

Abstract
Two AAA family ATPases, NSF and p97, have been implicated in membrane fusion during assembly and inheritance of organelles of the secretory pathway. We have now investigated the roles of AAA ATPases in membrane fusion during assembly of the peroxisome, an organelle outside the classical secretory system. Here, we show that peroxisomal membrane fusion in the yeast Yarrowia lipolytica requires two AAA ATPases, Pex1p and Pex6p. Release of membrane- associated Pex1p and Pex6p drives the asymmetric priming of two fusion partners. The next step, peroxisome docking, requires release of Pex1p from one partner. Subsequent fusion of the peroxisomal membranes is independent of both Pex1p and Pex6p.
Keywords: microbody, biogenesis, organelle assembly, in vitro reconstitution, peroxin
Introduction
All membrane fusion reactions occurring during assembly and inheritance of organelles of the secretory pathway depend on two ATPases of the AAA protein family, N-ethylmaleimide-sensitive factor (NSF) (Sec18p) and/or p97/VCP (Cdc48p) (Patel and Latterich 1998). Heterotypic fusion of secretory vesicles to their target membranes (Rothman 1994) and homotypic fusion of yeast vacuoles (Haas and Wickner 1996) require NSF or its yeast orthologue, Sec18p. Homotypic fusion of ER membranes in yeast is mediated by Cdc48p (Latterich et al. 1995), while reassembly of Golgi cisternae involves NSF and p97/VCP (Acharya et al. 1995; Rabouille et al. 1995). According to the SNARE hypothesis (Rothman 1994), recruitment of NSF and soluble NSF attachment proteins (SNAPs) to the complex formed by vesicle- and target membrane–specific SNAP receptors (v-SNAREs and t-SNAREs, respectively) yields the SNARE complex. ATP hydrolysis by NSF leads to disassembly of the SNARE complex, directly driving fusion of vesicle and target membranes (Söllner et al. 1993; Rothman 1994). However, there is evidence indicating that NSF acts at ATP-dependent pre- or postfusion steps, rather than in membrane fusion itself (Nichols and Pelham 1998; Ungermann et al. 1998), by disassembling futile SNARE complexes on the same membrane. This permits either formation of productive SNARE complexes between opposing membranes at a prefusion step (Nichols et al. 1997; Ungermann et al. 1998) or recycling of v-SNAREs to vesicles without t-SNAREs at a postfusion step (Nichols and Pelham 1998; Schwarz 1999). NSF and p97 may also have multiple roles in membrane fusion and/or different roles in fusing different types of membranes. While the ATPase activity of NSF is required for intercisternal Golgi transport (Whiteheart et al. 1994), the role for NSF during postmitotic Golgi membrane fusion may be distinct from its ATPase-dependent ability to disrupt SNARE pairs (Müller et al. 1999). Moreover, whereas v- and t-SNAREs suffice to mediate mixing of membrane and luminal contents in some liposome fusion assays (Weber et al. 1998), in other assays, NSF and p97 alone without SNARE proteins are apparently sufficient for rapid membrane fusion (Otter-Nilsson et al. 1999).
Recently, we demonstrated that two distinct AAA ATPases, Pex1p and Pex6p, are required for fusion of peroxisomes in the yeast Yarrowia lipolytica (Titorenko et al. 2000). Fusion between two populations of small peroxisomal vesicles, P1 and P2, has been reconstituted in vitro and shown to be inhibited by antibodies to either ATPase (Titorenko et al. 2000). These findings provide us with an experimental tool with which to study the mechanism of peroxisomal membrane fusion and to analyze the role of Pex1p and Pex6p in this process. This, in turn, creates the opportunity to compare the mechanisms of membrane fusion for the peroxisome, an organelle outside the classical secretory system, to the basic principles of membrane fusion established for organelles of the secretory pathway.
Materials and Methods
Strains and Reagents
The Yarrowia lipolytica wild-type strain E122 (MatA ura3-302 leu2-270 lys8-11) and mutant strains pex5KO (MatA ura3-302 leu2-270 lys8-11 pex5::LEU2) (Szilard et al. 1995) and pex6KO (MatA ura3-302 leu2-270 lys8-11 pex6::LEU2) (Titorenko and Rachubinski 1998); media, growth conditions, and antibodies to thiolase (THI), Pex1p and Pex6p (Titorenko et al. 2000); and production of Fab fragments of IgGs (Haas and Wickner 1996) have been previously described.
Subcellular Fractionation and Peroxisome Isolation
Lysed and homogenized spheroplasts (Szilard et al. 1995) were subfractionated by a multistep differential centrifugation procedure (Titorenko et al. 1998) to yield the postnuclear supernatant (PNS), 20,000-g pellet and supernatant (20KgP and 20KgS, respectively), and the 200,000-g pellet and supernatant (200KgP and 200KgS, respectively) subcellular fractions. To purify P1 and P2 peroxisomes, the 200KgP fraction was subjected to isopycnic centrifugation on a discontinuous sucrose gradient, followed by recovery of peak fractions containing P1 and P2 by flotation on a multistep sucrose density gradient (Titorenko et al. 2000).
In Vitro Assay of Association of Pex1p and Pex6p with Peroxisomes
l[35S]methionine–labeled P1 from wild-type cells or l[35S]methionine–labeled P2 from pex5KO cells were incubated individually in T99 buffer (15 mM MES, pH 6.0, 100 mM KCl, 50 mM potassium acetate, 3 mM MgCl2, 2 mM magnesium acetate, 250 mM sorbitol, 40 mM creatine phosphate, and 10 U/ml creatine kinase) in the presence or absence of 1 mg/ml of cytosol from unlabeled wild-type cells, 1 mM ATP, or 1 mM ATPγS. Cytosol was prepared in 15 mM MES, pH 6.0, containing 250 mM sorbitol (Rexach and Schekman 1991). After a 10-min incubation at 26°C, peroxisomes were pelleted by centrifugation at 100,000 g for 8 min at 4°C in a Beckman TLA120.2 rotor. Pex1p and Pex6p were immunoprecipitated under denaturing conditions (Szilard et al. 1995) from pellet and supernatant fractions. Immunoprecipitates were resolved by SDS-PAGE and visualized by fluorography.
In Vitro Peroxisome Fusion Assay
P1 peroxisomes from wild-type cells and P2 peroxisomes from pex5KO cells containing the precursor form of the peroxisomal matrix protein thiolase (pTHI) labeled with l-[35S]methionine were incubated at 26°C in T99 buffer with or without cytosol (1 mg/ml) from unlabeled wild-type cells and 1 mM ATP, either before or after mixing of P1 and P2. Anti-Pex1p and anti–Pex6p Fab were added to concentrations of 40 and 70 μg/ml, respectively. After each incubation step, peroxisomes were repelleted by centrifugation. At the end of the 90-min reaction, individual samples were analyzed by immunoprecipitation of both pTHI and the mature form of thiolase (mTHI) under denaturing conditions, SDS-PAGE, and fluorography (Titorenko et al. 2000). Fluorograms were quantitated by densitometry, and the percent conversion of pTHI to mTHI as a measure of fusion was calculated. Pex6p was depleted from cytosol by immunoaffinity chromatography with antibodies covalently coupled to protein A–Sepharose (Szilard et al. 1995).
Results
We first examined the dynamics of association of Pex1p and Pex6p with peroxisomes during peroxisome fusion. In wild-type Y. lipolytica cells, Pex1p was exclusively membrane-associated and primarily in a high speed (200,000 g) pelletable organelle fraction (200KgP; Fig. 1 A). The 200KgP is enriched for small peroxisomal vesicles, P1 and P2 (Titorenko et al. 2000). Pex6p was both cytosolic (200KgS) and membrane-associated (Fig. 1 A). Pex1p associated with both P1 and P2, whereas Pex6p localized to P2 only (Fig. 1 B). Pex1p and Pex6p are peripheral membrane proteins on the cytosolic surface of P1 (Pex1p) and P2 (both proteins) (Titorenko et al. 2000).
Figure 1.

Localization of Pex1p and Pex6p in vivo and during peroxisome fusion in vitro. Pex1p and Pex6p were detected by immunoblotting the indicated subcellular fractions (A) or the P1 and P2 peroxisomes (B) isolated from wild-type cells. (C) 35S-labeled P1 or 35S-labeled P2 was incubated individually with or without unlabeled wild-type cytosol, ATP, or ATPγS, as indicated. After a 10-min incubation at 26°C, peroxisomes were pelleted. Pex1p and Pex6p were immunoprecipitated under denaturing conditions from the pellet (P) and supernatant (S) fractions. (D) Unlabeled P1 and 35S-labeled P2 were incubated individually with unlabeled wild-type cytosol and ATP. After a 10-min incubation at 26°C, P1 and P2 were pelleted, resuspended in T99 buffer, and mixed. After a 10-min incubation at 26°C with or without unlabeled wild-type cytosol, ATP or ATPγS, peroxisomes were pelleted. Pex1p and Pex6p were immunoprecipitated under denaturing conditions from the pellet (P) and supernatant (S) fractions. Immunoprecipitates in C and D were resolved by SDS-PAGE and visualized by fluorography. (E) 35S-labeled (lanes 1 and 3) or unlabeled (lanes 2 and 4) P1 were supplemented with unlabeled cytosol (lanes 1 and 3) or with unlabeled cytosol containing labeled Pex1p that had been released from P1 after a 10-min incubation with cytosol and ATP (lanes 2 and 4). (F) 35S-labeled (lanes 1 and 3) or unlabeled (lanes 2 and 4) P2 was supplemented with 35S-labeled (lanes 2 and 4) or unlabeled (lanes 1 and 3) wild-type cytosol. (G) 35S-labeled (lanes 1 and 3) or unlabeled (lanes 2 and 4) P2 and unlabeled P1 (all samples) were incubated individually with unlabeled cytosol and ATP. After a 10-min incubation at 26°C, P1 and P2 were pelleted, resuspended in T99 buffer, and mixed. Samples were supplemented with unlabeled cytosol (lanes 1 and 3) or with unlabeled cytosol containing labeled Pex1p that had been released from P2 after a 10-min incubation with P1, cytosol, and ATP (lanes 2 and 4). Aliquots of samples in E–G, incubated with or without ATP or ATPγS, were taken at the times indicated. Peroxisomes were pelleted, and P1- or P2-associated 35S-labeled Pex1p or Pex6p was detected as in C and D.
Incubating P1 alone with cytosol and ATP caused the release of Pex1p from the membrane (Fig. 1 C, lanes 1 and 2). Pex1p was not released if cytosol, ATP, or both components were omitted, or if ATPγS (a nonhydrolyzable analogue of ATP) replaced ATP (Fig. 1 C, lanes 3–10). Therefore, release of Pex1p from P1 requires cytosol and ATP hydrolysis but not the presence of P2. In contrast, Pex1p was not released from P2 under any condition in the absence of P1 (Fig. 1 C, lanes 1 and 2), whereas Pex6p release from P2 required cytosol and ATP hydrolysis but not the presence of P1 (Fig. 1 C). Importantly, incubation of P1 and P2 individually with cytosol and ATP, followed by their mixing and further incubation with cytosol and ATP, led to the complete release of Pex1p from P2 (Fig. 1 D, lanes 1 and 2). Therefore, release of Pex1p from P2 requires cytosol and ATP hydrolysis (Fig. 1 D, lanes 3–10) and is dependent on the presence of P1.
Incubation of [35S]methionine-labeled P1 with unlabeled cytosol and ATP resulted in the complete release of labeled Pex1p from P1 by 5 min (Fig. 1 E, lane 1). When unlabeled P1 was incubated with ATP and unlabeled cytosol containing labeled Pex1p, which had been released from P1 in the above reaction, none of the labeled Pex1p was recruited back to P1 (Fig. 1 E, lane 2). Release of labeled Pex1p from P1 was prevented by the substitution of ATPγS for ATP (Fig. 1 E, lane 3). Also, labeled Pex1p, which had been released from P1 after a 10-min incubation with cytosol and ATP, did not bind P1 in the presence of ATPγS (Fig. 1 E, lane 4). Therefore, Pex1p does not return to the surface of P1 peroxisomes after being released from P1 in a cytosol- and ATP hydrolysis–dependent manner.
Incubation of [35S]methionine-labeled P2 with unlabeled cytosol and ATP led to complete release of labeled Pex6p from P2 by 5 min (Fig. 1 F, lane 1). When unlabeled P2 was incubated with ATP and labeled cytosol, the labeled Pex6p trafficked to P2 from the cytosol and returned to the cytosol after 3 min (Fig. 1 F, lane 2). Substitution of ATPγS for ATP prevented both release of Pex6p from P2 and trafficking of Pex6p from the cytosol to P2 (Fig. 1 F, lanes 3 and 4, respectively). Thus, Pex6p shuttles between the cytosol and the surface of P2 peroxisomes in a cytosol- and ATP hydrolysis–dependent manner.
Incubation of unlabeled P1 and labeled P2 individually with unlabeled cytosol and ATP, followed by their mixing and further incubation with unlabeled cytosol and ATP, led to the complete loss of labeled Pex1p from P2 by 5 min (Fig. 1 G, lane 1). This cytosol-, ATP hydrolysis–, and P1-dependent release of Pex1p from P2 was irreversible. Indeed, when unlabeled P1 and P2 were first incubated individually with unlabeled cytosol and ATP, then mixed and incubated with ATP and unlabeled cytosol (containing labeled Pex1p that had been released from P2 in the above reaction), the labeled Pex1p did not return to the surface of P2 peroxisomes (Fig. 1 G, lane 2). Therefore, unlike P2-associated Pex6p, Pex1p does not undergo a binding/release cycle between the cytosol and P2.
Addition of anti–Pex1p Fab or anti–Pex6p Fab was shown to inhibit the in vitro fusion of P1 and P2 peroxisomes (Titorenko et al. 2000). Fusion was inhibited if isolated P1 was treated with anti–Pex1p Fab (Fig. 2, lane 2) or if isolated P2 was treated with either anti–Pex1p or anti–Pex6p Fab (Fig. 2, lanes 5 and 6) before their mixing and incubation in the fusion reaction. Therefore, P1 and P2 undergo priming (activation) before docking (a stable, specific interaction, see results below), and priming is independent of physical contact between partners and commits both partners to docking. Anti–Pex1p Fab did not inhibit fusion when added to P1 previously incubated with cytosol and ATP (Fig. 2, lane 8). These data, combined with the fact that cytosol- and ATP hydrolysis–dependent release of Pex1p from P1 can occur before mixing with P2 (Fig. 1 C), suggest that priming of P1 requires P1-associated Pex1p but not contact with P2, and is driven by cytosol- and ATP hydrolysis–dependent irreversible release of Pex1p from P1.
Figure 2.
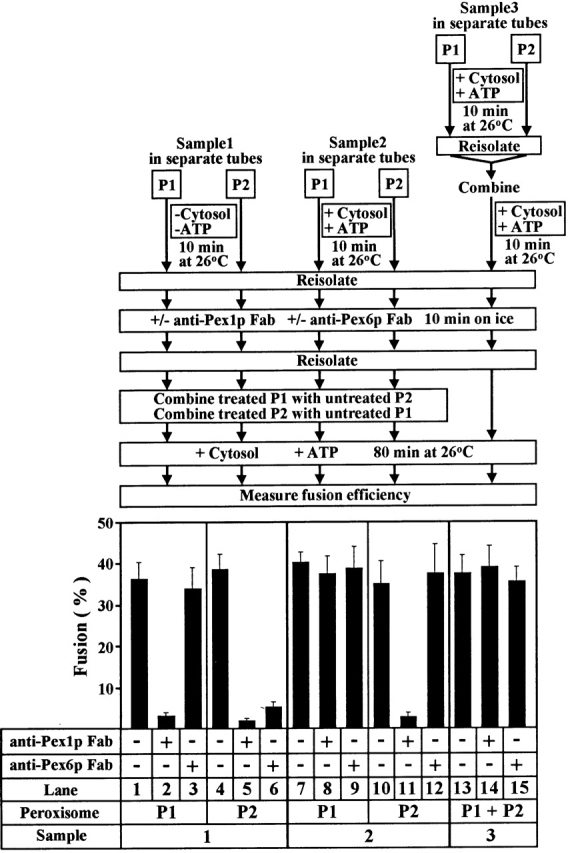
Effects of anti–Pex1p Fab and anti–Pex6p Fab on peroxisome priming, docking, and fusion. P1 from wild-type cells and P2 from pex5KO cells containing the precursor form of the peroxisomal matrix protein thiolase (pTHI) labeled with [35S]methionine were incubated with or without unlabeled wild-type cytosol and ATP, as shown. Anti–Pex1p Fab and anti–Pex6p Fab were added to concentrations of 40 and 70 μg/ml, respectively. Samples were analyzed by immunoprecipitation of both pTHI and mTHI, SDS-PAGE, and fluorography. Fluorograms were quantitated by densitometry, and the percent conversion of pTHI to mTHI (fusion) was calculated.
We next analyzed the role of Pex6p in priming P2. Treatment of P2 with anti–Pex6p Fab before, but not after, incubation with cytosol and ATP inhibited fusion (Fig. 2, lanes 6 and 12, respectively). Pex6p shuttles between the cytosol and P2 peroxisomes in a cytosol- and ATP hydrolysis–dependent manner (Fig. 1 F). When the reversible trafficking of Pex6p to P2 was prevented by using cytosol from a pex6 deletion mutant strain (pex6KO), fusion was reduced (Fig. 3, Reaction 1). This reduction could be reversed by incubating P2 directly with wild-type cytosol and ATP (Fig. 3, Reaction 2) but not with Pex6p-depleted wild-type cytosol and ATP (Fig. 3, Reaction 3). Therefore, priming of P2 requires P2-associated Pex6p, and is driven by an ATP hydrolysis–dependent, binding release cycle for Pex6p between the cytosol and P2. The observed inhibition of peroxisome fusion by treatment of P2 with anti–Pex1p Fab before mixing with P1 and incubation in the fusion reaction (Fig. 2, lane 5) does not conclusively demonstrate an involvement of P2-associated Pex1p in priming P2. Because Pex1p remains associated with P2 after priming is complete (Fig. 1 C), a role for Pex1p in priming cannot be uncoupled from its role in peroxisome docking.
Figure 3.

Priming of P2 for docking and fusion is driven by an ATP hydrolysis–dependent binding release cycle for cytosolic Pex6p and P2. P2 from pex5KO cells containing 35S-labeled pTHI were treated as indicated, combined with untreated P1 from wild-type cells, and incubated further as shown. Samples, taken at the indicated times, were analyzed by immunoprecipitation of both pTHI and mTHI under denaturing conditions, SDS-PAGE, and fluorography. Fusion efficiency was quantitated as in Fig. 2.
Priming of P1 and P2 peroxisomes before their physical contact (see above) commits both fusion partners to their subsequent docking, followed by fusion itself (Titorenko et al. 2000). We define peroxisome docking as the stable and specific interaction between P1 and P2 that occurs after their mixing in the in vitro fusion reaction, leads to the formation of a stable docking complex of buoyant density intermediate to the densities of P1 and P2 (Titorenko et al. 2000), and commits P1 and P2 to subsequent fusion. While priming (Fig. 2) and docking (Titorenko et al. 2000) of P1 and P2 in vitro were essentially complete after 10 min of incubation, in vitro fusion itself had a lag period of ∼10 min and reached steady state after 90–120 min of incubation (Titorenko et al. 2000). To evaluate the role of P2-associated Pex1p in docking P1 and P2, P1 and P2 were first primed individually by incubation with cytosol and ATP, and then treated with anti–Pex1p Fab either before or after their mixing. Anti–Pex1p Fab inhibited fusion when added to P2 before mixing (Fig. 2, lane 11) but not after mixing (Fig. 2, lane 14); i.e., under conditions allowing complete release of Pex1p from P2 (Fig. 1 C) and complete docking of P1 and P2 (Titorenko et al. 2000). Therefore, both P2-associated Pex1p and ATP hydrolysis– and cytosol-dependent release of Pex1p from P2 are required to dock P1 and P2. Neither Pex1p nor Pex6p is required for fusion itself, as incubation of mixed P1 and P2 with cytosol and ATP caused complete release of Pex1p and Pex6p (Fig. 1C and Fig. D), and led to complete docking of P1 and P2 (Titorenko et al. 2000) and made subsequent fusion resistant to anti–Pex1p Fab and anti–Pex6p Fab (Fig. 2, lanes 14 and 15).
Peroxisome fusion requires cytosol and ATP hydrolysis (Titorenko et al. 2000). We next evaluated the requirement for cytosol and ATP hydrolysis at the different steps of fusion. P1 and P2, which were isolated after being primed and docked, fused efficiently in a subsequent incubation, even in the absence of ATP or cytosol or both components (Fig. 4 A). Fusion was also unaffected by the substitution of ATPγS for ATP (Fig. 4 A). Therefore, the peroxisome fusion event itself requires neither cytosol nor ATP hydrolysis.
Figure 4.
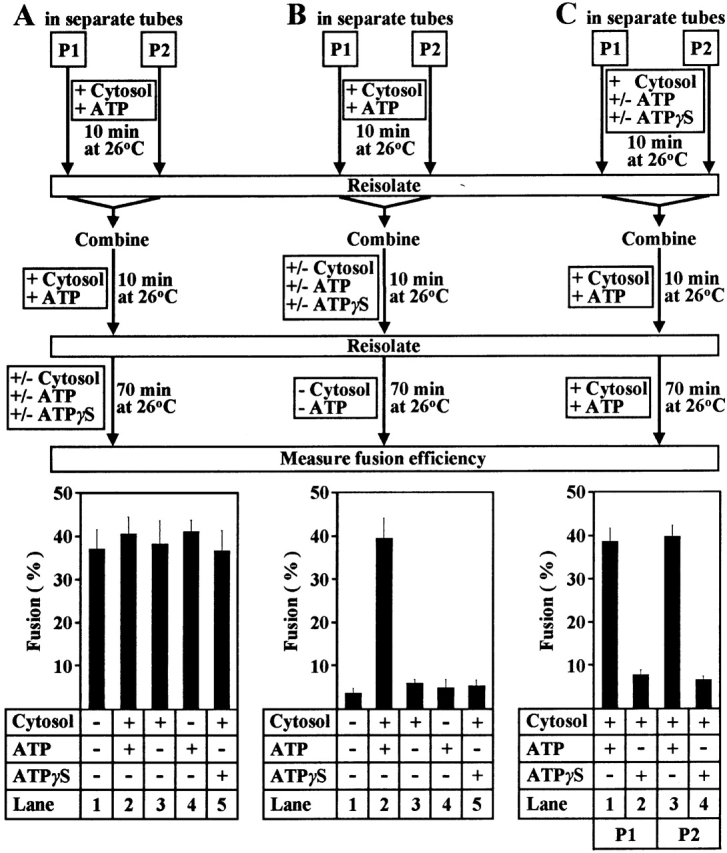
Peroxisome priming and docking, but not fusion itself, require cytosol and ATP hydrolysis. P1 (from wild-type cells) and P2 (from pex5KO cells containing 35S-labeled pTHI) were incubated in the presence or absence of wild-type unlabeled cytosol, ATP or ATPγS, and were further treated as indicated. Samples were analyzed by immunoprecipitation of pTHI and mTHI under denaturing conditions, SDS-PAGE, and fluorography. Fluorograms were quantitated by densitometry. The percent conversion of pTHI to mTHI (fusion) is presented.
We next studied the requirement for cytosol and ATP hydrolysis in peroxisome docking. P1 and P2, individually primed with cytosol and ATP, were mixed and incubated with or without cytosol and ATP or in the presence of ATPγS. Peroxisomes were reisolated and incubated in the absence of cytosol and ATP. Under these conditions, the efficiency of peroxisome docking is the limiting factor in fusion, because P1 and P2 were primed individually, and fusion itself requires neither cytosol nor ATP. Only primed peroxisomes that were mixed and incubated with cytosol and ATP underwent efficient fusion (Fig. 4 B, lane 2). Primed peroxisomes that were mixed and incubated without ATP, cytosol, or both components, or incubated with cytosol and ATPγS, did not fuse (Fig. 4 B). Therefore, docking of P1 and P2 requires both cytosol and ATP hydrolysis.
We evaluated the requirement for ATP hydrolysis in peroxisome priming. P1 and P2 that were incubated individually with cytosol and ATPγS were incompetent for subsequent docking and fusion (Fig. 4 C, lanes 2 and 4, respectively). Therefore, priming of P1 and P2 requires ATP hydrolysis. As reported above, ATP hydrolysis during priming drives cytosol-dependent release of membrane-bound Pex1p from P1 and Pex6p from P2 and stimulates the binding/release cycle between cytosolic Pex6p and P2. Therefore, ATP hydrolysis–driven priming of P1 and P2 for docking requires cytosolic proteins, including the cytosolic form of Pex6p.
Fig. 5 presents a model for Pex1p- and Pex6p-assisted peroxisome fusion. Although fusion between P1 and P2 involves vesicles from the same compartment type (peroxisome), the protein compositions of the fusion partners are dissimilar (Titorenko et al. 2000). Therefore, fusion between P1 and P2 is pseudoheterotypic. Peroxisome fusion is a multistep process that includes priming, docking, and fusion events. Before priming, Pex1p is associated with P1, while Pex1p and Pex6p are bound to the outer surface of P2. ATP hydrolysis, possibly by Pex1p on P1 and by Pex1p and/or Pex6p on P2, triggers cytosol-dependent release of Pex1p from P1 and of Pex6p from P2. Release of P1-associated Pex1p and P2-associated Pex6p activates peroxisomes for docking and fusion. Priming of P1 and P2 does not require contact between them but does require cytosolic proteins. One cytosolic protein needed to prime P2 is the cytosolic form of Pex6p. Pex6p shuttles between the cytosol and the surface of P2 in a cytosol- and ATP hydrolysis–dependent manner. P2-associated Pex1p remains bound to the peroxisome during priming. Docking of primed P1 and P2 requires P2-associated Pex1p, and is driven by ATP hydrolysis– and cytosol-dependent release of Pex1p from P2. Fusion of P1 and P2 finally leads to the formation of larger and more dense peroxisomes, P3 (Titorenko et al. 2000). Fusion itself does not require Pex1p, Pex6p, cytosolic proteins, or ATP hydrolysis.
Figure 5.

A model for peroxisome fusion in Y. lipolytica. (P1 and P2) Two distinct forms of small peroxisomal vesicles. (P3) Peroxisome resulting from the fusion of P1 and P2. (1 and 6) Pex1p and Pex6p, respectively. (asterisks) Primed P1 and P2 activated for docking. See Results for details.
Discussion
We have studied membrane fusion during assembly of the peroxisome, an organelle outside the secretory system. Our findings suggest that membrane fusion in the secretory system may be fundamentally different from peroxisome membrane fusion. Fusion in the secretory pathway requires recycling of components of the fusion machinery for further rounds of fusion (Jahn and Südhof 1999). In contrast, in vitro and in vivo fusion between two populations of small peroxisomal vesicles, P1 and P2, leads to the formation of P3 peroxisomes, which in vivo convert to mature peroxisomes in a multistep assembly pathway in Y. lipolytica (Titorenko et al. 2000). Accordingly, mixing of the membrane and luminal contents of P1 and P2 peroxisomes includes only one round of fusion and may, as a consequence, not require recycling of components of the membrane fusion machinery.
Here, we show that the mixing of the membrane and luminal contents of peroxisomes in vitro depends on two AAA ATPases, Pex1p and Pex6p, whose role in membrane fusion recently has been demonstrated (Titorenko et al. 2000). Of note, deletions of the genes for two previously identified ATPases of the AAA protein family, Sec18p and Cdc48p, are lethal in yeast (Kaiser and Schekman 1990; Fröhlich et al. 1991), as they affect membrane fusion events required for the assembly and inheritance of vitally important organelles of the secretory system. In contrast, deletions of the genes encoding Pex1p and Pex6p are conditional in yeast and affect only growth on carbon sources whose utilization requires assembly and extensive proliferation of functionally intact peroxisomes (Erdmann et al. 1991; Spong and Subramani 1993; Heyman et al. 1994; Titorenko and Rachubinski 1998). This finding strongly suggests that neither Pex1p nor Pex6p is essential for the membrane fusion reactions involved in the assembly and inheritance of organelles of the secretory system.
Our data indicate that the roles of Pex1p and Pex6p in priming and docking peroxisomes for fusion are different from those of previously identified AAA ATPases in other membrane fusion reactions. First, priming of P1 and P2 for docking is asymmetric; i.e., P1 peroxisomes are primed by cytosol-dependent and ATP hydrolysis–triggered release of Pex1p, whereas P2 peroxisomes are primed by cytosol-dependent and ATP hydrolysis–triggered release of Pex6p. Possibly, P2-associated Pex1p is also involved in priming P2. This contrasts with vacuole priming by Sec17p (α-SNAP) and Sec18p (NSF), which is symmetric; i.e., both fusion partners are primed by the combined action of these two proteins (Mayer and Wickner 1997; Ungermann et al. 1998). Second, peroxisome docking requires P2-associated Pex1p, whereas neither Pex1p nor Pex6p needs to associate with primed P1 to achieve docking. Therefore, docking of P1 and P2 peroxisomes is asymmetric, in contrast to the tethering and docking of yeast vacuoles (Ungermann et al. 1998). Notably, in contrast to Pex1p, no AAA ATPase has been previously implicated in the docking of fusion partners. How Pex1p functions in peroxisome docking remains to be elucidated. Further analysis of the functions of Pex1p and Pex6p and the identification of other molecular players involved in peroxisome fusion will provide greater insight into the general mechanisms regulating membrane fusion outside the secretory system.
Acknowledgments
We thank Dr. Paul Melançon (Department of Cell Biology, University of Alberta, Edmonton, Alberta, CA) for helpful discussion.
This work was supported by grant MT-15131 from the Medical Research Council (MRC) of Canada to R.A. Rachubinski. R.A. Rachubinski is an MRC Senior Scientist and an International Research Scholar of the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: 20KgP, 20,000-g pellet; 20KgS, 20,000-g supernatant; 200KgP, 200,000-g pellet; 200KgS, 200,000-g supernatant (cytosol); mTHI, mature form of THI; NSF, N-ethylmaleimide–sensitive factor; PNS, postnuclear supernatant; pTHI, precursor form of THI; SNAP, soluble NSF attachment protein; SNARE, SNAP receptor; THI, thiolase.
References
- Acharya U., Jacobs R., Peters J.-M., Watson N., Farquhar M.G., Malhotra V. The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell. 1995;82:895–904. doi: 10.1016/0092-8674(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Erdmann R., Wiebel F.F., Flessau A., Rytka J., Beyer A., Fröhlich K.-U., Kunau W.-H. PAS1, a yeast gene required for peroxisome biogenesis, encodes a member of a novel family of putative ATPases. Cell. 1991;64:499–510. doi: 10.1016/0092-8674(91)90234-p. [DOI] [PubMed] [Google Scholar]
- Fröhlich K.-U., Fries H.W., Rudiger M., Erdmann R., Botstein D., Mecke D. Yeast cell cycle protein CDC48p shows full-length homology to the mammalian protein VCP and is a member of a protein family involved in secretion, peroxisome formation and gene expression. J. Cell Biol. 1991;114:443–453. doi: 10.1083/jcb.114.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A., Wickner W. Homotypic vacuole fusion requires Sec17p (yeast α-SNAP) and Sec18p (yeast NSF) EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3296–3305. [PMC free article] [PubMed] [Google Scholar]
- Heyman J.A., Monosov E., Subramani S. Role of the PAS1 gene of Pichia pastoris in peroxisome biogenesis. J. Cell Biol. 1994;127:1259–1273. doi: 10.1083/jcb.127.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R., Südhof T.C. Membrane fusion and exocytosis. Annu. Rev. Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- Kaiser C.A., Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell. 1990;61:723–733. doi: 10.1016/0092-8674(90)90483-u. [DOI] [PubMed] [Google Scholar]
- Latterich M., Fröhlich K.-U., Schekman R. Membrane fusion and the cell cycleCdc48p participates in the fusion of ER membranes. Cell. 1995;82:885–893. doi: 10.1016/0092-8674(95)90268-6. [DOI] [PubMed] [Google Scholar]
- Mayer A., Wickner W. Docking of yeast vacuoles is catalyzed by the ras-like GTPase Ypt7 after symmetric priming of Sec18p (NSF) J. Cell Biol. 1997;136:307–317. doi: 10.1083/jcb.136.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J.M.M., Rabouille C., Newman R., Shorter J., Freemont P., Schiavo G., Warren G., Shima D.T. An NSF function distinct from ATPase-dependent SNARE disassembly is essential for Golgi membrane fusion. Nat. Cell Biol. 1999;1:335–340. doi: 10.1038/14025. [DOI] [PubMed] [Google Scholar]
- Nichols B.J., Pelham H.R.B. SNAREs and membrane fusion in the Golgi apparatus. Biochim. Biophys. Acta. 1998;1404:9–31. doi: 10.1016/s0167-4889(98)00044-5. [DOI] [PubMed] [Google Scholar]
- Nichols B.J., Ungermann C., Pelham H.R.B., Wickner W., Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Otter-Nilsson M., Hendriks R., Pecheur-Huet E.-I., Hoekstra D., Nilsson T. Cytosolic ATPases, p97 and NSF, are sufficient to mediate rapid membrane fusion. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:2074–2083. doi: 10.1093/emboj/18.8.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S., Latterich M. The AAA teamrelated ATPases with diverse functions. Trends Cell Biol. 1998;8:65–71. [PubMed] [Google Scholar]
- Rabouille C., Levine T.P., Peters J.-M., Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995;82:905–914. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- Rexach M.F., Schekman R.W. Distinct biochemical requirements for the budding, targeting, and fusion of ER-derived transport vesicles. J. Cell Biol. 1991;114:219–229. doi: 10.1083/jcb.114.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman J.E. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Schwarz T.L. NSF is up to new tricks. Nat. Cell Biol. 1999;1:E141–E143. doi: 10.1038/14090. [DOI] [PubMed] [Google Scholar]
- Söllner T., Bennett M.K., Whiteheart S.W., Scheller R.H., Rothman J.E. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Spong A.P., Subramani S. Cloning and characterization of PAS5a gene required for peroxisome biogenesis in the methylotrophic yeast Pichia pastoris . J. Cell Biol. 1993;123:535–548. doi: 10.1083/jcb.123.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard R.K., Titorenko V.I., Veenhuis M., Rachubinski R.A. Pay32p of the yeast Yarrowia lipolytica is an intraperoxisomal component of the matrix protein translocation machinery. J. Cell Biol. 1995;131:1453–1469. doi: 10.1083/jcb.131.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko V.I., Rachubinski R.A. Mutants of the yeast Yarrowia lipolytica defective in protein exit from the endoplasmic reticulum are also defective in peroxisome biogenesis. Mol. Cell. Biol. 1998;18:2789–2803. doi: 10.1128/mcb.18.5.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko V.I., Smith J.J., Szilard R.K., Rachubinski R.A. Pex20p of the yeast Yarrowia lipolytica is required for the oligomerization of thiolase in the cytosol and for its targeting to the peroxisome. J. Cell Biol. 1998;142:403–420. doi: 10.1083/jcb.142.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko V.I., Chan H., Rachubinski R.A. Fusion of small peroxisomal vesicles in vitro reconstructs an early step in the in vivo multistep peroxisome assembly pathway of Yarrowia lipolytica . J. Cell Biol. 2000;147:29–43. doi: 10.1083/jcb.148.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C., Sato K., Wickner W. Defining the functions of trans-SNARE pairs. Nature. 1998;396:543–548. doi: 10.1038/25069. [DOI] [PubMed] [Google Scholar]
- Weber T., Zemelman B.V., McNew J.A., Westermann B., Gmachl M., Parlati F., Söllner T.H., Rothman J.E. SNAREpinsminimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Whiteheart S.W., Rossnagel K., Buhrow S.A., Brunner M., Jaenicke R., Rothman J.E. N-ethylmaleimide–sensitive fusion proteina trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J. Cell Biol. 1994;126:945–954. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]