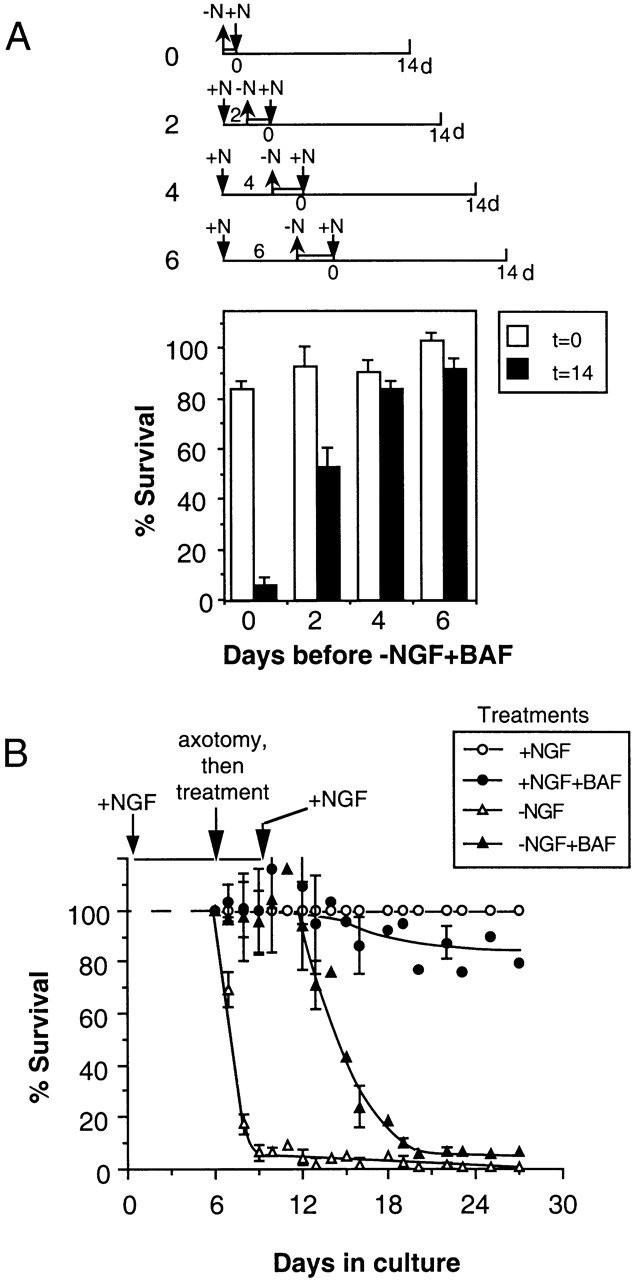
Neuritotomy converts the rescue phenotype of intact 6-DIV neurons into that of 0-DIV. (A) Neurons were cultured for 0, 2, 4, or 6 d in the presence of 50 ng/ml NGF before being deprived of NGF in the presence of 100 μM BAF. After >90% of neurons deprived of NGF in the absence of BAF died by apoptosis (24, 43, 48, and 60 h for 0-, 2-, 4-, and 6-d cultures, respectively), the medium was replaced with medium containing 100 ng/ml NGF, and cultures were counted immediately (t = 0), and again after 14 d (t = 14). Results show the percentage of survival after normalization to survival of cultures treated continuously with NGF (mean ± SD of quadruplicate wells). (B) Purified SCG neurons were aggregated by plating them overnight in suspension in the presence of NGF and then cultured as islets on a laminin substrate. After 6 d, neurites were cut with a glass needle, and cell bodies were collected, trypsinized, and plated as a single cell suspension in the presence or absence of NGF and/or BAF as indicated in the legend. After 3 d, when >95% of the NGF-deprived neurons had died by apoptosis, NGF was added to all the cultures and survival was followed for 22 d. Results show mean ± SD from three independent experiments.
One feature that distinguishes 0-DIV neurons from 6-DIV neurons is that the former undergo axotomy during their preparation. Therefore, we examined whether the phenotype typical of 0-DIV neurons could be induced in 6-DIV neurons by axotomizing them in vitro. To perform this experiment, aggregates of purified 0-DIV neurons were prepared and cultured for 6-DIV. Neuritotomy was performed by cutting the neurites close to the cell bodies, and single cell suspensions were prepared and cultured in the presence or absence of NGF and/or BAF (see Materials and Methods for details). Fig. 8 B shows that when neuritotomized 6-DIV neurons were plated in the presence of NGF they survived, just like 0-DIV neurons, and when the neurons were plated in the absence of NGF they died (apoptotically, data not shown) over 3 d. This rate of death was very similar to that of intact, NGF-deprived 6-DIV neurons and was much slower than the death of NGF-deprived 0-DIV neurons, showing that, in this respect, the neuritotomized 6-DIV neurons were not converted to the 0-DIV phenotype. However, when the neuritotomized 6-DIV neurons were treated with BAF during the 3-d period of NGF deprivation and returned to NGF, none of the neurons were rescued by NGF. Moreover, like 0-DIV neurons, BAF-treated/NGF-deprived neuritotomized neurons, which had been returned to NGF, also lost their mitochondria. Fig. 9 shows EM micrographs of a typical 6-DIV–neuritotomized neuron that was cultured either in the presence of NGF (A) for a further 6 d, or was BAF-treated/NGF-deprived for 3 d and then cultured in the presence of NGF for another 3 d (B). No mitochondrial profiles were detected in the latter neurons although the Golgi apparatus, ER, and nucleus were all intact. A substantial amount of autophagy can be observed. Fig. 9 C shows that all of the neurons cultured continuously in the presence of NGF contained >10 mitochondria per section. However, no mitochondria were detected in 72% of cell sections from 6-DIV neurons that had been neuritotomized, then BAF-treated/NGF-deprived for 3 d and then returned to NGF for 3 d. Thus, neuritotomy of 6-DIV neurons causes them to acquire similar properties to those of 0-DIV neurons, suggesting that a signal is induced by injury which fundamentally alters the order of steps that lead to death commitment.
Figure 9.
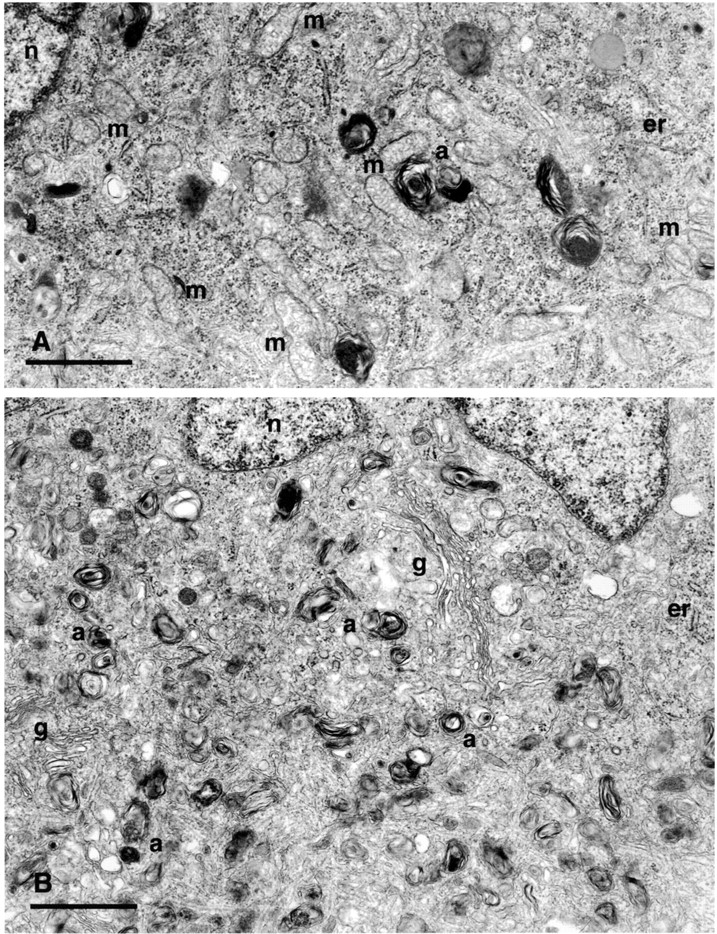

Neuritotomized 6-DIV neurons that are BAF-treated and NGF-deprived lose their mitochondria. (A) 6-DIV SCG neurons were neuritotomized as described in Fig. 8 and cultured in the presence of NGF, or BAF-treated in the absence of NGF for 3 d. NGF was added to the BAF-treated/NGF-deprived neurons for 3 d, after which neurons were fixed for EM. (A) 6-DIV–neuritotomized neuron treated with NGF for a further 6 d after axotomy; (B) 6-DIV neuritotomized neuron treated with BAF during 3 d of NGF deprivation followed by NGF addition for 3 d. m, mitochondria; g, Golgi apparatus; n, nucleus; a, autophagic bodies. Note the complete absence of mitochondria in B. (C) 23 individual cell sections of neurons from each treatment were visualized under the EM viewer, and the number of mitochondria per section was counted. Data were sorted into 12 groups (sections containing 0–10 or >10 mitochondria) and plotted as a percentage of neurons containing 0–10 or >10 mitochondria per cell section.
Axotomized Neurons Are Competent-to-Die in Response to Cyt c but Are Not Committed to Die
One possible way in which axotomy might induce a change in the order of steps that lead to death commitment is by altering the neuron's responses to death signals. It has been shown that intact 6-DIV neurons do not die in response to microinjection of cyt c unless they have undergone a prior period of NGF deprivation, even when the amounts injected are 250-fold higher than the endogenous levels (Neame et al. 1998). Therefore, it was suggested that for cytosolic cyt c to induce apoptosis, the cells have to reach a stage which was termed “competence-to-die” (Deshmukh and Johnson 1998). To test whether axotomy might induce competence to die, we introduced cyt c into both 0-DIV and neuritotomized 6-DIV neurons by trituration (see Materials and Methods). Fig. 10 shows that bovine cyt c (triturated to levels that were 12-fold above those of the endogenous cyt c) caused rapid apoptosis in both sets of neurons, 47 and 43% of neurons, respectively, dying by apoptosis within 4 h despite the continuous presence of NGF (Fig. 10 B). In contrast, yeast cyt c (which does not activate caspases; Ellerby et al. 1997) had no apoptotic effect (<3% apoptosis in both sets of neurons), confirming that trituration does not damage the neuronal responses to NGF (Nobes and Tolkovsky 1995; Nobes et al. 1996). Most importantly, the addition of BAF during the trituration abrogated apoptosis in both 0-DIV and neuritotomized 6-DIV cells (7 and 2.5% apoptosis, respectively). Fig. 10 A shows that neurons containing bovine cyt c died by typical apoptosis (a), whereas the nuclei of neurons triturated with yeast cyt c (b), or bovine cyt c in the presence of BAF (c) were normal. To demonstrate that BAF was capable of inhibiting apoptotic death sufficiently to allow long-term rescue by NGF, neurons were triturated with bovine cyt c in the presence of BAF and immediately cultured in the presence of NGF. The majority of neurons in which BAF had prevented apoptosis after cyt c trituration survived at least 13 d (e), which is similar to neurons triturated with yeast cyt c (Fig. 10 A, f), whereas neurons that had been triturated with bovine cyt c alone had died (Fig. 10 A, d). Since 0-DIV and neuritotomized 6-DIV neurons undergo rapid apoptosis in response to bovine cyt c, they are, by definition, competent-to-die. We, therefore, propose that axotomy/neuritotomy advances the order of events in the death signaling pathway by rapidly inducing competence-to-die. Therefore, the delayed competence-to-die described previously is not a stable property of 6-DIV neurons. Furthermore, neurons that are competent-to-die in response to cytosolic cyt c are not necessarily committed to die since NGF can rescue these neurons as long as BAF is present to prevent caspase activation. Hence, BAF is just as capable of suppressing death commitment after cyt c release in 0-DIV neurons as it is in intact 6-DIV neurons. These findings support the idea that an aspect of NGF deprivation which is distinct from cyt c release is the cause of irreversible mitochondrial dysfunction.
Discussion
In this paper, we have investigated the mechanisms underlying death commitment in SCG neurons. We show that NGF-dependent SCG neurons can differ in the mechanisms that commit them to die after NGF deprivation (summarized in Fig. 11). In 0-DIV neurons, the time course of rescue by NGF after NGF deprivation is determined by caspase-independent processes, since similar amounts of rescue by NGF are achieved irrespective of whether apoptosis execution has been prevented by the pan-caspase inhibitor BAF. In contrast, in intact 6-DIV SCG neurons, death commitment is dependent on caspase activity since NGF can rescue neurons that have been NGF-deprived for several days as long as caspases are inhibited by BAF (Deshmukh et al. 1996). The mechanisms determining death commitment appear to depend on the cellular context in which death signals function since axotomy of 6-DIV neurons resets the mechanism of death commitment from being caspase-dependent to being caspase-independent while growth in NGF drives the change in the other direction.
Figure 11.

Axotomy alters the sequence of events leading to the death commitment point. In intact 6-DIV neurons, NGF deprivation induces competence-to-die and mitochondrial changes resulting in cytochrome c release, which together activate caspases that result in apoptosis. BAF can inhibit caspases before death commitment and NGF addition reengages protein synthesis, allowing energy levels to be restored, and cells to grow. Thus, in the presence of BAF, mitochondria are still intact and caspases are not activated by cyt c release. In axotomized 6-DIV neurons or 0-DIV neurons, which are axotomized during preparation, the death commitment point is advanced and is now independent of caspase activation. This point of commitment occurs because NGF deprivation in conjunction with axotomy now produces irreparable mitochondrial dysfunction. Despite sustained activity of survival signaling kinases induced by NGF, these neurons fail to engage net protein synthesis; they cannot repair protein loss or reverse cyt c degradation, and within 3 d, all the mitochondria are eliminated from the cells, which die slowly by starvation. We propose to name this process “limoktonia.”
These data raise two major questions: (1) what are the mechanisms underlying death commitment point in the caspase-independent mode of death; and (2) what are the effects of axotomy on the cell death pathways?
Irreversible Mitochondrial Dysfunction Causes Failure of Coupling between NGF Signaling and Rescue of Axotomized SCG Neurons
In seeking the answer to the first question, we needed to eliminate both the possibility that BAF treatment is simply ineffective in blocking death commitment in axotomized neurons, and that Trk, the NGF receptor, merely fails to signal when NGF is added after a period of NGF deprivation and BAF treatment. We ruled out the first possibility by showing that BAF was capable of preventing both death and death commitment of 0-DIV neurons when death was induced rapidly by injection of bovine cyt c (Fig. 10). Indeed, with NGF, these neurons grew robustly, showing that BAF can inhibit cyt c–mediated caspase activation sufficiently to allow rescue of axotomized neurons as long as neurons do not undergo a period of NGF deprivation. We ruled out the second possibility by showing that NGF induced long-term activation of Akt and ERK1/2 (Fig. 3), which are two signals that mediate the survival of SCG neurons (Crowder and Freeman 1998; Philpott et al. 1997; Anderson and Tolkovsky 1999; Xue et al. 2000). In fact, activation levels were no different than those found in neurons maintained continually with NGF for 1 d or to those found in neurons maintained for the first day with CPTcAMP (which supports survival without activating ERK1/2 or Akt), showing that neither the intensity of signaling nor its duration were impaired.
Despite appropriate survival signaling in response to NGF, axotomized neurons that were not rescued by NGF failed to grow neurites and increase in size (Fig. 4 and Fig. 5), unlike similarly treated established cultures which dramatically increase in cell size upon readdition of NGF (Deshmukh et al. 1996; Franklin and Johnson 1998). Clearly, recovery of anabolic metabolism was impaired since net protein synthesis and ATP levels in NGF-deprived, BAF-treated neurons grown with NGF for 3 d were ∼20% of that observed in neurons that had not been NGF-deprived. However, similar losses of size and protein synthesis were observed in established neurons during long-term NGF deprivation in the presence of BAF (Deckwerth and Johnson 1993; Deshmukh et al. 1996); yet, these cells recovered with NGF treatment. Further analysis showed that the mitochondria of intact 6-DIV neurons regained cyt c within 1 d of addition of NGF to BAF treated/NGF-deprived neurons (similar to Martinou et al. 1999), but 0-DIV neurons failed to recover any cyt c protein (Fig. 6), keeping the mitochondria metabolically inert. When we examined the mitochondria of 0-DIV neurons by electron microscopy to see whether we could detect any signs of structural damage, we were surprised to find that mitochondria were virtually eliminated from the neurons within 3 d of NGF addition to BAF-treated/NGF-deprived neurons. In transition, some of the mitochondria had altered shapes (as do those described by Martinou et al. 1999), but none of the gross swelling seen in late stages of apoptosis or condensation was observed. The loss of mitochondria appeared to be specific since there was no apparent structural deficiency in any other organelle. The changes in mitochondria were not due to the addition of NGF, per se, since the same changes in the neurons occurred when they were maintained in the presence of BAF but without NGF addition (data not shown). These data are, to our knowledge, the first report of complete disappearance of mitochondria from intact cells following apoptotic signals. Lemasters et al. 1998 has suggested that autophagy is used by cells as a protective mechanism to eliminate mitochondria that have open permeability transition pores, and, therefore, dysfunctional inner membrane function. Whether this is the phenomenon we are seeing is not clear since we do not observe mitochondrial swelling before the mitochondria disappear. Interestingly, when SCG neurons overexpressing Bcl-2 are subjected to NGF deprivation and BAF treatment, they do not lose cyt c from the mitochondria nor do they eliminate their mitochondria (Xue, L., unpublished data). These data show that for NGF deprivation to provoke the orderly elimination of mitochondria, some damage to the mitochondria must take place. We are currently testing the possibility that the signal is allied to an irreversible drop in mitochondrial membrane potential (Neame et al. 1998). Whatever the mechanism may be, it is clear that NGF cannot arrest the loss of mitochondria. Thus, as described in Fig. 11, we propose that the irreversible mitochondrial change that is induced by NGF deprivation (and which signals mitochondrial elimination) is the single most important event that induces caspase-independent death commitment. Because of this damage, NGF fails to rescue the neurons.
Axotomy Resets the Mechanism of Death Commitment from Being Caspase-dependent to Being Caspase-independent
Having established that 0-DIV and 6-DIV neurons become committed to die at different biochemical points along a common, cyt c–dependent apoptotic pathway (Deshmukh and Johnson 1998; Neame et al. 1998; Fletcher, G.C., data not shown), we went on to explore the reasons why 0-DIV and 6-DIV neurons behaved differently. We wished to test whether the recent axotomy undergone by the newly isolated neurons during extraction was responsible for these changes, and/or whether the cell bodies of 6-DIV neurons had acquired novel properties. We found that neurons cultured with NGF before NGF deprivation undergo a gradual shift from the 0-DIV to the 6-DIV phenotype, with ∼50% rescue by NGF occurring in neurons that had been cultured for 2 d with NGF before NGF deprivation and BAF cotreatment. Interestingly, this time course is very similar to the time course of disappearance of c-Jun protein, whose expression (but not phosphorylation) is elevated in 0-DIV SCG neurons during their preparation (Virdee et al. 1997). c-Jun has been suggested to be necessary for apoptosis in SCG neurons (Estus et al. 1994; Ham et al. 1995; Eilers et al. 1998), but is clearly not sufficient since 0-DIV neurons expressing c-Jun are rescued by NGF. However, if c-Jun must be phosphorylated to participate in neuronal death (Eilers et al. 1998), then preexistence of c-Jun could advance the mechanism of apoptosis by providing a ready substrate for JNK, which is activated within hours of NGF deprivation (Virdee et al. 1997). Death via the JNK pathway has been proposed to be mediated by upregulation of the Fas ligand (LeNiculescu et al. 1999), but our preliminary data does not support activation of the Fas pathway as a cause of death in 0-DIV SCG neurons (which already express high amounts of Fas ligand; Tolkovsky, A.M., unpublished data).
Conversely, NGF induces protective mechanisms such as the antiapoptotic protein FLICE inhibitory protein (Raoul et al. 1999; Wiese et al. 1999), which could delay apoptosis induced by several death receptors. NGF also induces strong neurite outgrowth, which could account for the altered cellular behavior. To test this possibility, we cultured SCG neurons for 6 d, and reexposed them to the processes of axotomy and separation used during isolation of 0-DIV neurons. Unlike their intact 6-DIV counterparts, these axotomized 6-DIV neurons reverted their behavior to that of 0-DIV neurons, in that after NGF deprivation and BAF treatment, they could not be rescued. However, interestingly, their intrinsic rate of death after NGF deprivation was similar to that of intact 6-DIV neurons (50% time of death ∼25 h) compared with 0-DIV neurons (50% time of death ∼15 h; Edwards and Tolkovsky 1994), thus, supporting our previous evidence that the slower rate of death of older and/or more established cultures is not due to a reservoir of signaling intermediates provided by the neurites, but is rather an intrinsic property of cell bodies that increases with neuron age. Whatever the mechanism leading to the slower rate of apoptosis onset (which could be the FLICE inhibitory protein), it clearly does not contribute to the mechanism that defines the death commitment point since neuritotomized 6-DIV apoptosed slowly, yet they switched from having a caspase-dependent to a caspase-independent mode of death commitment.
Death Commitment and Competence-to-Die Are Separable Events
Since the death commitment point of axotomized 6-DIV neurons reverted to being caspase-independent, like 0-DIV neurons, we speculated that axotomy may make these cells competent-to-die and that competence to die and caspase-independent death commitment may be associated. Axotomized cells were indeed competent-to-die in response to cytosolic cyt c; thus, axotomy can mimic a period of NGF deprivation (Deshmukh and Johnson 1998) and sensitize established neurons to cyt c release. However, since axotomized neurons containing exogenous cyt c can be rescued by NGF in the presence of BAF (Fig. 9), death commitment and competence-to-die must be two separate events. Further, since these BAF-treated cells are both competent-to-die and contain cytosolic cyt c but can be rescued, another event must be required for death commitment. We postulate that death commitment arises because of irreversible mitochondrial dysfunction resulting from NGF deprivation.
The inhibition of caspases has been proposed as a potential strategy to prevent cell loss arising from pathology and trauma (Schulz et al. 1999; Kumar 1999). If caspase inhibition is to be a successful strategy, it must inhibit both apoptosis and death commitment of cells. While caspase inhibition can clearly prevent apoptosis, it is still not clear whether it can prevent the eventual loss of cells. Indeed, mechanisms of death other than apoptosis have been proposed to cause neurodegenerative disease (Perry et al. 1998; Migheli et al. 1999; Selznick et al. 1999). Our data suggest that the inhibition of the apoptotic pathway in some damaged neurons may merely prevent the removal of these cells by apoptosis, and that the cells will be lost nonetheless by a mechanism involving the selective loss of the mitochondria, and energy starvation. The relevance of these observations to axotomized systems in vivo remains to be studied.
In summary, our data suggest that the main difference between 0-DIV and established cultures of SCG neurons is related neither to the age of the neurons, nor to their altered responsiveness to NGF, nor to the differences in the ability of BAF to prevent caspase activity. Rather, it appears that the difference is due to the signals produced by axotomy, which render the neurons competent-to-die prematurely and induce a major shift in death commitment point which, together with NGF deprivation, causes irreversible damage to the mitochondria. Our preliminary data suggest that cell death by mitochondrial loss is widespread in cultured cells. We propose to name this nonnecrotic, nonapoptotic mode of death “limoktonia” (limoktonia, pronounced with emphasis on “nia,” which is the Greek term for starvation).
Acknowledgments
We are grateful to Molly Sheldon for her excellent technical assistance, and thank Dr. Jeremy Skepper (Department of Anatomy, University of Cambridge, Cambridge, UK) for his help with the confocal microscopy and EM, Dora Kemp (McDonald Institute, Cambridge, UK), and John Bashford, Adrian Newman, and Ian Bolton (all three from Department of Anatomy), at the Audio Visual Media Group of the Anatomy Department for help with the figures. We are indebted to Lisa Fagg and especially to Wojtek Rakowicz for scrutinizing the manuscript and to members of our lab for many helpful discussions.
This work was supported by a BBSRC CASE Studentship funded in part by Novartis Pharma (to G.C. Fletcher), a Medical Research Council Industrial Collaborative Studentship funded in part by Merck, Sharpe & Dohme (to S.K. Passingham), and by a Wellcome Trust Programme grant (to A.M. Tolkovsky and L. Xue).
Footnotes
Abbreviations used in this paper: BAF, t-butoxy carbonyl-Asp(O-methyl)-CH2F; CPTcAMP, 8-(4-chloropheny thio) cAMP; cyt c, cytochrome c; fmk, fluoromethylketone; MTT, 3-[4,5 dimethylthiazol-2-phenyl]-2,5-diphenyl-tetrazolium; SCG, superior cervical ganglia; zVAD.fmk, benzyloxyonyl-Val-Ala-Asp(O-methyl)-CH2F; 0-DIV, newly isolated SCG neurons; 6-DIV, 6-day cultured SCG neurons.
References
- Anderson C.N.G., Tolkovsky A.M. A role for MAPK/ERK in sympathetic neuron survivalprotection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J. Neurosci. 1999;19:664–673. doi: 10.1523/JNEUROSCI.19-02-00664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun J.S., Novak R., Herzog K.H., Bodner S.M., Cleveland J.L., Tuomanen E.I. Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat. Med. 1999;5:298–302. doi: 10.1038/6514. [DOI] [PubMed] [Google Scholar]
- Buckmaster A., Nobes C.D., Edwards S.N., Tolkovsky A.M. Nerve growth-factor is required for induction of c-fos immunoreactivity by serum, depolarization, cyclic-AMP or trauma in cultured rat sympathetic neurons. Eur. J. Neurosci. 1991;3:698–707. doi: 10.1111/j.1460-9568.1991.tb00855.x. [DOI] [PubMed] [Google Scholar]
- Buttgreit F., Brand M.D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995;312:183–187. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary P., Ahmed F., Quebada P., Sharma S.C. Caspase inhibitors block the retinal ganglion cell death following optic nerve transection. Mol. Brain Res. 1999;67:36–45. doi: 10.1016/s0169-328x(99)00032-7. [DOI] [PubMed] [Google Scholar]
- Chen J., Nagayama T., Jin K.L., Stetler R.A., Zhu R.L., Graham S.H., Simon R.P. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J. Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Deshmukh M., D'Costa A., Demaro J.A., Gidday J.M., Shah A., Sun Y.L., Jacquin M.F., Johnson E.M., Holtzman D.M. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder R.J., Freeman R.S. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J. Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowder R.J., Freeman R.S. The survival of sympathetic neurons promoted by potassium depolarization, but not by cyclic AMP, requires phosphatidylinositol 3-kinase and Akt. J. Neurochem. 1999;73:466–475. [PubMed] [Google Scholar]
- Deckwerth T., Johnson E.M. Temporal analysis of events associated with programmed cell-death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M. Evidence of a novel event during neuronal deathdevelopment of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Vasilakos J., Deckwerth T.L., Lampe P.A., Johnson E.M. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J. Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton R.M., Deckwerth T.L., Parsadanian A.S., Johnson E.M. Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturationa phenotype indistinguishable from Bax deletion. J. Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.N., Tolkovsky A.M. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J. Cell Biol. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A., Whitfield J., Babij C., Rubin L.L., Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J. Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellerby H.M., Martin S.J., Ellerby L.M., Naiem S.S., Rabizadeh S., Salvesen G.S., Casiano C.A., Cashman N.R., Green D.R., Bredesen D.E. Establishment of a cell-free system of neuronal apoptosiscomparison of premitochondrial, mitochondrial, and postmitochondrial phases. J. Neurosci. 1997;17:6165–6178. [PMC free article] [PubMed] [Google Scholar]
- Estus S., Zaks W.J., Freeman R.S., Gruda M., Bravo R., Johnson E.M., Jr. Altered gene expression in neurons during programmed cell deathidentification of c-jun as necessary for neuronal apoptosis. J. Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K., Zhu J.M., Namura S., Shimizu-Sasamata M., Endres M., Ma J.Y., Dalkara T., Yuan J.Y., Moskowitz M.A. Prolonged therapeutic window for ischemic brain damage caused by delayed caspase activation. J. Cereb. Blood Flow Metab. 1998;18:1071–1076. doi: 10.1097/00004647-199810000-00003. [DOI] [PubMed] [Google Scholar]
- Franklin J.L., Johnson E.M. Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J. Cell Biol. 1998;142:1313–1324. doi: 10.1083/jcb.142.5.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoli P.A., Bondurant M.C. Function of caspases in regulating apoptosis caused by erythropoietin deprivation in erythroid progenitors. J. Cell. Physiol. 1999;178:133–143. doi: 10.1002/(SICI)1097-4652(199902)178:2<133::AID-JCP2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Ham J., Babij C., Whitfield J., Pfarr C.M., Lallemand D., Yaniv M., Rubin L.L. A c-jun dominant-negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- Hara H., Friedlander R.M., Gagliardini V., Ayata C., Fink K., Huang Z.H., Shimizu-Sasamata M., Yuan J.Y., Moskowitz M.A. Inhibition of interleukin 1 β-converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch T., Marchetti P., Susin S.A., Dallaporta B., Zamzami N., Marzo I., Geuskens M., Kroemer G. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene. 1997;15:1573–1581. doi: 10.1038/sj.onc.1201324. [DOI] [PubMed] [Google Scholar]
- Johnson M.D., Kinoshita Y., Xiang H., Ghatan S., Morrison R.S. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J. Neurosci. 1999;19:2996–3006. doi: 10.1523/JNEUROSCI.19-08-02996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermer P., Klocker N., Bahr M. Long-term effect of inhibition of ced 3-like caspases on the survival of axotomized retinal ganglion cells in vivo. Exp. Neurol. 1999;158:202–205. doi: 10.1006/exnr.1999.7094. [DOI] [PubMed] [Google Scholar]
- Kumar S. Regulation of caspase activation in apoptosisimplications in pathogenesis and treatment of disease. Clin. Exp. Pharmacol. Physiol. 1999;26:295–303. doi: 10.1046/j.1440-1681.1999.03031.x. [DOI] [PubMed] [Google Scholar]
- Leist M., Single B., Castoldi A.F., Kuhnle S., Nicotera P. Intracellular adenosine triphosphate (ATP) concentrationa switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters J.J., Nieminen A.L., Qian T., Trost L.C., Elmore S.P., Nishimura Y., Crowe R.A., Cascio W.E., Bradham C.A., Brenner D.A., Herman B. The mitochondrial permeability transition in cell deatha common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- LeNiculescu H., Bonfoco E., Kasuya Y., Claret F.X., Green D.R., Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol. Cell. Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.S., Kim C.N., Jemmerson R., Wang X.D. Induction of apoptotic programme in cell free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Martinou I., Desagher S., Eskes R., Antonsson B., Andre E., Fakan S., Martinou J.C. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy N.J., Whyte M.K.B., Gilbert C.S., Evan G.I. Inhibition of Ced-3/ICE–related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesner P.W., Bible K.C., Martins L.M., Kottke T.J., Srinivasula S.M., Svingen P.A., Chilcote T.J., Basi G.S., Tung J.S., Krajewski S. Characterization of caspase processing and activation in HL-60 cell cytosol under cell-free conditionsnucleotide requirement and inhibitor profile. J. Biol. Chem. 1999;274:22635–22645. doi: 10.1074/jbc.274.32.22635. [DOI] [PubMed] [Google Scholar]
- Migheli A., Atzori C., Piva R., Tortarolo M., Girelli M., Schiffer D., Bendotti C. Lack of apoptosis in mice with ALS. Nat. Med. 1999;9:966–967. doi: 10.1038/12381. [DOI] [PubMed] [Google Scholar]
- Miller T.M., Moulder K.L., Knudson C.M., Creedon D.J., Deshmukh M., Korsmeyer S.J., Johnson E.M. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame S.J., Rubin L.L., Philpott K.L. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C.D., Tolkovsky A.M. Neutralizing anti-p21ras Fabs suppress rat sympathetic neuron survival induced by NGF, LIF, CNTF and cyclic AMP. Eur. J. Neurosci. 1995;7:344–350. doi: 10.1111/j.1460-9568.1995.tb01069.x. [DOI] [PubMed] [Google Scholar]
- Nobes C.D., Reppas J.B., Markus A., Tolkovsky A.M. Active p21Ras is sufficient for rescue of rat sympathetic neurons. Neuroscience. 1996;70:1067–1079. doi: 10.1016/0306-4522(95)00420-3. [DOI] [PubMed] [Google Scholar]
- Perry G., Nunomura A., Smith M.A. A suicide note from Alzheimer disease neurons? Nat. Med. 1998;8:897–898. doi: 10.1038/nm0898-897. [DOI] [PubMed] [Google Scholar]
- Philpott K.L., McCarthy M.J., Klippel A., Rubin L.L. Activated phosphatidylinositol 3-kinase and Akt kinase promote survival of superior cervical neurons. J. Cell Biol. 1997;139:809–815. doi: 10.1083/jcb.139.3.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C., Henderson C.E., Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J. Cell Biol. 1999;147:1049–1061. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierle G.S., Hansson O., Leist M., Nicotera P., Widner H., Brundin P. Caspase inhibition reduces apoptosis and increases survival of nigral transplants. Nat. Med. 1999;5:97–100. doi: 10.1038/4785. [DOI] [PubMed] [Google Scholar]
- Schulz J.B., Weller M., Moskowitz M.A. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann. Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Selznick L.A., Holtzman D.M., Han B.H., Gokden M., Srinivasan A.N., Johnson E.M., Roth K.A. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J. Neuropath. Exp. Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- Stefanis L., Park D.S., Friedman W.J., Greene L.A. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J. Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian R.V., Quinlan C., Trautz S., Hackett M.C., Mankovich J.A., Banach D., Ghayer T., Brady K.D., Wong W.W. Substrate specificities of caspase family proteases. J. Biol. Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Villa P.G., Henzel W.J., Sensenbrenner M., Henderson C.E., Pettmann B. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J. Cell Sci. 1998;111:713–722. doi: 10.1242/jcs.111.6.713. [DOI] [PubMed] [Google Scholar]
- Virdee K., Tolkovsky A.M. Activation of p44 and p42 MAP kinases is not essential for the survival of rat sympathetic neurons. Eur. J. Neurosci. 1995;7:2159–2169. doi: 10.1111/j.1460-9568.1995.tb00637.x. [DOI] [PubMed] [Google Scholar]
- Virdee K., Bannister A.J., Hunt S.P., Tolkovsky A.M. Comparison between the timing of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurones. J. Neurochem. 1997;69:550–561. doi: 10.1046/j.1471-4159.1997.69020550.x. [DOI] [PubMed] [Google Scholar]
- Virdee K., Xue L., Hemmings B.A., Goemans C., Heumann R., Tolkovsky A.M. Nerve growth factor–induced PKB/Akt activity is sustained by phosphoinositide 3-kinase dependent and independent signals in sympathetic neurons. Brain Res. 1999;837:127–142. doi: 10.1016/s0006-8993(99)01643-1. [DOI] [PubMed] [Google Scholar]
- Wiese S., Digby M.R., Gunnersen J.M., Gotz R., Pei G., Holtmann B., Lowenthal J., Sendtner M. The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat. Neurosci. 1999;2:978–983. doi: 10.1038/14777. [DOI] [PubMed] [Google Scholar]
- Xiang H., Kinoshita Y., Knudson C.M., Korsmeyer S.J., Schwartzkroin P.A., Morrison R.S. Bax involvement in p53-mediated neuronal cell death. J. Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L., Fletcher G.F., Tolkovsky A.M. Autophagy is activated by apoptotic signalling in sympathetic neuronsan alternative mechanism of death execution. Mol. Cell. Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- Xue L., Murray J.H., Tolkovsky A.M. The Ras/PI3K and Ras/ERK pathways function as independent survival modules each of which inhibits a distinct apoptotic signalling pathway in sympathetic neurons. J. Biol. Chem. 2000;275:8817–8824. doi: 10.1074/jbc.275.12.8817. [DOI] [PubMed] [Google Scholar]
- Yakovlev A.G., Knoblach S.M., Fan L., Fox G.B., Goodnight R., Faden A.I. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J. Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]