


Abstract
APOBEC3G (A3G), a host protein that inhibits HIV-1 reverse transcription and replication in the absence of Vif, displays cytidine deaminase and single-stranded (ss) nucleic acid binding activities. HIV-1 nucleocapsid protein (NC) also binds nucleic acids and has a unique property, nucleic acid chaperone activity, which is crucial for efficient reverse transcription. Here we report the interplay between A3G, NC and reverse transcriptase (RT) and the effect of highly purified A3G on individual reactions that occur during reverse transcription. We find that A3G did not affect the kinetics of NC-mediated annealing reactions, nor did it inhibit RNase H cleavage. In sharp contrast, A3G significantly inhibited all RT-catalyzed DNA elongation reactions with or without NC. In the case of (−) strong-stop DNA synthesis, the inhibition was independent of A3G's catalytic activity. Fluorescence anisotropy and single molecule DNA stretching analyses indicated that NC has a higher nucleic acid binding affinity than A3G, but more importantly, displays faster association/disassociation kinetics. RT binds to ssDNA with a much lower affinity than either NC or A3G. These data support a novel mechanism for deaminase-independent inhibition of reverse transcription that is determined by critical differences in the nucleic acid binding properties of A3G, NC and RT.
INTRODUCTION
Human APOBEC3G (A3G) is a host cytidine deaminase, which was first identified by Sheehy et al. (1) as the cellular factor that blocks HIV-1 replication in the absence of the viral Vif protein. Cellular expression of A3G results in its incorporation into vif-deficient HIV-1 particles, whereas its presence in WT virions is dramatically reduced by Vif-induced degradation via the ubiquitination-proteosome pathway (2).
Initial studies suggested that the deamination activity of A3G contributes to its antiviral activity and is associated with G to A hypermutation (3–6). However, more recent results indicate that a deaminase-independent mechanism might also be involved in A3G's antiviral activity: (i) Deamination activity is not absolutely correlated with antiviral activity against HIV-1 (7–15); (ii) Several reports (though not all) indicate that hepatitis B virus may be inhibited by A3G without significant detection of G to A hypermutation (16–18) and (iii) Other APOBEC proteins block replication of mouse mammary tumor virus (19) and several retrotransposons (14,20–24) in the absence of editing activity.
Prior to the discovery of A3G, it was already known that Δvif viruses produced in ‘nonpermissive’ cells (PBMCs or certain T-cell lines, e.g. H9) were 100- to 1000-fold less infectious than WT (25–27) and were deficient in their ability to complete reverse transcription (28–31). Moreover, similar results were obtained in endogenous reverse transcription assays (30,32,33). More recent analysis of HIV-1-infected cells expressing A3G or A3F has confirmed the initial observations (5,10,13,15,34–38).
Efficient and specific reverse transcription depends on the viral nucleocapsid protein (NC), which functions as a nucleic acid chaperone (39–43). This means that NC can catalyze nucleic acid conformational rearrangements that lead to the most thermodynamically stable structures (44). Like NC, A3G has two zinc finger domains and binds nucleic acids (45). However, whereas A3G has a strong preference for binding single-stranded (ss) nucleic acids (6,12), NC binds ss or double-stranded (ds) RNA and DNA (46) [for further information on NC's nucleic acid binding properties, see Ref. (47)]. The two proteins also differ in their effects on virus replication: NC acts as a positive factor, whereas A3G is an inhibitor in the absence of Vif.
We previously reported the successful preparation of highly purified, catalytically active A3G expressed in a baculovirus system and demonstrated that the availability of a pure protein (without contamination by other proteins, either host or viral) was invaluable for rigorous analysis of the biochemical properties of A3G (12). In the course of this study, we unexpectedly found that A3G does not interfere with NC binding to ssRNA (and vice versa) (12). This suggested that inhibition of reverse transcription by A3G is likely to be unrelated to an effect on NC chaperone function.
To test this hypothesis and to probe the mechanism that might be involved, we took advantage of defined biochemical assay systems that we have developed over the years for studies on viral DNA synthesis (48–51). Thus, using our highly purified A3G as well as purified NC and RT, we investigated the effect of A3G on a series of reconstituted reactions that occur during reverse transcription. This allowed us to perform an independent analysis of individual steps in the pathway, which is not possible in cell-based systems. We found that A3G inhibited all reverse transcriptase (RT)-catalyzed DNA elongation reactions, but not RNase H activity or NC's ability to promote annealing. These observations could be explained by critical differences in the binding properties of NC, A3G and RT, as measured by single-molecule DNA stretching and fluorescence anisotropy (FA). Our findings are unique and provide strong support for a novel mechanism that could account for the observed deaminase-independent A3G-mediated antiviral activity.
MATERIALS AND METHODS
Materials
Purified 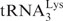 from human placenta was obtained from Bio S&T (Lachine, Quebec, Canada). DNA and RNA oligonucleotides were purchased from Lofstrand (Gaithersburg, MD), Integrated DNA Technologies (Coralville, IA), Oligos Etc., Inc. (Wilsonville, OR). [γ-32P]ATP (3000 Ci/mmol) and [α-32P]dCTP (6000 Ci/mmol) were purchased from GE Healthcare (Piscataway, NJ). HIV-1 RT was obtained from Worthington Biochemical Corp. (Lakewood, NJ). Calf intestinal phosphatase, T4 polynucleotide kinase, and Vent DNA polymerase were obtained from New England Biolabs (Beverly, MA). SUPERaseIn, an RNase inhibitor, was purchased from Ambion, Inc. (Austin, TX). Recombinant wild-type HIV-1 NC (55-amino-acid form) was a generous gift from Dr Robert Gorelick and was prepared as described previously (52,53). Recombinant enzymatically active A3G and the deaminase-deficient A3G mutant (C291S) were expressed in a baculovirus expression system and purified as previously described (12). A3G preparations were confirmed to be free from contamination with RNases (data not shown) and no RNA degradation was apparent in any of the experiments (e.g. see Figure 2A).
from human placenta was obtained from Bio S&T (Lachine, Quebec, Canada). DNA and RNA oligonucleotides were purchased from Lofstrand (Gaithersburg, MD), Integrated DNA Technologies (Coralville, IA), Oligos Etc., Inc. (Wilsonville, OR). [γ-32P]ATP (3000 Ci/mmol) and [α-32P]dCTP (6000 Ci/mmol) were purchased from GE Healthcare (Piscataway, NJ). HIV-1 RT was obtained from Worthington Biochemical Corp. (Lakewood, NJ). Calf intestinal phosphatase, T4 polynucleotide kinase, and Vent DNA polymerase were obtained from New England Biolabs (Beverly, MA). SUPERaseIn, an RNase inhibitor, was purchased from Ambion, Inc. (Austin, TX). Recombinant wild-type HIV-1 NC (55-amino-acid form) was a generous gift from Dr Robert Gorelick and was prepared as described previously (52,53). Recombinant enzymatically active A3G and the deaminase-deficient A3G mutant (C291S) were expressed in a baculovirus expression system and purified as previously described (12). A3G preparations were confirmed to be free from contamination with RNases (data not shown) and no RNA degradation was apparent in any of the experiments (e.g. see Figure 2A).
Figure 2.

Effect of A3G on -primed (−) SSDNA synthesis. (A) Time course of annealing to RNA UL244. Reactions were performed in the absence or presence of NC and A3G, as indicated by the headings at the top of the gel. The positions of the RNA UL244 template and the annealed RNA duplex are shown on the right. (B) The percentage of annealed product was calculated by dividing the amount of annealed RNA by the sum of annealed plus unannealed RNA, multiplied by 100. Symbols: no NC/no A3G (filled circles); +NC/no A3G (open squares); +NC/+hdA3G (open circles); and +NC/+A3G (open triangles). (C) A /RNA 244 complex was extended by HIV-1 RT in the absence (lane 1) or presence of hdA3G (lanes 2–4) or A3G (lanes 5–7). The positions of (−) SSDNA and initial pause products at bases +1, +3 and +5 are shown on the right. A3G concentrations: lane 1, 0 nM; lanes 2 and 5, 20 nM; lanes 3 and 6, 40 nM; lanes 4 and 7, 80 nM.
Methods
Plasmid construction
All plasmid sequences were derived from the HIV-1 pNL4-3 clone (54). Plasmid pUL (viral insert from nt 566 to 1419) was constructed from the previously described pRUG plasmid (51).
Preparation of RNA
The DNA templates for in vitro RNA transcription were derived from plasmid pRUG [for transactivation response element (TAR) RNA, nucleotide(s) (nt) 1–59] or pUL (for RNA UL244, nt 113–244). DNA fragments containing both the T7 promoter and the DNA sequence equivalent to the desired vRNA were amplified by PCR using Vent DNA polymerase and the following primers: forward primer (5′-ccaatgcttaatcagtgaggc), located at the start of the amp gene in pUL; reverse primers, 5′-gtcctgcgtcgagagatc (RNA UL244) and 5′-gggttccctagttagccaga (TAR RNA). The DNA fragments were gel-purified and transcribed using an Ambion MEGAscript kit (Ambion Inc., Austin, TX). Gel-purified RNAs were dephosphorylated by calf intestinal phosphatase and were then labeled at their 5′ ends with [γ-32P]ATP, using T4 polynucleotide kinase (55). Unincorporated nucleotides were removed by passing the reaction mixture through NucAway Spin Columns (Ambion). RNA 244 (51) and acceptor RNA 148 (49) were prepared as described.
Reverse transcription assays
Reaction components are given for 20 µl unit reactions, which were scaled up as needed. SUPERaseIn at a final concentration of 0.5 U/µl was added to all RNA-containing reaction mixtures. Incubation/preincubation was always at 37°C. Six percent native or denaturing polyacrylamide gels were used for annealing or extension assays, respectively, except as noted. Radioactivity was quantified by using a Typhoon PhosphorImager and ImageQuant software.
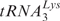 annealing to viral RNA (vRNA)
annealing to viral RNA (vRNA)
Reaction mixtures contained buffer (50 mM Tris–HCl (pH 8.0), 75 mM KCl, 0.1 mM MgCl2, 1 mM DTT), 0.2 pmol of purified human placental and 0.1 pmol of 5′ 32P-labeled RNA UL244 and were incubated in the absence or presence of A3G (80 nM) with or without HIV-1 NC (7 nt/NC, 0.2 µM). Prior to loading on the gel, the samples were treated with Proteinase K (0.5 mg/ml).
(−)Strong-Stop DNA [(−) SSDNA] synthesis
Template RNA 244 (0.1 pmol) and 0.1 pmol each of human or 32P-labeled D18 primer, [complementary to the 18-nt primer-binding site (PBS)] were heat-annealed as described (51). A3G or heat-denatured (hd)A3G was then added to buffer designated as ‘reaction buffer’ (50 mM Tris–HCl (pH 8.0), 75 mM KCl, 7 mM MgCl2, 1 mM DTT) and the mixture was preincubated for 5 min. Primer extension was initiated by adding HIV-1 RT (0.1 pmol) and 50 µM each of dATP, dTTP and dGTP plus 10 µCi of [α-32P]dCTP () or 50 µM each of all four dNTPs (D18).
Annealing and minus-strand transfer
Minus-strand annealing was performed as described previously (56) in the presence or absence of A3G (80 nM) with or without HIV-1 NC (3.5 nt/NC, 0.4 µM). For minus-strand transfer assays, reaction mixtures contained the components present in annealing reactions as well as HIV-1 RT (0.4 pmol), all four dNTPs (each at 50 µM), and 1 mM MgCl2.
Initiation of plus-strand DNA synthesis
The assay was performed using a modified version of a previous protocol (57). Briefly, the 15-nt polypurine tract (PPT) primer (0.2 pmol) was heat-annealed to a 35-nt minus-strand DNA template (0.1 pmol) and the hybrid was preincubated with or without A3G (80 nM) for 5 min in reaction buffer (see above). DNA synthesis was initiated by adding HIV-1 RT (0.4 pmol), all four dNTPs (each at 0.5 µM), and [α-32P]dCTP (20 µCi), which results in an internally labeled DNA product. Samples were loaded on a 15% denaturing gel. Since RNase H cleavage removes the annealed 15-nt PPT RNA, the DNA product is 20 nt.
Plus-strand transfer
The assay was performed as described previously (50) except that the final concentrations of 5′ 32P-labeled (+) SSDNA donor (50 nt) and minus-strand DNA acceptor (48 nt) were 5 nM, all four dNTPs were at 50 µM each, and HIV-1 RT was 20 nM. Reactions were incubated in the presence or absence of NC (3.5 nt/NC, 0.14 µM), with or without A3G (80 nM).
Single molecule DNA stretching
Purified bacteriophage lambda DNA (48 500 bp) was labeled on its 3′ ends with biotin and single DNA molecules were captured between two streptavidin-coated, 5 µm diameter polystyrene beads using a dual beam optical tweezers instrument (58,59). The 16.5 µm contour length DNA molecule was stretched in 100 nm steps using a piezoelectric flexure translation stage (Melles Griot, Carlsbad, CA) to reveal the DNA force-extension curve, as described previously (58). After stretching a single DNA molecule in DNA stretching buffer (10 mM HEPES, pH 7.5, 50 mM Na+, at 20°C), and verifying that a single molecule was present, the buffer solution was exchanged for a solution containing the same buffer with a fixed protein concentration. The protein exchange procedure was then repeated for different protein concentrations in order to determine the effect of protein on DNA stretching behavior. The transition width and hysteresis were analyzed as described previously (60) for 3 or more DNA molecules.
FA experiments
Equilibrium binding of HIV-1 NC, HIV-1 RT and human A3G to a 6-carboxyfluorescein (FAM)-labeled 20-nt ssDNA oligonucleotide (5′-FAM-JL587D, 5′FAM-CTTCTTTGGGAGTGAATTAG-3′) was examined using FA. The HPLC-purified oligonucleotide 5′-FAM-JL587D was purchased from TriLink Biotechnologies (San Diego, CA). FA measurements were performed on an Analyst AD plate reader system (Molecular Devices, Sunnyvale, CA) using Corning 3676 low-volume 384-well black non-binding surface polystyrene plates. Reaction mixtures contained 20 nM 5′-FAM-JL587D, varying concentrations of NC, RT or A3G, and buffer consisting of 50 mM Tris–HCl (pH 8.0), 75 mM KCl, 7 mM MgCl2 and 1 mM DTT. Samples were excited at 485 nm and the emission intensities at 530 nm from the parallel and perpendicular planes were measured. Apparent equilibrium dissociation constants (Kd) were determined by plotting the FA signal, A, as a function of protein concentration, C. The data were fit assuming 1:1 oligomer:protein binding using the expression (61,62):
where
is the fraction of oligonucleotides bound, D is the oligonucleotide strand concentration, and AB and AF are the anisotropy values of the fully bound and unbound oligonucleotides, respectively. R is the ratio of the fluorescence intensity of saturated bound oligonucleotide relative to free oligonucleotide, which accounts for changes in fluorescence intensity upon protein binding (46,47,61).
RESULTS
Effect of A3G on primer placement and (−) SSDNA synthesis
To determine the mechanism by which A3G inhibits HIV-1 DNA synthesis, we analyzed the effect of A3G on single steps in the reverse transcription pathway (Figure 1) (43). Annealing and DNA synthesis were assayed in a series of reconstituted model systems. Our goal was to determine whether A3G interfered with the nucleic acid chaperone activity of NC, the catalytic activity of RT, or both during these reactions. Note that in most of the assays, the concentration of A3G did not exceed 80 nM, since A3G precipitates at high concentrations (12).
Figure 1.

Schematic diagram of the events in reverse transcription. Step 1. Reverse transcription is initiated by a cellular tRNA primer (, in the case of HIV-1), following annealing of the 3′ 18 nt of the tRNA to the 18-nt PBS near the 5′ end of the genome. RT catalyzes synthesis of (−) SSDNA, which contains copies of the R sequence and the unique 5′ genomic sequence (U5). Step 2. As the primer is extended, the RNase H activity of RT degrades the genomic RNA sequences that have been reverse transcribed. Step 3. (−) SSDNA is transferred to the 3′ end of vRNA (minus-strand transfer). Step 4. Elongation of minus-strand DNA and RNase H degradation continue. Plus-strand synthesis is initiated by the 15-nt PPT immediately upstream of the unique 3′ genomic sequence (U3). Step 5. RT copies the u3, u5 and r regions in minus-strand DNA, as well as the 3′ 18 nt of the tRNA primer, thereby reconstituting the PBS. The product formed is termed (+) SSDNA. Step 6. RNase H removal of the tRNA and PPT primers from minus- and plus-strand DNAs, respectively. Step 7. Plus-strand transfer, facilitated by annealing of the complementary PBS sequences at the 3′ ends of (+) SSDNA and minus-strand DNA, is followed by circularization of the two DNA strands and displacement synthesis. Step 8. Minus- and plus-strand DNAs are elongated, resulting in a linear dsDNA with a long terminal repeat (LTR) at each end. vRNA is shown by an open rectangle and minus-and plus-strand DNAs are shown by black and gray rectangles, respectively. The tRNA primer is represented by a short open rectangle (3′ 18 nt of the tRNA) attached to a ‘clover-leaf’ (remaining tRNA bases). Minus- and plus-strand sequences are depicted in lower and upper case, respectively. The very short white rectangles represent fragments produced by RNase H cleavage of genomic RNA. Adapted from reference (43) with permission from Elsevier.
To evaluate the effect of A3G on the first step in reverse transcription (primer placement), we investigated the time course of NC-dependent annealing of human to the PBS in a short vRNA template (RNA UL244) (Figure 2A and B). In the absence of NC and A3G, no annealing was detected. As shown in Figure 2B, the rates of annealing in the presence of NC were similar for reactions with and without A3G and the end point values at 64 min were 70% and 66%, respectively. Addition of NC and hdA3G resulted in a slightly enhanced rate of annealing, but the end point value was very close to the values with and without native A3G. These results demonstrate that A3G did not interfere with NC-mediated formation of the vRNA–tRNA complex.
Primer placement is followed by extension of and synthesis of (−) SSDNA (Figure 1, steps 1 and 2). To measure extension alone, the tRNA was first heat annealed to the vRNA 244 template (51); the (−) SSDNA product formed by addition of RT was internally labeled (Figure 2C) (51). In the absence of A3G (lane 1) or in the presence of hdA3G (lanes 2–4), equivalent amounts of the 258-nt full-length product [(−) SSDNA attached to ] and pause products (including the initial +1, +3 and +5 DNAs) were detected. However, in the presence of increasing amounts of native A3G (20–80 nM) (lanes 5–7), the amounts of fully extended product and pause products were greatly reduced; with 80 nM A3G, only the +1 and +5 DNAs could be detected (lane 7). Furthermore, some of the pause products observed in the presence of A3G differed from those made in the absence of the inhibitor (compare lanes 2–4 with lanes 5 and 6). Taken together, these results indicate that native A3G strongly inhibited tRNA-primed (−) SSDNA synthesis.
To investigate the kinetics of (−) SSDNA synthesis in the presence of increasing amounts of A3G, we used 5′ 32P-labeled D18 in place of tRNA (Figure 3A). This allowed us to obtain quantitative data, which were plotted as the percent of (−) SSDNA in total DNA products [% (−) SSDNA] versus Time (Figure 3B). In accord with the tRNA experiment Figure 2C), A3G reduced (−) SSDNA synthesis in a dose-dependent manner (Figure 3A and B). For example, at 64 min, the relative amount of (−) SSDNA synthesized in the 80 nM reaction was decreased by 24-fold compared with the minus A3G value. In addition, as also shown in Figure 2C, we observed changes in the pausing pattern in A3G-containing reactions, which were accentuated with increasing concentrations of A3G (Figure 3A). Interestingly, mapping these sites on the RNA 244 template structure (51) indicated that the pause sites occurred near or within ss regions in the template (see Figure 3C, which was derived from the gel data in Figure S1). This is consistent with A3G's well-documented, strong preference for binding to ss nucleic acids (6,12).
Figure 3.

Effect of A3G on (−) SSDNA synthesis primed by D18. (A) Time course of (−) SSDNA synthesis in reactions containing 32P-labeled D18 and RNA 244 in the presence of increasing concentrations of A3G. Positions of (−) SSDNA and D18 are shown on the right. (B) Graph of percent (−) SSDNA formed plotted versus incubation time. The percentage of (−) SSDNA product was calculated by dividing the amount of (−) SSDNA by the total amount of DNA, multiplied by 100. Symbols: 0 nM (filled circles); 20 nM (open squares); 40 nM (open circles); and 80 nM A3G (open triangles). (C) Mapping of pause sites on the RNA 244 template in A3G-containing reactions was based on the data shown in Figure S1. The arrows point to the pause sites.
The data in Figures 2 and 3 demonstrate that A3G dramatically suppressed (−) SSDNA synthesis primed by either or D18. The results with D18 also suggest that the reduction of tRNA-primed (−) SSDNA synthesis by A3G was not due to an altered configuration of the tRNA/vRNA initiation complex. Interestingly, when 6-fold less RNA template was used, 20 nM of A3G could completely inhibit (−) SSDNA synthesis (data not shown). This indicates that A3G inhibition of (−) SSDNA synthesis is dependent on the ratio of A3G:RNA.
It was also of interest to determine whether deaminase activity is required for the A3G inhibitory effect on (−) SSDNA synthesis. To address this question, we used purified deaminase-deficient A3G C291S protein (12) and then compared the effect of WT A3G or C291S on D18-primed (−) SSDNA synthesis. As shown in Figure 4, the rates and extents of (−) SSDNA synthesis in the presence of WT or mutant A3G were virtually the same, in accord with the observation that WT and C291S proteins have similar Kd values for binding to ss nucleic acids (12). The data of Figure 4 are significant and demonstrate that the inhibitory effect of A3G on (−) SSDNA synthesis is independent of A3G deaminase activity in our assay system.
Figure 4.

A3G inhibition of (−) SSDNA synthesis in the absence of deaminase activity. Symbols: no A3G (filled circles); WT A3G (80 nM) (open squares) and A3G C291S (80 nM) (open triangles).
Effect of A3G on minus-strand transfer reactions
Synthesis of a full-length minus-strand copy of the vRNA genome is achieved by transfer of (−) SSDNA to the 3′ end of vRNA (‘acceptor RNA’) followed by RT-catalyzed extension of the annealed DNA (Figure 1, step 3). This process (minus-strand transfer) is facilitated by base pairing of the complementary repeat regions at the 3′ ends of the nucleic acid substrates and is dependent on RNase H cleavage of vRNA sequences annealed to (−) SSDNA (43).
In Figure 5A, we show the time course of RNase H cleavage with the heat-annealed 59-bp TAR RNA/DNA hybrid, which was incubated with and without NC, in the absence or presence of A3G. Reactions containing A3G appeared to have slightly faster cleavage rates. However, the overall cleavage pattern was the same under all four conditions. These results indicated that A3G does not interfere with RNase H cleavage and is consistent with A3G's limited ability to bind to an RNA–DNA hybrid (6,12).
Figure 5.

Effect of A3G on minus-strand transfer reactions. (A) Effect of A3G on the time course of RNase H cleavage in the absence or presence of NC. 32P-labeled TAR RNA (0.1 pmol) and TAR DNA (0.2 pmol) were heat annealed and the hybrid was incubated at 37°C in reaction buffer (see above) with 0.4 pmol HIV-1 RT with or without NC (7 nt/NC, 0.1 µM), with or without A3G (80 nM). Samples were loaded on a 15% denaturing gel. Positions of the major cleavage products are indicated on the right. (B) Time course of annealing of 32P-labeled DNA 128 to RNA 148 incubated in the absence or presence of A3G (80 nM) with or without NC (3.5 nt/NC, 0.4 µM). Symbols: no NC/+A3G (filled circles); +NC/no A3G (open squares); and +NC/+A3G (open circles). (C) Schematic diagram illustrating the minus-strand transfer assay system. The R homology is 94 nt; U5 and U3 are 34 and 54 nt, respectively. (D) Graph of percent transfer product plotted versus incubation time. To quantify the percentage of strand transfer, the amount of transfer product was divided by the total amount of DNA, multiplied by 100. Symbols: no NC/no A3G (filled circles); no NC/+A3G (open squares); +NC/no A3G (open circles); and +NC/+A3G (open triangles).
To assay the effect of A3G on minus-strand transfer, we first measured NC-mediated annealing using DNA 128 and acceptor RNA 148 (see schematic diagram in Figure 5C) in the absence or presence of A3G (Figure 5B). NC is required to transiently destabilize the complementary TAR RNA and DNA structures within the repeat region, before hybrid formation can occur (43). Interestingly, A3G had only a minimal effect on annealing in the presence of NC. The rates were fairly similar with and without A3G and there was only a small reduction (11%) in the extent of the reaction when A3G was added. In a control reaction, we found that the extent of annealing minus NC, plus A3G was ∼10% (data not shown), in accord with the value obtained in the absence of NC and A3G (63).
We also assayed minus-strand transfer with the complete system, which depends upon both annealing and RT-catalyzed elongation reactions (Figure 5C). The percent of total DNA present as the 182-nt strand transfer product (% Transfer Product) was quantified and plotted versus Time (Figure 5D). As we showed previously (49,64,65), NC significantly enhanced minus-strand transfer. However, when A3G was added, NC-mediated strand transfer was dramatically reduced (Figure 5D). Note that in the absence of NC, the amount of transfer product made was extremely small and little effect of A3G was observed.
Viewed collectively, the results of Figure 5 strongly suggest that A3G inhibited minus-strand transfer by blocking RT-catalyzed DNA elongation. In contrast, A3G did not significantly interfere with NC-facilitated annealing of (−) SSDNA to acceptor RNA. These findings parallel the results obtained for primer placement (Figure 2A and B) and tRNA-primed synthesis of (−) SSDNA (Figure 2C).
Effect of A3G on (+) SSDNA synthesis and plus-strand transfer
While elongation of minus-strand DNA is being completed, the PPT RNA primer initiates synthesis of a short DNA termed (+) SSDNA (Figure 1, steps 4 and 5) (43). To determine whether A3G interferes with this step, we used a simple oligonucleotide assay that was previously developed in our laboratory (48,57). The 15-nt PPT was heat-annealed to a 35-nt minus-strand DNA template and the hybrid was then incubated with RT, which catalyzed the extension and subsequent removal of the PPT. The amount of 20-nt DNA product formed in reactions with and without A3G was quantified and plotted (Figure 6A).
Figure 6.

Effect of A3G on PPT initiation and plus-strand transfer. (A) Time course of PPT-primed plus-strand DNA synthesis. The 15-nt PPT RNA was heat-annealed to a 35-nt minus-strand DNA template and was then extended by HIV-1 RT. The 20-nt DNA product was internally labeled with [α-32P]dCTP in the absence (filled circles) and presence (open squares) of A3G (80 nM). The amount of 20-nt DNA was plotted as Relative Extension (%) versus Time (min), where 100% represents the end point value for the ‘no A3G’ reaction. (B) Time course of plus-strand transfer. The percentage of 80-nt plus-strand DNA product was calculated as described in the legend to Figure 5D. Symbols: no NC/no A3G (filled circles); +NC/no A3G (open circles); and +NC/+A3G (open triangles).
The results demonstrated that A3G reduced both the rate and extent of (+) SSDNA synthesis by 2- to 3-fold. The effect of A3G on this reaction was lower than what we observed for (−) SSDNA synthesis (Figure 3A and B) or minus-strand transfer (Figure 5D). This is expected since long templates were used for those assays. By contrast, in the experiment shown in Figure 6A, only one or at most two molecules of A3G can bind to the available 20-nt ss region in the template (12,66). Nevertheless, it is clear from the data that A3G was also able to inhibit RNA-primed DNA-dependent DNA polymerization by RT.
The plus-strand transfer reaction, like minus-strand transfer, consists of two steps: (i) NC-mediated annealing of the complementary PBS sequences in (+) SSDNA and minus-strand acceptor DNA; and (ii) RT-catalyzed elongation of both the plus- and minus-strand DNAs to yield a double-stranded DNA product (Figure 1, steps 6–8) (43). In our assay, only the plus-strand 32P-labeled 80-nt DNA is detected. In the absence of A3G, NC stimulated plus-strand transfer by 3-fold (Figure 6B), as reported previously (50). When A3G was added, reduction of synthesis was observed beginning at 10–15 min (e.g. at 15 min, by ∼4-fold), whereas the extent of the reaction at 120 min was decreased by 2-fold. These results showed that A3G inhibited DNA-dependent DNA polymerization.
Taken together, the in vitro data demonstrate that A3G interfered with all of the elongation reactions catalyzed by RT. In contrast, A3G was shown to have virtually no inhibitory effect on NC-mediated annealing reactions.
Single molecule DNA stretching and FA-binding measurements
In an effort to understand these results, the nucleic acid binding properties of A3G, NC and RT were investigated using single molecule DNA stretching and FA.
When optical tweezers are used to stretch single DNA molecules by applying forces approaching 60 pN (Figure 7), a force-induced melting transition occurs, in which dsDNA is converted to ssDNA (59). In the absence of protein, this transition occurs over a very narrow force range due to the cooperative melting of DNA. Saturating levels of HIV-1 NC result in a significant increase in the width of the force-induced melting transition (46), which correlates with NC's relatively efficient chaperone activity. In addition, the reversibility of DNA stretch/relax curves in the presence of NC (i.e., DNA stretch curves show very little hysteresis), suggests that NC has a fast nucleic acid binding on/off rate and is therefore capable of rapidly switching between dsDNA and ssDNA bound states (60).
Figure 7.

Examples of lambda DNA stretching (continuous) and relaxation (dashed) curves. (A) A3G. 0 nM (black); 150 nM (red). (B) NC. 0 nM (black); 10 nM (red). All stretching experiments were conducted at 20°C in 10 mM HEPES, pH 7.5 and 50 mM Na+.
To probe the nucleic acid binding properties of A3G, we stretched single lambda DNA molecules in the presence of varying amounts of A3G (Figure 7A; data not shown) and characterized the transition width and hysteresis. The transition width increased from 3.7 ± 0.2 pN in the absence of protein to 18.3 ± 2.4 pN at protein saturation (∼90 nM A3G and above). Examination of the stretching and relaxation curves showed that there was significant hysteresis (Figure 7A), which increased with A3G concentration, reaching a value of ▵Ghysteresis = 0.56 ± 0.02 kcal/mol per bp at 150 nM A3G, as compared to ▵Ghysteresis = 0.22 ± 0.06 kcal/mol per bp for saturated HIV-1 NC binding (Figure 7B) (60). The greater hysteresis in the presence of A3G reflects the inability of DNA to reanneal due to the presence of bound protein, which dissociates more slowly than the relaxation step time of ∼1 s. In other experiments, RT was found not to have any measurable effect on lambda DNA stretching (Wang,F. and Williams,M.C., unpublished data).
The apparent binding affinities of HIV-1 NC, RT and human A3G to a 20-mer ssDNA oligonucleotide, 5′-FAM-JL587D, are given in Table 1 (also see Figure S2). HIV-1 NC binds to the 20-mer DNA with an approximately 3-fold greater affinity than human A3G. Both A3G and HIV-1 NC bind ssDNA with a significantly higher affinity (8- and 22-fold, respectively) than HIV-1 RT. These studies suggested that A3G should compete very effectively with RT for binding to ssDNA, but would not readily displace NC. Taken together with the stretching data, these results help to explain why A3G inhibits RT-catalyzed elongation reactions, but fails to impact NC-mediated annealing.
Table 1.
Apparent binding affinities of HIV-1 NC, HIV-1 RT and A3G to 20-mer DNA oligonucleotide (5′-FAM-JL587D)
| Kd (nM)a | |
|---|---|
| HIV-1 NC | 84.1 ± 7.8 |
| HIV-1 RT | 1840 ± 390 |
| A3G | 238 ± 95 |
aKd values were determined from three independent experiments. The error determinations represent the standard deviation.
DISCUSSION
Previous studies demonstrated that A3G inhibits reverse transcription during infection with Δvif HIV-1 (28–31), with effects on synthesis of both early and late products (5,10,13,15,35–37). In the present study, for the first time, we show how A3G affects each individual step in reverse transcription (Figure 1), using a series of well-defined, reconstituted assay systems and highly purified A3G, RT and NC proteins. The results demonstrate that A3G inhibits elongation by HIV-1 RT directly, and not by blocking NC's nucleic acid chaperone activity (43). These findings are consistent with our previous result showing that NC and A3G do not compete for binding to RNA (12) and taken together, represent strong evidence that at a physical and functional level, NC and A3G do not interfere with each other's activities.
More specifically, we report that A3G inhibits (−) SSDNA and (+) SSDNA synthesis, minus- and plus-strand DNA transfer, and elongation of minus- and plus-strand DNAs. Using a deaminase-deficient A3G mutant, we also show that (−) SSDNA synthesis was inhibited in the absence of A3G's enzymatic activity (Figure 4). Additionally, during (−) SSDNA synthesis in the presence of A3G, we find that RT pauses at unique sites, which map to bases in or near ss regions in the RNA template structure (Figure 3C; also see Figure S1), consistent with A3G's preference for binding to ss nucleic acids (6,12). Since a direct interaction between A3G and RT could not be detected in pull-down assays (data not shown), the results further suggest that A3G binding to the template physically blocks RT movement along the template. It should be noted that A3G has a more profound effect on DNA-primed DNA polymerase activity than the ss-binding protein T4 gene 32. Thus, in a primer extension assay with a DNA template, gene 32 protein inhibited minus-strand DNA synthesis at a concentration of 400 nM. In contrast, an A3G effect was already observed at a concentration of 12.5 nM and by 50 nM, the DNA product was no longer detectable (data not shown).
Systematic study of A3G inhibition of individual reverse transcription reactions is not amenable to investigation with more complex cell-based assays. However, PCR analysis has indicated that during infection, A3G inhibits synthesis of late DNA products to a greater extent than early products (15,35,37). Conceivably, successive inhibition at each step has a cumulative effect. Recently, Guo et al. (35) showed that reduction of early reverse transcripts in A3G-expressing cells infected with Δvif virions is correlated with decreased tRNA priming in vitro. The authors suggested that an NC–A3G interaction might be inhibiting viral DNA synthesis. However, it was not determined whether A3G affected annealing and/or subsequent tRNA extension. In a new in vitro study, this group reports that in the presence of NC, A3G reduced annealing by a little less than 2-fold (67). The reason for the difference between these results and our data showing that A3G does not inhibit NC-mediated primer placement (but has a strong effect on primer extension) (Figures 2 and 3) in all likelihood reflects significant differences in the experimental conditions for annealing with respect to the nt/NC ratio and the solution ionic strength.
Recently, it has been reported that A3G (15,37) and A3F (15) inhibit HIV-1 integration as well as reverse transcription. A3G and A3G C291S could each be specifically coimmunoprecipitated with NC and integrase (IN) present in HIV-1 Δvif virions; coimmunoprecipitation of A3F with virion-associated IN could also be shown (15). Sequence analysis of 2-LTR circle junction clones from unintegrated DNA synthesized in the presence of A3G showed that in some cases, DNA at the U5 end had an additional 6 RNA bases derived from the 3′ terminus of . This suggests that A3G causes a defect in the tRNA removal step that limits plus-strand transfer and ultimately integration (37). Since we present definitive data showing that A3G does not inhibit RNase H cleavage of RNA in a preformed hybrid (Figure 5A), we suggest that the aberrant cleavage observed in vivo results from A3G binding to site(s) on the tRNA primer and/or the ssDNA template, thereby interfering with synthesis of plus-strand DNA (Figure 6A) and formation of a proper substrate for tRNA removal. Based on their results, Mbisa et al. (37) have made a similar proposal.
It was of interest to determine whether the A3G concentrations in the present study were within the physiological range. A recent report showed that only 7 (±4) molecules of A3G per virion are incorporated into vif-deficient HIV-1 produced from human PBMCs, indicating that only a few molecules of A3G are sufficient to inhibit HIV-1 replication (68). Interestingly, even the highest concentrations of A3G used in our experiments (up to 80 nM) are well below the estimated concentration of A3G in the virion (13 ± 8 µM). This value was obtained by assuming that the virus is a sphere of radius 60 nm (69).
Despite the high estimated concentration of A3G in the virus, the expected ratio of A3G:total nt of genomic RNA in vivo is much lower than the ratio that is typically used in vitro. However, RNA is highly folded and rarely ss (39). Indeed, the vast majority of vRNA is likely to be involved in secondary and tertiary interactions (70–72). Since A3G binds poorly to dsRNA (6,12), the effective ratio of A3G:ss nt in vivo is higher than predicted on the basis of total nt of RNA. Thus, the preferential binding of A3G to ssRNA (6,12) and the slow rate of A3G dissociation (Figure 7) could still result in A3G-induced inhibition of reverse transcription during infection.
The interpretation of the results presented here follows from our studies on the nucleic acid binding properties of NC, A3G and RT. Thus, the DNA stretching data in the presence of A3G show an increase in the force-induced melting transition width, suggesting that it is capable of binding to both ss and dsDNA. However, relative to NC, considerably higher concentrations of A3G are required to observe changes in the shape of the DNA stretching curve. Moreover, in sharp contrast to the small hysteresis observed with NC (Figure 7B), which decreases with increasing protein concentration (60), significant hysteresis is observed with A3G (Figure 7A). In this case, the amount of hysteresis increases when the protein concentration is elevated (data not shown). Similar behavior is observed with the ssDNA-binding protein T7 gene 2.5 (73), whose primary role in DNA replication is to stabilize ssDNA.
The DNA stretching results indicate that A3G binds preferentially to ssDNA and is unable to rapidly switch between binding to ssDNA and dsDNA. This finding can help to explain A3G's inhibition of reverse transcription, which requires rapid access of RT to the ssDNA or RNA template. Similarly, the fact that HIV-1 NC does not interfere with DNA synthesis is consistent with its ability to rapidly adjust to different binding states. The stretching data complement the FA–binding measurements, which show that the ssDNA binding order of the three proteins is NC>A3G>>RT (Table 1; Figure S2). Taken together, these data explain why NC is not readily displaced by A3G.
A major issue in research on A3G's antiviral effect is the question of whether this activity is deaminase-dependent or -independent (or both), but there is still no clear consensus. There are data favoring the dependent mechanism, as shown in several studies of G to A hypermutation (3,4,6) and in some reports on reverse transcription in Δvif HIV-1-infected cells (15,37). However, there is also accumulating evidence indicating that both types of mechanisms might be involved in A3G's effect on reverse transcription in vivo (13,15). Our studies do not exclude a requirement for editing, but based on biochemical and biophysical analysis, provide support for a novel molecular mechanism that could account for deaminase-independent inhibition of reverse transcription and virus replication that is observed in vivo.
In summary, our findings demonstrate for the first time that A3G can inhibit RT-catalyzed elongation in a deaminase-independent manner without interfering with NC-mediated chaperone activity. These results suggest that A3G has an intrinsic effect on viral DNA synthesis, which is independent of the replication steps following reverse transcription. Interplay of the equilibrium and kinetic differences between HIV-1 NC, A3G and RT with respect to their nucleic acid binding interactions is likely to be a major determinant of deaminase-independent A3G inhibition of RT-catalyzed DNA extension.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
ACKNOWLEDGEMENTS
We thank Dr Robert J. Gorelick for his generous gift of recombinant NC protein and Dr Alan Rein for valuable discussion and critical reading of the manuscript. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Child Health and Human Development (J.G.L.), the NIH Intramural AIDS Targeted Antiviral Program (A.M.G.), an NIH Kirschstein-NRSA Individual Fellowship, F32 GM072396 (K.S.M.), NIH grant GM065056 (K.M.F.), and NSF MCB-0238190 and NIH GM072462 grants (M.C.W.). Funding to pay the Open Access publication charges for the article was provided by the Intramural Research Program of the NIH, National Institute of Child Health and Human Development.
Conflict of interest statement. None declared.
REFERENCES
- 1.Sheehy A.M., Gaddis N.C., Choi J.D., Malim M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 2.Ehrlich E.S., Yu X.F. Lentiviral Vif: viral hijacker of the ubiquitin-proteasome system. Int. J. Hematol. 2006;83:208–212. doi: 10.1532/IJH97.06013. [DOI] [PubMed] [Google Scholar]
- 3.Harris R.S., Bishop K.N., Sheehy A.M., Craig H.M., Petersen-Mahrt S.K., Watt I.N., Neuberger M.S., Malim M.H. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 4.Lecossier D., Bouchonnet F., Clavel F., Hance A.J. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 5.Mangeat B., Turelli P., Caron G., Friedli M., Perrin L., Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 6.Yu Q., König R., Pillai S., Chiles K., Kearney M., Palmer S., Richman D., Coffin J.M., Landau N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 7.Shindo K., Takaori-Kondo A., Kobayashi M., Abudu A., Fukunaga K., Uchiyama T. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J. Biol. Chem. 2003;278:44412–44416. doi: 10.1074/jbc.C300376200. [DOI] [PubMed] [Google Scholar]
- 8.Navarro F., Bollman B., Chen H., König R., Yu Q., Chiles K., Landau N.R. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333:374–386. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Newman E.N., Holmes R.K., Craig H.M., Klein K.C., Lingappa J.R., Malim M.H., Sheehy A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005;15:166–170. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 10.Bishop K.N., Holmes R.K., Malim M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opi S., Takeuchi H., Kao S., Khan M.A., Miyagi E., Goila-Gaur R., Iwatani Y., Levin J.G., Strebel K. Monomeric APOBEC3G is catalytically active and has antiviral activity. J. Virol. 2006;80:4673–4682. doi: 10.1128/JVI.80.10.4673-4682.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwatani Y., Takeuchi H., Strebel K., Levin J.G. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes R.K., Koning F.A., Bishop K.N., Malim M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 14.Holmes R.K., Malim M.H., Bishop K.N. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Luo K., Wang T., Liu B., Tian C., Xiao Z., Kappes J., Yu X.F. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 2007;81:7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turelli P., Mangeat B., Jost S., Vianin S., Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- 17.Rosler C., Kock J., Kann M., Malim M.H., Blum H.E., Baumert T.F., von Weizsacker F. APOBEC-mediated interference with hepadnavirus production. Hepatology. 2005;42:301–309. doi: 10.1002/hep.20801. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen D.H., Gummuluru S., Hu J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007;81:4465–4472. doi: 10.1128/JVI.02510-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okeoma C.M., Lovsin N., Peterlin B.M., Ross S.R. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007;445:927–930. doi: 10.1038/nature05540. [DOI] [PubMed] [Google Scholar]
- 20.Bogerd H.P., Wiegand H.L., Doehle B.P., Lueders K.K., Cullen B.R. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bogerd H.P., Wiegand H.L., Hulme A.E., Garcia-Perez J.L., O'Shea K.S., Moran J.V., Cullen B.R. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl Acad. Sci. USA. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H., Lilley C.E., Yu Q., Lee D.V., Chou J., Narvaiza I., Landau N.R., Weitzman M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 23.Muckenfuss H., Hamdorf M., Held U., Perkovic M., Lower J., Cichutek K., Flory E., Schumann G.G., Munk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 24.Stenglein M.D., Harris R.S. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 2006;281:16837–16841. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- 25.Fisher A.G., Ensoli B., Ivanoff L., Chamberlain M., Petteway S., Ratner L., Gallo R.C., Wong-Staal F. The sor gene of HIV-1 is required for efficient virus transmission in vitro. Science. 1987;237:888–893. doi: 10.1126/science.3497453. [DOI] [PubMed] [Google Scholar]
- 26.Strebel K., Daugherty D., Clouse K., Cohen D., Folks T., Martin M.A. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature. 1987;328:728–730. doi: 10.1038/328728a0. [DOI] [PubMed] [Google Scholar]
- 27.Fouchier R.A., Simon J.H., Jaffe A.B., Malim M.H. Human immunodeficiency virus type 1 Vif does not influence expression or virion incorporation of gag-, pol-, and env-encoded proteins. J. Virol. 1996;70:8263–8269. doi: 10.1128/jvi.70.12.8263-8269.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sova P., Volsky D.J. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type 1. J. Virol. 1993;67:6322–6326. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Schwedler U., Song J., Aiken C., Trono D. vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goncalves J., Korin Y., Zack J., Gabuzda D. Role of Vif in human immunodeficiency virus type 1 reverse transcription. J. Virol. 1996;70:8701–8709. doi: 10.1128/jvi.70.12.8701-8709.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon J.H., Malim M.H. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J. Virol. 1996;70:5297–5305. doi: 10.1128/jvi.70.8.5297-5305.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dettenhofer M., Cen S., Carlson B.A., Kleiman L., Yu X.F. Association of human immunodeficiency virus type 1 Vif with RNA and its role in reverse transcription. J. Virol. 2000;74:8938–8945. doi: 10.1128/jvi.74.19.8938-8945.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dornadula G., Yang S., Pomerantz R.J., Zhang H. Partial rescue of the Vif-negative phenotype of mutant human immunodeficiency virus type 1 strains from nonpermissive cells by intravirion reverse transcription. J. Virol. 2000;74:2594–2602. doi: 10.1128/jvi.74.6.2594-2602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J., Potash M.J., Volsky D.J. Functional domains of APOBEC3G required for antiviral activity. J. Cell. Biochem. 2004;92:560–572. doi: 10.1002/jcb.20082. [DOI] [PubMed] [Google Scholar]
-
35.Guo F., Cen S., Niu M., Saadatmand J., Kleiman L. Inhibition of -primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 2006;80:11710–11722. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaiser S.M., Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J. Virol. 2006;80:875–882. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mbisa J.L., Barr R., Thomas J.A., Vandegraaff N., Dorweiler I.J., Svarovskaia E.S., Brown W.L., Mansky L.M., Gorelick R.J., et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007;81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y., Guo F., Cen S., Kleiman L. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology. 2007;365:92–100. doi: 10.1016/j.virol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 39.Herschlag D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995;270:20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 40.Darlix J.L., Lapadat-Tapolsky M., de Rocquigny H., Roques B.P. First glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Mol. Biol. 1995;254:523–537. doi: 10.1006/jmbi.1995.0635. [DOI] [PubMed] [Google Scholar]
- 41.Rein A., Henderson L.E., Levin J.G. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem. Sci. 1998;23:297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 42.Cristofari G., Darlix J.L. The ubiquitous nature of RNA chaperone proteins. Prog. Nucleic Acid Res. Mol. Biol. 2002;72:223–268. doi: 10.1016/s0079-6603(02)72071-0. [DOI] [PubMed] [Google Scholar]
- 43.Levin J.G., Guo J., Rouzina I., Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 44.Tsuchihashi Z., Brown P.O. DNA strand exchange and selective DNA annealing promoted by the human immunodeficiency virus type 1 nucleocapsid protein. J. Virol. 1994;68:5863–5870. doi: 10.1128/jvi.68.9.5863-5870.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarmuz A., Chester A., Bayliss J., Gisbourne J., Dunham I., Scott J., Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 46.Cruceanu M., Urbaneja M.A., Hixson C.V., Johnson D.G., Datta S.A., Fivash M.J., Stephen A.G., Fisher R.J., Gorelick R.J., et al. Nucleic acid binding and chaperone properties of HIV-1 Gag and nucleocapsid proteins. Nucleic Acids Res. 2006;34:593–605. doi: 10.1093/nar/gkj458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fisher R.J., Fivash M.J., Stephen A.G., Hagan N.A., Shenoy S.R., Medaglia M.V., Smith L.R., Worthy K.M., Simpson J.T., et al. Complex interactions of HIV-1 nucleocapsid protein with oligonucleotides. Nucleic Acids Res. 2006;34:472–484. doi: 10.1093/nar/gkj442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell M.D., Levin J.G. Sequence and structural determinants required for priming of plus-strand DNA synthesis by the human immunodeficiency virus type 1 polypurine tract. J. Virol. 1996;70:5288–5296. doi: 10.1128/jvi.70.8.5288-5296.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo J., Henderson L.E., Bess J., Kane B., Levin J.G. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J. Virol. 1997;71:5178–5188. doi: 10.1128/jvi.71.7.5178-5188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu T., Guo J., Bess J., Henderson L.E., Levin J.G. Molecular requirements for human immunodeficiency virus type 1 plus-strand transfer: analysis in reconstituted and endogenous reverse transcription systems. J. Virol. 1999;73:4794–4805. doi: 10.1128/jvi.73.6.4794-4805.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwatani Y., Rosen A.E., Guo J., Musier-Forsyth K., Levin J.G. Efficient initiation of HIV-1 reverse transcription in vitro. Requirement for RNA sequences downstream of the primer binding site abrogated by nucleocapsid protein-dependent primer-template interactions. J. Biol. Chem. 2003;278:14185–14195. doi: 10.1074/jbc.M211618200. [DOI] [PubMed] [Google Scholar]
- 52.Wu W., Henderson L.E., Copeland T.D., Gorelick R.J., Bosche W.J., Rein A., Levin J.G. Human immunodeficiency virus type 1 nucleocapsid protein reduces reverse transcriptase pausing at a secondary structure near the murine leukemia virus polypurine tract. J. Virol. 1996;70:7132–7142. doi: 10.1128/jvi.70.10.7132-7142.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee N., Gorelick R.J., Musier-Forsyth K. Zinc finger-dependent HIV-1 nucleocapsid protein-TAR RNA interactions. Nucleic Acids Res. 2003;31:4847–4855. doi: 10.1093/nar/gkg679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adachi A., Gendelman H.E., Koenig S., Folks T., Willey R., Rabson A., Martin M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo J., Wu W., Yuan Z.Y., Post K., Crouch R.J., Levin J.G. Defects in primer-template binding, processive DNA synthesis, and RNase H activity associated with chimeric reverse transcriptases having the murine leukemia virus polymerase domain joined to Escherichia coli RNase H. Biochemistry. 1995;34:5018–5029. doi: 10.1021/bi00015a013. [DOI] [PubMed] [Google Scholar]
- 56.Guo J., Wu T., Bess J., Henderson L.E., Levin J.G. Actinomycin D inhibits human immunodeficiency virus type 1 minus-strand transfer in in vitro and endogenous reverse transcriptase assays. J. Virol. 1998;72:6716–6724. doi: 10.1128/jvi.72.8.6716-6724.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powell M.D., Ghosh M., Jacques P.S., Howard K.J., Le Grice S.F., Levin J.G. Alanine-scanning mutations in the “primer grip” of p66 HIV-1 reverse transcriptase result in selective loss of RNA priming activity. J. Biol. Chem. 1997;272:13262–13269. doi: 10.1074/jbc.272.20.13262. [DOI] [PubMed] [Google Scholar]
- 58.McCauley M., Hardwidge P.R., Maher L.J., III, Williams M.C. Dual binding modes for an HMG domain from human HMGB2 on DNA. Biophys. J. 2005;89:353–364. doi: 10.1529/biophysj.104.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCauley M.J., Williams M.C. Mechanisms of DNA binding determined in optical tweezers experiments. Biopolymers. 2007;85:154–168. doi: 10.1002/bip.20622. [DOI] [PubMed] [Google Scholar]
- 60.Cruceanu M., Gorelick R.J., Musier-Forsyth K., Rouzina I., Williams M.C. Rapid kinetics of protein-nucleic acid interaction is a major component of HIV-1 nucleocapsid protein's nucleic acid chaperone function. J. Mol. Biol. 2006;363:867–877. doi: 10.1016/j.jmb.2006.08.070. [DOI] [PubMed] [Google Scholar]
- 61.Lundblad J.R., Laurance M., Goodman R.H. Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol. Endocrinol. 1996;10:607–612. doi: 10.1210/mend.10.6.8776720. [DOI] [PubMed] [Google Scholar]
- 62.Lakowicz J.R. Principles of Fluorescence Spectroscopy. New York: Kluwer Academic/Plenum; 1999. [Google Scholar]
- 63.Guo J., Wu T., Anderson J., Kane B.F., Johnson D.G., Gorelick R.J., Henderson L.E., Levin J.G. Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus- and plus-strand transfer. J. Virol. 2000;74:8980–8988. doi: 10.1128/jvi.74.19.8980-8988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hong M.K., Harbron E.J., O’Connor D.B., Guo J., Barbara P.F., Levin J.G., Musier-Forsyth K. Nucleic acid conformational changes essential for HIV-1 nucleocapsid protein-mediated inhibition of self-priming in minus-strand transfer. J. Mol. Biol. 2003;325:1–10. doi: 10.1016/s0022-2836(02)01177-4. [DOI] [PubMed] [Google Scholar]
- 65.Heilman-Miller S.L., Wu T., Levin J.G. Alteration of nucleic acid structure and stability modulates the efficiency of minus-strand transfer mediated by the HIV-1 nucleocapsid protein. J. Biol. Chem. 2004;279:44154–44165. doi: 10.1074/jbc.M401646200. [DOI] [PubMed] [Google Scholar]
- 66.Chelico L., Pham P., Calabrese P., Goodman M.F. APOBEC3G DNA deaminase acts processively 3′ –> 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006;13:392–399. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
-
67.Guo F, Cen S, Niu M, Yang Y, Gorelick RJ, Kleiman L. The interaction of APOBEC3G with HIV-1 nucleocapsid inhibits annealing to viral RNA. J. Virol. 2007;81:11322–11331. doi: 10.1128/JVI.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu H., Chertova E., Chen J., Ott D.E., Roser J.D., Hu W.S., Pathak V.K. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology. 2007;360:247–256. doi: 10.1016/j.virol.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 69.Briggs J.A., Wilk T., Welker R., Kräusslich H.G., Fuller S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003;22:1707–1715. doi: 10.1093/emboj/cdg143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Darlix J.L., Schwager M., Spahr P.F., Bromley P.A. Analysis of the secondary and tertiary structures of Rous sarcoma virus RNA. Nucleic Acids Res. 1980;8:3335–3354. doi: 10.1093/nar/8.15.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murti K.G., Bondurant M., Tereba A. Secondary structural features in the 70S RNAs of Moloney murine leukemia and Rous sarcoma viruses as observed by electron microscopy. J. Virol. 1981;37:411–419. doi: 10.1128/jvi.37.1.411-419.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berkhout B., van Wamel J.L. The leader of the HIV-1 RNA genome forms a compactly folded tertiary structure. RNA. 2000;6:282–295. doi: 10.1017/s1355838200991684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shokri L., Marintcheva B., Richardson C.C., Rouzina I., Williams M.C. Single molecule force spectroscopy of salt-dependent bacteriophage T7 gene 2.5 protein binding to single-stranded DNA. J. Biol. Chem. 2006;281:38689–38696. doi: 10.1074/jbc.M608460200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.