

Abstract
Cellularization of the Drosophila embryo is a specialized form of cytokinesis that results in the formation of a polarized epithelium. The mechanisms of membrane growth during cytokinesis are largely unknown. It is also unclear whether membrane growth and polarization represent distinct processes that occur simultaneously or whether growth of the membrane is involved in the emergence of polarity. Using a combination of surface labeling and particles tracking techniques, we monitored the dynamics of marked membrane regions during cellularization. We find that the major source of membrane is intracellular, rather than in the form of a plasma membrane reservoir. Depolymerization of microtubules inhibits the export of a newly synthesized transmembrane protein from the Golgi apparatus to the plasma membrane and simultaneously blocks membrane growth. Membrane insertion occurs in a defined sequence at specific sites, first apical, then apical–lateral. Diffusion of the membrane appears insufficient to compete with the massive local insertion of new membrane. We thus identify a tightly regulated scheme of polarized membrane insertion during cellularization. We propose that such a mechanism could participate in the progressive emergence of apical–basal polarity.
Keywords: cytokinesis, polarity, membrane growth, epithelial cells , cellularization
Introduction
An important challenge in cell biology is to understand the mechanisms underlying the establishment and maintenance of cell polarity. Many different cell types show a polarized organization reflected in the asymmetric distribution of proteins and lipids in the plasma membrane and the asymmetric organization of the microtubule and actin cytoskeleton. This organization is key to their functional specialization. For example, it is essential during polar bud growth in the yeast Saccharomyces cerevisiae, polarized growth during axon formation, and the transport of nutrients across endothelial barriers. Some cases involve the imposition of polarity on previously existing membranes whereas others are associated with new membrane growth. In spite of obvious differences in the process, many components of the polarization pathway are shared across species and the mechanisms have been the subject of intense cell biological and biochemical investigations (for a review see Drubin and Nelson 1996). However, it remains a challenge to integrate the available information into a dynamic picture where the behavior of organelles and the cytoskeleton is monitored as polarity arises and where the hierarchy of decisions is reconstructed through the discovery of key regulators.
Cellularization of the Drosophila blastoderm is a remarkable process where the formation of a polarized epithelium is coupled to the process of cytokinesis. It offers an opportunity to understand the mechanisms of membrane growth during furrowing. It also allows one to ask whether membrane growth can contribute to the emergence of membrane polarity or whether these represent distinct processes. The first 13 nuclear divisions of the Drosophila embryo occur in a syncytium, resulting in 6,000 peripheral nuclei located beneath the plasma membrane. During cellularization, the membrane surface increases ∼25-fold, invaginates between the nuclei, and ultimately yields 6,000 epithelial cells 30 µm tall. Most studies so far have focussed on the cytoskeletal rearrangements that control cellularization (Schejter et al. 1992; Foe et al. 1993). A systematic search for genes zygotically required for cellularization (Merrill et al. 1988; Wieschaus and Sweeton 1988) led to the discovery of three essential loci that control the integrity of the actin cytoskeleton at the invagination front called the furrow canal (FC): serendipity-alpha (sry-α) (Schweisguth et al. 1990), nullo (Rose and Wieschaus 1992), and bottleneck (bnk) (Schejter and Wieschaus 1993a). This screen supported the view that the global contraction of the actomyosin network at the leading edge of the furrow provides the force necessary to pull down the membrane similar to standard cytokinesis (for a review see Glotzer 1997). An alternative model proposes that the polarized distribution of microtubules might exert a force pushing the membrane and forcing it inward (Foe et al. 1993). These models constitute the current framework for our understanding of cellularization. However, neither model addresses the origin of the membrane required to accommodate the massive increase in membrane surface during cellularization. An attractive idea proposes that much, if not all of the membrane, exists as a reservoir in the form of villous projections of the plasma membrane (Fullilove and Jacobson 1971; Turner and Mahowald 1976). Alternatively, membrane growth could derive from the secretory pathway (Loncar and Singer 1995). In both models the membrane has hitherto only been viewed as a passive aspect of furrowing, whether it is pulled downward by the actomyosin network or pushed inward by microtubules.
At the onset of gastrulation, the resultant epidermal cells have the hallmarks of polarized cells with adherens junctions separating the apical and basal–lateral domains. It is still unknown how this polarity is established. It may occur during cleavage of the embryo when the membrane grows and invaginates between the nuclei or, following the classical example of epithelial Madin-Darby canine kidney (MDCK) cells, it could occur after cellularization when contacts between cells trigger the formation of adherens and septate junctions. Nonpolarized MDCK cells use E-cadherin–based cell–cell contacts as an external cue to position and to trigger the assembly of adherens junctions (Drubin and Nelson 1996). Adherens junctions in turn act as a fence to isolate two membrane domains that are subsequently the site of specific membrane targeting. Proteins travelling along the biosynthetic pathway are sorted at the level of the trans-Golgi network and directed to either the apical or the basal–lateral membranes. In addition, the endosomal compartment receives and redirects lipids and proteins to their apical or basal–lateral destinations. Thus, the adherens junctions serve both as a trigger of polarization and a device to maintain polarity. In neurons, the axonal and somatodentridical membranes are thought to be the analogue of the apical and basal–lateral membranes on the basis of similar protein targeting profiles (Dotti and Simons 1990). Vectorial cytoplasmic flow occurs at sites of future membrane growth and precedes axon formation (Bradke and Dotti 1997). Despite these similarities, neurons are devoid of adherens junctions and must resort to different strategies to maintain the integrity of adjacent membrane domains. Intramembrane diffusion barriers exist and involve interactions between the membrane and the underlying actin and spectrin cytoskeleton (Winckler et al. 1999). The most obvious difference between neurons and MDCK cells is membrane growth, which is extensive in the former and minimal in the latter.
The work presented here uses Drosophila to address whether the polarization of epithelial cells can be governed by membrane growth under situations of rapid membrane mobilization. In other words, is polarity reflected in the regulated insertion of proteins and associated lipids at different locations as the membrane grows, or does polarity appear subsequently when nonpolarized cells interact in a manner akin to MDCK cells? To address these issues, we developed techniques that allow the visualization of the flux of membrane proteins in living embryos during cellularization. We compared these dynamic patterns to the distribution of a newly synthesized transmembrane protein in stage-fixed embryos. We conclude that growth of the plasma membrane stems from the remobilization of ER- and/or Golgi-derived membrane populations that insert at precise locations in a regulated manner. The localized membrane delivery we identify in our experiments is such that lectin-labeled membrane patches and microbeads bound to an intact membrane are consistently displaced away from the sites of insertion. This suggests a mechanism in which polarization of the plasma membrane might be inherently linked to the polarized pattern of membrane growth during embryonic cleavage.
Materials and Methods
Fly Strains
OregonR stocks were used as wild-type controls in all of our experiments. To generate embryos zygotically deficient for Neur from mothers with a wild-type Neur dosage, we crossed C(3)se females with males heterozygous for Df(3L)81k19.
Surface Label of Living Embryos with Wheat Germ Agglutinin
Embryos were collected at 25°C on agar plates after 30-min collections. Embryos were aged 90 min at 25°C, dechorionated in 30% bleach for 2 min, rinsed with water, and lined up on a strip of agar. Embryos were then transferred on a coverslip covered with a thin layer of glue. After dehydration, the embryos were covered with S700 halocarbon oil where they continued to develop normally.
At the desired stage, embryos were injected with lectin as follows. Wheat germ agglutinin (WGA) coupled to the Alexa488 fluorochrome (Molecular Probes, Inc.) was kept as a 1 mg/ml stock solution in PBS at −20°C. WGA was thawed, diluted 1:3–1:5 in PBS, and centrifuged at 14,000 rpm for 10 min. It was loaded in a fine capillary needle and injection was conducted in the perivitelline space at 50% egg length (further information available upon request from the authors at tlecuit@ molbio.princeton.edu). The coverslip was mounted on an inverted Zeiss LSM510 confocal microscope where embryonic development proceeds normally. All images were processed using Adobe Photoshop® software.
Coating, Injection, and Tracking Fluorescent Microspheres in Living Embryos
0.5-μm YellowGreen fluorescent carboxylated microspheres (Polysciences, Inc.) were coated with WGA–Alexa488 using the carbodiimide kit for covalent coupling (Polysciences, Inc.). WGA coated beads were diluted in PBS and injected in the vitelline space and followed under the confocal microscope. YellowGreen fluorescent beads were detected using a relatively large pinhole to be reliably tracked in 2-μm optical slices. The maximum area of contact between the beads and the membrane was calculated to be 0.25 × 0.25 × 3.14 = 0.19 μm2. The membrane surface at the onset of phase 3 was largely underestimated, with the approximation to a smooth cylinder 6.5 μm in diameter and 5 μm high: 2 × 3.25 × 3.14 × 5 = 102 μm. This gave a minimum estimate of the relative surface of ∼1/537.
Antibody Staining
Antibody staining against Neurotactin (Neur), Armadillo (Arm), and myosin required heat fixation, which was described in Peifer et al. 1994. For embryos preinjected with colcemid, most of the halocarbon oil was removed manually; what remained was desolved in heptane. The heptane was replaced with a boiling NaCl buffer (68 mM NaCl, 0.03% Triton X-100) for 5–10 s. Injected embryos cannot be devitellinized with methanol/heptane and the vitelline membrane was removed instead with a sharp tungsten needle. Other fixations for antibody staining to Discs-lost (Dlt), Discs-large (Dlg), and β-tubulin were done using standard protocols with 15% formaldehyde in PBS/heptane for 15 min, followed with methanol popping except for injected embryos.
In all cases, antibody staining was performed in BBT (PBS/0.1% BSA/0.1% Tween 20), except for an initial blocking step with 10% BSA in PBS and 0.1% Tween 20. Primary antibodies were incubated overnight at 4°C with the following dilutions: rabbit anti-Dlt 1:1,000 (Bhat et al. 1999); rabbit anti-Dlg 1:200; rabbit anti-Myo 1:4,000 (generous gift of C. Fields); monoclonal mouse anti-Neur (BP106) 1:5 (Hybridoma Bank); monoclonal mouse anti-Arm 1:50; rabbit anti-Arm 1:200 (Peifer et al. 1994); mouse anti–β-tubulin 1:500 (Sigma Aldrich), rabbit anti-βCOP 1:200 (Ripoche et al. 1994). Subsequent reactions were performed at room temperature. Preadsorbed secondary antibodies coupled to Alexa488, Alexa546 (Molecular Probes, Inc.), and Cy3 were used at 1:500. The DNA dye Hoechst was used at 0.1 μg/ml for 5 min at the end of the staining protocols. Embryos were mounted in Aqua Poly Mount (Polysciences, Inc.).
Colcemid Injections
Dechorionated embryos were lined up and dehydrated as described above. Colcemid (Sigma-Aldrich) was stored as a 5 mM stock solution at 4°, diluted in water to 1.25 mM. Injection with an Eppendorf transjector allowed a reproducible result with an estimated final concentration ∼12–25 μM around the middle of the embryo. The effect was uniform around the circumference of the embryo at the injection site. Lower concentrations of colcemid showed variable effects and microtubules were not depolymerized reproducibly (data not shown). Injection was performed at 50% egg length at 16–18°C.
Results
Four Phases in the Formation of a Polarized Epithelium
Cleavage of the syncytial blastoderm begins on entry into interphase of the 14th nuclear cycle and lasts ∼70 min at 20°C. Living embryos can be followed with differential interferance contrast (DIC) microscopy as shown in Fig. 1. Immediately after mitosis 13 nuclei, reappear as spheres 5 μm in diameter beneath the plasma membrane (Fig. 1 A, arrow). The cell surface above each nucleus protrudes in a dome shape known as a somatic bud, a feature of the membrane during interphase 9–14 (Foe and Alberts 1983). During the cellularization process that follows, four phases can be observed, each characterized by a distinct feature of membrane invagination. Phase 1 lasts 10 min and results in the formation of the FC, a structure corresponding to the tip of the invaginating membrane. The nuclei also start to elongate basally. In the next three phases, each 20 min long, the FC moves inward at an increasing rate. In phase 2, the speed is very slow and the cellularization front remains in an almost steady position 5 μm basal to the surface of the embryo (Fig. 1B and Fig. C, arrow). During this phase, the nuclei complete their elongation. In phase 3, invagination becomes more obvious, although still at a relatively slow rate until it reaches the basal part of the nuclei (Fig. 1 D, arrow). The invagination rate then abruptly increases by a factor of two in phase 4 and in the next 20 min gives rise to cells 35-μm tall (Fig. 1 E). Previous subdivision of the process into slow and fast phases corresponds to phases 2/3 and 4, respectively (Foe and Alberts 1983).
Figure 1.

Video time-lapse recording of a living embryo at 20°C with DIC optics (left, A–E). Confocal section through fixed embryos stained with antibodies against Dlt (red) or Arm (green) or with Hoechst (blue) (right, F–I). All embryos are viewed from the side at the same scale (bar, 35 μm), except for F′ and G′ (bar, 5 μm). The black/white images on the right of F′ and G′ show the embryos from above. (A and B) Phase 1. The nuclei are spherical (arrow) and sit beneath the plasma membrane (arrowhead). The FC is visible within 10 min (B, arrow). (B and C) Phase 2. The FC hardly moves inward in 20 min, while the nuclei complete their elongation. (C and D) Phase 3. Slow invagination of the membrane until the FC reaches the basal part of the nuclei. (D and E) Phase 4. Fast invagination. The columnar epithelium is 35 µm tall. (F) Same stage as A (onset of phase 1). Arm (green) and Dlt (red) are localized at the membrane between the nuclei in a broad flat area, viewed at higher magnification below (F′, left in red, middle in white). Note the scattered distribution of Arm (F′, right). (G) Same stage as B (onset of phase 2). Arm now concentrates in thin strips (G′, right inset) and Dlt confines to a narrow ring (G′, middle). From the side (G′, left), Dlt is restricted to the FC (arrow) and Arm marks the apical boundary of the FC (arrowhead). (H) Same stage as D (onset of phase 4). Arm is still localized at the basal junction (arrow) and starts to accumulate apically at low levels (arrowhead). Dlt persists in the FC. (I) At the end of cellularization, Arm accumulates in the presumptive apical junction (arrow) and still localizes to the basal junction. Dlt is associated with the basal membrane.
The initial stages of cellularization can be followed with greater accuracy using a membrane-attached PDZ-containing protein, Dlt (Bhat et al. 1999), that ultimately localizes to the FC. At the beginning of phase 1, before nuclear elongation, Dlt is distributed in a broad flat area of the plasma membrane between the nuclei and is absent from the somatic buds (red, Fig. 1F and Fig. F′, right, viewed from top). These flat areas of the plasma membrane have been described using electron microscopy (Fullilove and Jacobson 1971). In embryos fixed at slightly later stages, the staining was restricted to a small teardrop-shaped domain, if viewed from the side (Fig. 1G and Fig. G′, left, arrow), or a ring if viewed from the top (Fig. 1 G′, middle) that corresponds to the FC. This transition occurs as the nuclear elongation begins and corresponds to the time when the FC becomes visible in transmitted light (Fig. 1 B). Dlt accumulation at the FC persists through cellularization as the FC moves basally (Fig. 1H and Fig. I). In conjunction with the initial transition in Dlt distribution, a junctional complex forms immediately apical to the FC. Arm/β-catenin is localized at the membrane through its interaction with DE-cadherin that marks the adherens junctions (Barth et al. 1997). Initially, Arm is detected in scattered spots straddling the Dlt localization domain (Fig. 1 F′, right). Shortly after, Arm concentrates into narrow strips immediately apical to the Dlt domain at the boundary between the FC and the apical membrane (Fig. 1 G, and 1 G′, left [arrowhead] and right). Arm persists in this location until the end of cellularization (Fig. 1H and Fig. I) and marks a basal junctional complex that also contains DE-cadherin (Hunter and Wieschaus 2000). At the onset of phase 4, when invagination accelerates, a new zone of Arm accumulation builds up at the apical–lateral membrane. It is barely visible at the beginning (Fig. 1 H, arrowhead), but becomes conspicuous and broad as phase 4 proceeds (Fig. 1 I) and marks the future mature apical adherens junction that subsequently forms when gastrulation begins (Muller and Wieschaus 1996). In conclusion, several proteins become restricted to distinct regions of the plasma membrane as it grows inward. This shows that polarization of the plasma membrane, rather than beginning when cellularization is completed, is progressive and concomitant to the process of membrane growth.
The Furrow Canal Is a Distinct Membrane Region Formed in Phase 1
To understand how the polarization of the membrane arises and how to characterize the sites of new membrane insertion, we developed a labeling technique in living embryos. We labeled the plasma membrane by injecting the fluorescent lectin WGA in the perivitelline space of a living embryo. Under physiological conditions, WGA–Alexa488 is a heterodimer that selectively binds to N-acetylglucosamine and N-acetylneuraminic acid (sialic acid) residues found on numerous membrane glycoproteins. When WGA–Alexa488 is injected, only a very small area of plasma membrane is labeled and occurs within seconds after injection, effectively generating a localized pulse of labeled membrane. No WGA is detected in a free unbound form in the vitelline space. This contrasts with other fluorescent lectins, such as soybean agglutinin, which poorly binds to the membrane, diffuses around the entire circumference of the embryo, and remains unbound in the vitelline space (data not shown). We then used this labeling technique to characterize membrane synthesis, the rationale being that if new intracellular membrane is inserted, it will be seen as unlabeled membrane that either dilutes or displaces “old” labeled membrane. We first injected WGA–Alexa488 during phase 1. Initially, WGA is detected over the entire surface of the somatic buds (Fig. 2 A, top), reflecting the numerous villous projections observed in electron microscopy (Fullilove and Jacobson 1971; Turner and Mahowald 1976). By the end of phase 1, WGA is no longer detected in the regions above the nuclei (Fig. 2 A, dashed line), suggesting that this membrane is rapidly renewed by incorporation of unlabeled membrane from the interior. WGA label is still visible between the nuclei where it forms interconnected rings whose positions correspond to the future site of membrane invagination. This label coincides with the FC (Fig. 2 C, arrowheads) and remains associated with the cellularization front as it moves inward (Fig. 2 B). The basal displacement of this staining during cellularization confirms the subdivision of cellularization into four distinct phases based on the rate of membrane invagination (Fig. 1). Several time points are shown in different colors in Fig. 2 B. In phase 2 (+12′ and +14′), the label barely moves within 2 min. In phase 3 however, it moves inward of ∼1 µm within 2 min (+33′ and +35′). The label is still detectable late in phase 4 and moves much faster (+56′ and +58′).
Figure 2.

Confocal sections through living embryos after injection with WGA–Alexa488. All pictures show the embryo from the side. N, nuclei. The embryos were grown at 25°C, a temperature that increases the rate of invagination compared with 20°C as shown in Fig. 1. (A and B) WGA label of the same embryo at different time points shown in different colors. At time 0 (A), WGA is broadly distributed at the plasma membrane surface. 14 min later, WGA is only detected between nuclei, and the apical membrane (dashed line) is unlabeled. This staining persists throughout invagination (B) and is shown 2 min apart in phase 2 (+12′ and +14′), phase 3 (+33′ and +35′) and phase 4 (+56′ and +58′). (C) Another embryo where WGA (arrowhead, top) colocalizes with the FC (bottom, arrowhead). (D) Scheme illustrating the changing distribution of the initial label (red) from its apical distribution to the FC. The green membrane represents the newly inserted unlabeled membrane. Bar, 5 μm.
We conclude from these observations that two adjacent membrane regions form in phase 1 that show distinct behaviors (Fig. 2 D): the surface above the nuclei shows a rapid flux or turnover, whereas the FC defines a more stable membrane region that persists throughout the course of cellularization. Our data do not distinguish whether the persistent high levels of staining in the FC in phase 1 reflect only low turnover rates in that structure compared with the apical membrane or the active relocalization of the label from the apical membrane to the FCs. With the caveat that WGA may alter the normal behavior of the plasma membrane, these data suggest that the FC forms as a separate membrane region. Consistent with this view, note that Dlt becomes restricted to and remains in the FC throughout cellularization (Fig. 1) and that a basal junction with high levels of Arm forms at the boundary between the FC and the adjacent membrane of the somatic bud (Fig. 1). This junction may be required to isolate the FC from the rest of the plasma membrane.
Apical Insertion of New Membrane and Lateral Transfer of Old Membrane
The FC can actually only be labeled during phase 1. WGA–Alexa488 injected during phase 2 appears to be excluded from the canal (Fig. 3B and Fig. B′, arrowhead), although it binds uniformly to the remainder of the embryo's surface (Fig. 3 A, purple). As membrane invaginates at a slow speed during phase 3, the label is gradually depleted from the apical membrane while the lateral label persists and the size of the labeled domain even expands (Fig. 3 A, blue; compare double arrow at time 0 and +18′). The parallel nature of these processes suggests that membrane initially on the apical surface may be transferred laterally as cellularization proceeds, continuing with what happens during phase 1.
Figure 3.

Confocal sections through living embryos at 25°C after injection with WGA–Alexa488. (A) Injection during phase 2 and observation during phase 3 at time 0 (purple, middle) and 18 min later (blue, bottom). Double arrow marks the growing lateral labeled membrane region. (B and B′) As in A at time 0. The basal extent of the label (B, arrow) is adjacent to, but excluded from the FC (B′, arrowhead). A nucleus (N) is outlined. (C) Injection during late phase 3 and observation during phase 4 at time 0 (red, middle) and 12 min later. The arrowhead points to a gap of unlabeled membrane appearing apical–laterally while the basal–lateral labeled membrane (double arrow) translocates basally. (D) WGA–Alexa488 (green, 1st label) was injected in phase 2, transferred laterally during phase 3 similarly to A. At the onset of phase 4, WGA–rhodamine (red) was injected. The embryo is shown shortly after. (E) Diagram summarizing the data. The apical and lateral membrane in early phase 3 (green) is transferred laterally as new membrane is inserted apically (red, middle). New insertion apical–laterally (yellow) follows in phase 4 (bottom) as the green region moves basally in register with the FC (black).
The edge of the lateral labeled patches maintain their association with the invaginating FC, and the labeled regions behave as though they simply accompany the inward growth of the membrane until the end of cellularization. In fact, the original most intensely labeled membrane regions move basally at the same speed as the FCs through phases 3 and 4. This suggests that there is no obvious incorporation of unlabeled membrane at the invagination front or that if it occurred it would not contribute to the growth of the plasma membrane. Instead, new membrane incorporation appears to occur predominantly apically and accounts for the bulk of the membrane growth during phase 3.
Lateral Insertion of New Membrane Becomes Predominant in Phase 4
The rate of membrane invagination greatly increases in phase 4. We find that the pattern of membrane insertion is very different from that characterized for the preceding stages. When WGA label is injected at the end of phase 3, fluorescence is initially detected both apically and laterally (Fig. 3 C, red and middle at time 0). In contrast to the earlier stages, the apical membrane maintains its label, though intensity slightly decreases, as cellularization proceeds. A gap of unlabeled membrane emerges in the apical portion of the lateral region (arrowhead in Fig. 3 C, 12 min after time 0). This gap grows in size and the lateral membrane previously labeled at time 0 is displaced basally and maintains its association with the FC (not shown). We conclude that the point of new unlabeled membrane insertion changes during phase 4, shifting from predominantly apical to predominantly lateral. Given the mild reduction of the apical membrane label, we conclude that apical insertion still persists, but our data show that the major site of insertion is lateral.
The changes in distribution of the labeled lectin at the membrane surface do not reflect a change in distribution of the WGA receptors under either normal or experimental conditions. For instance, if lateral transfer of WGA during phase 3 stemmed from increasing levels of WGA receptors laterally as a result of local changes in membrane composition, injection of WGA at the end of phase 3 would predominantly label the lateral membrane. However, our data show that the ability of WGA to bind the apical membrane appears constant at any time during cellularization (compare Fig. 3A and Fig. C, at time 0). Clearance of fluorescent WGA from apical regions and lateral transfer could also be due to a redistribution of the WGA receptors induced by the binding of the WGA ligand. To control that the apical membrane still contains the WGA receptors, we sequentially labeled the membrane with WGA conjugated with different fluorochromes. First, we injected WGA–Alexa488 during phase 3, leading to the lateral transfer of that label as described in Fig. 3 A. At the onset of phase 4, WGA–rhodamine was injected in the same region of the embryo. As shown in Fig. 3 D, this second label (red) is predominantly apical and tapers off along the lateral membrane, while the first label (green) is mostly in the basal lateral membrane. These experiments argue that the dynamic changes in WGA are unlikely due to the natural or induced redistribution of the WGA receptors.
In conclusion, throughout the course of cellularization, unlabeled membrane is constantly incorporated into labeled plasma membrane. This incorporation occurs in a defined sequence and a specific pattern, first apical, then apical–lateral. Note that the membrane miscibility appears limited under conditions where WGA is bound to the plasma membrane.
Displacement of Microspheres on the Membrane Surface
To further test this idea, we probed the dynamic properties of the plasma membrane in a situation where it is unaffected. We followed the movement of 0.5-μm microspheres bound to an intact plasma membrane. These beads were coated with WGA so they bind the membrane very rapidly upon injection in the vitelline space. The maximum area of contact between the bead and the membrane (0.19 μm2) is much smaller than the total plasma membrane surface area at the onset of phase 3 (at least 100 μm2; see Materials and Methods). Therefore, the beads can be considered as small passive patches representing 1/550th of the total membrane when invagination begins and even less later on. Such beads are thus predicted to follow the global movement of membrane populations they are imbedded in. In particular, if membrane mixing is important and creates a constant homogeneous state in the membrane, the locally delivered new membrane will effectively distribute evenly around the bead and, over time, the movement of the bead will be random. However, if membrane mixing is limited, new inserted membrane should consistently displace both the recipient membrane and the beads away from the sites of insertion, thus creating anisotropic growth of the membrane.
We injected WGA-coated fluorescent 0.5-μm beads in the vitelline space of living embryos at different stages. The dilution was such that, on average, less than one bead bound the membrane of a forming cell, to avoid any possible interaction between beads. During phases 1 or 2, beads are seen in the FC a few minutes after injection and remain at the invaginating front through phases 3 and 4. Fig. 4 A shows a bead at time 0 in red and at the end of phase 4 in green (the arrowhead points to the FC). This behavior is observed in 96% of the cases (n = 55) when beads are injected at early stages (Fig. 4 D, top, blue). The remaining 4% represent beads located just above the FC which also accompany the movement of the FC. When injected at the beginning of phase 3, the beads rapidly bind the incipient lateral membrane. Fig. 4B and Fig. B′, shows two representative examples where the left panel shows the DIC image at the initial time point: the arrowhead and the small arrows point to the initial positions of the FC and the beads, respectively. The right panel shows the positions of the beads at different time points: the blue bead indicates the initial location (arrows), and subsequent locations are in red and green. The beads are displaced away from the apical sites of membrane insertion and move in register with the advancing FC (Fig. 4B and Fig. B′). 89% of the beads (n = 122) have such a behavior and are eventually located along the basal portion of the lateral membrane (Fig. 4 D, green shaded area). The remaining 11% are found in the medial portion of the lateral membrane (Fig. 4 D, white area). During phase 4, when insertion becomes predominantly apical–lateral, a different situation is observed. Beads injected at the onset of phase 4 remain in the apical region of the membrane, above the insertion site, in 88% of cases (n = 56) (Fig. 4 D, red shaded area) and don't accompany the rapid movement of the invaginating membrane. Fig. 4 C shows the position of three beads at the end of cellularization; the two arrowheads show the extent of membrane growth during that time window.
Figure 4.
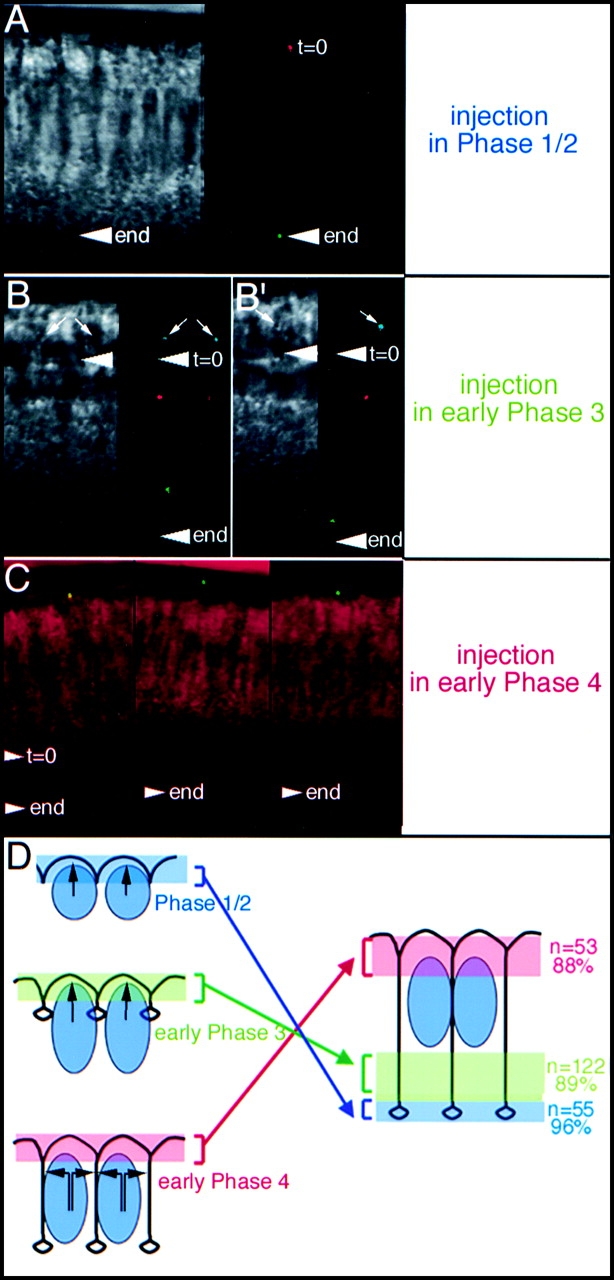
Injection of beads in phase 1/2. (A) Right panel shows a bead at time 0 (red) and the end of phase 4 (green). Left panel shows the DIC image corresponding to the final time point. The arrowhead shows the FC position. (B and B′) Injection in early phase 3. The arrowhead in all images shows the position of the FC at time 0 and the end of phase 4 (end). The DIC image (left) shows the initial time points and the beads (right) are shown in a color-coded fashion: blue at time 0, red later, and green at the end. Note, the beads have accompanied the movement of the FC. The apparent brightness of the beads varies as their position with respect to the confocal optical slices changes during cellularization. (C) Injection in early phase 4. The beads (green) are shown at the end of cellularization together with the DIC image (red). The arrowheads show the relative movement of the FC during the time lapse. (D) Summarizes the observations. Beads are displaced within membrane regions in accordance with the times of injection. Black arrows indicate the sites of membrane insertion.
Our data show that the microspheres have a reproducible nonrandom mobility and are displaced away from insertion sites with their final distribution depending on the time when they bind the membrane: the most basally located beads were injected earlier than more apically located beads. Given the relatively small size of the beads with respect to the surrounding membrane surface, the movement of the beads most likely reveals the behavior of the supporting membrane as a whole. We therefore infer that membrane growth appears anisotropic, mainly occurring at insertion sites. This situation might reveal that diffusion in the plane of the membrane, although effective, is not enough to cancel the effect of massive membrane insertion in a defined site.
Insertion of Neurotactin, a Newly Synthesized Transmembrane Protein
The labeling experiments suggest that membrane growth is mostly accounted for by a reservoir of intracellular membrane originating from the secretory pathway. To assess the contribution of ER and Golgi-based membranes to cellularization, we used antibodies to Neur, a transmembrane protein whose synthesis during cellularization requires de novo RNA synthesis in late syncytial embryos (Fig. 5 A). We followed the localization of Neur along the biosynthetic pathway. Neur is first detected in early phase 2 during nuclear elongation after the basal junction and FCs have formed (Fig. 5 B). At this point, the membrane has not started to grow and most of Neur is localized in punctate structures in the apical and basal cytoplasm (Fig. 5 B, arrowheads). The number and intensity of punctate Neur structures increase in the basal cytoplasm during phase 3 (Fig. 5 C, bracket), suggesting an increased level of synthesis. We compared the cytoplasmic localization of Neur to that of ER and Golgi markers. The ER resident chaperone BiP is concentrated in an apical perinuclear region (Fig. 5 F, arrowhead), and extends at lower levels in an intricate web into the basal cytoplasm (Fig. 5 F, bracket). In contrast to mammalian cells, the Golgi apparatus of insects is dispersed (Ripoche et al. 1994). The cis-Golgi marker βCOP (Oprins et al. 1993; Ripoche et al. 1994) is detected mostly in the basal cytoplasm in punctate structures at the beginning (Fig. 5 G) and the end of cellularization (Fig. 5 H). Therefore, Neur accumulates in a basal region of the cytoplasm where ER and Golgi are found in close association and might exchange vesicles. Neur and βCOP punctate distributions only rarely overlap (Fig. 5 I, arrowheads), although a spatial relationship exists (arrows). This situation may reflect the presence of Neur in regions of the Golgi apparatus devoid of βCOP, or in areas of the ER in close proximity to the cis-Golgi region. We cannot distinguish among these possibilities at present.
Figure 5.

Confocal sections through fixed embryos stained with an antibody to Neur (white in B–E and green in I), βCOP (white in G and H; red in I), BiP (F), and myosin (red in A and D). N, nuclei. (A) Neur is not expressed in an embryo zygotically mutant for Neur (arrowhead). A control embryo of similar age is shown (arrow). (B–E) Neur is initially found in the cytoplasm in punctate structures (arrowheads in B). As levels build up in the cytoplasm (bracket), Neur accumulates in the membrane during phase 3 (C, arrowhead, and D). Neur is absent from the FC shown in red with an antibody to myosin (D, inset). In phase 4 (E), Neur is localized apically and laterally but at higher levels in the apical–lateral surface (arrowhead). (F) BiP is concentrated in the apical perinuclear region (arrowhead) and extends to lower levels in the basal cytoplasm (bracket). (G and H) βCOP marks the cis-Golgi membranes in the basal cytoplasm during cellularization. Note its presence in punctate structures. (I) Double staining for βCOP (red) and Neur (green). Colocalization appears in yellow (arrowheads). Note the cases of close association (arrows). (J) Embryo labeled with WGA at the end of phase 3 and viewed in late phase 4. The site of unlabeled membrane insertion during phase 4 (arrowhead) coincides well with the zone of increased Neur staining (Fig. 4 E, arrowhead). Arrows point to the FC.
During phase 3, Neur also becomes visible along the invaginating lateral membrane (Fig. 5 C, arrowhead). Towards the end of phase 3, Neur levels increase in the lateral membrane, at the expense of a reduced cytoplasmic localization below the nuclei (compare Fig. 5C and Fig. D). It is also detected on the apical surface, but is absent from the FC (marked using an antibody to myosin in Fig. 5 D, inset, red). This latter point is in agreement with our finding that the FC is a membrane region set aside very early before Neur can be detected in the embryo. At the end of cellularization, membrane staining of Neur is even stronger, especially in the apical regions of the lateral membrane (Fig. 5 E, arrowhead). The distribution of Neur is eventually asymmetric, with higher levels coinciding with the zone of late apical–lateral membrane insertion (region 3) observed in WGA labeling experiments. Fig. 5 J shows an embryo labeled with WGA in late phase 3 and viewed at the end of cellularization to illustrate this parallel: the arrowhead points to the zone of unlabeled membrane insertion, which appears to coincide with the zone of increased Neur staining (compare Fig. 5E and Fig. J, arrowheads).
In conclusion, the transmembrane protein Neur is induced during cellularization and localized in the secretory pathway in proximity to cis-Golgi membranes. Neur is continuously inserted to the growing plasma membrane. These observations further support the view that new membrane originating from the secretory pathway is constantly used as raw material for membrane growth.
Microtubules Mediate the Mobilization of Secretory Membranes to the Growing Plasma Membrane
Neur must be transported over a large distance of ∼20 μm from the Golgi apparatus to the sites of insertion in the plasma membrane. Blocking this transport should immediately stall cellularization if the flux of membrane from the Golgi apparatus is the primary source of new membrane for cellularization. As shown in Fig. 6, a good candidate for the mechanism underlying an apical movement of Golgi membranes is microtubular transport. Microtubule asters are located apical to the nuclei beneath the plasma membrane (Fig. 6 A, arrowhead) and very long microtubules extend basally up to 30 μm deep inside the embryo (Fig. 6A and Fig. B) well within the area where the bulk of the cis-Golgi membranes are detected (Fig. 6 A) and where Neur accumulates in punctate structures (Fig. 5 C).
Figure 6.

Wild-type embryos (A and B) and embryos injected with colcemid during phase 2 (C–F) observed 30 min later. Embryos were fixed and stained with antibodies to β-tubulin (A and B, green and white, respectively), βCOP (A, B, E, and F, red), BiP (D, red), Neur (C, D, and F, green), Arm (C, red), or with Hoechst (blue). N, nuclei. (A and B) Microtubules extend basally where the bulk of the Golgi apparatus is concentrated at the beginning (A) and end (B) of cellularization. Arrowheads point to the FC. (C) Neur (green) accumulates in the basal cytoplasm in abundant punctate structures and is not detected in the plasma membrane marked with Arm (red). (D) BiP (red and white) is concentrated in the usual apical perinuclear location and is excluded from the basal cytoplasm where Neur (green) is concentrated. (E) βCOP is in the usual form of punctate structures in the basal cytoplasm. (F) Higher magnification view shows that βCOP (red) and Neur (green) either colocalize (arrowheads) or are associated (arrows).
Depolymerization of microtubules by injection of colchicine or colcemid at the onset of cellularization blocks membrane invagination (Foe and Alberts 1983). The effect appears very specific to phases 1, 2, and the beginning of 3, since only a moderate or no reduction in the rate of membrane invagination is detected later in phase 3 and 4 (data not shown). Since Neur is continuously delivered to the growing plasma membrane under normal conditions, we examined the effect of colcemid on Neur distribution. We injected colcemid in embryos at the onset of cellularization, fixed them 30 min later, when cellularization would normally reach completion, and stained them with Neur antibodies. Under these conditions, Neur is virtually absent from the plasma membrane and accumulates at abnormally high levels in punctate structures in the basal cytoplasm reminiscent of those seen under normal conditions (Fig. 6 C). Furthermore, we could not detect any obvious disorganization of the secretory apparatus under these conditions: βCOP is still detected in basal punctate structures (compare Fig. 6E and A, or Fig. 5 G) and BiP remains in its usual apical location (Fig. 6 D), although we noticed a mild reduction in the basal extent of the BiP signal in colcemid-treated embryos. The exclusion of BiP from the area where Neur accumulates and the similarity between the punctate structures seen with βCOP and Neur (Fig. 6 F) suggest that the Neur punctate structures seen in colcemid-treated embryos are most likely cis-Golgi–associated membranes.
Although we cannot exclude additional roles for microtubules, these data establish a link between plasma membrane growth and export of Neur from Golgi-associated membranes. This requirement further establishes that the membrane needed for cellularization originates from intracellular organelles.
Discussion
Using novel techniques to follow the dynamics of the plasma membrane in living embryos, we explored the modalities of membrane growth during cellularization.
We find that cellularization involves the remobilization of an intracellular membrane reservoir and its regulated insertion at well-defined sites in a precise sequence, first apical, then apical–lateral. This unexpected control of membrane delivery during cleavage of the embryo may contribute to the initiation and slow emergence of apical–basal polarity as the epithelium forms.
Cellularization Involves the Remobilization of an Intracellular Membrane Reservoir
The origin of the membrane required for cytokinesis and for cellularization, in particular, has been a long-standing unresolved issue. We conclude that the major reservoir of membrane is intracellular, based on the following observations. First, at any time during cellularization, the previously labeled plasma membrane incorporates membrane that is unlabeled and therefore must originate from inside the cell. Second, Neur, a transmembrane protein that we showed is induced de novo during cellularization, accumulates in Golgi-associated membranes and is continuously transferred to the growing plasma membrane. When microtubules are depolymerized, cellularization immediately stalls and Neur accumulates in abnormally abundant punctate structures of the basal cytoplasm reminiscent of those seen under normal conditions. The exclusion of BiP staining from this region and the presence of βCOP punctate structures in close proximity or colocalizing with Neur argue that Neur is properly exported from the ER to the Golgi apparatus and accumulates in Golgi-associated membranes when microtubules are depolymerized. This is in contrast to mammalian cells, where ER to Golgi export is a microtubule-dependent process and may identify a special feature of the ER and Golgi apparatus organization in early Drosophila embryos. Taken together, our data argue that ER and Golgi membranes constitute the major raw material for plasma membrane growth during cellularization. This is at odds with the idea that membrane grows through the simple redistribution of apical membrane and that the villous projections constitute the major reservoir for invagination (Fullilove and Jacobson 1971).
Membrane is normally made in the ER and passes through the Golgi apparatus constitutively on its way to the plasma membrane. Given the relatively constant amount of membranes stained with either BiP or βCOP antibodies from cycle 9–14 embryos (not shown), it is probable that the intracellular membrane reservoir is made long before cellularization begins and that cellularization involves the regulated mobilization of this membrane pool to the plasma membrane. It will be interesting to see which steps of the secretory pathway are regulated to accommodate the need for rapid membrane export. Moreover, the shift from a slow rate to a fast rate of invagination suggests that during cellularization regulation of membrane export changes. The existence of a mutant that results in a specific defect in membrane growth during phases 1–3, but where phase 4 (fast phase) proceeds normally (Merrill et al. 1988), supports this view and may provide an entry point into this regulation.
Insertion of New Membrane as an Active Mechanism during Cytokinesis?
A surprising result is that the majority of the membrane accounting for growth and invagination is not targeted to the front of the invaginating membrane, which is in contradiction to a report from Loncar and Singer 1995. The appeal of the latter model was that it provided a simple mechanism for directional growth of the membrane. Loncar and Singer 1995 showed electron micrographs where vesicles line up and fuse ahead of the FC, suggesting that the membrane might grow by lateral aggregation of individual vesicles. Our data contradict this scenario. First, Neur is not detected in the FC although its levels build up progressively in the apical and lateral membranes. Second, through our lectin label experiments we show that the FC is a separate membrane region formed in the first 10 min of cellularization (phase 1) and sealed from the adjacent membrane by the presence of a basal junction that accumulates high levels of Arm. Moreover, the membrane is targeted apically in phase 3 or laterally in phase 4 and labeled membrane basal to these insertion sites move basally with the FC. Similarly, microspheres bound to the basal lateral membrane consistently accompany the FC movement. This leads us to conclude that the majority of the new membrane is inserted either apically or apico–laterally, but not at the FC. How might apical or even lateral insertion of membrane contribute to the basal translocation of the FC? Interestingly, our data show that the recipient lateral membrane moves basally in phases 3 and 4 as if pushed away from the insertion point. We propose that insertion of the membrane might play an active role in the invagination process and force the membrane to grow. Directionality could stem from the active role of the contractile actomyosin network that exerts a tension at the tip of the growing membrane (Schejter and Wieschaus 1993b) or from the pushing force of the microtubules (Foe et al. 1993). However, since in colcemid-treated embryos cellularization proceeds in phase 4 (Foe et al. 1993; our unpublished results), we favor a model in which insertion of the membrane generates the important propulsive force, one that is guided and stabilized by an actomyosin network.
A Link between Membrane Growth and Polarization during Cytokinesis?
Different lines of evidence also suggest that polarization of the plasma membrane might be inherently linked to the process of membrane growth during cleavage of the Drosophila blastoderm. Membrane insertion does not occur at random and uniformly over the entire surface of the plasma membrane. Rather, new membrane is delivered at well-defined sites that change from apical to apical–lateral at different phases of cellularization. This change coincides with a sudden twofold increase in the rate of membrane invagination from phase 3 to 4. Therefore, during cleavage of the Drosophila embryo, membrane trafficking is regulated in a way that allows polarized membrane delivery. Genetic analysis of syntaxin1, a target membrane protein termed t-SNARE, in Drosophila cellularizing embryos (Burgess et al. 1997) and during cytokinesis of the early Caenorhabditis elegans embryo (Jantsch-Plunger and Glotzer 1999), have shown that membrane fusion events are required for cytokinesis. However, these studies could not identify where membrane delivery occured and whether it was polarized. Moreover, it is believed that t-SNAREs do not directly provide spatial cues for membrane delivery since, in budding yeast, the t-SNARE Sec9p/SNAP-25 is localized over the whole cell surface of the forming bud (Brennwald et al. 1994), although membrane delivery only occurs at the tip of the bud. Since then, it has been shown in Saccharomyces cerevisiae (TerBush and Novick 1995) and in MDCK cells (Grindstaff et al. 1998) that spatial specificity for membrane delivery might be provided by components of a conserved protein complex termed exocyst, and in particular Sec6/Sec8 (for a review see Hsu et al. 1999). Sec6 and Sec8 localize to the sites of membrane delivery in contrast to t-SNAREs. Genetic analysis of sec6 and sec8 for which orthologues have been found in Drosophila (Lloyd et al. 2000) might be more revealing than earlier reports on syntaxin. In particular, it should be possible to ask whether the change in the site of membrane insertion from phase 3 to 4 involves the recruitment of D-sec6 and D-sec8 to a new location, or if two independent sets of molecules are involved. In the light of our data, it is tantalizing to propose that the scheme of polarized membrane delivery we identify could participate in the emerging polarization of the plasma membrane.
Two independent sets of experiments argue that membrane miscibility might be insufficient to compete with the effect of massive local membrane delivery, thus lending support to our model. At any time during cellularization, WGA-labeled membrane regions located basal to the sites of membrane delivery move in register with the advancing front of invagination and segregate from the newly inserted membrane as if displaced away from the sites of insertion. We do not think this is a simple effect of WGA binding to the membrane, as a similar conclusion was drawn when we tracked WGA-coated microspheres bound to an otherwise intact plasma membrane. The microbeads serve as landmarks on the plasma membrane and reveal its dynamic properties. Isolated beads move away from the sites of membrane insertion and follow the inward movement of the FC. This situation would not occur if diffusion in the plane of the membrane was enough to cancel the effect of local membrane delivery. Together, these experiments argue that the history of membrane insertion is partly recapitulated in the spatial arrangements of distinct membrane regions, the most basal being also the oldest, generally speaking. This would provide a powerful mechanism to polarize the forming epithelium if the composition of the newly inserted membrane from the ER and Golgi apparatus changes as a function of time through the progressive induction of new transmembrane proteins. Each set of transmembrane proteins might in turn recruit specific groups of membrane-bound proteins and further define each region as a compositionally unique membrane domain. We studied the transmembrane protein Neur and found that its localization in phase 4 reflects the scheme of apico–lateral membrane insertion in agreement with our model. Neur is absent from the FC, a membrane region that is formed before induction of Neur. Neur is also localized at higher levels in the apical–lateral sites of membrane insertion.
Such a model could explain how a prepolarity initiated during cellularization would subsequently participate in recruitment of proteins required for the formation of a mature apical adherens junction. The apical adherens junction is only effective in tightly connecting adjacent cells and serving as a diffusion barrier after cellularization is completed and when gastrulation begins. At this stage, Arm/β-catenin is concentrated in a tight apical belt (Muller and Wieschaus 1996), with E-cadherin, Dlt (Bhat et al. 1999), and Dlg, another PDZ-domain containing protein (Woods et al. 1996; Hough et al. 1997). All these proteins are required for the integrity of the mature adherens junction. Interestingly, the early signs of junction assembly can be tracked back to phase 4 of cellularization when Arm and Dlg become progressively localized to a broad region of the apical lateral membrane (Fig. 1, and our unpublished observations) together with βH- and β-spectrins (Thomas and Williams 1999). This broad polarized localization could reflect the insertion of new membrane in this region, as we show (Fig. 3 and Fig. 5). Shortly after the completion of phase 4, Dlg and Arm become restricted to the typical apical beltlike localization pattern and Dlt is recruited in the mature junction (our unpublished observations).
To conclude, our data show that cellularization, a specialized form of cytokinesis, is characterized by a precise scheme of membrane growth and insertion that accompanies the emergence of cell polarity. Cellularization may use a novel strategy to polarize the membrane, which we suggest occurs through the sequential insertion of new membrane at different sites and is supported by the fact that membrane mixing between the recipient and newly inserted membrane populations may not be enough to compete with the overwhelming insertion of new membrane. It will be interesting to see whether this pertains to other situations of massive membrane growth such as axon formation. Our findings also open the interesting possibility that membrane insertion might actively participate in furrowing during cytokinesis.
Acknowledgments
We are very grateful to G. Waters, T. Vogt, Y. Ahmed, and R. Hoang for insightful and useful comments on the manuscript; and to M. Bhat and H. Bellen for the antibody to Dlt, C. Fields for the antibody to myosin, and V. Ripoche and V. Malhotra for the antibody to βCOP. We also thank J. Goodhouse for help with confocal microscopy.
E. Wieschaus is an investigator of the Howard Hughes Medical Institute. T. Lecuit was supported by the Howard Hughes Medical Institute, the Philippe Foundation, and a long-term fellowship from the Human Frontier Science Program Organization.
Footnotes
Abbreviations used in this paper: Arm, Armadillo; DIC, differential interference contrast; Dlg, Discs-large; Dlt, Discs-lost; FC, furrow canal; MDCK, Madin-Darby canine kidney; Neur, Neurotactin; WGA, wheat germ agglutinin.
References
- Barth A.I., Nathke I.S., Nelson W.J. Cadherins, catenins and APC proteininterplay between cytoskeletal complexes and signaling pathways. Curr. Opin. Cell Biol. 1997;9:683–690. doi: 10.1016/s0955-0674(97)80122-6. [DOI] [PubMed] [Google Scholar]
- Bhat M.A., Izaddoost S., Lu Y., Cho K.O., Choi K.W., Bellen H.J. Discs Lost, a novel multi-PDZ domain protein, establishes and maintains epithelial polarity. Cell. 1999;96:833–845. doi: 10.1016/s0092-8674(00)80593-0. [DOI] [PubMed] [Google Scholar]
- Bradke F., Dotti C.G. Neuronal polarityvectorial cytoplasmic flow precedes axon formation. Neuron. 1997;19:1175–1186. doi: 10.1016/s0896-6273(00)80410-9. [DOI] [PubMed] [Google Scholar]
- Brennwald P., Kearns B., Champion K., Keranen S., Bankaitis V., Novick P. Sec9 is a SNAP-25-like component of a yeast SNARE complex that may be the effector of Sec4 function in exocytosis. Cell. 1994;79:245–258. doi: 10.1016/0092-8674(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Burgess R.W., Deitcher D.L., Schwarz T.L. The synaptic protein syntaxin1 is required for cellularization of Drosophila embryos. J. Cell Biol. 1997;138:861–875. doi: 10.1083/jcb.138.4.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti C.G., Simons K. Polarized sorting of viral glycoproteins to the axon and dendrites of hippocampal neurons in culture. Cell. 1990;62:63–72. doi: 10.1016/0092-8674(90)90240-f. [DOI] [PubMed] [Google Scholar]
- Drubin D.G., Nelson W.J. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Foe, V.E., G.M. Odell, and B.A. Edgar. 1993. Mitosis and morphogenesis in the Drosophila embryo: point and counterpoint. The Development of Drosophila melanogaster. Vol. I. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 149–300.
- Foe V.E., Alberts B.M. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J. Cell Sci. 1983;61:31–70. doi: 10.1242/jcs.61.1.31. [DOI] [PubMed] [Google Scholar]
- Fullilove S.L., Jacobson A.G. Nuclear elongation and cytokinesis in Drosophila montana . Dev. Biol. 1971;26:560–577. doi: 10.1016/0012-1606(71)90141-2. [DOI] [PubMed] [Google Scholar]
- Glotzer M. Cytokinesis. Curr. Biol. 1997;7:R274–R276. doi: 10.1016/s0960-9822(06)00135-7. [DOI] [PubMed] [Google Scholar]
- Grindstaff K.K., Yeaman C., Anandasabapathy N., Hsu S.C., Rodriguez-Boulan E., Scheller R.H., Nelson W.J. Sec6/8 complex is recruited to cell–cell contacts and specifies transport vesicle delivery to the basal–lateral membrane in epithelial cells. Cell. 1998;93:731–740. doi: 10.1016/s0092-8674(00)81435-x. [DOI] [PubMed] [Google Scholar]
- Hough C.D., Woods D.F., Park S., Bryant P.J. Organizing a functional junctional complex requires specific domains of the Drosophila MAGUK Discs large. Genes Dev. 1997;11:3242–3253. doi: 10.1101/gad.11.23.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.C., Hazuka C.D., Foletti D.L., Scheller R.H. Targeting vesicles to specific sites on the plasma membranethe role of the sec6/8 complex. Trends Cell Biol. 1999;9:150–153. doi: 10.1016/s0962-8924(99)01516-0. [DOI] [PubMed] [Google Scholar]
- Hunter C., Wieschaus E. Regulated expression of nullo is required for the formation of distinct apical and basal adherens junctions in the Drosophila blastoderm. J. Cell Biol. 2000;150:391–401. doi: 10.1083/jcb.150.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch-Plunger V., Glotzer M. Depletion of syntaxins in the early Caenorhabditis elegans embryo reveals a role for membrane fusion events in cytokinesis. Curr. Biol. 1999;9:738–745. doi: 10.1016/s0960-9822(99)80333-9. [DOI] [PubMed] [Google Scholar]
- Lloyd T.E., Verstreken P., Ostrin E.J., Phillippi A., Lichtarge O., Bellen H.J. A genome-wide search for synaptic vesicle cycle proteins in Drosophila . Neuron. 2000;26:45–50. doi: 10.1016/s0896-6273(00)81136-8. [DOI] [PubMed] [Google Scholar]
- Loncar D., Singer S.J. Cell membrane formation during the cellularization of the syncytial blastoderm of Drosophila . Proc. Natl. Acad. Sci. USA. 1995;92:2199–2203. doi: 10.1073/pnas.92.6.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill P.T., Sweeton D., Wieschaus E. Requirements for autosomal gene activity during precellular stages of Drosophila melanogaster. Development. 1988;104:495–509. doi: 10.1242/dev.104.3.495. [DOI] [PubMed] [Google Scholar]
- Muller H.A., Wieschaus E. Armadillo, bazooka, and stardust are critical for early stages in formation of the zonula adherens and maintenance of the polarized blastoderm epithelium in Drosophila . J. Cell Biol. 1996;134:149–163. doi: 10.1083/jcb.134.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprins A., Duden R., Kreis T.E., Geuze H.J., Slot J.W. Beta-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J. Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M., Sweeton D., Casey M., Wieschaus E. wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development. 1994;120:369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- Ripoche J., Link B., Yucel J.K., Tokuyasu K., Malhotra V. Location of Golgi membranes with reference to dividing nuclei in syncytial Drosophila embryos. Proc. Natl. Acad. Sci. USA. 1994;91:1878–1882. doi: 10.1073/pnas.91.5.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose L.S., Wieschaus E. The Drosophila cellularization gene nullo produces a blastoderm-specific transcript whose levels respond to the nucleocytoplasmic ratio. Genes Dev. 1992;6:1255–1268. doi: 10.1101/gad.6.7.1255. [DOI] [PubMed] [Google Scholar]
- Schejter E.D., Rose L.S., Postner M.A., Wieschaus E. Role of the zygotic genome in the restructuring of the actin cytoskeleton at the cycle-14 transition during Drosophila embryogenesis. Cold Spring Harb. Symp. Quant. Biol. 1992;57:653–659. doi: 10.1101/sqb.1992.057.01.071. [DOI] [PubMed] [Google Scholar]
- Schejter E.D., Wieschaus E. Bottleneck acts as a regulator of the microfilament network governing cellularization of the Drosophila embryo Cell 75 1993. 373 385a [DOI] [PubMed] [Google Scholar]
- Schejter E.D., Wieschaus E. Functional elements of the cytoskeleton in the early Drosophila embryo Annu. Rev. Cell Biol 9 1993. 67 99b [DOI] [PubMed] [Google Scholar]
- Schweisguth F., Lepesant J.A., Vincent A. The serendipity alpha gene encodes a membrane-associated protein required for the cellularization of the Drosophila embryo. Genes Dev. 1990;4:922–931. doi: 10.1101/gad.4.6.922. [DOI] [PubMed] [Google Scholar]
- TerBush D.R., Novick P. Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae . J. Cell Biol. 1995;130:299–312. doi: 10.1083/jcb.130.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G.H., Williams J.A. Dynamic rearrangement of the spectrin membrane skeleton during the generation of epithelial polarity in Drosophila . J. Cell Sci. 1999;112:2843–2852. doi: 10.1242/jcs.112.17.2843. [DOI] [PubMed] [Google Scholar]
- Turner F.R., Mahowald A.P. Scanning electron microscopy of Drosophila embryogenesis. 1. The structure of the egg envelopes and the formation of the cellular blastoderm. Dev. Biol. 1976;50:95–108. doi: 10.1016/0012-1606(76)90070-1. [DOI] [PubMed] [Google Scholar]
- Wieschaus E., Sweeton D. Requirements for X-linked zygotic gene activity during cellularization of early Drosophila embryos. Development. 1988;104:483–493. doi: 10.1242/dev.104.3.483. [DOI] [PubMed] [Google Scholar]
- Winckler B., Forscher P., Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- Woods D.F., Hough C., Peel D., Callaini G., Bryant P.J. Dlg protein is required for junction structure, cell polarity, and proliferation control in Drosophila epithelia. J. Cell Biol. 1996;134:1469–1482. doi: 10.1083/jcb.134.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]