

Abstract
Myogenic differentiation is a highly orchestrated, multistep process that is coordinately regulated by growth factors and cell adhesion. We show here that integrin-linked kinase (ILK), an intracellular integrin– and PINCH-binding serine/threonine protein kinase, is an important regulator of myogenic differentiation. ILK is abundantly expressed in C2C12 myoblasts, both before and after induction of terminal myogenic differentiation. However, a noticeable amount of ILK in the Triton X-100–soluble cellular fractions is significantly reduced during terminal myogenic differentiation, suggesting that ILK is involved in cellular control of myogenic differentiation. To further investigate this, we have overexpressed the wild-type and mutant forms of ILK in C2C12 myoblasts. Overexpression of ILK in the myoblasts inhibited the expression of myogenic proteins (myogenin, MyoD, and myosin heavy chain) and the subsequent formation of multinucleated myotubes. Furthermore, mutations that eliminate either the PINCH-binding or the kinase activity of ILK abolished its ability to inhibit myogenic protein expression and allowed myotube formation. Although overexpression of the ILK mutants is permissive for the initiation of terminal myogenic differentiation, the myotubes derived from myoblasts overexpressing the ILK mutants frequently exhibited an abnormal morphology (giant myotubes containing clustered nuclei), suggesting that ILK functions not only in the initial decision making process, but also in later stages (fusion or maintaining myotube integrity) of myogenic differentiation. Additionally, we show that overexpression of ILK, but not that of the PINCH-binding defective or the kinase-deficient ILK mutants, prevents inactivation of MAP kinase, which is obligatory for the initiation of myogenic differentiation. Finally, inhibition of MAP kinase activation reversed the ILK-induced suppression of myogenic protein expression. Thus, ILK likely influences the initial decision making process of myogenic differentiation by regulation of MAP kinase activation.
Keywords: integrin, PINCH, MAP kinase, myogenin, myotubes
Introduction
Terminal myogenic differentiation is a highly orchestrated, multistep process that is controlled by environmental cues including growth factors and the extracellular matrix (McDonald et al. 1995; Sastry and Horwitz 1996; Wewer and Engvall 1996; Durbeej et al. 1998; Gullberg et al. 1998; Burkin and Kaufman 1999). For example, it has been well described that differentiation of myoblasts plated on appropriate extracellular matrix proteins (e.g., collagens) into multinucleated myotubes can be induced by depriving the cells of growth factors (Weintraub 1993; Lassar et al. 1994; Olson and Klein 1994; Andres and Walsh 1996). During this process, the activity of MAP kinase is downregulated and expression of myogenic transcription factors such as myogenin is upregulated, followed by expression of other muscle-specific proteins such as myosin heavy chain (MHC) and subsequent formation of multinucleated myotubes (Lassar et al. 1994; Andres and Walsh 1996; Bennett and Tonks 1997).
Extensive studies over the last one and a half decades have demonstrated crucial roles of cell adhesion receptors, including integrins, in the regulation of terminal myogenic differentiation (McDonald et al. 1995; Sastry and Horwitz 1996; Gullberg et al. 1998; Burkin and Kaufman 1999). In genetic model systems such as Drosophila and Caenorhabditis elegans, it has been well documented that integrins are involved in sarcomere formation and stabilization or muscle cell attachment (Volk et al. 1990; Gettner et al. 1995; Martin-Bermudo and Brown 1996; Bloor and Brown 1998; Bunch et al. 1998; Gullberg et al. 1998; Prokop et al. 1998). In vertebrates, a number of integrins are expressed in muscle cells, and the expression level, subtype, and activation state of the integrins are precisely regulated during myogenesis (Boettiger et al. 1995; Sastry and Horwitz 1996; Gullberg et al. 1998; Burkin and Kaufman 1999). Antibody ligation of specific β1 integrins inhibits vertebrate myoblast differentiation (Menko and Boettiger 1987; Rosen et al. 1992). Furthermore, gene transfer experiments have demonstrated that myoblast differentiation and proliferation are regulated by β1 integrins (Sastry et al. 1996) and this regulation is achieved through the β1 integrin cytoplasmic domain (Sastry et al. 1999). Studies using chimeric transgenic mice that were α5 integrin −/−: +/+ showed that the α5 −/− cells were able to contribute to skeletal muscle, but the myofibers were unstable, resulting in a form of muscular dystrophy (Taverna et al. 1998). Similar results showing a mild muscular dystrophy were obtained with a targeted deletion of the α7 integrin chain (Mayer et al. 1997), and mutations in the human integrin α7 gene lead to a congenital myopathy (Hayashi et al. 1998). Thus, integrins function in terminal myogenic differentiation by participating in both the initial decision making process and the later morphogenic processes such as cell fusion and maintaining the integrity of myotubes.
Integrin-linked kinase (ILK) is a focal adhesion serine/threonine protein kinase that interacts with β1 integrins through the COOH-terminal domain (Hannigan et al. 1996; Dedhar et al. 1999) and PINCH, an adaptor protein comprising five LIM domains, through the NH2-terminal ankyrin (ANK) repeat domain (Tu et al. 1999; Wu 1999). The ILK-binding site has been mapped to the COOH-terminal zinc finger, which is located within the first LIM domain of PINCH (Li et al. 1999a; Wu 1999). The ILK–PINCH interaction is required for proper subcellular localization of ILK (Li et al. 1999a; Wu 1999). Furthermore, it may also connect ILK with components of the growth factor and small GTPase signaling pathways via other PINCH-binding proteins such as Nck-2 (Tu et al. 1998; Wu 1999). Recent biochemical and functional studies have indicated that ILK serves as a mediator in integrin-mediated signal transduction (Hannigan et al. 1996; Radeva et al. 1997; Delcommenne et al. 1998; Novak et al. 1998; Wu et al. 1998; Troussard et al. 1999; Tu et al. 1999; Wu 1999). In this study, we have investigated the roles and potential mechanisms of ILK in cellular control of terminal myogenic differentiation.
Materials and Methods
Antibodies
Mouse monoclonal anti-ILK antibody 65.1 was generated as previously described (Li et al. 1999a). Mouse monoclonal anti-FLAG antibody M5 was from Eastman Kodak Co. Mouse monoclonal antimyogenin F5D and anti–MyoD 5.8A were from PharMingen. Hybridoma for monoclonal anti–myosin heavy chain (MHC) antibody MF20 was obtained from the Developmental Studies Hybridoma Bank. Rabbit polyclonal anti-FAK antibody C-20 was purchased from Santa Cruz Biotechnology, Inc. Antiphosphotyrosine antibody RC20:HRPO was purchased from Transduction Laboratories. Rabbit polyclonal anti-p44/42 MAPK and anti–phospho-p44/42 MAPK (Thr202/Tyr204) antibodies were purchased from New England BioLabs. HRP-conjugated goat anti–mouse IgG and goat anti–rabbit IgG were purchased from Jackson ImmunoResearch Laboratories.
Cell Culture and Myogenic Differentiation
Mouse C2C12 myoblasts (American Type Culture Collection) were maintained at subconfluent densities in growth medium (GM) consisting of DME (Life Technologies) supplemented with 10% FBS (Sigma Chemical Co.). To induce terminal myogenic differentiation, cells were seeded into collagen I–coated plates (Becton Dickinson), and were shifted to differentiation medium (DM) consisting of DME supplemented with 2% horse serum (Sigma Chemical Co.) when the cells reached 70–80% confluence.
Construction and Transfection of Wild-type and Mutant Forms of FLAG-tagged ILK
To create NH2-terminal FLAG epitope-tagged proteins, cDNAs encoding the mouse wild-type ILK (residues 1–452) and the PINCH-binding defective ANK1 deletion mutant (residues 66–452, referred to as ΔANK1), respectively, were cloned into a mammalian expression vector pFLAG-CMV-2 (Eastman Kodak Co.) as described previously (Li et al. 1999a). A cDNA encoding the human kinase–deficient ILK mutant (referred to as KD) containing a single mutation (Glu359 → Lys) was PCR amplified from the recombinant plasmid GH31R (Novak et al. 1998; Wu et al. 1998) and cloned into pFLAG-CMV-2 using EcoRI-SalI sites.
To generate stable transfectants, C2C12 cells were cotransfected with pFLAG-CMV-2 vectors containing ILK, ΔANK1, or KD cDNA, or pFLAG-CMV-2 vector lacking the ILK sequence as a control, and pcDNA3 (Invitrogen) carrying a neomycin-resistant marker at a ratio of 10:1, using the LipofectAMINE PLUS reagent (Life Technologies). C2C12 cells expressing the FLAG-tagged wild-type and mutant forms of ILK were selected with 1 mg/ml of G418 (Life Technologies) and cloned as described previously (Li et al. 1999b). A total of five FLAG-ILK–expressing clones (C27, E1.3, F6.2, F31, and F41), three FLAG-ΔANK1–expressing clones (G2, G24, and G43), and three FLAG-KD–expressing clones (B38, E20, and H15) were isolated independently. The cells were maintained in culture medium containing 200 μg/ml of G418.
Immunoblotting
Cells were washed twice with PBS and lysed in ice-cold RIPA extraction buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 1% Triton X-100, 0.25% sodium deoxycholate, 2 mM EDTA, and 2 mM EGTA) containing protease inhibitors 4-(2-aminoethyl)-benzenesulfonyl fluoride (0.2 mM), 10 μg/ml aprotinin, 1 μg/ml pepstatin, and 5 μg/ml leupeptin (Sastry et al. 1999) unless otherwise specified. The cell lysates were clarified by centrifugation at 10,000 g for 15 min. Protein concentration of the clarified lysates was determined using bicinchoninic acid (BCA) protein assay reagents (Pierce Chemical Co.). Proteins (5–15 μg) were resolved by SDS-PAGE and transferred onto Immobilon-P membranes (Millipore). The membranes were blocked with TBST buffer (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Tween 20) containing 5% nonfat dry milk and incubated with primary antibodies (0.5–1 μg/ml) as specified in each experiment. After three washes with TBST, the membranes were incubated with appropriate secondary antibodies (1:10,000 dilution) and washed, and the bound antibodies were detected with SuperSignal chemiluminescent substrate (Pierce Chemical Co.).
For immunoblotting analysis of phospho-p44/42 MAPK, cells cultured in GM or DM for 3 h were lysed in the RIPA buffer containing protease inhibitors (as described above) and phosphatase inhibitors (30 mM sodium pyrophosphate, 100 mM NaF, 2 mM sodium orthovanadate). An equal amount (8 μg/lane) of the cell lysates was separated on 8% SDS-PAGE gels. After blotting, the membranes were blocked with 2% BSA in TBST. Active MAPK was detected with an anti–phospho(Thr202/Tyr204)-p44/42 MAPK antibody that specifically recognizes the active forms of MAPK (New England BioLabs). Duplicate membranes were analyzed for the total MAPK with an anti-p44/42 MAPK polyclonal antibody (New England BioLabs). The membranes were reprobed with an anti-FAK polyclonal antibody (C-20) to confirm equal loading of proteins.
For immunoblotting analysis of ILK, parental C2C12 cells were cultured in GM or induced to differentiate in DM for 6 d and harvested. Half of the cells was lysed in PBS, pH 7.4, containing 1% Triton X-100 and protease inhibitors 4-(2-aminoethyl)-benzenesulfonyl fluoride (0.2 mM), 10 μg/ml aprotinin, 1 μg/ml pepstatin, and 5 μg/ml leupeptin. The other half was lysed in PBS, pH 7.4, containing 1% SDS and the protease inhibitors. An equal amount (10 μg/lane) of the cell lysates was separated on 10% SDS-PAGE gels, and ILK was detected by immunoblotting with a monoclonal anti-ILK antibody 65.1 (Li et al. 1999a).
For myogenin and MyoD immunoblotting, cells cultured in GM or induced to differentiate in DM for 4 d were lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% SDS, 2 mM EDTA, 2 mM EGTA) containing protease inhibitors as described above. An equal amount (15 μg/lane) of the cell lysates was separated on 10% SDS-PAGE gels. The membranes were first immunoblotted with an antimyogenin mAb F5D or anti-MyoD mAb 5.8A, and were reprobed with anti-MHC mAb F20, anti-FLAG mAb M5, or anti-FAK polyclonal antibody C-20 as specified in each experiment.
Immunoprecipitation
Cells cultured in GM or in DM for 4 d were lysed in the RIPA buffer containing protease inhibitors and phosphatase inhibitors as described above. The cell lysates (300 μg) were mixed with 10 μl of polyclonal anti-FAK antibody C-20 (2 μg) in a final volume of 500 μl. The samples were incubated at 4°C for 2 h with continuous agitation. 10 μl of UltraLink immobilized protein G (Pierce Chemical Co.) was added and incubated at 4°C for an additional 2 h. The beads were pelleted gently and washed four times with RIPA buffer. The precipitated proteins were released from the beads by boiling in 60 μl of SDS-PAGE sample buffer for 5 min. Equal volumes of the samples were loaded onto SDS-PAGE. Total FAK protein and the tyrosine-phosphorylated FAK were detected by immunoblotting with anti-FAK antibody C-20 and antiphosphotyrosine antibody RC20:HRPO (Transduction Laboratories), respectively.
Inhibition of p44/42 MAPK Activity
The activation of MAPK in C2C12 cells was inhibited by treatment of the cells with specific MEK inhibitor PD98059 (New England BioLabs) based on a previously described method (Sastry et al. 1999). In brief, ILK-overexpressing and parental C2C12 cells were cultured in GM or DM in the presence or absence of 25 μM PD98059 for a period of time, as specified in each experiment, and lysed with the RIPA buffer. MAPK activation and myogenin expression were analyzed by immunoblotting with anti–phospho(Thr202/Tyr204)-p44/42 MAPK antibody and antimyogenin antibody F5D, respectively, as described above.
Nuclear Staining
Cells were cultured in GM or DM for 4 d in a 24-well tissue culture plate. The cells were rinsed with PBS, fixed with 4% paraformaldehyde solution in PBS for 20 min at room temperature, and incubated with permeabilization solution (0.1% Triton X-100 and 0.1% sodium citrate) for 2 min at 4°C. After rinsing with PBS, the cells were incubated with 20 μg/ml of Hoechst 33258 (Sigma Chemical Co.) in PBS for 5 min and observed under a fluorescence microscope.
Results
ILK Regulates Terminal Myogenic Differentiation
To begin to investigate the roles of ILK in regulation of terminal myogenic differentiation, we analyzed the cellular levels of ILK in C2C12 myoblasts before and after induction of myogenic differentiation by immunoblotting with a monoclonal anti-ILK antibody. Abundant ILK was detected in Triton X-100 lysates of C2C12 myoblasts cultured in growth medium (Fig. 1, lane 1). Additional slower migrating bands, which could represent either detergent-resistant ILK-containing complexes or other ILK-related proteins (Li et al. 1999a), were also detected in the C2C12 lysates (Fig. 1). After switching to differentiation medium for 6 d, the amount of ILK in the Triton X-100 lysates of C2C12 myoblasts was noticeably reduced (Fig. 1, compare lane 2 with lane 1). The reduction of the ILK level in the Triton X-100 lysates accompanying terminal myogenic differentiation could result from a decrease of overall cellular ILK or, alternatively, from a more selective reduction of ILK in the Triton X-100–soluble fractions including membrane and cytosolic fractions. To test this, we extracted the total cellular proteins from the C2C12 cells with SDS. Immunoblotting analyses of the SDS extracts indicated that the overall cellular level of ILK was not decreased after induction of terminal myogenic differentiation (Fig. 1, lanes 3 and 4). Taken together, these results indicate that the amount of ILK in the Triton X-100–soluble subcellular fractions, but not the overall cellular level of ILK, was decreased accompanying terminal myogenic differentiation.
Figure 1.

The amount of ILK in Triton X-100–soluble fractions is reduced during terminal myogenic differentiation. Mouse C2C12 myoblasts were cultured in growth medium (GM) and induced to differentiate by switching to the differentiation medium (DM). The cells were extracted with lysis buffers containing 1% Triton X-100 or 1% SDS as described in Materials and Methods. ILK in the Triton X-100 (lanes 1 and 2) or SDS (lanes 3 and 4) extracts were analyzed by immunoblotting with a monoclonal anti-ILK antibody 65.1.
The correlation between the downregulation of ILK level in the Triton X-100–soluble fractions and the terminal myogenic differentiation suggests that ILK is likely involved in the regulation of myogenic differentiation. To investigate this, we overexpressed an epitope (FLAG)-tagged ILK in C2C12 myoblasts. C2C12 myoblasts were transfected with an expression vector containing the full-length ILK coding sequence under the CMV promoter (pFLAG-ILK). The FLAG-ILK transfectants were selected with G418 and cloned as described previously (Li et al. 1999b). A total of five FLAG-ILK–expressing C2C12 clones (C27, E1.3, F6.2, F31, and F41) were independently obtained. The expression of FLAG-ILK in the C2C12 cells before and after the induction of myogenic differentiation was confirmed by immunoblotting (Fig. 2 a, lanes 6–15). No FLAG-ILK was detected in the parental C2C12 cells (Fig. 2 a, lanes 1–5) or the vector-only control transfectants (Fig. 2 a, lanes 16–20).
Figure 2.

Effect of ILK on myogenin expression. Lysates (15 μg protein/lane) from cells cultured in growth medium (day 0) or in differentiation medium (DM) for 1, 2, 3, or 4 d were immunoblotted with anti-FLAG mAb M5 (a), antimyogenin mAb F5D (b), and anti-FAK polyclonal antibody C-20 (c). (lanes 1–5) Parental C2C12 cells; (lanes 6–10) FLAG-ILK–expressing clone E1.3; (lanes 11–15) FLAG-ILK–expressing clone C27; (lanes 16–20) vector-only control transfectants. Similar results were obtained with all five independently isolated FLAG-ILK–expressing clones.
One of the critical events at the initiation of terminal myogenic differentiation is induction of myogenin, which is a member of the MyoD family of skeletal muscle–specific, basic Helix-Loop-Helix transcription factors (Lassar et al. 1994). As expected, myogenin was not detected in C2C12 cells grown in growth medium (Fig. 2 b, lanes 1, 6, 11, and 16). Myogenin expression was induced in the parental C2C12 cells and the vector-only control transfectants after they were shifted to differentiation medium (Fig. 2 b, lanes 1–5 and 16–20). By contrast, the induction of myogenin expression was almost completely inhibited in C2C12 cells overexpressing FLAG-ILK (Fig. 2 b, lanes 6–15). In additional experiments, we have found that overexpression of FLAG-ILK also significantly reduced the expression of MyoD, another member of the MyoD family of basic Helix-Loop-Helix transcription factors (data not shown). Overexpression of FLAG-ILK did not affect FAK expression under either the growth or differentiation condition (Fig. 2 c), suggesting that ILK selectively regulates the expression of the muscle-specific transcription factors.
We next analyzed the effect of ILK overexpression on MHC, another marker for terminal myogenic differentiation. Abundant myosin heavy chain was expressed by the parental C2C12 cells and the vector-only transfectants after they were shifted to the differentiation medium (Fig. 3 a, lanes 2 and 4). By marked contrast, no MHC was detected in cells overexpressing FLAG-ILK, either before or after induction of differentiation (Fig. 3 a, lanes 5–8). Equal protein loading was confirmed by probing the same membrane with a polyclonal anti-FAK antibody (Fig. 3 b). Thus, overexpression of ILK inhibits the expression of MHC as well as that of myogenin and MyoD, indicating that ILK plays a crucial role in the regulation of myogenic protein expression.
Figure 3.
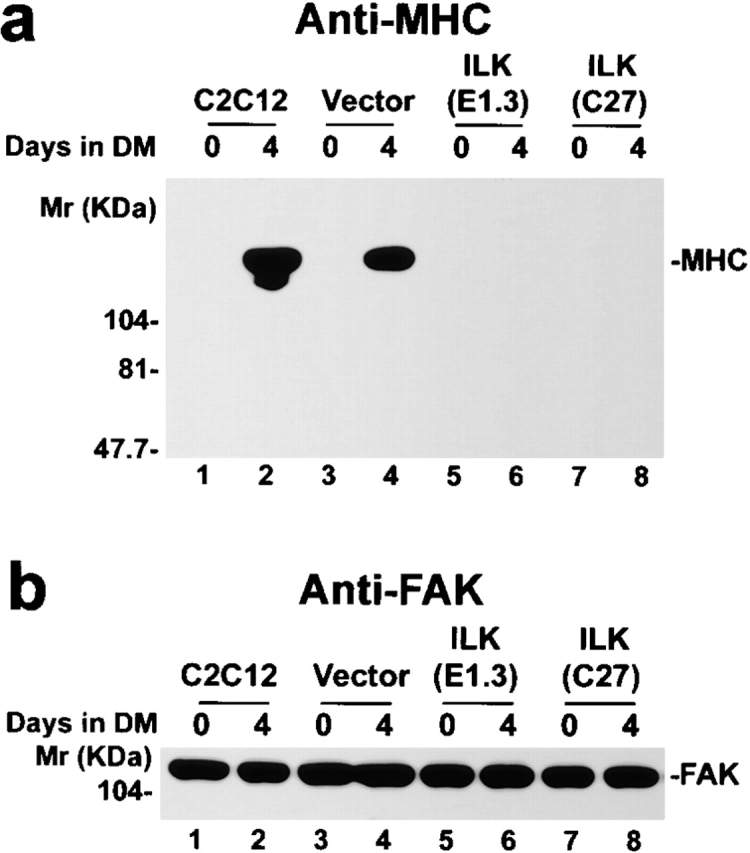
Effect of ILK on myosin heavy chain expression. Lysates (15 μg protein/lane) from cells cultured in GM (day 0) or DM for 4 d were immunoblotted with anti-MHC mAb F20 (a) and anti-FAK antibody C-20 (b). (lanes 1 and 2) Parental C2C12 cells; (lanes 3 and 4) the vector-only control transfectants; (lanes 5 and 6) FLAG-ILK–overexpressing clone E1.3; (lanes 7 and 8) FLAG-ILK–overexpressing clone C27. Similar results were obtained with all five independently isolated FLAG-ILK–expressing clones.
After induction of myogenic protein expression, C2C12 cells undergo extensive cell fusion, resulting in the formation of multinucleated myotubes (Andres and Walsh 1996). As expected, abundant multinucleated myotubes were detected in both the parental C2C12 cells and the vector-only control transfectants 4 d after induction of differentiation (Fig. 4b and Fig. d). In marked contrast, cells that overexpress FLAG-ILK failed to form multinucleated myotubes under identical experimental conditions (Fig. 4f and Fig. h). Thus, consistent with an inhibitory role of ILK in myogenic protein expression, overexpression of FLAG-ILK in C2C12 myoblasts suppresses the formation of multinucleated myotubes.
Figure 4.
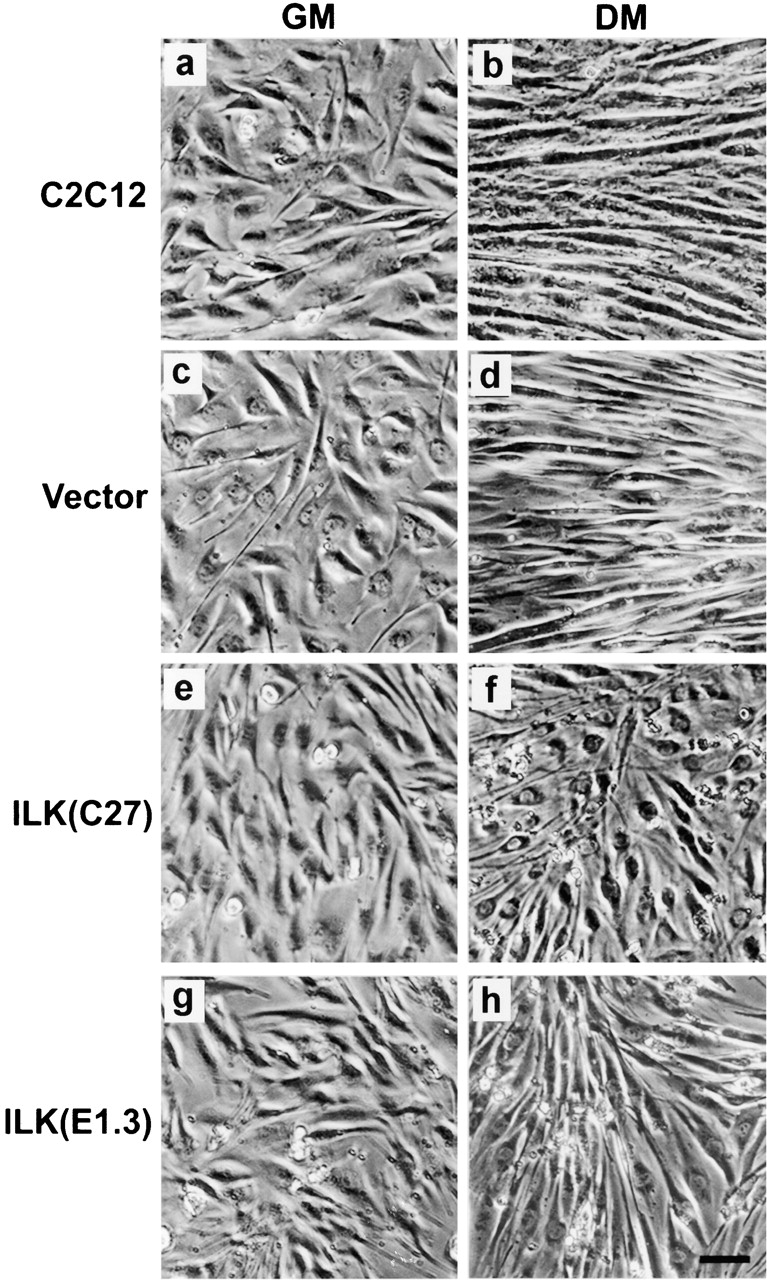
Effect of ILK on myotube formation. Parental C2C12 cells (a and b), the vector-only control transfectants (c and d), the FLAG-ILK–overexpressing clones C27 (e and f), and E1.3 (g and h) were grown in GM (left) and switched to DM for 4 d (right). Abundant myotubes were observed in C2C12 (b) and the vector control (d), but not in FLAG-ILK–expressing clones C27 (f) and E1.3 (h), after induction of differentiation. Similar results were obtained with all five independently isolated FLAG-ILK–expressing clones. Bar, 100 μm.
The Kinase Activity of ILK Is Required for Suppression of Myogenic Differentiation
ILK contains four ankyrin repeats at the NH2 terminus and a protein kinase catalytic domain at the COOH terminus (Hannigan et al. 1996; Li et al. 1997; Dedhar et al. 1999; Wu 1999). To test whether the ILK kinase catalytic activity is involved in suppression of myogenic differentiation, we expressed a FLAG-tagged kinase-deficient (KD) ILK point mutant (Novak et al. 1998; Wu et al. 1998), in which the highly conserved Glu359 within the ILK catalytic domain was substituted with lysine, in C2C12 cells. The expression of FLAG-KD in the transfectants before and after induction of myogenic differentiation was confirmed by immunoblotting (Fig. 5 a, lanes 5–8). Overexpression of the kinase-deficient ILK mutant, unlike that of the wild-type ILK (Fig. 5b and Fig. c, lane 4), did not inhibit the expression of myogenin (Fig. 5 b, lanes 6 and 8), MyoD (data not shown), or MHC (Fig. 5 c, lanes 6 and 8). Equal protein loading was confirmed by probing the same membranes with a polyclonal anti-FAK antibody (Fig. 5 d). Thus, ablation of the kinase activity relieves the inhibition on the expression of myogenin, MyoD, and MHC, indicating that ILK inhibits myogenic protein expression, at least in part, through its catalytic activity.
Figure 5.
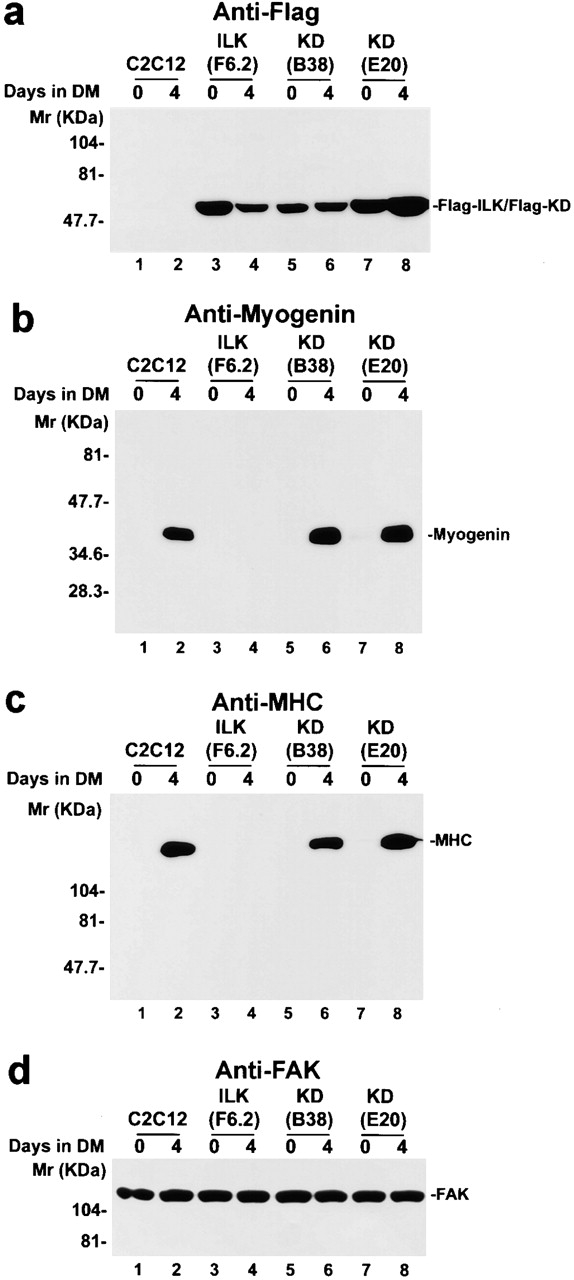
Overexpression of the kinase-deficient ILK mutant fails to inhibit myogenic protein expression. Parental C2C12 cells (lanes 1 and 2), FLAG-ILK–expressing clone F6.2 (lanes 3 and 4), and FLAG-KD–expressing clones B38 (lanes 5 and 6) and E20 (lanes 7 and 8) were cultured in GM (day 0) or DM for 4 d. Cell lysates (15 μg protein/lane) were immunoblotted with anti-FLAG mAb M5 (a), antimyogenin mAb F5D (b), anti-MHC mAb MF20 (c), and anti-FAK antibody C-20 (d). Similar results were obtained with all three independently isolated FLAG-KD–overexpressing clones.
Role of the PINCH-binding Activity of ILK in the Suppression of Myogenic Differentiation
The NH2-terminal ankyrin repeat domain of ILK mediates interaction with PINCH (Tu et al. 1999), an adaptor protein comprising five LIM domains (Rearden 1994; Wu 1999). To assess whether PINCH binding plays a role in suppression of myogenic differentiation, we expressed a FLAG-tagged PINCH-binding defective ILK mutant (ΔANK1; Li et al. 1999a), in which the first ankyrin repeat is deleted, in C2C12 cells. Expression of FLAG-ΔANK1 (Fig. 6 a, lanes 9–12) in the transfectants, but not in the parental C2C12 or the vector-only control (Fig. 6 a, lanes 1–4), before and after induction of differentiation was confirmed by immunoblotting. After induction of myogenic differentiation, the C2C12 cells that express the PINCH-binding defective ILK mutant (Fig. 6, lanes 10 and 12), like the parental C2C12 cells (Fig. 6, lane 2) or the vector-only transfectants (Fig. 6, lane 4), expressed myogenin (Fig. 6 b) and MHC (Fig. 6 c). In parallel control experiments, as expected, the induction of myogenin and myosin heavy chain was inhibited in cells expressing FLAG-ILK (Fig. 6b and Fig. c, lane 8) but not in those expressing the kinase-deficient ILK mutant (Fig. 6b and Fig. c, lane 6). Analysis of MyoD expression revealed that overexpression of the PINCH-binding defective ILK mutant, unlike that of the wild-type ILK, did not decrease MyoD expression (data not shown). Taken together, these results suggest that in addition to the kinase catalytic activity, the PINCH-binding activity is most likely also required for the suppression of myogenic protein expression.
Figure 6.
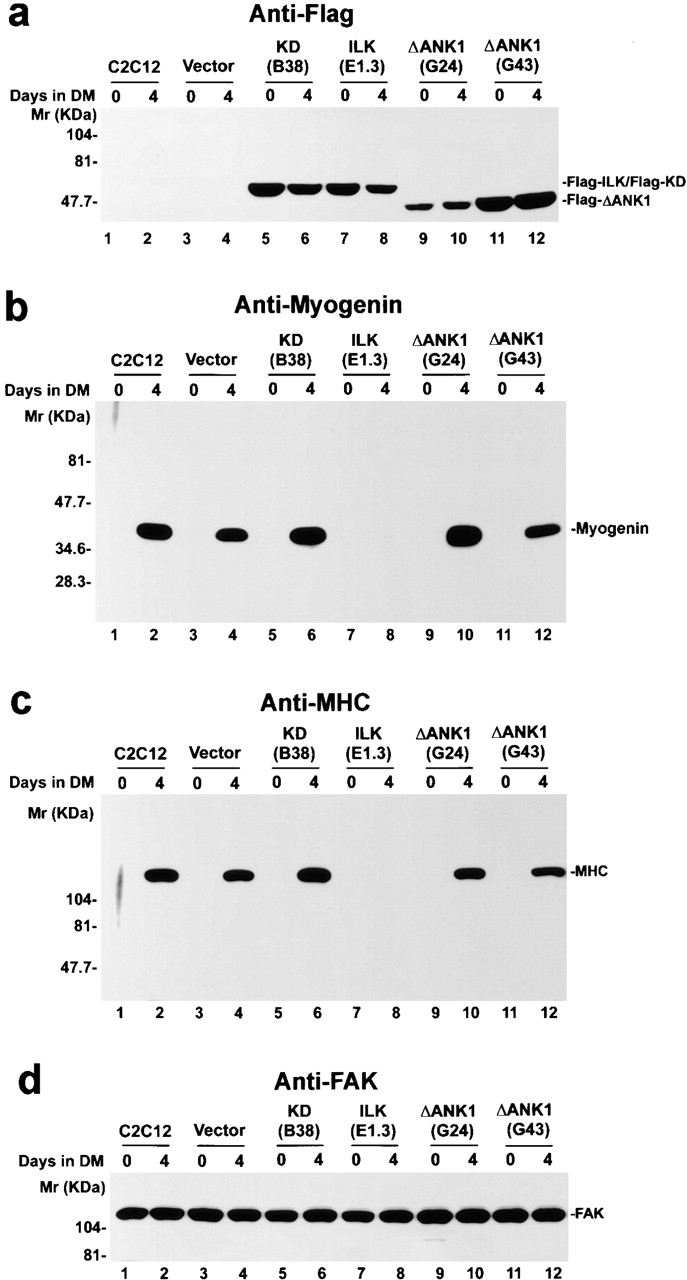
Overexpression of the PINCH-binding defective ILK mutant fails to inhibit myogenic protein expression. Parental C2C12 cells (lanes 1 and 2), the vector control (lanes 3 and 4), FLAG-KD–expressing clone B38 (lanes 5 and 6), FLAG-ILK–expressing clone E1.3 (lanes 7 and 8), and FLAG-ΔANK1–expressing clones G24 (lanes 9 and 10) and G43 (lanes 11 and 12) were cultured in GM (day 0) or DM for 4 d. Cell lysates (15 μg protein/lane) were immunoblotted with anti-FLAG mAb M5 (a), antimyogenin mAb F5D (b), anti-MHC mAb MF20 (c), and anti-FAK antibody C-20 (d). Similar results were obtained with all three independently isolated FLAG-ΔANK1–overexpressing clones.
Expression of the PINCH-binding Defective or Kinase-deficient ILK Mutants Resulted in the Formation of Myotubes with Abnormal Morphology
Extracellular matrix and growth factors not only control the expression of myogenic transcription factors and other myogenic proteins, but also influence cell fusion and organization of myotubes. Previous studies have suggested that cell adhesion receptors including integrins are involved in cell fusion and organization of myotubes (Menko and Boettiger 1987; Volk et al. 1990; Rosen et al. 1992; Boettiger et al. 1995; Mayer et al. 1997; Durbeej et al. 1998; Gullberg et al. 1998; Taverna et al. 1998; Burkin and Kaufman 1999; Montanaro et al. 1999; Tachibana and Hemler 1999). To assess whether ILK plays a role in the myogenic morphogenesis, we analyzed myotube formation by C2C12 cells overexpressing the kinase-deficient and PINCH-binding defective ILK mutants. The results showed that C2C12 cells overexpressing the kinase-deficient mutant (Fig. 7c and Fig. d) or the PINCH-binding defective mutant (Fig. 7e and Fig. f), unlike those overexpressing FLAG-ILK (Fig. 7g and Fig. h), were able to form multinucleated myotubes after induction of differentiation. However, the myotubes derived from the cells overexpressing the mutant forms of ILK frequently exhibited an abnormal morphology (Fig. 7, c–f). In contrast to myotubes derived from the parental C2C12 (Fig. 7 a) or the vector-only transfectants (Fig. 7 b), in which nuclei were well aligned along the myotubes, we have observed in cells overexpressing the ILK mutants many giant myotubes in which nuclei were clustered (Fig. 7, c–f). These results suggest that ILK, in addition to influencing the initial decision making process, may also play a role in the later stages (cell fusion or organization of myotubes) of terminal myogenic differentiation.
Figure 7.
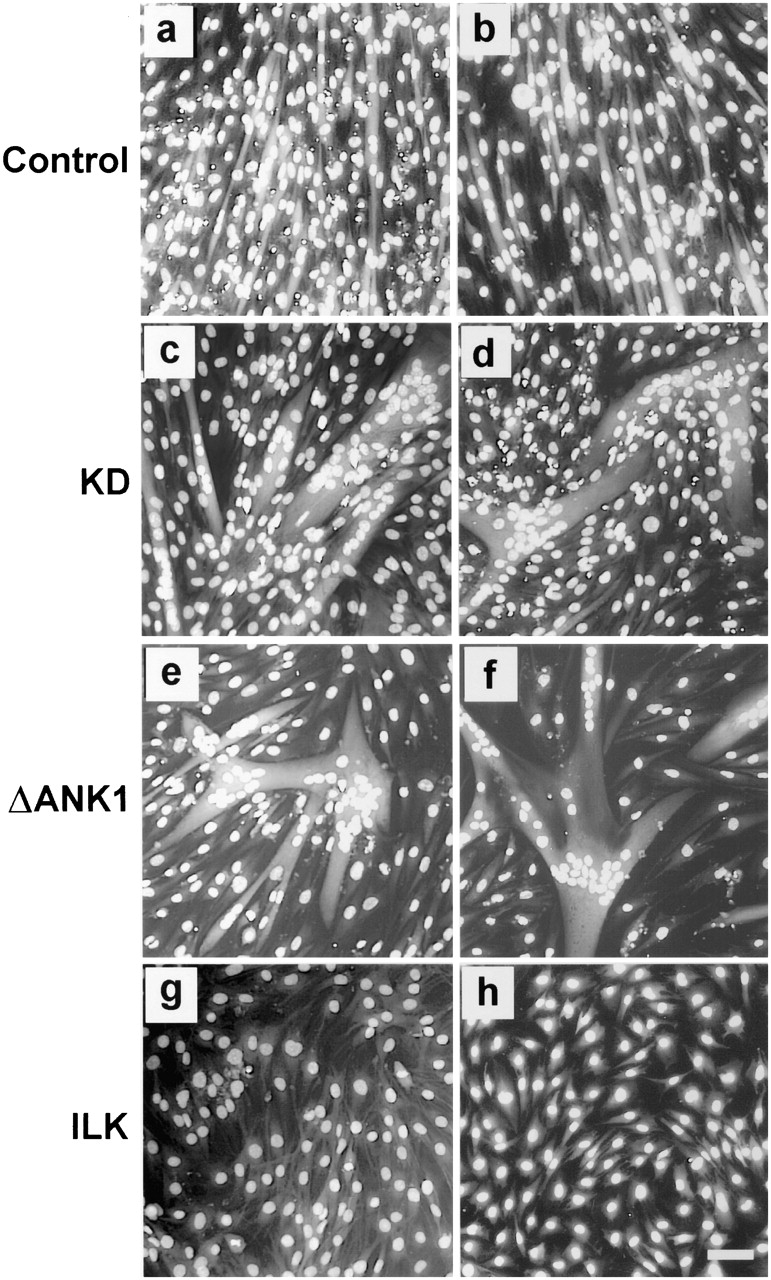
Effect of the kinase-deficient and the PINCH-binding defective ILK mutants on myogenic morphogenesis. Parental C2C12 cells (a), the vector-only control transfectants (b), FLAG-KD–expressing clones B38 (c) and E20 (d), FLAG-ΔANK1–expressing clones G24 (e) and G43 (f), and FLAG-ILK–expressing clones C27 (g) and F41 (h) were cultured in DM for 4 d, fixed, and stained with Hoechst 33258. Multinucleated myotubes were detected in C2C12 (a), vector control (b), and the ILK mutant–expressing cells (c–f) but not the FLAG-ILK–expressing cells (g and h). Giant myotubes with clustered nuclei were evident in FLAG-KD– and FLAG-ΔANK1–expressing cells (c–f). Similar results were obtained with all independently isolated FLAG-KD–overexpressing and FLAG-ΔANK1–overexpressing clones. Bar, 250 μm.
ILK Influences Initiation of Terminal Myogenic Differentiation through Regulation of p44/42 MAP Kinase (Erk1 and Erk2) Activation
Integrins control the terminal myogenic differentiation, at least in part, by regulation of MAP kinase activation (Sastry et al. 1999). Consistent with previous studies (Bennett and Tonks 1997), the amounts of active forms of p44/42 MAP kinases (Erk1 and Erk2) in the parental C2C12 cells (Fig. 8 a, lanes 1 and 2) and the vector-only control cells (Fig. 8 a, lanes 7 and 8) were decreased upon induction of terminal myogenic differentiation. By contrast, the amount of active forms of p44/42 MAP kinases (Erk1 and Erk2) in C2C12 cells overexpressing the wild-type ILK remained high after shifting to differentiation medium (Fig. 8 a, lanes 3–6). Probing the same samples with an anti-p44/42 MAP kinase antibody showed that the total protein level of the p44/42 MAP kinases was not altered by overexpression of ILK or induction of differentiation (Fig. 8 b, lanes 1–8). Equal protein loading was confirmed by immunoblotting with an anti-FAK antibody (Fig. 8 c, lanes 1–8). Because the downregulation of MAP kinase activity is required for myoblasts to initiate terminal myogenic differentiation (Bennett and Tonks 1997; Sastry et al. 1999), these results suggest that ILK suppresses myogenic differentiation, at least in part, by preventing inactivation of p44/42 MAP kinases.
Figure 8.
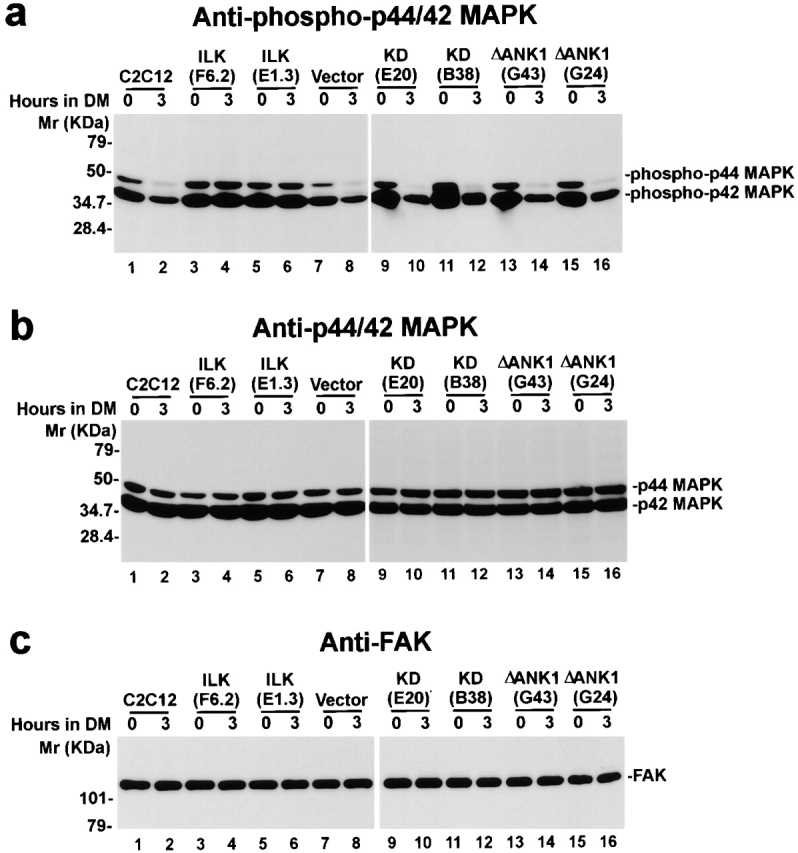
Regulation of p44/42 MAP kinase activation by ILK. Parental C2C12 cells (lanes 1 and 2), FLAG-ILK–overexpressing clones F6.2 (lanes 3 and 4) and E1.3 (lanes 5 and 6), the vector-only control transfectants (lanes 7 and 8), FLAG-KD–overexpressing clones E20 (lanes 9 and 10) and B38 (lanes 11 and 12), and FLAG-ΔANK1–overexpressing clones G43 (lanes 13 and 14) and G24 (lanes 15 and 16) were cultured in GM (0 h) or shifted to DM for 3 h. Cell lysates (8 μg protein/lane) were immunoblotted with an anti–phospho(Thr202/Tyr204)-p44/42 MAPK antibody that specifically recognizes the active forms of MAPK (a), an anti-p44/42 MAPK that recognizes total p44/42 MAPK proteins (b) and an anti-FAK antibody (c), respectively.
To further analyze the mechanism by which ILK regulates myogenic differentiation, we examined the effects of overexpression of the kinase-deficient or the PINCH-binding defective ILK mutants on activation of p44/42 MAP kinases. In contrast to C2C12 cells overexpressing the wild-type ILK (Fig. 8 a, lanes 3–6), the amounts of the active forms of p44/42 MAP kinases in C2C12 cells overexpressing the ILK mutants (Fig. 8 a, lanes 9–16) were downregulated after shifting to differentiation medium. The total protein level of the p44/42 MAP kinases was not altered by overexpression of the ILK mutants (Fig. 8 b, lanes 9–16). Equal protein loading was further confirmed by immunoblotting with an anti-FAK antibody (Fig. 8 c, lanes 9–16). These results indicate that ablation of the kinase activity or the PINCH-binding activity of ILK eliminated its ability to regulate MAP kinase activation. Because neither the kinase-deficient mutant nor the PINCH defective mutant inhibits terminal myogenic differentiation, these results provide additional evidence suggesting that p44/42 MAP kinases serve as downstream effectors of ILK in the regulation of terminal myogenic differentiation. In contrast to the major difference in MAP kinase activation, FAK activation, as indicated by the tyrosine phosphorylation level of FAK, in the ILK-overexpressing C2C12 cells, parental C2C12 cells and the vector-only transfectants, did not differ under either growth (Fig. 9 a) or differentiation (Fig. 9 b) condition, suggesting that ILK likely regulates MAP kinase activation via a pathway independent of FAK activation. This result is consistent with previous findings showing that the FAK tyrosine phosphorylation level is not altered during the α5β1 integrin–mediated suppression of myogenic differentiation (Sastry et al. 1999).
Figure 9.
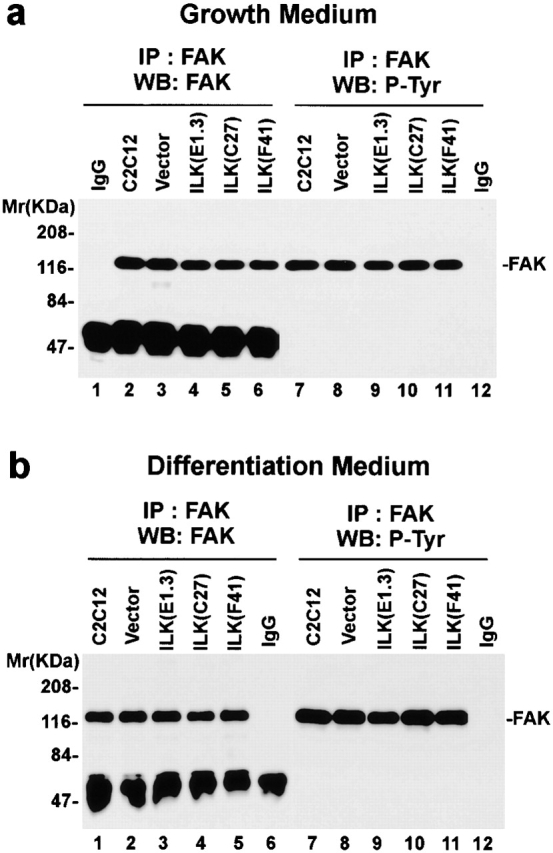
Tyrosine phosphorylation of FAK is not affected by ILK. Parental C2C12 cells, the vector control transfectants, and FLAG-ILK–expressing clones E1.3, C27, and F41 were cultured in GM (a) or in DM (b) for 4 d. FAK was immunoprecipitated from cell lysates with a rabbit polyclonal anti-FAK antibody C-20 or irrelevant rabbit IgG as indicated in the figure. Total FAK protein (lanes 1–6) and the tyrosine-phosphorylated FAK (lanes 7–12) in the immunoprecipitates were detected by immunoblotting with anti-FAK antibody C-20 and an HRP-conjugated antiphosphotyrosine antibody RC20:HRPO, respectively.
We reasoned that if ILK indeed suppresses myogenic differentiation through sustaining MAP kinase activation, inactivation of MAP kinase should reverse the ILK-induced suppression of terminal myogenic differentiation. To test this, we treated the cells with PD98059, a MEK inhibitor that specifically inhibits MAP kinase activation (Alessi et al. 1995). As expected, p44/42 MAP kinases were inactivated in parental C2C12 cells after mitogen deprivation, either in the absence or presence of the MEK inhibitor (Fig. 10 a, lanes 1–3). In ILK-overexpressing C2C12 cells, whereas p44/42 MAP kinases remained active after mitogen deprivation in the absence of the MEK inhibitor (Fig. 10 a, lanes 5, 8, and 11), the amounts of active forms of p44/42 MAP kinases were significantly reduced in the presence of the MEK inhibitor (Fig. 10 a, lanes 6, 9, and 12). Thus, activation of p44/42 MAP kinases that were induced by ILK overexpression was effectively inhibited by the specific MEK inhibitor PD98059, indicating that ILK activates p44/42 MAP kinases through MEK. In further supporting a key role of MAP kinase in the ILK-induced suppression of myogenic differentiation, inhibition of MAP kinase reversed the ILK-induced suppression of myogenin (Fig. 10 b, lanes 6, 9, and 12) and myosin heavy chain (data not shown) expression. In control experiments, myogenin was readily detected in parental C2C12 cells after mitogen deprivation, either in the absence or presence of the MEK inhibitor (Fig. 10 b, lanes 2 and 3). We conclude from these results that ILK influences the initial decision making process of myogenic differentiation by regulation of MAP kinase activation.
Figure 10.

Inactivation of MAPK reverses the ILK-induced suppression of myogenin expression. Parental C2C12 cells (lanes 1–3) and FLAG-ILK–expressing clones C27 (lanes 4–6), F6.2 (lanes 7–9), and E1.3 (lanes 10–12) were grown in GM or in DM in the absence (−) or presence (+) of the specific MEK inhibitor PD98059 as indicated in the figure. Cells were harvested 24 h (a) or 4 d (b) after PD98059 treatment. The active forms of MAPK and myogenin were detected by immunoblotting with an anti–phospho(Thr202/Tyr204)-p44/42 MAPK antibody (a) and an antimyogenin antibody F5D (b), respectively.
Discussion
How myoblastic cells control terminal myogenic differentiation is a fascinating and clinically important question. Although it has been well established that growth factors and extracellular matrix proteins, through interactions with their cell-surface receptors, provide crucial signals controlling terminal myogenic differentiation (McDonald et al. 1995; Sastry and Horwitz 1996; Wewer and Engvall 1996; Durbeej et al. 1998; Gullberg et al. 1998; Burkin and Kaufman 1999), the intracellular events transducing the signals are not completely understood. In this study, we have identified ILK as an important regulator in the initial decision making process of myogenic differentiation. A correlation between downregulation of the ILK level in the Triton X-100–soluble fractions and terminal myogenic differentiation was observed. In further experimental studies, we found that overexpression of ILK effectively inhibits the expression of myogenic transcription factors and suppresses the subsequent myotube formation. The inhibition of myogenic differentiation requires both the PINCH-binding and the kinase activities of ILK, suggesting that the relative amount, subcellular localization, and the kinase activity of ILK are crucial elements in the cellular regulation of terminal myogenic differentiation.
The finding that ILK functions in the initial decision making process of terminal myogenic differentiation is consistent with recent studies by Sastry et al. 1999 who have demonstrated that overexpression of the β1 integrin cytoplasmic domain inhibits terminal myogenic differentiation. ILK was initially identified based on its interaction with the β1 integrin cytoplasmic domain (Hannigan et al. 1996). ILK is present in cell–matrix adhesion sites (Li et al. 1999a). Furthermore, the kinase activity of ILK can be activated by integrin-mediated cell adhesion to fibronectin (Delcommenne et al. 1998). In a recent study, we have found that MIBP, a muscle-specific β1 integrin binding protein, is critically involved in the regulation of myogenic differentiation (Li et al. 1999b). Because both ILK (Hannigan et al. 1996) and MIBP (Li et al. 1999b) interact with the β1 cytoplasmic domain, it is attractive to propose that ILK works in concert with MIBP and other integrin-proximal proteins such as FAK and paxillin (Sastry et al. 1999) in transducing signals from β1 integrins to downstream targets leading to the suppression of terminal myogenic differentiation.
A key downstream target of integrin-mediated regulation of terminal myogenic differentiation is MAP kinase (Sastry et al. 1999). Overexpression of the β1 integrin cytoplasmic domain enhances MAP kinase activation, which maintains the myoblasts in a proliferative, undifferentiated state (Sastry et al. 1999). Inhibition of MAP kinase activation, on the other hand, relieves the integrin-mediated suppression of myogenic differentiation (Sastry et al. 1999). Thus, inactivation of MAP kinases is an essential event in integrin-mediated regulation of myoblast cell cycle withdrawal and initiation of terminal myogenic differentiation (Sastry et al. 1999). In this study, we have demonstrated that overexpression of ILK, but not that of the PINCH-binding defective or the kinase-deficient ILK mutant, resulted in a sustained activation of MAP kinases (Erk1 and Erk2). Furthermore, inhibition of MAP kinase activation reverses the ILK-induced suppression of myogenic differentiation. These results provide strong evidence for the notion that ILK is an important component of the integrin signaling pathway that regulates MAP kinase activation and, ultimately, the decision of proliferation versus differentiation. Because MAP kinase activation is critically involved in cell cycle progression through the G1 phase (Bottazzi et al. 1999; Roovers et al. 1999), a process that is corporately regulated by growth factors and integrins (Assoian 1997; Schwartz 1997; Howe et al. 1998; Giancotti and Ruoslahti 1999), the finding that ILK enhances MAP kinase activation is also consistent with recent observations that overexpression of ILK in epithelial cells promotes anchorage-independent cell cycle progression (Radeva et al. 1997) and tumor formation (Wu et al. 1998).
In addition to demonstrating a prominent role in the initial decision making process of terminal myogenic differentiation, our results suggest that ILK may also play a role in the later stages of myogenic differentiation, namely modulation of cell fusion or maintaining the integrity of myotubes. Overexpression of the PINCH-binding defective or the kinase-deficient ILK mutants, which is permissive for the initiation of myogenic differentiation, resulted in the formation of myotubes with altered morphology (giant myotubes containing clustered nuclei). ILK is a multidomain protein with several distinct biochemical activities including integrin-binding, PINCH-binding, and catalysis of serine/threonine phosphorylation (Dedhar et al. 1999; Wu 1999). Thus, ILK mutants, in which one of the activities (e.g., PINCH-binding or kinase activity) is ablated, could function as dominant negative inhibitors of endogenous ILK. Indeed, a dominant negative inhibitory effect of the kinase-deficient ILK mutant in ILK signaling has been observed in previous studies (Delcommenne et al. 1998; Troussard et al. 1999). A role of ILK in the modulation of myogenic morphogenesis is further supported by previous studies showing that alterations in the expression or functions of β1 integrins, to which ILK binds (Hannigan et al. 1996), resulted in abnormal muscle structure. For example, dystrophic muscles with giant muscle fibers or increased numbers of nuclei per fiber with altered position and size have been observed in α5 integrin (++;−−) chimeric mice (Taverna et al. 1998) and mice lacking α7 integrin (Mayer et al. 1997). Treatment of myoblasts with an antibody that alters α5β1 integrin function also results in the formation of myotubes with an altered morphology (e.g., myotubes with clustered nuclei; Boettiger et al. 1995). The similar effects of ILK and the β1 integrins on myogenic morphogenesis strongly suggest that ILK functions in this process through, at least in part, modulation of integrin signaling. Recent studies in C. elegans have provided strong genetic evidence for a critical role of ILK and its binding partner PINCH in integrin functions during muscle development. Deficiency in β-integrin/pat-3 results in a specific developmental arrest phenotype termed Pat (paralyzed and arrested elongation at the twofold stage), which is caused by a dysfunction of body wall muscles (Gettner et al. 1995). The loss of expression of either ILK/pat-4 (Mackinnon, A.C., and B. Williams, personal communication) or PINCH/unc97 (Hobert et al. 1999) causes a similar body wall muscle–defective Pat phenotype. The dual functions of ILK in myogenesis suggest that ILK may play a crucial role in the regulation of normal muscle regeneration as well as pathological conditions, such as muscular dystrophies or other myopathies.
Acknowledgments
We would like to thank Drs. Shoukat Dedhar (Jack Bell Research Centre and University of British Columbia) for human ILK constructs, Richard Mayne for valuable discussion, and Benjamin Williams (University of Illinois at Urbana-Champaign) for sharing results before publication.
This work was supported by the National Institutes of Health grant DK54639 (to C. Wu) and research project grant No. 98-220-01-CSM from the American Cancer Society (to C. Wu). C. Wu is a V Foundation Scholar.
Footnotes
Abbreviations used in this paper: ANK, ankyrin; BCA, bicinchoninic acid; DM, differentiation medium; FAK, focal adhesion kinase; GM, growth medium; ILK, integrin-linked kinase; KD, kinase deficient; MAP, mitogen-activated protein; MHC, myosin heavy chain.
References
- Alessi D., Cuenda A., Cohen P., Dudley D., Saltiel A. PD098059 is a specific inhibitor of the activation of mitogen-activated protein kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Andres V., Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J. Cell Biol. 1996;132:657–666. doi: 10.1083/jcb.132.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assoian R.K. Anchorage-dependent cell cycle progression. J. Cell Biol. 1997;136:1–4. doi: 10.1083/jcb.136.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett A.M., Tonks N.K. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- Bloor J.W., Brown N.H. Genetic analysis of the Drosophila alphaPS2 integrin subunit reveals discrete adhesive, morphogenetic and sarcomeric functions. Genetics. 1998;148:1127–1142. doi: 10.1093/genetics/148.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger D., Enomoto-Iwamoto M., Yoon H.Y., Hofer U., Menko A.S., Chiquet-Ehrismann R. Regulation of integrin alpha5beta1 affinity during myogenic differentiation. Dev. Biol. 1995;169:261–272. doi: 10.1006/dbio.1995.1142. [DOI] [PubMed] [Google Scholar]
- Bottazzi M.E., Zhu X., Bohmer R.M., Assoian R.K. Regulation of p21(cip1) expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol. 1999;146:1255–1264. doi: 10.1083/jcb.146.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch T.A., Graner M.W., Fessler L.I., Fessler J.H., Schneider K.D., Kerschen A., Choy L.P., Burgess B.W., Brower D.L. The PS2 integrin ligand tiggrin is required for proper muscle function in Drosophila . Development. 1998;125:1679–1689. doi: 10.1242/dev.125.9.1679. [DOI] [PubMed] [Google Scholar]
- Burkin D.J., Kaufman S.J. The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 1999;296:183–190. doi: 10.1007/s004410051279. [DOI] [PubMed] [Google Scholar]
- Dedhar S., Williams B., Howell P.L., Hannigan G. Integrin linked kinase (ILK)a regulator of integrin and growth-factor signaling. Trends Cell Biol. 1999;9:319–323. doi: 10.1016/s0962-8924(99)01612-8. [DOI] [PubMed] [Google Scholar]
- Delcommenne M., Tan C., Gray V., Rue L., Woodgett J., Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA. 1998;95:11211–11216. doi: 10.1073/pnas.95.19.11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M., Henry M.D., Campbell K.P. Dystroglycan in development and disease. Curr. Opin. Cell Biol. 1998;10:594–601. doi: 10.1016/s0955-0674(98)80034-3. [DOI] [PubMed] [Google Scholar]
- Gettner S.N., Kenyon C., Reichardt L.F. Characterization of beta pat-3 heterodimers, a family of essential integrin receptors in C. elegans . J. Cell Biol. 1995;129:1127–1141. doi: 10.1083/jcb.129.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Gullberg D., Velling T., Lohikangas L., Tiger C.F. Integrins during muscle development and in muscular dystrophies. Front. Biosci. 1998;3:D1039–D1050. doi: 10.2741/a344. [DOI] [PubMed] [Google Scholar]
- Hannigan G.E., Leung-Hagesteijn C., Fitz-Gibbon L., Coppolino M.G., Radeva G., Filmus J., Bell J.C., Dedhar S. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1-integrin-linked protein kinase. Nature. 1996;379:91–96. doi: 10.1038/379091a0. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.K., Chou F.L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- Hobert O., Moerman D.G., Clark K.A., Beckerle M.C., Ruvkun G. A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans . J. Cell Biol. 1999;144:45–57. doi: 10.1083/jcb.144.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A., Aplin A.E., Alahari S.K., Juliano R.L. Integrin signaling and cell growth control. Curr. Opin. Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- Lassar A.B., Skapek S.X., Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr. Opin. Cell Biol. 1994;6:788–794. doi: 10.1016/0955-0674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Li F., Liu J., Mayne R., Wu C. Identification and characterization of a mouse protein kinase that is highly homologous to human integrin-linked kinase. Biochim. Biophys. Acta. 1997;1358:215–220. doi: 10.1016/s0167-4889(97)00089-x. [DOI] [PubMed] [Google Scholar]
- Li F., Zhang Y., Wu C. Integrin-linked kinase is localized to cell-matrix focal adhesions but not cell-cell adhesion sites and the focal adhesion localization of integrin-linked kinase is regulated by the PINCH-binding ANK repeats J. Cell Sci. 112 1999. 4589 4599a [DOI] [PubMed] [Google Scholar]
- Li J., Mayne R., Wu C. A novel muscle-specific beta1 integrin binding protein (MIBP) that modulates myogenic differentiation J. Cell Biol. 147 1999. 1391 1397b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Bermudo M.D., Brown N.H. Intracellular signals direct integrin localization to sites of function in embryonic muscles. J. Cell Biol. 1996;134:217–226. doi: 10.1083/jcb.134.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer U., Saher G., Fassler R., Bornemann A., Echtermeyer F., von der Mark H., Miosge N., Poschl E., von der Mark K. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat. Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- McDonald K.A., Horwitz A.F., Knudsen K.A. Adhesion molecules and skeletal myogenesis. Semin. Dev. Biol. 1995;6:105–116. [Google Scholar]
- Menko A.S., Boettiger D. Occupation of the extracellular matrix receptor, integrin, is a control point for myogenic differentiation. Cell. 1987;51:51–57. doi: 10.1016/0092-8674(87)90009-2. [DOI] [PubMed] [Google Scholar]
- Montanaro F., Lindenbaum M., Carbonetto S. α-Dystroglycan is a laminin receptor involved in extracellular matrix assembly on myotubes and muscle cell viability. J. Cell Biol. 1999;145:1325–1340. doi: 10.1083/jcb.145.6.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A., Hsu S.C., Leung-Hagesteijn C., Radeva G., Papkoff J., Montesano R., Roskelley C., Grosschedl R., Dedhar S. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc. Natl. Acad. Sci. USA. 1998;95:4374–4379. doi: 10.1073/pnas.95.8.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E.N., Klein W.H. bHlh factors in muscle developmentdead lines and commitments, what to leave in and what to leave out. Genes Dev. 1994;8:1–8. doi: 10.1101/gad.8.1.1. [DOI] [PubMed] [Google Scholar]
- Prokop A., Martin-Bermudo M.D., Bate M., Brown N.H. Absence of PS integrins or laminin A affects extracellular adhesion, but not intracellular assembly, of hemiadherens and neuromuscular junctions in Drosophila embryos. Dev. Biol. 1998;196:58–76. doi: 10.1006/dbio.1997.8830. [DOI] [PubMed] [Google Scholar]
- Radeva G., Petrocelli T., Behrend E., Leung-Hagesteijn C., Filmus J., Slingerland J., Dedhar S. Overexpression of the integrin-linked kinase promotes anchorage-independent cell cycle progression. J. Biol. Chem. 1997;272:13937–13944. doi: 10.1074/jbc.272.21.13937. [DOI] [PubMed] [Google Scholar]
- Rearden A. A new LIM protein containing an autoepitope homologous to “senescent cell antigen.”. Biochem. Biophys. Res. Commun. 1994;201:1124–1131. doi: 10.1006/bbrc.1994.1822. [DOI] [PubMed] [Google Scholar]
- Roovers K., Davey G., Zhu X., Bottazzi M.E., Assoian R.K. alpha5beta1 integrin controls cyclin D1 expression by sustaining mitogen-activated protein kinase activity in growth factor-treated cells. Mol. Biol. Cell. 1999;10:3197–3204. doi: 10.1091/mbc.10.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen G.D., Sanes J.R., LaChance R., Cunningham J.M., Roman J., Dean D.C. Roles for the integrin VLA-4 and its counter receptor VCAM-1 in myogenesis. Cell. 1992;69:1107–1109. doi: 10.1016/0092-8674(92)90633-n. [DOI] [PubMed] [Google Scholar]
- Sastry S.K., Horwitz A.F. Adhesion-growth factor interactions during differentiationan integrated biological response. Dev. Biol. 1996;180:455–467. doi: 10.1006/dbio.1996.0319. [DOI] [PubMed] [Google Scholar]
- Sastry S.K., Lakonishok M., Thomas D.A., Muschler J., Horwitz A.F. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J. Cell Biol. 1996;133:169–184. doi: 10.1083/jcb.133.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry S.K., Lakonishok M., Wu S., Truong T.Q., Huttenlocher A., Turner C.E., Horwitz A.F. Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J. Cell Biol. 1999;144:1295–1309. doi: 10.1083/jcb.144.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.A. Integrins, oncogenes, and anchorage independence. J. Cell Biol. 1997;139:575–578. doi: 10.1083/jcb.139.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana I., Hemler M.E. Role of transmembrane 4 superfamily (TM4SF) proteins CD9 and CD81 in muscle cell fusion and myotube maintenance. J. Cell Biol. 1999;146:893–904. doi: 10.1083/jcb.146.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna D., Disatnik M.H., Rayburn H., Bronson R.T., Yang J., Rando T.A., Hynes R.O. Dystrophic muscle in mice chimeric for expression of α5 integrin. J. Cell Biol. 1998;143:849–859. doi: 10.1083/jcb.143.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troussard A.A., Tan C., Yoganathan T.N., Dedhar S. Cell extracellular matrix interactions stimulate the AP-1 transcription factor in an integrin linked kinase (ILK) and glycogen synthase kinase-3 dependent manner. Mol. Cell. Biol. 1999;19:7420–7427. doi: 10.1128/mcb.19.11.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y., Li F., Goicoechea S., Wu C. The LIM-only protein PINCH directly interacts with integrin-linked kinase and is recruited to integrin-rich sites in spreading cells. Mol. Cell. Biol. 1999;19:2425–2434. doi: 10.1128/mcb.19.3.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y., Li F., Wu C. Nck-2, a novel Src homology2/3-containing adaptor protein that interacts with the LIM-only protein PINCH and components of growth factor receptor kinase signaling pathways. Mol. Biol. Cell. 1998;9:3367–3382. doi: 10.1091/mbc.9.12.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk T., Fessler L.I., Fessler J.H. A role for integrin in the formation of sarcomeric cytoarchitecture. Cell. 1990;63:525–536. doi: 10.1016/0092-8674(90)90449-o. [DOI] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesisredundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Wewer U.M., Engvall E. Merosin/laminin-2 and muscular dystrophy. Neuromuscul. Disord. 1996;6:409–418. doi: 10.1016/s0960-8966(96)00384-7. [DOI] [PubMed] [Google Scholar]
- Wu C. Integrin-linked kinase and PINCHpartners in regulation of cell-extracellular matrix interaction and signal transduction. J. Cell Sci. 1999;112:4485–4489. doi: 10.1242/jcs.112.24.4485. [DOI] [PubMed] [Google Scholar]
- Wu C., Keightley S.Y., Leung-Hagesteijn C., Radeva G., Coppolino M., Goicoechea S., McDonald J.A., Dedhar S. Integrin-linked protein kinase regulates fibronectin matrix assembly, E-cadherin expression, and tumorigenicity. J. Biol. Chem. 1998;273:528–536. doi: 10.1074/jbc.273.1.528. [DOI] [PubMed] [Google Scholar]