

Abstract
To test the role of ER luminal environment in apoptosis, we generated HeLa cell lines inducible with respect to calreticulin and calnexin and investigated their sensitivity to drug-dependent apoptosis. Overexpression of calreticulin, an ER luminal protein, resulted in an increased sensitivity of the cells to both thapsigargin- and staurosporine-induced apoptosis. This correlated with an increased release of cytochrome c from the mitochondria. Overexpression of calnexin, an integral ER membrane protein, had no significant effect on drug-induced apoptosis. In contrast, calreticulin-deficient cells were significantly resistant to apoptosis and this resistance correlated with a decreased release of cytochrome c from mitochondria and low levels of caspase 3 activity. This work indicates that changes in the lumen of the ER amplify the release of cytochrome c from mitochondria, and increase caspase activity, during drug-induced apoptosis. There may be communication between the ER and mitochondria, which may involve Ca2+ and play an important role in conferring cell sensitivity to apoptosis. Apoptosis may depend on both the presence of external apoptosis-activating signals, and, as shown in this study, on an internal factor represented by the ER.
Keywords: apoptosis, calreticulin, endoplasmic reticulum, calcium-binding protein
Introduction
The ER plays a critical role in a variety of process including the maintenance of intracellular Ca2+ homeostasis, synthesis, posttranslational modification and folding of membrane associated, secreted and integral membrane proteins. In virtually all non-muscle cells, storage, release, and uptake of Ca2+ are regulated by proteins of the ER. Ca2+ in the lumen of the ER is either free or bound to luminal proteins. It is released from the ER through the inositol 1,4,5-trisphosphate (InsP3) receptor/Ca2+ channel and taken up from the cytosol by the sarcoplasmic/endoplasmic Ca2+-ATPase (SERCA) (Pozzan et al. 1994). Many diverse cellular functions are regulated by alterations in intracellular Ca2+ concentration, including secretion, contraction-relaxation, cell motility, cytoplasmic and mitochondrial metabolism, protein synthesis and folding, gene expression, and cell cycle progression (Pozzan et al. 1994). Evidence now indicates that changes in intracellular Ca2+ homeostasis also play a role in modulation of apoptosis (Oppenheim 1991; Barres et al. 1992; Preston and Berlin 1992; Baffy et al. 1993; Lam et al. 1994; Distelhorst et al. 1996; Marin et al. 1996; McConkey et al. 1996; Reynolds and Eastman 1996; Szalai et al. 1999), a naturally occurring form of cell death which is important for proper development and homeostasis in many tissues (Jacobson et al. 1997; Nagata 1997).
Calreticulin is a major Ca2+-binding chaperone found in the lumen of the ER (Michalak 1996; Krause and Michalak 1997; Meldolesi and Pozzan 1998; Michalak et al. 1999). It is also involved in the regulation of intracellular Ca2+ homeostasis, steroid-sensitive gene expression and cell adhesion (Krause and Michalak 1997; Michalak et al. 1999). Calreticulin shares amino acid sequence identity with calnexin, an integral ER membrane chaperone (Bergeron et al. 1994; Hammond and Helenius 1995). Calnexin and calreticulin are both chaperones believed to play a critical role in quality control processes during protein synthesis and folding (Bergeron et al. 1994; Hammond and Helenius 1995; Helenius et al. 1997; Spiro et al. 1996; Vassilakos et al. 1998; Ihara et al. 1999; Saito et al. 1999). Calnexin may also play a role in control of Ca2+ homeostasis (John et al. 1998) Recently, John et al. 1998 reported that calreticulin and calnexin may interact with SERCA2b resulting in a lower capacity for Ca2+ transport by the Ca2+-ATPase.
In this study, we have investigated the possible role of the ER in modulation of apoptosis. We altered the expression of calreticulin and calnexin, ER luminal and integral membrane proteins, and then initiated apoptosis. We show that increased expression of the ER luminal protein calreticulin increased cell sensitivity to apoptosis, whereas overexpression of calnexin, an integral ER membrane protein, had no significant effect on the drug-induced apoptosis. Furthermore, we show that calreticulin-deficient cells, from calreticulin knockout mice, were significantly resistant to staurosporine-induced apoptosis. This resistance to apoptosis was accompanied by reduced cytochrome c release from mitochondria and lower caspase 3 enzyme activity. Our findings suggest that the ER, via specific components of its luminal environment, may play an important role in the modulation of cell sensitivity of apoptosis.
Materials and Methods
Materials
[3H]thymidine and 45Ca2+ were from Amersham Pharmacia Biotech. HeLa Tet-On cells, pTRE and pTK-Hyg plasmids, fetal bovine serum for the Tet system, doxycycline, hygromycin B, and FITC-labeled Annexin-V were purchased from CLONTECH Laboratories, Inc. DME, fetal bovine serum, trypsin-EDTA, l-glutamine, penicillin, streptomycin, G418 (Geneticin) and RPMI 1640 were purchased from GIBCO BRL. Staurosporine, bradykinin and thapsigargin were from Sigma. Fura-2/AM was from Molecular Probes. In Situ Cell Death Detection for the TUNEL assay (TdT-mediated dUTP nick end labeling) was from Boehringer. Restriction endonuclease and DNA-modifying enzymes were obtained from Boehringer, GIBCO BRL, and Bio/Can Scientific. Goat anti-calreticulin antibodies were prepared as described previously (Milner et al. 1991). Rabbit anti-calnexin antibodies were purchased from StressGen. FITC-conjugated secondary antibodies were from Bio/Can Scientific. Anti-cytochrome c antibody (7H8.2C12) was from PharMingen. Ac-DEVD-AFC was from Biomol. All chemicals were of the highest grade available.
Plasmids
cDNA encoding full-length rabbit calreticulin and canine calnexin was subcloned into the pTRE plasmid to generate pTRE-CRT and pTRE-CNX expression vectors, respectively. These vectors were used for generation of the Tet-On–inducible cell lines. The nucleotide sequence of all cDNAs was confirmed by the DNA Sequencing Laboratory in the Department of Biochemistry, using an Applied Biosystems DNA sequencer (model 373A). Plasmid DNA was purified by QIAGEN column chromatography.
Plasmids and Generation of the Tet-On Calreticulin and Calnexin Cell Lines
The Ca2+-phosphate method was used for transfection of the HeLa Tet-On cells, using 40 μg plasmid DNA (pTRE-CRT or pTRE-CNX) and 2 μg selection plasmid (pTK-Hyg). Transfected cells were selected for growth in the presence of 200 μg hygromycin B/ml culture medium. Single colonies of the hygromycin B–resistant cells were tested for doxycycline (Dox)-dependent expression of calreticulin and calnexin, by Western blotting with anti-calreticulin or anti-calnexin antibodies. Two cell lines, which showed highly inducible expression of calreticulin or calnexin, were selected for this study. These cell lines were designated KN1 for inducible expression of calreticulin, and KNX2 for inducible expression of calnexin.
Cell Culture
A1.1 cells (cloned cell line derived from mouse T cells) were grown in RPMI medium supplemented with 5% fetal calf serum, 100 μM 2-mercaptoethanol, 2 mM l-glutamine, 10 mM Hepes, pH 7.5, and 50 U penicillin, 50 μg streptomycin, and 5 μg gentamycin per milliliter. A1.1 cells (5 × 106) were incubated in the presence of 5 μM synthetic oligodeoxynucleotides in 1 ml RPMI medium per well. The following oligodeoxynucleotides were used: anti-sense oligodeoxynucleotide 5′-CCG CCC CGG CCC GCC ATG CTC CTT TCG GTG-3′ including an ATG initiation codon (underlined), and scrambled oligodeoxynucleotide 5′-GAC ACG AAC GAC CAG CGA GGA G-3′. Synthetic oligodeoxynucleotides and sequencing primers were made in the DNA Sequencing Laboratory in the Department of Biochemistry, using an Applied Biosystems 394 DNA/RNA synthesizer. To initiate glucocorticoid-dependent apoptosis, cells were incubated for 12 h in RPMI containing 5% charcoal-treated fetal calf serum, followed by incubation for 24 h in RPMI containing 25–500 nM dexamethasone (Dex) or RPMI alone.
HeLa cells and Tet-On cells were maintained in DME supplemented with 10% fetal bovine serum, 100 U penicillin/ml, 100 μg DME streptomycin/ml, and 2 mM l-glutamine. DME was also supplemented with 10% tetracycline-negative fetal bovine serum for Tet system. 2 μg Dox/ml was added into the culture medium to induce expression of calreticulin or calnexin in Tet-On cell lines.
Mouse embryonic fibroblasts (MEF) were isolated from calreticulin-deficient crt −/− and wild-type embryos. crt −/− and wild-type embryos were dissociated, washed, trypsinized for 30 min, and cultured in 6-well tissue-culture plates. To generate immortal MEF cell lines, cells were transfected with a pSV-7 vector encoding the SV-40 large T antigen (Conzen and Cole 1995). After 2 wk in culture individual colonies of immortalized MEF were picked and used for experiments. Cells were maintained in DME containing 20% fetal calf serum. To induce apoptosis, cells were incubated with 2 μM, 100 nM, or 10 nM staurosporine for 90 min. This treatment was followed with Annexin-V binding assay, which was carried out as recommended by the manufacturer and always including propidium staining assay.
Immunoblotting and Immunocytochemistry
Cells were washed three times with ice-cold PBS and then lysed as described by (Mery et al. 1996). Proteins were separated by SDS-PAGE (Laemmli 1970) and transferred to nitrocellulose membrane (Towbin et al. 1979). Goat anti-calreticulin and rabbit anti-calreticulin (affinity-purified CRT283) were prepared as described earlier (Milner et al. 1991; Michalak et al. 1996). Rabbit anti-ERp57 antibodies, a generous gift of D. Thomas (Genetics Group, Biotechnology Research Institute, National Research Council of Canada, Montrèal, Canada), were used at 1:1,000 (Zapun et al. 1998). Immunoblotting was carried out as described by (Nakamura et al. 1995). For analysis of cytochrome c release from mitochondria, preparation of extracts and immunoblotting was carried out as described by (Bossy-Wetzel et al. 1998; Bossy-Wetzel and Green 1999). Indirect immunofluorescence of KN1 and KNX2 cells was carried out as described by (Opas et al. 1991). In brief, KN1 and KNX2 cells were plated on coverslips and cultured in the presence or absence of 2 μg Dox/ml for 24 h. Cells were washed with PBS and fixed with 4% paraformaldehyde. Coverslips were mounted in Vinol 205S and examined with a Bio-Rad Laboratories confocal fluorescence microscope (model MRC-600) equipped with a krypton/argon laser.
Apoptosis Assay
For the Annexin-V binding assay, cells were plated on 6-cm dishes at 5 × 103/cm2 and incubated for 24 h in the presence or absence of 2 μg Dox/ml. To induce apoptosis, KN1 and KNX2 cells were incubated for 90 min in the presence of 2 μM staurosporine or for 24 h in the presence of 200 nM thapsigargin. Calreticulin-deficient and wild-type MEF were incubated with 2 μM staurosporine for 4 h. The cells were washed with PBS, harvested, and incubated with FITC-labeled Annexin-V as recommended by the manufacturer, and analyzed by flow cytometry. For the TUNEL assay, cells were plated on 10-cm dishes at 5 × 103/cm2 and incubated for 24 h with or without 2 μg Dox/ml. Staurosporine (10 nM, 100 nM, or 2 μM) was added for 4 h, or 4 h 30 min, to induce apoptosis. Alternatively, some cells were treated with UV for 1 min or with 10 or 100 μM etoposide to induce apoptosis. The cells were washed with PBS, harvested and incubated with the TUNEL reaction mixture, as recommended by the manufacturer. This was followed by flow cytometry or fluorescence microscopy. Caspase 3 activity was measured by the DEVD-AFC cleavage assay (Bossy-Wetzel and Green 1999). 50 μg protein extract was diluted in a buffer containing 200 μM Ac-DEVD-AFC, 20 mM Pipes, pH 7.2, 100 mM NaCl, 1 mM EDTA, 0.1% Chaps, 10% sucrose, and 10 mM DTT (caspase buffer). The kinetics of substrate cleavage were measured at 37°C under constant agitation using a spectrofluorometer at λex = 400 nm and λem at 505 nm. Apoptosis was also quantified by staining cells with eosin and hematoxylin (Bossy-Wetzel et al. 1998). Apoptotic cells were counted using a light microscope. Apoptosis was detected by cell shrinkage, chromatin condensation and nuclear fragmentation.
Ca2+ Measurements
In KN1 cells, the total Ca2+ in ER Ca2+ stores was estimated using 45Ca2+ (10 μCi/ml) as described earlier (Mery et al. 1996). For measurement of cytoplasmic Ca2+concentration ([Ca2+]c), cells (1.5 × 106/ml) were loaded with the fluorescent Ca2+ indicator fura-2/AM (2 μM) taking precautions to avoid dye sequestration as described by (Mery et al. 1996). Fura-2 fluorescence was measured at λex = 340 nm. To measure changes in cytoplasmic Ca2+concentration, cells were stimulated with 1 μM thapsigargin or with 200 nM bradykinin (Mery et al. 1996; Waser et al. 1997).
Miscellaneous
Protein assays were carried out as described by (Bradford 1976). All recombinant DNA techniques were conducted according to standard protocols. Transgenic mice expressing the GFP reporter gene under control of the calreticulin promoter were generated as described (Mesaeli et al. 1999). Mouse embryos were dissected, fixed and prepared for fluorescence microscopy (Mesaeli et al. 1999). Images were reconstructed using the Adobe Photoshop program.
Results
Apoptosis is an active and genetically controlled process of eliminating unwanted cells during development, tissue turnover and metamorphosis (Jacobson et al. 1997). While studying transactivation of the calreticulin promoter and expression of the protein during mouse embryogenesis we noticed that the calreticulin promoter was highly active in several tissues including cells destined for apoptosis during mouse development. We monitored transactivation of the calreticulin promoter by detection of the fluorescent signal obtained from calreticulin promoter-driven expression of the green fluorescent protein (GFP) reporter gene in transgenic mice (Mesaeli et al. 1999). Fig. 1A and Fig. B, shows section of a developing eye (A) and a limb bud (B) of a 14.5-d-old transgenic mouse embryo. The highest fluorescent signal, indicative of high expression of GFP, was found in the central retina (Fig. 1 A, R), the lens vesicle (Fig. 1 A, LV) and in the interdigital cells (Fig. 1 B, IC). It is well established that central retina and lens vesicle cells undergo apoptosis during normal development (Ganan et al. 1996; Zou and Niswander 1996). Apoptosis is also responsible for eliminating the cells between developing digits (Ganan et al. 1996; Zou and Niswander 1996). Furthermore, in preliminary biochemical experiments with A1.1 cells (a cloned cell line derived from T cells), we found that when we downregulated calreticulin, using anti-sense oligodeoxynucleotides (Fig. 2 A), there was a protective effect against Dex-induced apoptosis as assessed by determining the degree of DNA fragmentation (Fig. 2 B). Since calreticulin is involved in Ca2+ homeostasis these findings are in line with earlier studies which showed that Ca2+ release from ER may affect Dex-induced apoptosis in W7MG1 mouse lymphoma cells (Lam et al. 1993) or T lymphocytes (Jayaraman and Marks 1997). Taken together, these findings suggested to us that changes in expression of calreticulin, an ER luminal protein, may play a role in cell sensitivity to apoptosis induced by external stimuli.
Figure 1.
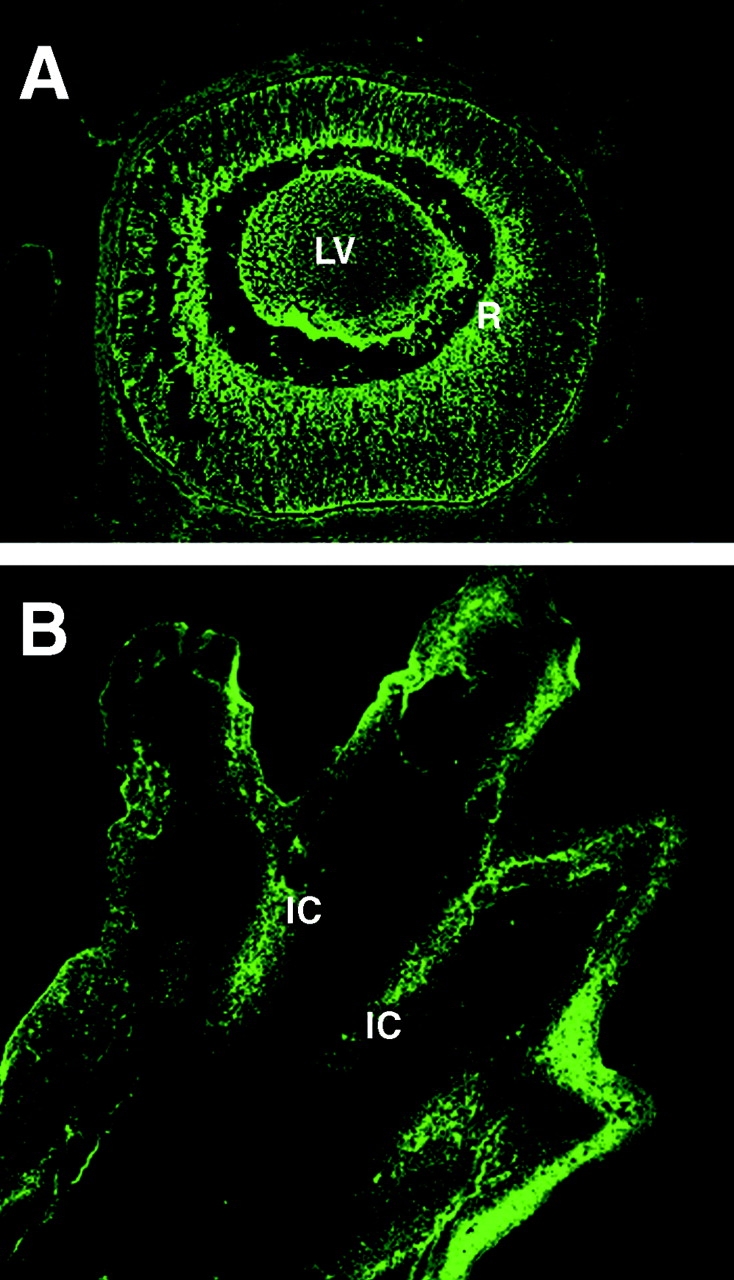
Activation of calreticulin promoter in developing eye and limb buds of transgenic mouse embryos. Transgenic mice expressing GFP under control of the calreticulin promoter were generated as described by Mesaeli et al. 1999. Activation of the calreticulin promoter was monitored by detection of the fluorescent signal obtained from calreticulin promoter-driven expression of the GFP reporter gene (Mesaeli et al. 1999). Mouse embryos (14.5 d old) were dissected out of the uterus and processed for fluorescence analysis using a confocal microscope (Mesaeli et al. 1999). (A) A section of a developing embryonic eye; (B) a section across developing limb bud. High activation of the calreticulin promoter was detected in central retina (R), lens vesicle (LV) and in interdigital cells (IC).
Figure 2.
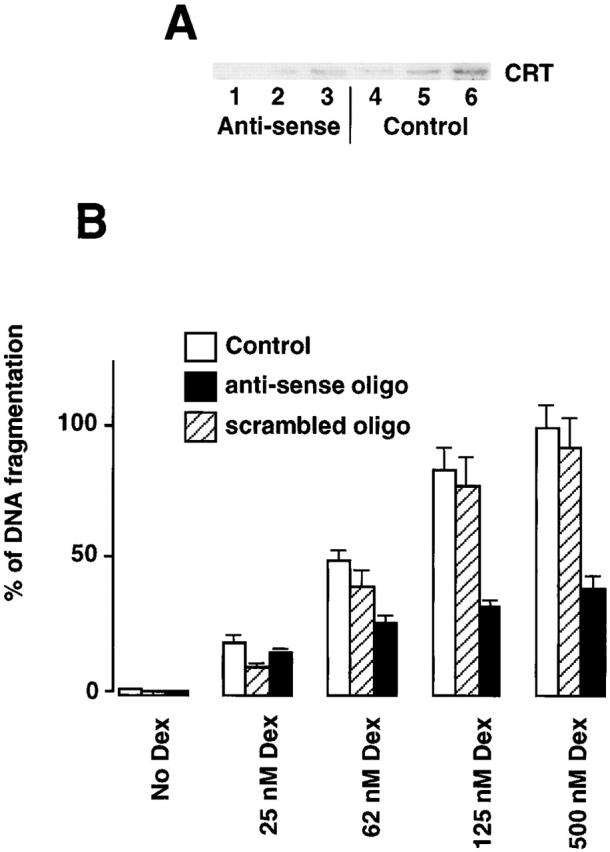
Calreticulin modulates dexamethasone-dependent apoptosis of A1.1 cells. (A) Western blot analysis of calreticulin in A1.1 cells incubated with calreticulin-specific anti-sense oligodeoxynucleotide. Cells were lysed with RIPA buffer, protein separated in SDS-PAGE, transferred to nitrocellulose membrane, and probed with goat anti-calreticulin antibodies. Lanes 1–3, 10, 20, and 30 μg of cell extract from anti-sense oligodeoxynucleotide-treated A1.1 cells, respectively; lanes 4–6, 10, 20, and 30 μg of cell extract from untreated A1.1 cells, respectively. CRT, calreticulin. (B) Downregulation of expression of calreticulin was regulated in A1.1 cells by incubation with specific anti-sense oligodeoxynucleotide followed by DNA fragmentation analysis. Apoptosis was induced with a different concentration of Dex. Open bar, control cells; hatched bar, cells treated with scrambled control oligodeoxynucleotide; full bar, cells treated with anti-sense oligodeoxynucleotide. Data are means ± SD of three independent experiments.
Expression of Calreticulin and Calnexin in Tet-On–inducible HeLa Cells
To investigate the role of calreticulin and calnexin on cell sensitivity to apoptosis, we generated Tet-On HeLa cell lines that are inducible with respect to calreticulin (designated KN1) and calnexin (designated KNX2). In Tet-On cells, gene expression is turned on when doxycycline (Dox) is added to the culture medium. Incubation of the KN1 and KNX2 cells with Dox resulted in 2.3 ± 0.2-fold (mean ± SD; n = 4) and 2.2 ± 0.2-fold (mean ± SD; n = 4) induction in the expression of calreticulin (Fig. 3 A) and calnexin (Fig. 3 B), respectively. As internal control we tested for expression of ERp57, an ER luminal chaperone, in KN1 and KNX2 cells. Fig. 3A and Fig. B, shows that Dox had no effect on expression of ERp57. Expression of other ER proteins including BiP, ERp72, protein disulfide isomerase, Grp94, SERCA2, and InsP3 receptor was also not affected by Dox (not shown). The doubling time of these cell lines was ∼20 h and was not affected by the addition of Dox. After induction of protein synthesis with Dox in KN1 and KNX2 cells calreticulin and calnexin were both localized to the ER (Fig. 3 C). There was no immunoreaction with anti-calreticulin or anti-calnexin antibodies in the cytoplasm, in the nucleus, or on the cell surface. This indicates that the Dox-dependent induction of calreticulin and calnexin resulted in an increased accumulation of these proteins in the ER and not in other intracellular compartments.
Figure 3.
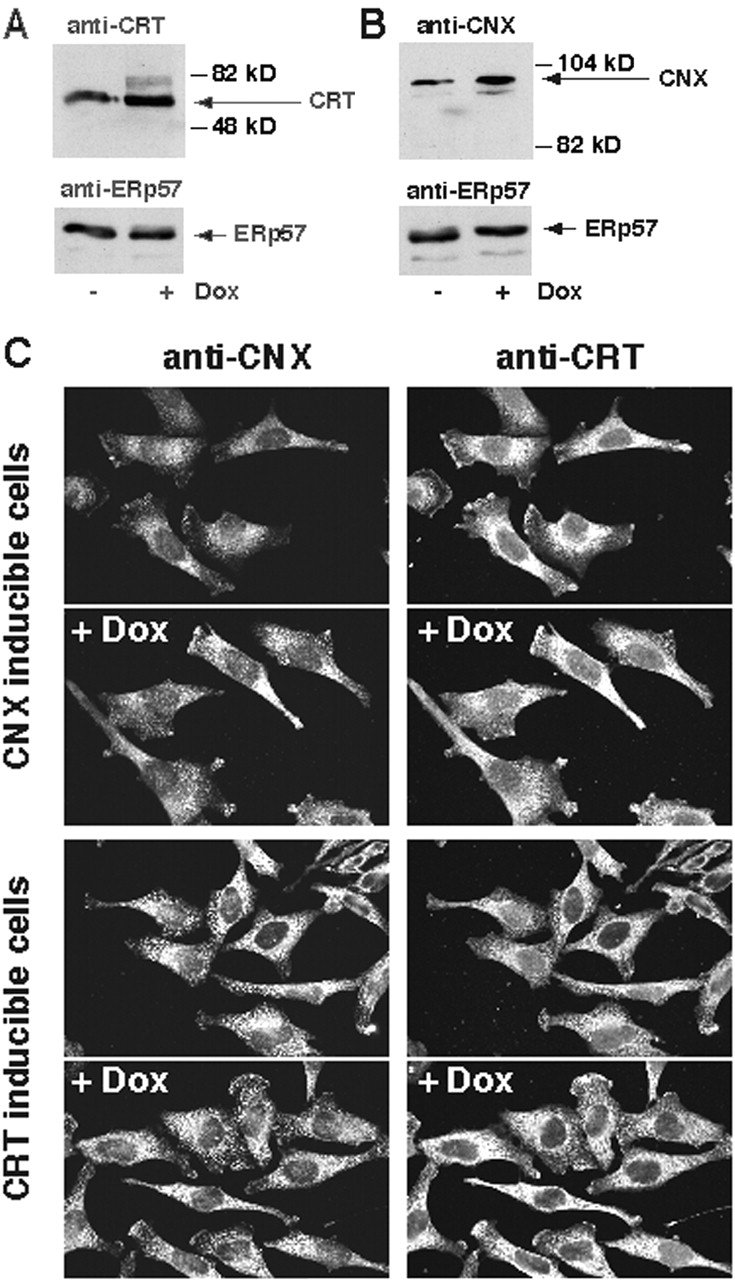
Expression of calreticulin and calnexin in Tet-On–inducible HeLa cell lines. Overexpression of calreticulin (A, KN1 cells) or calnexin (B, KNX2 cells) was induced by incubation of the cells in the presence of 2 μM Dox for 24 h. Cells were harvested, lysed with RIPA buffer. 10 μg of protein was separated in SDS-PAGE transferred to nitrocellulose membrane and probed with anti-calreticulin (A) or anti-calnexin (B) antibodies. Blots were normalized by probing with anti-ERp57 antibodies. Addition of Dox to KN1 or KNX2 cells resulted in 2.3 ± 0.2-fold (mean ± SE; n = 4) and 2.2 ± 0.2-fold (mean ± SE; n = 4) induction in the expression of calreticulin (A) and calnexin (B), respectively, as estimated by densitometry. The positions of molecular markers are indicated. (C) Localization of calreticulin and calnexin in KN1 (calreticulin inducible) and KNX2 (calnexin inducible). (Top) Localization of calreticulin and calnexin in the calnexin Tet-On KNX2 (calnexin inducible). (Bottom) Localization of calreticulin and calnexin in the calreticulin Tet-On KN1 cells (calreticulin inducible). To induce expression of calreticulin and calnexin, KN1 and KNX2 cells were treated with Dox for 24 h, respectively (+Dox). Both calreticulin and calnexin localized predominantly to an ER-like intracellular network. CRT, calreticulin; CNX, calnexin.
Induction of Apoptosis in KN1 and KNX2 Cells
Staurosporine (Raff et al. 1993; Bertrand et al. 1994; Jacobson et al. 1994) and thapsigargin (Lam et al. 1994) both induce apoptosis. To investigate the possible involvement of calreticulin and ER in apoptosis, we treated KN1 and KNX2 cells with Dox to induce overexpression of calreticulin and calnexin, respectively. We then incubated the cells with either thapsigargin or staurosporine, and measured apoptosis by Annexin-V binding or TUNEL assay. By itself, Dox did not induce apoptosis in HeLa cells, mock transfected control cells, or KN1 and KNX2 cells (Fig. 4 and Fig. 5, HeLa-On). When thapsigargin was used to induce apoptosis, we found that cells overexpressing calreticulin were more sensitive. As shown in Fig. 4, after treatment with thapsigargin, Annexin-V binding was greater in cells that were overexpressing calreticulin. Fig. 5 A shows that after treatment with staurosporine, Annexin-V binding was also increased in Dox-treated KN1 cells as compared with untreated KN1 cells. This shows that the cells overexpressing calreticulin were more sensitive to staurosporine. Similar results were obtained with 2 μM and 10 nM staurosporine (data not shown). In contrast, in KNX2 cells overexpressing calnexin, a small and statistically insignificant reduction in sensitivity to both thapsigargin (Fig. 4) and staurosporine (Fig. 5 A) was observed. These results indicate that the overexpression of calnexin did not affect apoptosis.
Figure 4.

Thapsigargin-dependent induction of apoptosis in cells overexpressing calreticulin and calnexin. KN1 and KNX2 cells were incubated with 2 μg Dox/ml (filled bars) for 24 h to induce expression of calreticulin and calnexin, respectively. Cells were treated with thapsigargin followed by Annexin-V binding assay. HeLa-On, mock transfected Tet-On HeLa cells. (Open bars) Cells not incubated with Dox; filled bars, Dox-treated cells. 100% value corresponds to 10,000 cells. Data are means ± SD of three independent experiments. ** P < 0.001.
Figure 5.
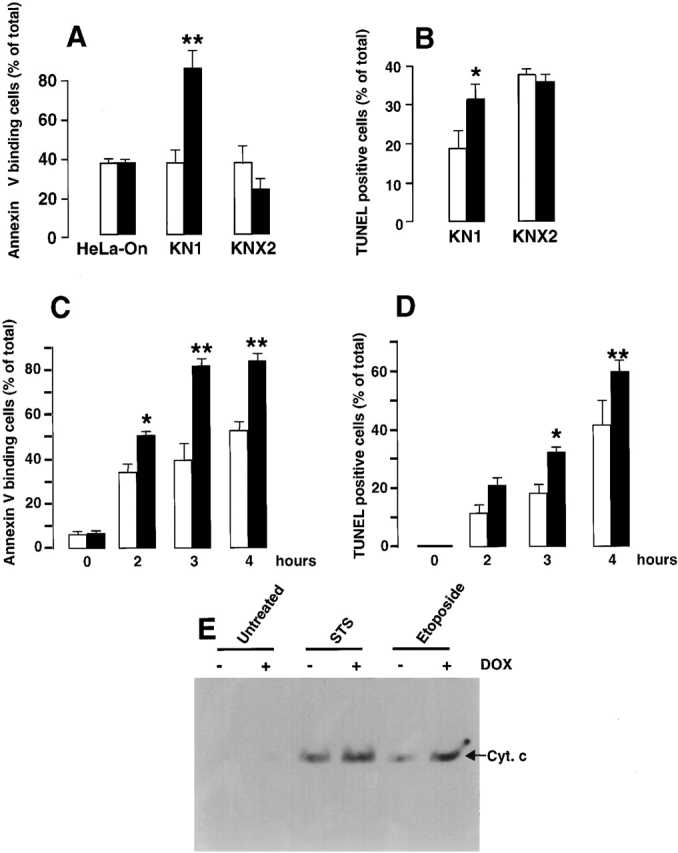
Induction of apoptosis in cells overexpressing calreticulin and calnexin. KN1 and KNX2 cells were incubated with 2 μg Dox/ml (filled bars) for 24 h to induce expression of calreticulin and calnexin, respectively. Cells were treated with staurosporine (A–D) followed by Annexin-V binding assay (A and C) or TUNEL analysis (B and E). In C and D KN1 cells were incubated with staurosporine for the time indicated in the figure followed by Annexin-V binding assay (C) or TUNEL analysis (D). Open bars, cells not incubated with Dox; filled bars, Dox-treated cells. 100% value corresponds to 10,000 cells. Data are means ± SD of three independent experiments. In E, KN1 cells were incubated with 2 μg Dox/ml (+) for 24 h to induce expression of calreticulin. To induce apoptosis control or Dox-treated cells were incubated with 100 nM staurosporine (STS) or 100 μM etoposide. Cytosolic extracts were prepared devoid of mitochondria, separated by SDS-PAGE and immunoblotted with anti-cytochrome c antibodies (Bossy-Wetzel and Green 1999). The position of cytochrome c (Cyt. c) is indicated by the arrow. ** P < 0.001 and * P < 0.005.
The sensitivity of KN1 and KNX2 cells to staurosporine was also evaluated by TUNEL analysis (Fig. 5 B). In keeping with the Annexin-V binding results described above, we found that Dox-dependent overexpression of calreticulin increased the sensitivity of KN1 cells to apoptosis (Fig. 5 B). After incubation with staurosporine, >30% of cells overexpressing calreticulin (Dox-treated KN1) were TUNEL positive, compared with only 18% of control cells. In contrast, Dox-dependent overexpression of calnexin did not affect the sensitivity of KNX2 cells to apoptosis. After treatment with staurosporine, the proportion of cells that were TUNEL positive was the same in Dox-treated and control cells.
We also carried out a time course analysis of staurosporine-induced apoptosis in KN1 cells overexpressing calreticulin (Fig. 5C and Fig. D). Increased sensitivity of KN1 cells overexpressing calreticulin to staurosporine-induced apoptosis was observed after 0–4 h of incubation with the drug (Fig. 5C and Fig. D). Time course for both Annexin-V binding (Fig. 5 C) and the appearance of TUNEL-positive cells (Fig. 5 D) were comparable indicating that overexpression of calreticulin likely affected the rate of apoptosis rather than the sequence of events associated with apoptosis.
Our results show that KN1 cells are more sensitive to apoptosis when they are overexpressing calreticulin. To determine whether or not apoptosis was occurring via similar pathways in control and test cells, we measured cytochrome c release from the mitochondria. Overexpression of calreticulin was induced with Dox, and then apoptosis was induced by treatment with staurosporine or etoposide. We prepared cell extracts, which were devoid of mitochondria, and then we assessed cytochrome c levels by immunoblotting. In KN1 cells treated with staurosporine or etoposide there was a detectable release of cytochrome c from the mitochondria (Fig. 5 E). In untreated KN1 cells there was no release of cytochrome c from the mitochondria (Fig. 5 E). In KN1 cells first incubated with Dox (overexpressing calreticulin) and then treated with staurosporine or etoposide, the release of cytochrome c from the mitochondria was consistently higher (Fig. 5 E, + Dox). To confirm the differences we had found in Annexin-V binding, TUNEL assay and cytochrome c release from mitochondria, we measured rates of DEVD cleavage (caspase activity) in cytosolic extracts from KN1 cells overexpressing calreticulin. Calreticulin expression was induced with Dox followed by a 60-min incubation with staurosporine to induce apoptosis. As expected the DEVD cleavage (caspase) activity was 1.75-fold higher in calreticulin overexpressing (Dox- and staurosporine-treated) KN1 cells than in the KN1 cells incubated with staurosporine alone.
Ca2+ Homeostasis in KN1 Cells
It is well documented that calreticulin plays a role in the regulation of intracellular Ca2+ homeostasis (Liu et al. 1994; Bastianutto et al. 1995; Camacho and Lechleiter 1995; Mery et al. 1996; Coppolino et al. 1997; Fasolato et al. 1998; John et al. 1998; Mesaeli et al. 1999). Therefore, we investigated the effect on Ca2+ homeostasis of overexpressing calreticulin in KN1 cells. First, we used equilibrium loading techniques to determine whether the Ca2+ content of intracellular Ca2+ stores was modified in KN1 cells that were overexpressing calreticulin (Mery et al. 1996). Control KN1 cells contained 70 ± 10 pmol of Ca2+/106 cells (mean ± SD; n = 3) whereas the KN1 cells treated with Dox (overexpressing calreticulin) contained 148 ± 12 pmol of Ca2+/106 cells (mean ± SD; n = 3). Similar increases in the Ca2+ content of intracellular Ca2+ stores have been reported for cells transiently (Bastianutto et al. 1995) and stably (Mery et al. 1996) transfected with a calreticulin expression vector.
Second, we used a Ca2+-sensitive fluorescent dye, fura-2, to investigate the effects of overexpressing calreticulin on [Ca2+]c in KN1 cells. Basal cytoplasmic Ca2+ concentration ([Ca2+]c) in control and Dox-treated KN1 cells was similar (∼130 ± 10 nM; mean ± SD; n = 3). To measure the Ca2+ associated with rapidly exchangeable intracellular Ca2+ stores, we used thapsigargin, an inhibitor of SERCA (Thastrup et al. 1990). When cells were treated with thapsigargin, the peak and duration of the [Ca2+]c elevations were comparable in control and Dox-treated KN1 cells (not shown). In contrast to this, we found that control and Dox-treated KN1 cells differed in their response to bradykinin. Bradykinin causes the release of Ca2+ from internal stores via activation of endogenous, agonist-dependent pathways. In both control and Dox-treated KN1 cells, bradykinin caused a rapid and transient increase in [Ca2+]c. However, the amplitude of this [Ca2+]c elevation was ∼1.5-fold greater in cells overexpressing calreticulin (620 ± 25 nM; mean ± SD; n = 3) than in control cells (350 ± 20 nM; mean ± SD; n = 3). It remains unclear why increased elevation of [Ca2+]c in KN1 cells overexpressing calreticulin was observed with bradykinin but not with thapsigargin. It is possible, that in the case of thapsigargin the slow Ca2+ release allows negative feedback mechanism such as extrusion of Ca2+ via the plasma membrane Ca2+-ATPase to obscure the differences in Ca2+ release in relation to the level of [Ca2+]c measurements (Mery et al. 1996).
In summary, we show that Dox-dependent overexpression of calreticulin in KN1 cells results in increased storage of Ca2+ in the ER and in elevated agonist-dependent Ca2+ release from internal stores. In contrast, we found no significant changes in total intracellular Ca2+ or in Ca2+ release in KNX2 cells overexpressing calnexin.
Induction of Apoptosis in Calreticulin-deficient Cells
Recently, we have generated a calreticulin knockout embryo (Mesaeli et al. 1999). For this study we established an immortalized calreticulin-deficient, MEF cell line (for details see Materials and Methods). Since crt −/− cells do not express calreticulin (Fig. 6 A) they provide an ideal tool to investigate the effects of calreticulin deficiency on apoptosis. Fig. 6 B compares staurosporine-induced apoptosis in wild-type cells (containing calreticulin) and calreticulin-deficient cells (crt −/−), as measured by Annexin-V binding. Interestingly, the calreticulin-deficient cells were significantly resistant to apoptosis (Fig. 6 B). We also performed a kinetic analysis of apoptosis induced by etoposide, staurosporine and UV (UVB; Fig. 6 C). Fig. 6 C shows that calreticulin-deficient cells were consistently more resistant to apoptosis compared with the wild-type cells.
Figure 6.
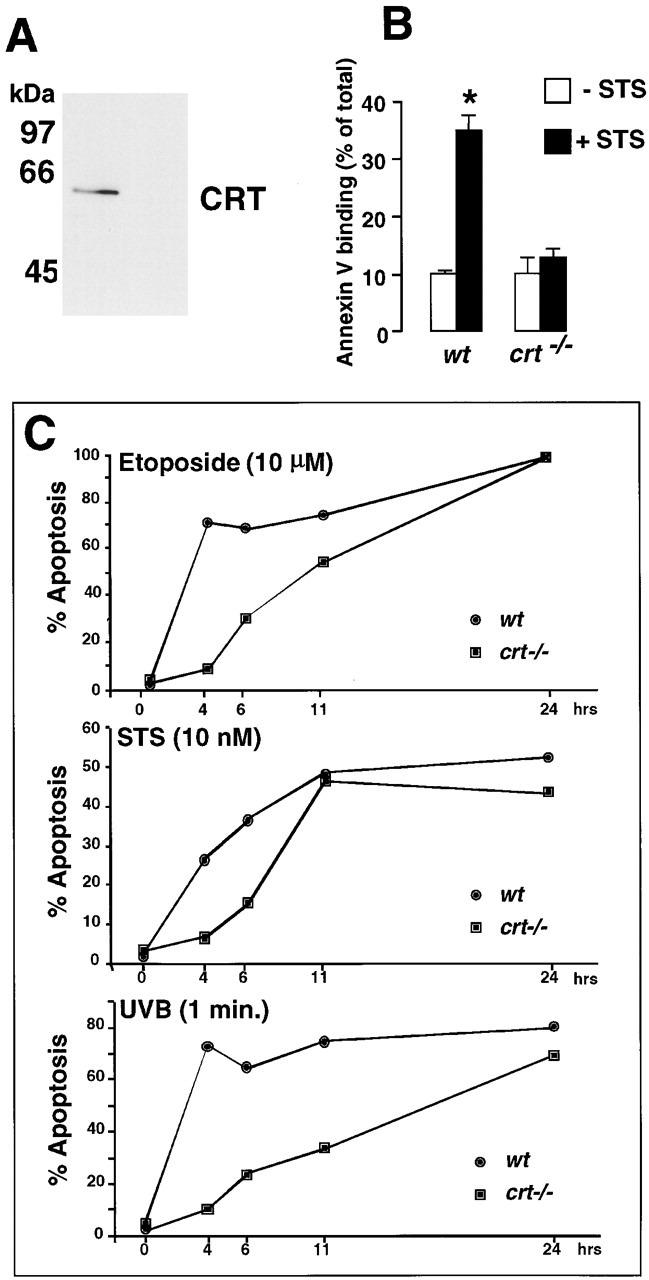
Induction of apoptosis in calreticulin-deficient cells. Wild-type (wt) and calreticulin deficient (crt−/−) mouse fibroblast cell lines were established as described in Materials and Methods. (A) Cells were lysed with RIPA buffer, protein separated in SDS-PAGE, transferred to nitrocellulose membrane and probed with the affinity-purified rabbit anti-calreticulin antibody. The location of calreticulin (CRT) and Bio-Rad Laboratories molecular mass markers are indicated. (B) Wild-type (wt) and calreticulin-deficient (crt−/−) mouse embryonic fibroblasts were treated for 30 min in the absence (open bars) or presence (filled bars) of staurosporine (STS) followed by Annexin-V binding assay as described under Materials and Methods. 100% value corresponds to 10,000 cells. Data are means ± SD of two independent experiments. In C, wild-type (wt) and calreticulin deficient (crt−/−) mouse embryonic fibroblasts were treated with 10 μM etoposide, 10 nM staurosporine (STS), or UV for 1 min. The percentage of apoptosis was determined by light microscopy of eosin and hematoxylin-stained cells as described in Materials and Methods. Statistically significant: * P < 0.001.
In parallel with these studies, we also tested the effects of calreticulin deficiency on Ca2+ homeostasis, using the Ca2+-sensitive fluorescent dye fura-2. We reported earlier (Mesaeli et al. 1999) that in wild-type and calreticulin-deficient MEF resting [Ca2+]c was similar (80 ± 15 nM; mean ± SD; n = 3). When cells were treated with thapsigargin the peak and duration of the [Ca2+]c elevations were comparable in control and calreticulin-deficient MEF (not shown; Mesaeli et al. 1999). In contrast, bradykinin-dependent Ca2+ release from intracellular Ca2+ stores was inhibited in calreticulin-deficient MEF (Mesaeli et al. 1999).
To determine whether apoptosis was occurring by similar pathways in wild-type and calreticulin-deficient cells, we measured cytochrome c release from the mitochondria. Wild-type and calreticulin-deficient MEF were treated with UV, etoposide or staurosporine, and then cytosolic extracts, devoid of mitochondria, were prepared and analyzed by immunoblotting. As expected, in untreated cells there was no release of cytochrome c from the mitochondria (Fig. 7 A, untreated). Treatment of wild-type MEF with UV, etoposide or staurosporine resulted in cytochrome c release from the mitochondria (Fig. 7 A, +/+). However, cytochrome c release from mitochondria of calreticulin-deficient cells, after treatment with UV, etoposide or staurosporine, was reduced (Fig. 7 A, −/−). During apoptosis, cytochrome c release from mitochondria activates caspase 9, which then cleaves and activates procaspase 3 (Srinivasula et al. 1998). To confirm the differences we had found in cytochrome c release from mitochondria, we measured rates of DEVD cleavage in cytosolic extracts, using the fluorometric substrate DEVD-AFC. We treated wild-type and calreticulin-deficient MEF with UV, etoposide or staurosporine, and then we prepared cytosolic extracts from the cells. No caspase activity was detected in untreated cells (Fig. 7 B, untreated). DEVD cleavage (caspase) activity was high in wild-type MEF treated with UV or etoposide (Fig. 7 B, wt), but was reduced in the calreticulin-deficient cells (Fig. 7 B, crt −/−). There was no detectable DEVDase activity in the staurosporine-treated crt −/− (Fig. 7 B). However, there was significant release of cytochrome c from mitochondria of crt −/− treated with staurosporine (Fig. 7 A). This apparent discrepancy is not surprising since mitochondrial cytochrome c release does not always depend on caspase activity in many apoptotic systems (Xiang et al. 1996; Yucheng et al. 2000).
Figure 7.
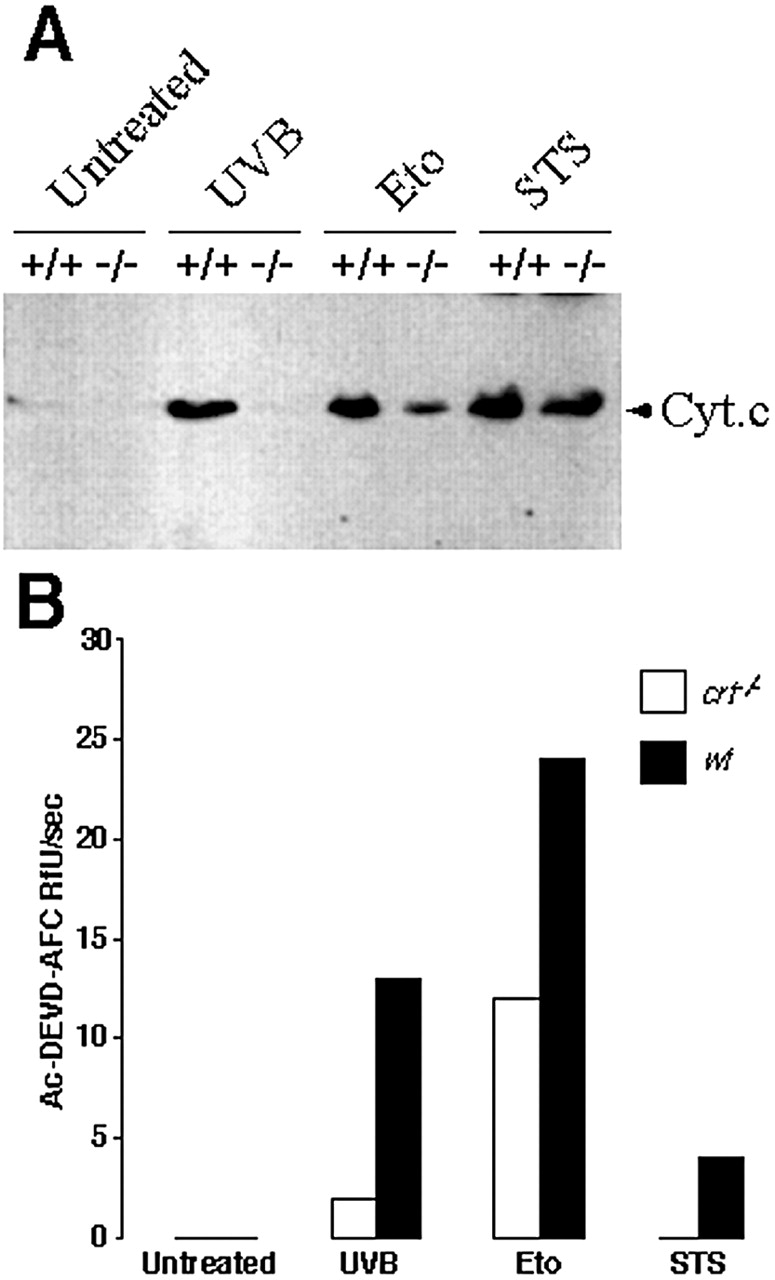
DEVD cleavage activity and cytochrome c release from mitochondria in calreticulin-deficient cells. In A, wild-type (+/+) and calreticulin-deficient (−/−) MEF were treated with UVB for 1 min, 10 μM etoposide, or 10 nM staurosporine (STS). 8 h after the treatment cytosolic extracts were prepared devoid of mitochondria, separated by SDS-PAGE, and immunoblotted with anti-cytochrome c antibodies (Bossy-Wetzel and Green 1999). The position of cytochrome c (Cyt. c) is indicated by the arrow. In B, cytosolic extracts from wild-type (wt) and calreticulin-deficient (crt−/−) MEF treated with UVB for 1 min, 10 μM etoposide, or 10 nM staurosporine (STS) were prepared and tested for DEVD-specific cleavage activity as described in Materials and Methods.
Discussion
In this study, we found that overexpression of calreticulin increased cell sensitivity to both thapsigargin-induced apoptosis and staurosporine-induced apoptosis. However, the overexpression of calnexin, another Ca2+-binding chaperone of the ER, had no significant effect on drug-induced apoptosis. Conversely, we found that cells lacking calreticulin were considerably resistant to drug-induced apoptosis. When we investigated Ca2+ homeostasis in cells overexpressing calreticulin, we found that bradykinin-dependent elevation of [Ca2+]c was increased. In contrast, in calreticulin-deficient cells bradykinin-dependent elevation of [Ca2+]c was inhibited (Mesaeli et al. 1999). We conclude that ER luminal environment in general and calreticulin in particular play a significant role in determining cell sensitivity to drug-induced apoptosis. This is likely due to calreticulin function as a regulator of Ca2+ homeostasis. Most importantly, we show that changes in the expression of calreticulin, which is resident in the lumen of the ER, affect the mitochondrial cytochrome c release/caspase signaling pathway of apoptosis.
Calreticulin is a ubiquitous Ca2+ binding protein, which is located in the ER and which has been implicated in such diverse functions as regulation of intracellular Ca2+ homeostasis, chaperone activity, gene expression and cell adhesion (Michalak et al. 1999). Given the diversity of its activities, there are many routes via which calreticulin might affect cell sensitivity to drug-induced apoptosis. In this study, we have shown that the lectin-like activity of calreticulin is probably not involved. The ER proteins calreticulin and calnexin share many chaperone functions; both are lectin-like chaperones which preferentially bind monoglucosylated oligosaccharides (Bergeron et al. 1994; Spiro et al. 1996; Helenius et al. 1997; Vassilakos et al. 1998) and misfolded proteins (Ihara et al. 1999; Saito et al. 1999). We found that altered expression of calnexin had no significant effect on cell sensitivity to apoptosis. It is, therefore, unlikely that calreticulin's chaperone activity shared with calnexin is involved in the observed changes in cell sensitivity to apoptosis. However, it cannot be ruled out that calreticulin chaperone activity towards a calreticulin-specific substrate may also play a role in modulation of cell sensitivity to apoptosis.
Calreticulin is a Ca2+ binding protein that modulates intracellular Ca2+ homeostasis (Liu et al. 1994; Bastianutto et al. 1995; Camacho and Lechleiter 1995; Mery et al. 1996; Coppolino et al. 1997; Fasolato et al. 1998; John et al. 1998; Mesaeli et al. 1999) and Ca2+ plays an important role in apoptosis (Preston and Berlin 1992; Baffy et al. 1993; Lam et al. 1994; Distelhorst et al. 1996; Marin et al. 1996; McConkey et al. 1996; Reynolds and Eastman 1996). Differential expression of calreticulin affects Ca2+ storage capacity of the ER, and most importantly, it modulates Ca2+ release from the ER (Liu et al. 1994; Bastianutto et al. 1995; Camacho and Lechleiter 1995; Mery et al. 1996; Coppolino et al. 1997; Fasolato et al. 1998; John et al. 1998; Mesaeli et al. 1999). For example we found that overexpression of calreticulin in the Tet-On–inducible system led to increased Ca2+ storage in the ER, and to increased elevation of [Ca2+]c in response to bradykinin. Furthermore, we have previously found that calreticulin-deficient MEF do not have a measurable InsP3-dependent Ca2+ release when stimulated with bradykinin (Mesaeli et al. 1999), indicating that calreticulin plays a role in modulating InsP3 receptor pathway. Camacho's group has proposed that calreticulin may modulate the function of the InsP3 receptor and/or SERCA (Camacho and Lechleiter 1995). Recently, John et al. 1998 reported that calreticulin may interact with SERCA2b, resulting in a lower transport capacity for the Ca2+-ATPase. We have shown that changes in Ca2+ homeostasis, particularly agonist-dependent Ca2+ release from the ER of calreticulin-deficient cells, correlate well with changes in cell sensitivity to staurosporine-induced apoptosis.
One most important implication of this current work is that changes in the lumen of the ER modify the mitochondrial cytochrome c/caspase apoptosis signaling pathway. For example, we found that, when cells are induced to undergo apoptosis, cytochrome c release from mitochondria and cytosolic caspase activity are both lower in calreticulin-deficient cells than in wild-type cells. Rizzuto et al. 1998 have shown that there are numerous close contacts between the ER and mitochondria, and that opening of the InsP3/Ca2+ channel in the ER affects Ca2+ homeostasis in mitochondria. Close proximity between ER and mitochondria may play a role in the regulation of Ca2+ signaling (Rizzuto et al. 1998) and cell sensitivity to apoptosis.
There is a growing body of evidence that the ER may play an important role in the apoptotic signaling processes, which lead to downstream activation of caspases and other proteases. For example, under normal conditions, Bcl-2 and Bcl-XL colocalize to the mitochondria and ER (Zhu et al. 1996; Ng et al. 1997; Ng and Shore 1998). Procaspase 8 and Bap31, a newly identified component in apoptotic signaling, are also localized to the ER (Ng et al. 1997; Ng and Shore 1998) indicating that Bap31 could influence Bcl-2, Bcl-XL and procaspase 8 (Ng et al. 1997). Bcl-2 protects cells from apoptosis and it is localized to ER and outer mitochondrial membrane (Lithgow et al. 1994; Zhu et al. 1996) suggesting that the protein may affect apoptosis by interfering with a specific function of the ER. Indeed, overexpression of Bcl-2 in either HeLa cells or mouse lymphoma A20 cells protects from apoptosis and results in reduced agonist-stimulated Ca2+ release from the thapsigargin-sensitive (ER) Ca2+ stores (Pinton et al. 2000; Foyouzi-Youseffi et al. 2000). In this study, we show that calreticulin-deficient cells are also resistant to apoptosis and, similarly to Bcl-2 overexpressers (Pinton et al. 2000), they have reduced agonist-stimulated Ca2+ release (Mesaeli et al. 1999). However, there is no change in the level of Bcl-2 in neither calreticulin deficient cells nor in cells overexpressing the protein (data not shown). Therefore, changes in cell sensitivity to apoptosis reported here are likely due to the modulation of expression of calreticulin and its effect on Ca2+ homeostasis. Furthermore, ER-associated InsP3 receptor/Ca2+ channel was recently identified as a new target for caspase 3 (Hirota et al. 1999). InsP3 receptor-mediated cytosolic Ca2+ spikes may also affect mitochondrial permeability during an apoptotic event (Szalai et al. 1999). Calreticulin expression is upregulated under these ER stress conditions (Llewellyn et al. 1996; Nguyen et al. 1996; Waser et al. 1997). It is not surprising, therefore, that changes in the ER luminal environment, described in this study, have profound effects on cell sensitivity to apoptosis. Our work suggests that calreticulin, and perhaps other proteins resident in the lumen of the ER, contributes to the regulation of cell sensitivity to apoptosis. Therefore, changes in ER-dependent Ca2+ homeostasis, and changes in expression of Ca2+ binding proteins in the ER, may confer sensitivity to apoptosis caused by diverse stimuli. Yuan's group have recently reported that caspase-12 is localized to the ER membrane and it is activated specifically by ER stress including disruption of Ca2+ homeostasis by thapsigargin or ionophore A23187 (Nakagawa et al. 2000). Therefore, it is also possible that stress in the ER due to, for example, differential expression of calreticulin can result in activation of apoptotic pathway(s) independent of mitochondria (Nakagawa et al. 2000).
Apoptosis plays an essential role during development (Jacobson et al. 1997). In many organs, cells are overproduced and then reduced by apoptosis to adjust their number. In vertebrate nervous system, for example, both neurons and oligodendrocytes are generated in excess, and up to half or more are eliminated by apoptosis (Oppenheim 1991; Barres et al. 1992). During the formation of embryonic eye many of central retina and lens vesicle cells are also eliminated by apoptosis (Ganan et al. 1996; Zou and Niswander 1996). The developing limb is a classical model where apoptosis has been acknowledged as a fundamental morphological mechanism responsible for the elimination of the cells between developing digits (Ganan et al. 1996; Zou and Niswander 1996). In this respect, as shown in Fig. 1, the calreticulin gene is highly activated in mouse embryo interdigital cells, in the central retina and in lens vesicle suggesting that changes in the expression of calreticulin, an ER luminal protein, may play a role in apoptosis during embryogenesis. Future studies are required to extend these preliminary observations and established correlation between calreticulin expression and periods of apoptosis during embryogenesis. An important point is that calreticulin is not a signal to initiate apoptosis, instead overexpression of the protein and/or changes in the ER luminal environment may alter cell sensitivity to a drug-induced apoptosis. This suggests that apoptosis may depend on both the presence of external apoptosis-activating signals, and, as shown in this study, on an internal factor represented by the ER. Alteration in the organization of ER and ER luminal environment are not initiators of apoptosis but they may be responsible for the modulation of cell susceptibility to apoptosis induced by the external stimuli.
Acknowledgments
We thank Ms. J. Chen for superb technical assistance. We thank D. Williams for calnexin cDNA.
This work was supported by grants (to M. Michalak, M. Opas, and R.C. Bleackley) from the Canadian Institutes of Health Research, from the Heart and Stroke Foundations of Alberta (to M. Michalak) and Ontario (to M. Opas), and from the National Institutes of Health (to D.R. Green). K. Nakamura is a fellow of the Alberta Heritage Foundation for Medical Research. R.C. Bleackley is a Canadian Institutes of Health Research Distinguished Scientist and a Medical Scientist of the Alberta Heritage Foundation for Medical Research. M. Michalak is a Canadian Institutes of Health Research Senior Scientist and a Medical Scientist of the Alberta Heritage Foundation for Medical Research.
Footnotes
Abbreviations used in this paper: Dox, doxycycline; Dex, dexamethasone; InsP3, inositol 1,4,5-trisphosphate; MEF, mouse embryonic fibroblast.
References
- Baffy G., Miyashita T., Williamson J.R., Reed J.C. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J. Biol. Chem. 1993;268:6511–6519. [PubMed] [Google Scholar]
- Barres B.A., Hart I.K., Coles H.S., Burne J.F., Voyvodic J.T., Richardson W.D., Raff M.C. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Bastianutto C., Clementi E., Codazzi F., Podini P., De Giorgi F., Rizzuto R., Meldolesi J., Pozzan T. Overexpression of calreticulin increases the Ca2+ capacity of rapidly exchanging Ca2+ stores and reveals aspects of their luminal microenvironment and function. J. Cell Biol. 1995;130:847–855. doi: 10.1083/jcb.130.4.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron J.J.M., Brenner M.B., Thomas D.Y., Williams D.B. Calnexina membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 1994;19:124–128. doi: 10.1016/0968-0004(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Bertrand R., Solary E., O'Connor P., Kohn K.W., Pommier Y. Induction of a common pathway of apoptosis by staurosporine. Exp. Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Green D.R. Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J. Biol. Chem. 1999;274:17484–17490. doi: 10.1074/jbc.274.25.17484. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer D.D., Green D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Camacho P., Lechleiter J.D. Calreticulin inhibits repetitive intracellular Ca2+ waves. Cell. 1995;82:765–771. doi: 10.1016/0092-8674(95)90473-5. [DOI] [PubMed] [Google Scholar]
- Conzen S.D., Cole C.N. The three transforming regions of SV40 T antigen are required for immortalization of primary mouse embryo fibroblasts. Oncogene. 1995;11:2295–2302. [PubMed] [Google Scholar]
- Coppolino M.G., Woodside M.J., Demaurex N., Grinstein S., St R., Arnaud, Dedhar S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature. 1997;386:843–847. doi: 10.1038/386843a0. [DOI] [PubMed] [Google Scholar]
- Distelhorst C.W., Lam M., McCormick T.S. Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene. 1996;12:2051–2055. [PubMed] [Google Scholar]
- Fasolato C., Pizzo P., Pozzan T. Delayed activation of the store-operated calcium current induced by calreticulin overexpression in RBL-1 cells. Mol. Biol. Cell. 1998;9:1513–1522. doi: 10.1091/mbc.9.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyouzi-Youseffi R., Arnaudeau S., Borner C., Kelley W.L., Tschopp J., Lew D.P., Demaurex N., Krause K.-H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 2000;97:5723–5728. doi: 10.1073/pnas.97.11.5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganan Y., Macias D., Duterque-Coquillaud M., Ros M.A., Hurle J.M. Role of TGF beta s and BMPs as signals controlling the position of the digits and the areas of interdigital cell death in the developing chick limb autopod. Development. 1996;122:2349–2357. doi: 10.1242/dev.122.8.2349. [DOI] [PubMed] [Google Scholar]
- Hammond C., Helenius A. Quality control in the secretory pathway. Curr. Opin. Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Helenius A., Trombetta E.S., Hebert D.N., Simons J.F. Calnexin, calreticulin and the folding of glycoproteins. Trends Cell Biol. 1997;7:193–200. doi: 10.1016/S0962-8924(97)01032-5. [DOI] [PubMed] [Google Scholar]
- Hirota J., Furuichi T., Mikoshiba K. Inositol 1,4,5-trisphosphate receptor type 1 is a substrate for caspase-3 and is cleaved during apoptosis in a caspase-3-dependent manner. J. Biol. Chem. 1999;274:34433–34437. doi: 10.1074/jbc.274.48.34433. [DOI] [PubMed] [Google Scholar]
- Ihara Y., Cohen-Doyle M.F., Saito Y., Williams D.B. Calnexin discriminates between protein conformational states and functions as a molecular chaperone in vitro . Mol. Cell. 1999;4:331–341. doi: 10.1016/s1097-2765(00)80335-4. [DOI] [PubMed] [Google Scholar]
- Jacobson W.D., Burne J.F., Raff M.C. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:1899–1910. doi: 10.1002/j.1460-2075.1994.tb06459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson W.D., Weil M., Raff M.C. Programmed cell death and animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Jayaraman T., Marks A.R. T cells deficient in inositol 1,4,5-trisphosphate receptor are resistant to apoptosis. Mol. Cell. Biol. 1997;17:3005–3012. doi: 10.1128/mcb.17.6.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John L.M., Lechleiter J.D., Camacho P. Differential modulation of SERCA2 isoforms by calreticulin. J. Cell Biol. 1998;142:963–973. doi: 10.1083/jcb.142.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause K.-H., Michalak M. Calreticulin. Cell. 1997;88:439–443. doi: 10.1016/s0092-8674(00)81884-x. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lam M., Dubyak G., Distelhorst C.W. Effect of glucocorticosteroid treatment on intracellular calcium homeostasis in mouse lymphoma cells. Mol. Endocrinol. 1993;7:686–693. doi: 10.1210/mend.7.5.8316252. [DOI] [PubMed] [Google Scholar]
- Lam M., Dubyak G., Chen L., Nunez G., Miesfeld R.L., Distelhorst C.W. Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl. Acad. Sci. USA. 1994;91:6569–6573. doi: 10.1073/pnas.91.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lithgow T., van Driel R., Bertram J.F., Strasser A. The protein product of the oncogene bcl-2 is a component of the nuclear envelope, the endoplasmic reticulum, and the outer mitochondrial membrane. Cell Growth. Differ. 1994;5:411–417. [PubMed] [Google Scholar]
- Liu N., Fine R.E., Simons E., Johnson R.J. Decreasing calreticulin expression lowers the Ca2+ response to bradykinin and increases sensitivity to ionomycin in NG-108-15 cells. J. Biol. Chem. 1994;269:28635–28639. [PubMed] [Google Scholar]
- Llewellyn D.H., Kendall J.M., Sheikh F.N., Campbell A.K. Induction of calreticulin expression in HeLa cells by depletion of the endoplasmic reticulum Ca2+ store and inhibition of N-linked glycosylation. Biochem. J. 1996;318:555–560. doi: 10.1042/bj3180555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin M.C., Fernandez A., Bick R.J., Brisbay S., Buja L.M., Snuggs M., McConkey D.J., von Eschenbach A.C., Keating M.J., McDonnell T.J. Apoptosis suppression by bcl-2 is correlated with the regulation of nuclear and cytosolic Ca2+ . Oncogene. 1996;12:2259–2266. [PubMed] [Google Scholar]
- McConkey D.J., Zhivotovsky B., Orrenius S. Apoptosis—molecular mechanisms and biomedical implications. Mol. Aspects Med. 1996;17:1–110. doi: 10.1016/0098-2997(95)00006-2. [DOI] [PubMed] [Google Scholar]
- Meldolesi J., Pozzan T. The endoplasmic reticulum Ca2+ storea view from the lumen. Trends Biochem Sci. 1998;23:10–14. doi: 10.1016/s0968-0004(97)01143-2. [DOI] [PubMed] [Google Scholar]
- Mery L., Mesaeli N., Michalak M., Opas M., Lew D.P., Krause K.-H. Overexpression of calreticulin increases intracellular Ca2+ storage and decreases store-operated Ca2+ influx. J. Biol. Chem. 1996;271:9332–9339. doi: 10.1074/jbc.271.16.9332. [DOI] [PubMed] [Google Scholar]
- Mesaeli N., Nakamura K., Zvaritch E., Dickie P., Dziak E., Krause K.-H., Opas M., MacLennan D.H., Michalak M. Calreticulin is essential for cardiac development. J. Cell Biol. 1999;144:857–868. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak M. Calreticulin. R.D. Landes Company; Austin: 1996. [Google Scholar]
- Michalak M., Burns K., Andrin C., Mesaeli N., Jass G.H., Busaan J.L., Opas M. Endoplasmic reticulum form of calreticulin modulates glucocorticoid-sensitive gene expression. J. Biol. Chem. 1996;271:29436–29445. doi: 10.1074/jbc.271.46.29436. [DOI] [PubMed] [Google Scholar]
- Michalak M., Corbett E.F., Mesaeli N., Nakamura K., Opas M. Calreticulinone protein, one gene, many functions. Biochem. J. 1999;344:281–292. [PMC free article] [PubMed] [Google Scholar]
- Milner R.E., Baksh S., Shemanko C., Carpenter M.R., Smillie L., Vance J.E., Opas M., Michalak M. Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth muscle sarcoplasmic reticulum and liver endoplasmic reticulum. J. Biol. Chem. 1991;266:7155–7165. [PubMed] [Google Scholar]
- Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A., Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Nakamura K., Endo F., Ueno T., Awata H., Tanoue A., Matsuda I. Excess copper and ceruloplasmin biosynthesis in long-term cultured hepatocytes from Long-Evans Cinnamon (LEC) rats, a model of Wilson disease. J. Biol. Chem. 1995;270:7656–7660. doi: 10.1074/jbc.270.13.7656. [DOI] [PubMed] [Google Scholar]
- Ng F.W., Shore G.C. Bcl-XL cooperatively associates with the Bap31 complex in the endoplasmic reticulum, dependent on procaspase-8 and Ced-4 adaptor. J. Biol. Chem. 1998;273:3140–3143. doi: 10.1074/jbc.273.6.3140. [DOI] [PubMed] [Google Scholar]
- Ng F.W., Nguyen M., Kwan T., Branton P.E., Nicholson D.W., Cromlish J.A., Shore G.C. p28 Bap31, a Bcl-2/Bcl-XL- and procaspase-8–associated protein in the endoplasmic reticulum. J. Cell Biol. 1997;139:327–338. doi: 10.1083/jcb.139.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.O., Capra J.D., Sontheimer R.D. Calreticulin is transcriptionally upregulated by heat shock, calcium and heavy metals. Mol. Immunol. 1996;33:379–386. doi: 10.1016/0161-5890(95)00149-2. [DOI] [PubMed] [Google Scholar]
- Opas M., Dziak E., Fliegel L., Michalak M. Regulation of expression and intracellular distribution of calreticulin, a major calcium binding protein of nonmuscle cells. J. Cell. Physiol. 1991;149:160–171. doi: 10.1002/jcp.1041490120. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pinton P., Ferrari D., Magalhaes P., Schulze-Osthoff K., Di Virgilio F., Pozzan T., Rizzuto R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2–overexpressing cells. J. Cell Biol. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzan T., Rizzuto R., Volpe P., Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Preston S.F., Berlin R.D. An intracellular calcium store regulates protein synthesis in HeLa cells, but it is not the hormone-sensitive store. Cell Calcium. 1992;13:303–312. doi: 10.1016/0143-4160(92)90065-z. [DOI] [PubMed] [Google Scholar]
- Raff M.C., Barres B.A., Burne J.F., Coles H.S., Ishizaki Y., Jacobson M.D. Programmed cell death and the control of cell survivallessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Reynolds J.E., Eastman A. Intracellular calcium stores are not required for Bcl-2-mediated protection from apoptosis. J. Biol. Chem. 1996;271:27739–27743. doi: 10.1074/jbc.271.44.27739. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Pinton P., Carrington W., Fay F.S., Fogarty K.E., Lifshitz L.M., Tuft R.A., Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Saito Y., Ihara Y., Leach M.R., Cohen-Doyle M.F., Williams D.B. Calreticulin functions in vitro as a molecular chaperone for both glycosylated and non-glycosylated proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:6718–6729. doi: 10.1093/emboj/18.23.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro R.G., Zhu Q., Bhoyroo V., Soling H.D. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver Golgi. J. Biol. Chem. 1996;271:11588–11594. doi: 10.1074/jbc.271.19.11588. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Ahmad M., Fernandes-Alnemri T., Alnemri E.S. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol. Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Szalai G., Krishnamurthy R., Hajnoczky G. Apoptosis driven by IP3-linked mitochondrial calcium signals. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O., Cullen P.J., Drobak B.K., Hanley M.R., Dawson A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheetsprocedure and some applications. Proc. Natl. Acad. Sci. USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilakos A., Michalak M., Lehrman M.A., Williams D.B. Oligosaccharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry. 1998;37:3480–3490. doi: 10.1021/bi972465g. [DOI] [PubMed] [Google Scholar]
- Waser M., Mesaeli N., Spencer C., Michalak M. Regulation of calreticulin gene expression by calcium. J. Cell Biol. 1997;138:547–557. doi: 10.1083/jcb.138.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., Chao D.T., Korsmeyer S.J. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA. 1996;93:14559–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yucheng K.L., Shelton J.M., Richardson J.A., Spencer E., Chen Z.J., Wang X., Williams R.S. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- Zapun A., Darby N.J., Tessier D.C., Michalak M., Bergeron J.J.M., Thomas D.Y. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J. Biol. Chem. 1998;273:6009–6012. doi: 10.1074/jbc.273.11.6009. [DOI] [PubMed] [Google Scholar]
- Zhu W., Cowie A., Wasfy G.W., Penn L.Z., Leber B., Andrews D.W. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- Zou H., Niswander L. Requirement for BMP signaling in interdigital apoptosis and scale formation. Science. 1996;272:738–741. doi: 10.1126/science.272.5262.738. [DOI] [PubMed] [Google Scholar]