

Abstract
Androgens play an important role in the growth of prostate cancer, but the molecular mechanism that underlies development of resistance to antiandrogen therapy remains unknown. Cyclin E has now been shown to increase the transactivation activity of the human androgen receptor (AR) in the presence of its ligand dihydrotestosterone. The enhancement of AR activity by cyclin E was resistant to inhibition by the antiandrogen 5-hydroxyflutamide. Cyclin E was shown to bind directly to the COOH terminus portion of the AB domain of the AR, and to enhance its AF-1 transactivation function. These results suggest that cyclin E functions as a coactivator of the AR, and that aberrant expression of cyclin E in tumors may contribute to persistent activation of AR function, even during androgen ablation therapy.
Keywords: cyclin E, androgen receptor, prostate cancer, coactivator, cell cycle
Introduction
Androgens play an important role in the development and progression of prostate cancer, one of the leading causes of cancer death in men (Parker et al. 1996). Prostate cancer initially is androgen-sensitive and responds well to androgen ablation therapy, which is the standard treatment for metastatic disease (Catalona 1994). However, treatment-resistant regrowth of tumors ensues within several years, and the molecular mechanism that underlies such resistance remains unknown. Androgens and estrogens regulate the growth of hormone-responsive prostate and breast cancer through the androgen receptor (AR) and the estrogen receptor, respectively, both of which belong to the nuclear receptor superfamily (Mangelsdorf et al. 1995). Mutations in the AR gene have been detected in metastatic hormone-independent prostate cancer (Taplin et al. 1995), and such mutations may contribute to the altered response of the receptor to the hormonal environment. However, mutation of the AR gene is a relatively rare event and cannot account for the adaptive response of most prostate tumors to endocrine therapy. Transfer of human surgical specimens to SCID mice has revealed that androgen-independent metastatic tumors are able to regain hormone responsiveness in a different host (Klein et al. 1997), an effect that might be mediated by cellular factors that modulate AR function in the nucleus.
The mitotic signal of androgens is thought to target ultimately the cell cycle machinery. Estrogens (Planas-Siva and Weinberg 1997; Prall et al. 1998) and androgens (Lu et al. 1997; Kokontis et al. 1998; Menjo et al. 1998) regulate the activity of cyclin-dependent kinases (Cdks) by modulating the expression of cyclins or Cdk inhibitors, thereby promoting progression through the G1–S transition of the cell cycle. In addition, evidence suggests that the estrogen receptor binds directly to cyclin D1 (Neuman et al. 1997; Zwijsen et al. 1997). Given the frequent overexpression and amplification of the cyclin D1 gene in breast cancer (Buckley et al. 1993; Keyomarsi and Pardee 1993), it is thought that cyclin D1 may activate the estrogen receptor in breast tumors in a Cdk-independent manner. However, little is known of whether the AR also binds directly to components of the cell cycle machinery and of how such an interaction might modulate the transactivation activity of the AR in prostate cancer. We now show that cyclin E potentiates the transactivation activity of the AR, and that this effect is mediated predominantly through the binding of cyclin E to the AB domain of the receptor.
Materials and Methods
Plasmids
Full-length cDNAs for human cyclin E, cyclin D1, cyclin A, or Cdk2 were inserted into the pcDNA3 vector and pVP16. A cDNA for mutant cyclin E (R130A) was kindly provided by K. Nakayama (Medical Institute of Bioregulation, Kyushu University, Japan). Full-length cDNAs for the human AR, estrogen receptor α (ERα), glucocorticoid receptor (GR), or progesterone receptor (PR) were inserted into the mammalian expression vector pSG5. The AB (amino acids 1–556) and EF (amino acids 624–918) domains of AR were cloned in the pGEX(4T-1) vector. The reporter constructs pARE2-tk-chloramphenicol acetyltransferase (CAT) and pBL-CAT8+/estrogen response element (ERE) were described previously (Tsai et al. 1989). GAL4-AR(AB) and GAL4-AR(EF) were constructed in the pM vector (CLONTECH Laboratories, Inc.).
Cell Culture, Transient Transfection, and CAT Assay
HeLa and MDAH041 cells were maintained in DME supplemented with 10% FBS, and LNCaP cells in RPMI 1640 supplemented with 10% FBS. 24 h before transfection, the culture medium was changed to DME (without phenol red) supplemented with 5% charcoal-treated FBS. Transfections were performed by the calcium phosphate precipitation technique with 8 μg of the androgen response element (ARE)–CAT reporter construct, 3 μg of β-galactosidase expression vector (as an internal control for differences in transfection efficiency), 1 μg of AR expression vector, and 8 μg of cyclin E expression vector. 2 μg of GAL4-AR(AB) or GAL4-AR(EF), 4 μg of VP16–cyclin E vector, and 8 μg of the 17M2-G–CAT reporter plasmid were used for mammalian two-hybrid analysis. 18 h after transfection, cells were rinsed in PBS and cultured in medium containing ligands or vehicle. After an additional 18 h, the cells were harvested and assayed for CAT and β-galactosidase activities. CAT activity was normalized on the basis of β-galactosidase activity.
Northern Blot Analysis
Northern blot analysis was performed as described previously (Takeyama et al. 1997). cDNA probes for prostate-specific antigen (PSA), AR, and β-actin were labeled with α[32P]dCTP by Random Primer DNA Labeling kit (TAKARA). Hybridization was performed according to the manufacturer's instructions. BAS1500 (Fuji) was used for densitometric analyses of mRNA bands.
Immunoprecipitation and Immunoblot Analysis
MDAH041 cells were transfected with expression vectors encoding cyclin E and either full-length AR, or the AB or EF domain of AR, cultured in the absence or presence of dihydrotestosterone (DHT), and lysed in IP buffer (150 mM NaCl, 2.5 mM EGTA, 1 mM EDTA, 0.1% Tween 20, 10% glycerol, 50 mM Hepes, pH 8.0, 2 μg/ml leupeptin, 100 μg/ml phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 20 μg/ml trypsin inhibitor. For immunoprecipitation, cell lysates (2 mg of protein) were incubated at 4°C with antibodies to cyclin E (Santa Cruz Biotechnology) and then with protein A–agarose beads (Boehringer). Bead-bound proteins were separated by SDS-PAGE on a 10% gel, transferred to nitrocellulose, and subjected to immunoblot analysis with anti-AR(AN1-15; Affinity Bioreagents; N-20 and C-19, Santa Cruz Biotechnology) antibodies. Immune complexes were detected with ECL reagents (Amersham Pharmacia Biotech).
GST Pull-down Assay
Glutathione S-transferase (GST), as well as GST fusion proteins containing AR(AB) or its deletion mutants were produced in Escherichia coli and purified with glutathione-Sepharose beads (Amersham Pharmacia Biotech). Cyclin E was synthesized in vitro in the presence of [35S]methionine with a reticulocyte lysate system (Promega). The 35S-labeled cyclin E was incubated in the presence of glutathione beads with GST or with GST fusion proteins. Bead-bound proteins were then analyzed by SDS-PAGE and autoradiography.
Results
Effect of Cyclin E on the Transactivation Activity of AR
To detect possible functional cross-talk between the AR and cell cycle regulators, we transfected MDAH041 human fibroblastic cells (which do not express an endogenous AR) with an AR expression vector, an ARE–CAT reporter construct, and expression vectors encoding cyclins D1, E, or A. Cyclin E increased the transactivation activity of the AR in the presence of DHT in a dose-dependent manner (Fig. 1 A). Cyclin E did not increase CAT activity in the absence of cotransfection with the AR expression vector (data not shown), indicating that this effect of cyclin E is mediated through the AR. This effect was specific to cyclin E; cyclin A did not affect CAT activity and cyclin D1 induced a slight decrease in CAT activity (Fig. 1 B). The amount of AR protein was not increased by overexpression of cyclin E (Fig. 1 B).
Figure 1.

Potentiation of the transactivation activity of the AR by cyclin E. A, MDAH041 cells were cotransfected with an AR expression vector, an ARE–CAT reporter plasmid, and the indicated amounts of a cyclin E expression vector. Cells were incubated in the presence of 1 nM DHT for 18 h and then assayed for CAT activity. Data in the top are expressed as relative CAT activity and are means (SD of triplicates from a representative experiment). The bottom shows the results of autoradiography. B, MDAH041 cells were cotransfected with an expression vector for AR, GR, or PR, an ARE–CAT reporter plasmid, and an expression vector for cyclins E, A, or D1. Cells were incubated in the absence or presence of 1 nM DHT (T), 1 nM dexamethasone (D), 1 nM progesterone (P), or 10 μM 5-OH-F for 18 h and then assayed for CAT activity (top). Cell lysates (20 μg of protein) were also subjected to immunoblot analysis with anti-AR(N-20). C, MDAH041 cells were cotransfected with an ERα expression vector and an ERE–CAT reporter plasmid in the absence or presence of an expression vector for cyclins A, D1, or E. Cells were incubated for 18 h in the absence or presence of 10 nM 17β-estradiol (E2) and then assayed for CAT activity. D, MDAH041cells were cotransfected with an AR expression vector and an ARE–CAT reporter plasmid in the absence or presence of an expression vector (3 μg) for wild-type (WT) or the R130A mutant of (Mut) cyclin E, or of an expression vector (5 μg) for wild-type (WT) or a kinase-inactive mutant (Mut) of Cdk2. Cells were incubated for 18 h in the presence of 1 nM DHT and in the absence or presence of 1 mM hydroxyurea (HU) or nocodazole (Noc; 0.1 μg/ml, only added during the final 8 h of incubation). Flow cytometry revealed that >70% of cells were arrested at G1–S or M phases by hydroxyurea and nocodazole, respectively (data not shown). E, LNCaP cells were transfected with a cyclin E expression vector or with the empty vector, and subsequently incubated for 18 h in the presence of 10 nM DHT. The relative abundance of PSA, AR, and β-actin mRNAs as revealed by densitometric scanning with the amount in cells not expressing exogenous cyclin E taken as 1.0 are indicated below each lane. The transfection efficiency was estimated as ∼30% by counting the number of β-galactosidase–positive cells.
Whereas the DHT-induced increase in CAT activity was blocked by incubation of cells not expressing exogenous cyclin with the antiandrogen, 5-hydroxyflutamide (5-OH-F), the DHT-dependent transactivation activity of the AR in cells expressing exogenous cyclin E was not completely inhibited only in cells incubated with a 10,000-fold molar excess of 5-OH-F (Fig. 1 B). Similar results were obtained in HeLa cells (data not shown).
Although the GR and PR act through the same DNA element as the AR (Cato et al. 1988), the potentiation of ARE-dependent transcription by cyclin E was specific to that mediated by the AR; cyclin E did not increase the transactivation activity of GR or PR in the presence of their ligands (Fig. 1 B). Exogenous cyclin E did not affect the estrogen-induced increase in transactivation activity of ERα (Fig. 1 C), whereas cyclin D1 increased ERα activity, as described previously (Neuman et al. 1997; Zwijsen et al. 1997).
Stimulation of the transactivation activity of the AR by cyclin E was also apparent when cells were blocked at G1–S or M phases of the cell cycle by treatment with hydroxyurea or nocodazole, respectively (Fig. 1 D), suggesting that this effect of cyclin E is independent of cell cycle progression. Overexpression of either wild-type or a kinase-inactive mutant of Cdk2 blocked the stimulation of AR activity by cyclin E, and a mutant cyclin E (R130A) that lacks the ability to bind Cdk2 (Nakayama et al. 2000) increased AR activity to the same extent as the wild-type protein (Fig. 1 D). Thus, this effect of cyclin E appears to be mediated by the free protein, rather than by the cyclin E–Cdk2 complex.
The effect of cyclin E on the induction of an endogenous androgen-responsive gene, PSA, was examined in human prostate tumor LNCaP cells (which express endogenous ARs). Exposure of these cells to DHT induced a 12-fold increase in the amount of PSA mRNA (data not shown). Exogenous cyclin E potentiated this effect of DHT on PSA gene expression by a factor of 2.7 (Fig. 1 E), without affecting the abundance of endogenous AR or β-actin mRNAs, supporting the notion that cyclin E acts as a coactivator of the AR.
Selective Activation of the AF-1 Function of AR by Cyclin E
The AR exhibits two transactivation functions that are mediated by ligand-independent (AF-1) and ligand-dependent (AF-2) activation domains located in the NH2-terminal AB region and in the COOH-terminal ligand-binding EF region, respectively (Langley et al. 1995; Doesburg et al. 1997). To localize the domain of AR that mediates the functional interaction with cyclin E, we performed a mammalian two-hybrid assay with either the AB or EF region of AR fused to the DNA-binding domain (DBD) of GAL4 and HeLa cells cotransfected with a GAL4-dependent reporter plasmid. The GAL4-AR(AB) construct alone exhibited a constitutive transactivation activity in the absence of DHT, reflecting its AF-1 function (Fig. 2 A). Expression of either cyclin E alone (Fig. 2 A) or a VP16–cyclin E fusion protein (data not shown) further increased CAT activity in cells expressing GAL4-AR(AB), but not GAL4-AR(EF). These results, together with the observation that cyclin E by itself, when fused to the GAL4 DBD, stimulated transcription (Fig. 2 B), suggest that cyclin E not only interacts with the AB domain of AR but also functions as a coactivator of the AR. Again, in these experiments, the abundance of the AR was not affected by overexpression of cyclin E (Fig. 2).
Figure 2.


Functional interaction of cyclin E with the AB domain of the AR. A, HeLa cells were cotransfected with 17M2-G-CAT, GAL4-AR(AB), or AR(EF), and an expression vector for cyclin E. Cells were incubated for 18 h in the absence or presence of 1 nM DHT and then assayed for CAT activity (top). Cell lysates (20 μg of protein) were also subjected to immunoblot analysis with anti-AR(N-20) or anti-AR(C-19) as indicated (bottom). B, The interaction between GAL4–cyclin E and VP16-AR(AB) was examined in a similar manner. Data in A and B are means (SD of triplicates from a representative experiment).
Binding of Cyclin E to the AB Domain of AR
To investigate further the physical association of cyclin E with the AR in vivo, we transfected MDAH041 cells with an expression vector for AR in the absence or presence of vectors encoding cyclins E, D1, or A. Cyclin E immunoprecipitates prepared from cells overexpressing full-length AR and cyclin E yielded an intense band of AR on immunoblot analysis, the broadness of which was increased by prior treatment of cells with DHT (Fig. 3 A). A faint AR band was also detected in the immunoprecipitates prepared from cells transfected with the AR vector alone, presumably reflecting association between endogenous cyclin E and recombinant AR; again, exposure of cells to DHT increased the broadness of the AR band, an effect likely attributable to AR phosphorylation (Blok et al. 1996). The AR was not detected in the cyclin A or cyclin D1 immunoprecipitates prepared from transfected cells incubated in the absence or presence of DHT (data not shown), indicating that the AR binds specifically to cyclin E in vivo.
Figure 3.
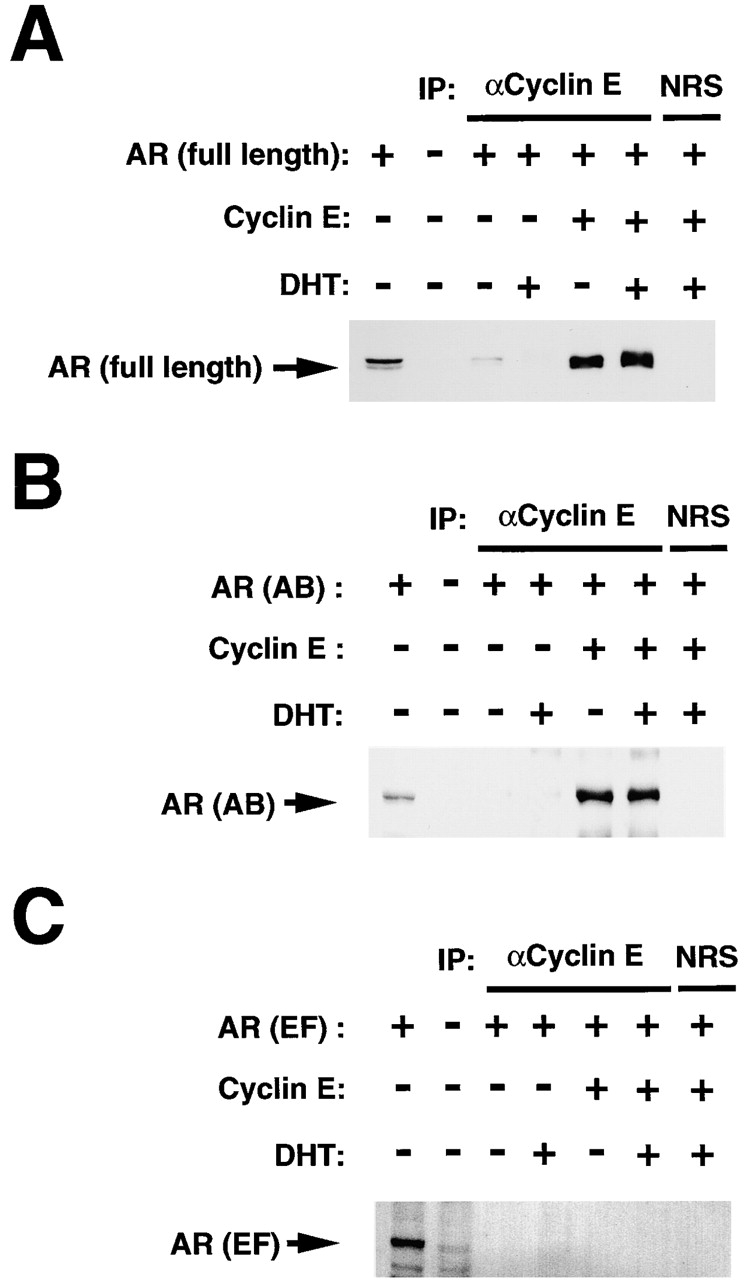
Physical association of cyclin E with the AB domain of AR in vivo. MDAH041 cells were cotransfected with expression vectors for cyclin E and for either full-length AR (A), the AB (B), or EF (C) regions of the AR. Cells were incubated for 18 h in the absence or presence of 1 nM DHT, lysed, and subjected to immunoprecipitation (IP) with antibodies to cyclin E or with normal rabbit serum (NRS) as a control. The resulting immunoprecipitates were then subjected to immunoblot analysis with anti-AR(N-20) (A and B) or anti-AR(C-19) (C). The first two lanes of each represent direct immunoblot analysis of cell lysates.
The region of AR that physically interacts with cyclin E in vivo was further examined by transfecting MDAH041 cells with expression vectors for cyclin E and for either the AB or EF region of AR. Cyclin E immunoprecipitates were then subjected to immunoblot analysis with antibodies specific for either the NH2-terminal or COOH-terminal region of AR, respectively. Whereas the AB domain of AR was coimmunoprecipitated with cyclin E (Fig. 3 B), the EF domain of AR was not (Fig. 3 C), indicating that cyclin E associates predominantly with the AB domain of AR. In these experiments, the abundance of the AB and EF fragments of the AR was not affected by overexpression of cyclin E (data not shown).
Finally, we examined whether cyclin E binds directly to the AB domain of AR and, if so, to which region of AR(AB) it binds. GST fusion proteins containing AR(AB) or various deletion mutants thereof (Fig. 4 A) were incubated in vitro with 35S-labeled cyclin E and the associated cyclin E was detected by SDS-PAGE and autoradiography. Cyclin E directly bound to NH2-terminally truncated mutants of AR(AB), but not to COOH-terminal truncation mutants (Fig. 4 D), indicating that the cyclin E-binding domain is located in the COOH-terminal portion of AR(AB).
Figure 4.

Localization of the cyclin E-binding domain within the AB region of AR. A, HeLa cells were cotransfected with a CAT reporter plasmid containing GAL4 binding sites and an expression vector encoding AR(AB) or its indicated deletion mutants fused to the GAL4 DBD, in the absence (open bars) or presence (closed bars) of an expression vector for cyclin E. Cells were subsequently lysed and assayed for CAT activity. Data are means (SD of triplicates from a representative experiment). B, Cell lysates (20 μg of protein) were also subjected to immunoblot analysis with antibodies to AR: top, anti-AR(N-20); bottom, anti-AR(AN1-15). C, HeLa cells were cotransfected with an ARE–CAT reporter construct and expression vectors for either full-length AR or the del.E mutant of AR, in the absence or presence of a cyclin E expression vector. Cells were incubated for 18 h in the absence or presence of 1 nM DHT and then assayed for CAT activity. Cell lysates (20 μg or protein) were also subjected to immunoblot analysis with anti-AR(C-19); the arrowheads indicate the positions of full-length AR and the del.E mutant. D, GST fusion proteins containing AR(AB) or its various deletion mutants were incubated with 35S-labeled cyclin E in vitro and bead-bound cyclin E was detected by SDS-PAGE and autoradiography.
In agreement with the results of the in vitro binding assay, cyclin E increased the transactivation activities of NH2-terminal truncation mutants of AR(AB), but not those of COOH-terminal truncation mutants (Fig. 4 A). Deletion of the region encompassing amino acids 419–556 (mutant del.13) markedly inhibited the ability of cyclin E to promote transcription (Fig. 4 A). The mutant AR proteins were expressed at a level similar to that of AR(AB) (Fig. 4 B). These results indicate that the binding of cyclin E to the COOH-terminal portion (amino acids 419–556) of AR(AB) is required for its ability to enhance the AF-1 function of the AR. Consistent with this notion, deletion of both the cyclin E-binding region and the transactivation domain from full-length AR (mutant del.E) prevented not only activation of AR by DHT, but also the stimulatory effect of cyclin E on AR function in the presence of DHT (Fig. 4 C).
Discussion
We have demonstrated a functional and physical interaction between cyclin E and the AR. Of the various cyclins tested, only cyclin E (not cyclins D1 or A) bound to the AR and increased its transactivation activity. Furthermore, complex formation with Cdk2 was not required for the interaction of cyclin E with the AR. Although the transactivation function of nuclear hormone receptors is modulated by phosphorylation (Kato et al. 1995), the AR was not phosphorylated by the cyclin E–Cdk2 complex in vitro (data not shown), rendering it unlikely that cyclin E overexpression enhances AR function as a result of phosphorylation by cyclin E–Cdk2. Also consistent with this notion are the observations that overexpression of a kinase-inactive mutant of Cdk2 inhibited the stimulatory effect of cyclin E and that a mutant cyclin E that lacks the ability to bind Cdk2 still enhanced AR function. Cyclin E also appears to interact specifically with the AR among the various nuclear hormone receptors tested; it thus did not affect ER, GR, or PR-mediated gene transcription. Finally, coimmunoprecipitation analysis with transfected cells and GST precipitation assays in vitro revealed that cyclin E associates predominantly with the AB domain of AR and that a cyclin E-binding domain is located in the COOH-terminal portion of AR(AB). The importance of this binding site in the enhancement of AR function by cyclin E was supported by the results of functional assays. To date, only a few coactivators have been shown to interact specifically with the AR and to enhance its transactivation function. The coactivator ARA70 interacts with the EF domain of AR in a ligand-specific manner and enhances its AF-2 function (Yeh and Chang 1996), whereas SNURF enhances not only AR-, GR-, and PR-mediated transactivation, but also basal transcription (Moilanen et al. 1998). Thus, cyclin E is a specific coactivator to be identified that binds directly to the AB domain of AR and enhances its AF-1 function.
Although the AR and estrogen receptor appear to interact with distinct cyclins, a functional and physical interaction between nuclear hormone receptors and components of the cell cycle machinery may represent a general mechanism by which the transactivation function of these receptors is regulated through recruitment of coactivators. From a clinical standpoint, our results suggest that the inappropriate activation of AR by cyclin E may underlie the development of resistance of prostate cancer. Thus, it would be of interest to study whether cyclin E is overexpressed in prostate cancer, especially in the transition to the hormone-independent stage.
Acknowledgments
We thank D. Morgan, M. Ohtsubo, and K. Nakayama for expression vectors, and J. Yanagisawa for discussion.
This work was supported in part by grants-in-aid for Cancer Research (to E. Ogata and S. Kato) and for Scientific Research on Priority Areas (to M. Nakanishi) from the Ministry of Education, Science, Sports and Culture of Japan.
Footnotes
Ayako Yamamoto and Yoshihiro Hashimoto contributed equally to this work.
Abbreviations used in this paper: AR, androgen receptor; ARE, androgen response element; CAT, chloramphenicol acetyltransferase; Cdk, cyclin-dependent kinase; DBD, DNA-binding domain; DHT, dihydrotestosterone; ERα, estrogen receptor α; GR, glucocorticoid receptor; GST, glutathione S-transferase; 5-OH-F, 5-hydroxyflutamide; PR, progesterone receptor; PSA, prostate-specific antigen.
References
- Blok L.J., de Ruiter P.E., Brinkmann A.O. Androgen receptor phosphorylation. Endocrine Res. 1996;3:197–219. doi: 10.3109/07435809609030508. [DOI] [PubMed] [Google Scholar]
- Buckley M.F., Sweeney K.J., Hamilton J.A., Sini R.L., Manning D.L., Nicholson R.I., de Fazio A., Watts C.K., Musgrove E.A., Sutherland R.L. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–2133. [PubMed] [Google Scholar]
- Catalona W.J. Management of cancer of the prostate. N. Engl. J. Med. 1994;331:996–1004. doi: 10.1056/NEJM199410133311507. [DOI] [PubMed] [Google Scholar]
- Cato A.C.B., Skroch P., Weinman J., Butkeraitis P., Ponta H. DNA sequences outside the receptor-binding sites differently modulate the responsiveness of the mouse mammary tumour virus promoter to various steroid hormones. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:1403–1410. doi: 10.1002/j.1460-2075.1988.tb02957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doesburg P., Kuil C.W., Berrevoets C.A., Steketee K., Faber P.W., Mulder E., Brinkmann A.O., Trapman J. Functional in vivo interaction between the amino-terminal, transactivation domain and the ligand binding domain of the androgen receptor. Biochemistry. 1997;36:1052–1064. doi: 10.1021/bi961775g. [DOI] [PubMed] [Google Scholar]
- Kato S., Endoh H., Masuhiro Y., Kitamoto T., Uchiyama S., Sasaki H., Masushige S., Gotoh Y., Nishida E., Kawashima H. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- Keyomarsi K., Pardee A.B. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc. Natl. Acad. Sci. USA. 1993;90:1112–1116. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein K.A., Reiter R.E., Redula J., Moradi H., Zhu X.L., Brothman A.R., Lamb D.J., Marcelli M., Belldegrun A., Witte O.N., Sawyers C.L. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat. Med. 1997;3:402–408. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- Kokontis J.M., Hay N., Liao S. Progression of LNCaP prostate tumor cells during androgen deprivationhormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol. Endocrinol. 1998;12:941–953. doi: 10.1210/mend.12.7.0136. [DOI] [PubMed] [Google Scholar]
- Langley E., Zhou Z.X., Wilson E.M. Evidence for an anti-parallel orientation of the ligand-activated human androgen receptor dimer. J. Biol. Chem. 1995;270:29983–29990. doi: 10.1074/jbc.270.50.29983. [DOI] [PubMed] [Google Scholar]
- Lu S., Tsai S.Y., Tsai M.-J. Regulation of androgen-dependent prostatic cancer cell growthandrogen regulation of CDK2, CDK4, and CKI p16 genes. Cancer Res. 1997;57:4511–4516. [PubMed] [Google Scholar]
- Mangelsdorf D.J., Thummel C., Beato M., Herrlich P., Schutz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P. The nuclear receptor superfamilythe second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menjo M., Kaneko Y., Ogata E., Ikeda K., Nakanishi M. Critical role for p27Kip1 in cell cycle arrest after androgen depletion in mouse mammary carcinoma cells (SC-3) Oncogene. 1998;17:2619–2627. doi: 10.1038/sj.onc.1202193. [DOI] [PubMed] [Google Scholar]
- Moilanen A.-M., Poukka H., Karvonen U., Hakli M., Janne O.A., Palvimo J.J. Identification of a novel RING finger protein as a coregulator in steroid receptor-mediated gene transcription. Mol. Cell. Biol. 1998;18:5128–5139. doi: 10.1128/mcb.18.9.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K., Nagahama H., Minamishima Y.A., Matsumoto M., Nakamichi I., Kitagawa K., Shirane M., Tsunematsu R., Tsukiyama T., Ishida N. Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman E., Ladha M.H., Lin N., Upton T.M., Miller S.J., DiRenzo J., Pestell R.G., Hinds P.W., Dowdy S.F., Brown M., Ewen M.E. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol. Cell. Biol. 1997;17:5338–5347. doi: 10.1128/mcb.17.9.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S.L., Tong T., Bolden S., Wingo P.A. Cancer statistics. CA Cancer J. Clin. 1996;46:5–27. doi: 10.3322/canjclin.46.1.5. [DOI] [PubMed] [Google Scholar]
- Planas-Siva M.D., Weinberg R.A. Estrogen-dependent cyclin E-Cdk2 activation through p21 redistribution. Mol. Cell. Biol. 1997;17:4059–4069. doi: 10.1128/mcb.17.7.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall O.W., Rogan E.M., Sutherland R.L. Estrogen regulation of cell cycle progression in breast cancer cells. J. Steroid Biochem. Mol. Biol. 1998;65:169–174. doi: 10.1016/s0960-0760(98)00021-1. [DOI] [PubMed] [Google Scholar]
- Takeyama K., Kitanaka S., Sato T., Kobori M., Yanagisawa J., Kato S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science. 1997;277:1827–1830. doi: 10.1126/science.277.5333.1827. [DOI] [PubMed] [Google Scholar]
- Taplin M.-E., Bubley G.J., Shuster T.D., Frantz M.E., Spooner A.E., Ogata G.K., Keer H.N., Balk D.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Tsai S.Y., Tsai M.-J., O'Malley B.W. Cooperative binding of steroid hormone receptors contributes to transcriptional synergism at target enhancer elements. Cell. 1989;57:443–448. doi: 10.1016/0092-8674(89)90919-7. [DOI] [PubMed] [Google Scholar]
- Yeh S., Chang C. Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc. Natl. Acad. Sci. USA. 1996;93:5517–5521. doi: 10.1073/pnas.93.11.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwijsen R.M.L., Wientjens E., Klompmaker R., van der Sman J., Bernards R., Michalides R.J. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]