

Abstract
The inoculation of MOG peptides into C57BL/6 mice induces CD4+ and CD8+ T cells, and recent work has shown that adoptive transfer of the latter population, after extensive in vitro stimulation, can cause EAE in naïve recipient mice. Herein, we have evaluated the incidence and severity of EAE, and the induction of CD4+ and CD8+ T cells, following MOG peptide inoculation of wt mice and of LMP-2KO mice that lack an intact immunoproteasome, a cytoplasmic organelle that is induced by chronic inflammation and that may be important for the presentation of MHC class I epitopes to CD8+ T cells. We report that EAE, evaluated by both clinical and histological criteria, is similar in LMP-2KO mice and wildtype C57B/6 mice (wt) in response to immunization with MOG peptides MOG35-55 and MOG40-54, suggesting that the immunoproteasome does not play a key role in the development of demyelinating disease. Furthermore, and consistent with previous reports, peptide-specific CD8+ T cells were barely detectable in the CNS of peptide-immunized mice, although peptide-specific CD4+ T cells were abundant. Therefore, we used a new technique to look for autoreactive CD8+ T cells in MOG peptide-immunized mice, and we report the identification of CD4+ and CD8+ T cells that, as late as 19 days after peptide injection, are actively producing IFNγ in vivo, in response to in vivo antigen contact.
Keywords: CD4+ T cell, CD8+ T cell, EAE, immunoproteasome
INTRODUCTION
Multiple sclerosis (MS) is the most common chronic demyelinating disease of the central nervous system (CNS). CNS infiltration by lymphocytes and macrophages is observed in association with MS lesions, and autoreactive T cells have been implicated as major mediators of pathology. The precise etiology of MS remains unclear, but the study of T cell-mediated myelin injury has yielded important advances in the understanding of MS pathogenesis and in treatment strategies (Steinman and Zamvil, 2006). Genetic susceptibility plays a part: consistent with its proposed T cell-mediated pathogenesis, MS has been linked with the receptors for IL2 and IL7 (The International Multiple Sclerosis Genetics Consortium, 2007); and also with various MHC class II alleles (Yaouanq et al., 1997; Lincoln et al., 2005; Yeo et al., 2007), implicating CD4+ T cells in this disease (Sawcer et al., 1996; Sawcer et al., 2005; Yeo et al., 2007). This finding is consistent with the presence of CD4+ T cells among the CNS immune cell infiltrates observed in MS brain tissues (Booss et al., 1983; McCallum et al., 1987), and is supported by the demonstration that CD4+ T cells are pathogenic in several animal models of autoimmune demyelination (reviewed, Delgado and Sheremata, 2006). However, clinical studies have reported that anti-CD4 monoclonal antibody therapy in MS patients does not prevent disease (Lindsey et al., 1994; van Oosten et al., 1997). Hence, CD4+ T cells may play a regulatory or modulatory role in this disease, rather than being the principal culprits; and detailed analyses of T lymphocytes in MS, including examination of brain tissue specimens, has determined that the majority of T cells are CD8+ (Booss et al., 1983; Traugott et al., 1983; Hauser et al., 1986a; Hauser et al., 1986b; McCallum et al., 1987). CD8+ T cells are found in MS lesions in the form of clonal populations, consistent with their having been locally expanded in response to a specific antigen (Babbe et al., 2000; reviewed, Steinman, 2001; Neumann et al., 2002). Antigen-specific CD8+ T cells are found in patients early in the course of MS (Jilek et al., 2007), and CD8+ T cells isolated from peripheral blood of MS patients contain clonally expanded populations that are specific for myelin antigens (Tsuchida et al., 1994; Dressel et al., 1997).
CD8+ T lymphocytes recognize short epitope peptides presented at the cell surface by MHC class I molecules. Oligodendrocytes express MHC class I, and its expression is increased in MS (Hoftberger et al., 2004); therefore, myelin-derived self epitopes on the surface of oligodendrocytes constitute one possible target for CD8+ T cells within the CNS, and studies have reported that myelin-specific CD8+ T cells can trigger the death of cultured human oligodendrocytes (Ruijs et al., 1993; Jurewicz et al., 1998). At first blush, neurons appear less likely targets for direct CD8+ T cell effects, because they express very low levels of MHC class I, and of other components of the class I antigen presentation pathway (Joly et al., 1991; Joly and Oldstone, 1992); however, MHC class I is up-regulated by neurons in response to injury or inflammation (Neumann et al., 1995; Neumann et al., 1997), potentially rendering them vulnerable to CD8+ T cell-mediated cytotoxicity. Indeed, blood-donor derived CD8+ cells can cause axon transection in cultured neurons (Medana et al., 2001). Thus, CD8+ lymphocytes also are implicated in the demyelination and pathogenesis of MS, but our understanding of the function of CD8+ T cells in the context of MS has been limited by the availability of appropriate animal models. We feel it important to point out that the mode of immunization may contribute to the predominance of CD4+ T cell-mediated effects in EAE models; in the vast majority of EAE models, antigen is delivered in the form of soluble proteins [+ adjuvants such as complete Freund’s adjuvant (CFA)] and – because of the differing natures of the MHC class I and class II antigen presentation pathways – this type of immunogen favors the induction of CD4+ T cells, rather than of CD8+ T cells. CD8+ T cells are most effectively induced by proteins that are synthesized in vivo; the proteins are degraded into short peptides by the proteasome, a cytoplasmic organelle, and these peptides are directed into the MHC class I antigen presentation pathway.
To try to better evaluate the role of CD8+ T cells in EAE, one can express various CNS proteins in vivo – for example, from live viruses or plasmid DNAs. Following in vivo administration of such materials, we and others have observed various effects on the recipients’ sensitivity to EAE: in some cases, disease is accelerated / exacerbated (Barnett et al., 1993); in others, it is ameliorated (Barnett et al., 1996; Wang et al., 1999). A role for CD8+ T cells in disease causation was implied by our observation that the detrimental (disease-sensitization) effects of a plasmid DNA encoding proteolipid protein (PLP) required fusion of ubiquitin to PLP (Theil et al., 2001); such ubiquitin fusion is known to enhance the priming of CD8+ T cells (Barry et al., 1995; Rodriguez et al., 1997; Tobery and Siliciano, 1997; Rodriguez et al., 1998). These data indirectly implicated CD8+ T cells in EAE pathogenesis, and more recent reports have shown clearly that CD8+ T cells can trigger disease. When adoptively transferred into naive mice, myelin-reactive CD8+ T cells taken from mice immunized with certain myelin oligodendrocyte glycoprotein (MOG) peptides have the capacity to induce EAE (Huseby et al., 2001; Sun et al., 2001; Sun et al., 2003; Ford and Evavold, 2005; Ford and Evavold, 2006). How might CD8+ T cells be involved in EAE pathogenesis?
Chronic inflammatory diseases such as EAE and MS (and many other autoimmune diseases) cause profound changes in MHC class I antigen presentation. Many of these changes are wrought by IFNγ, a key immunoregulatory protein. Most somatic cells express the IFNγ receptor (Stark et al., 1998), consistent with the pleiotropic effects of this cytokine. As well as upregulating the expression of MHC class I and several components of the antigen processing machinery, IFNγ also alters the proteasome. Proteasomes, found both in the cytosol and in the nucleus, are proteolytic organelles whose major function appears to be acting as the cell’s garbage disposal unit; they degrade cellular proteins – especially misfolded proteins (Schubert et al., 2000) – and are indispensable. The central core of the proteasome is a 20S barrel containing 28 proteins arranged as four stacked, concentric seven-member rings; the top & bottom rings are identical, each consisting of a multimer of 7 alpha subunits; and the two central rings are identical to each other, each comprising a multimer of 7 beta subunits. Only three of the 14 protein types found in the 20S core have catalytic activity (β1, β2, & β5). A 19S regulatory complex binds as a “cap” to the top & bottom of the 20S core, to generate the mature 26S organelle. More than a decade ago, it was shown that the protein constitution of the proteasome underwent an IFNγ-induced alteration during viral infections, with the constitutive proteasomes being, largely, replaced by modified structures that were termed immunoproteasomes (reviewed, Tanaka and Kasahara, 1998; Niedermann, 2002). Structural comparison of the two proteasomal types revealed changes in three of the 14 proteins; the three catalytic components of the constitutive proteasome were replaced by new proteins named LMP2, LMP7 & LMP10 (MECL1), which also have catalytic activity. These three new catalytic subunits in the immunoproteasome show a subtly-different cleavage preference, and therefore the immunoproteasome generates a distinct set of peptides (Monaco, 1992; Driscoll et al., 1993). Intriguingly, polymorphisms in LMP genes have been associated with certain autoimmune diseases, including ankylosing spondylitis (Maksymowych et al., 1994; Maksymowych et al., 1995; Maksymowych and Russell, 1995; Pryhuber et al., 1996; Maksymowych et al., 2000) although they have not been identified for MS or T1D (Liblau et al., 1993; van Endert et al., 1994). These observations led us to hypothesize that chronic inflammatory diseases result in widespread and long-term induction of the immunoproteasome, and that this would, in turn, lead to the presentation of a constellation of new self epitopes. Because the host’s CD8+ T cells have not previously encountered these epitopes, an autoreactive CD8+ T cell response will be mounted; and these new autoreactive cells will express IFNγ in response to neo-epitope contact, thereby perpetuating the pathological inflammatory process. This hypothesis provides an attractive explanation for the phenomenon of “epitope spreading”, in which the T cell response changes with time, with a gradual appearance of responses to an ever-widening array of self epitopes. This has been described for CD4+ T cells (Lehmann et al., 1992; Lehmann et al., 1993; Miller et al., 1997; Vanderlugt et al., 1998; Miller et al., 2001), for which it is ascribed to the release of self proteins, and their uptake by specialized antigen presenting cells, where they enter the MHC class II antigen presentation pathway. However, such a mechanism appears unlikely to occur for CD8+ T cells, which generally recognize epitopes which have been synthesized inside the presenting cell. In this study we (i) determine if an intact immunoproteasome is required for the development of MOG-EAE; (ii) evaluate the effects of immunoproteasome deficiency on MOG peptide-induced CD4+ and CD8+ T cell responses; and (iii) identify autoreactive CD4+ and CD8+ T cells that are actively synthesizing cytokines in vivo, in response to contact with cognate antigen.
MATERIALS & METHODS
Mice and Genotyping
LMP-2KO mice were originally bred onto the C57BL/6 background (van Kaer et al., 1994) and were a kind gift of Dr. F. V. Chisari (The Scripps Research Institute, La Jolla, CA), Dr. M. Gaczynska (University of Texas, San Antonio, TX), and Dr. L. van Kaer (Vanderbilt University, Nashville, TN). The wildtype C57BL/6 mice were obtained from a breeding colony maintained by The Scripps Research Institute. All procedures involving animals used in this study were approved by The Scripps Research Institute institutional animal care and use committee.
Myelin oligodendrocyte glycoprotein (MOG)-induced EAE
Experimental autoimmune encephalomyelitis was induced in mice by immunization as described previously (Crocker et al., 2006) using MOG peptides 35-55, 40-54, or 44-54 which have previously been utilized in the MOG EAE disease model, and in which CD8+ T cell responses have been implicated (Sun et al., 2001; Sun et al., 2003; Ford and Evavold, 2005; Ford and Evavold, 2006). The peptides were purchased or synthesized by Anaspec Inc.,San Jose, CA. Briefly, peptides were emulsified (1.5 mg/ml) in complete freund’s adjuvant (CFA; Sigma, St Louis, MO) containing 200ng/ml Mycobacterium tuberculosis (Difco, Detroit, MI). Emulsion was then deposited subcutaneously in the flanks of the hind legs (100μl/deposit). Additional animals of each genotype were immunized with an equivalent volume of CFA emulsion that did not contain MOG peptide. Each animal was administered 500ng Pertussis toxin i.p. (islet activating protein; List Biological Labs, Inc, Campbell, CA) at the time of immunization and again 48hrs later. Mice were evaluated daily for presentation of physical signs EAE using the following scoring system (Crocker et al., 2006) ; 0 = no signs; 0.5 = partial loss of tail tonicity; 1 = complete loss of tail tonicity; 2 = hind limb paresis; 3 = complete hind limb paralysis; 4 = forelimb paresis, quadriplegia or moribund; 5 = death due to EAE. Gross body weights were also monitored throughout the course of these experiments.
Histopathological analyses
Sagittal sections (6-8 μm thick) were prepared from spinal cord collected into formalin (10%) fixative and then paraffin-embedded. Tissues were then deparaffinized in xylenes (3×5min) followed by a graded series of alcohols (100,90,75%; 2×5 in each) before antigen retrieval by heat-treatment in citrate buffer (0.01mol/L; 25 min.), as described previously (Feuer et al., 2003). Sections were next blocked in 10% normal goat serum (NGS; Calbiochem, Temecula, CA) for one hour and then stained overnight at 4 °C to detect Iba-1, a marker of activated microglia, using anti-Iba-1 antibody (Wako, Osaka, Japan) diluted in 10% NGS. Immunostaining was visualized using anti-rabbit Rhodamine Red-X conjugated secondary antisera (diluted 1:200; Invitrogen, Carlsbad, CA) and images were collected digitally using a Zeiss Axiovert 200 microscope and AxioVision software.
Isolation of mononuclear cells from the CNS
Measurement of MOG peptide-specific CD4+ or CD8+ T-cell responses were performed by using leukocytes isolated from spinal cords of mice at 18 days following immunization (a time at which clinical EAE is most severe, see Crocker et al., 2006). As a control, cells were obtained from CFA-injected mice at the same time point post-inoculation. Mice were perfused intracardially with 10 ml saline to minimize contamination with blood-borne leukocytes, and brain/spinal cord tissues were collected into serum-free DMEM media. Single cell suspensions were prepared using a dounce homogenizer. Leukocytes were then purified away from ECM and other cellular debris by centrifugation using a 44% and 56% Percoll gradient.
Intracellular cytokine staining (ICCS)
The number and magnitude of MOG-specific CD4+ and CD8+ T cells was determined by ex vivo incubation of the leukocytes with the MOG peptide (1uM) used to evoke EAE. Interferon-gamma (IFNγ) production was measured using an anti-IFNγ antibody (e-Biosciences) as described previously (Whitmire et al., 2005; Crocker et al., 2006). Cell staining was analyzed using a BD Biosciences FacsCalibur and FlowJo software (Tree Star, Ashland, OR).
Using brefeldin A to identify T cells that have recently responded to in vivo antigen contact
Brefeldin A (BFA; cat# B6542, Sigma-Aldrich, St. Louis, MO) was prepared as a 20 mg/ml stock in DMSO. Immediately before in vivo injection, a 1:40 dilution (to 0.5 mg/ml) was made in PBS, and 500 μl of this solution (250 μg BFA) was injected intravenously. Six hours later the mice were sacrificed, their spleens were harvested and reduced to a single-cell suspension, and stained immediately (i.e., without ex vivo peptide stimulation) for T cell marker (CD4 or CD8) and for intracellular IFNγ.
RESULTS
Hypothesis: inflammation, the immunoproteasome, and CD8+ T cell epitope spreading
The hypothesis that was to be tested is outlined diagrammatically in Figure 1. A self protein is shown as being degraded into four peptides by the constitutive proteasome (upper image); one of the peptides (purple) triggers an autoreactive CD8+ T cell response which results in the production of IFNγ. (Note that, in the model system used herein, this triggering peptide is provide by direct injection). IFNγ also might be secreted by NK cells that have been activated by (for example) a variety of viral or bacterial insults, or by CD4+ T cells that are themselves responding to self antigen. Regardless of its source, the IFNγ acts on somatic cells, including APCs, via the near-ubiquitous IFNγ receptor, leading to the replacement of the constitutive proteasome by the immunoproteasome (lower image). Degradation of the same self protein by the immunoproteasome then results in a different constellation of peptides, some of which are new to the host immune system; this leads to the activation of a new population of autoreactive CD8+ T cells (orange), leading to epitope spreading, and perpetuating the chronic inflammatory process. This expansion of the epitope universe is immunoproteasome-dependent and so should be constrained by the absence of any one of the three proteins unique to the immunoproteasome. In this study we tested this hypothesis using mice that lack the gene for LMP-2 (LMP-2KO mice; see Materials and Methods).
Figure 1. Hypothesis: the immunoproteasome may be central to CD8+ T cell epitope spreading.
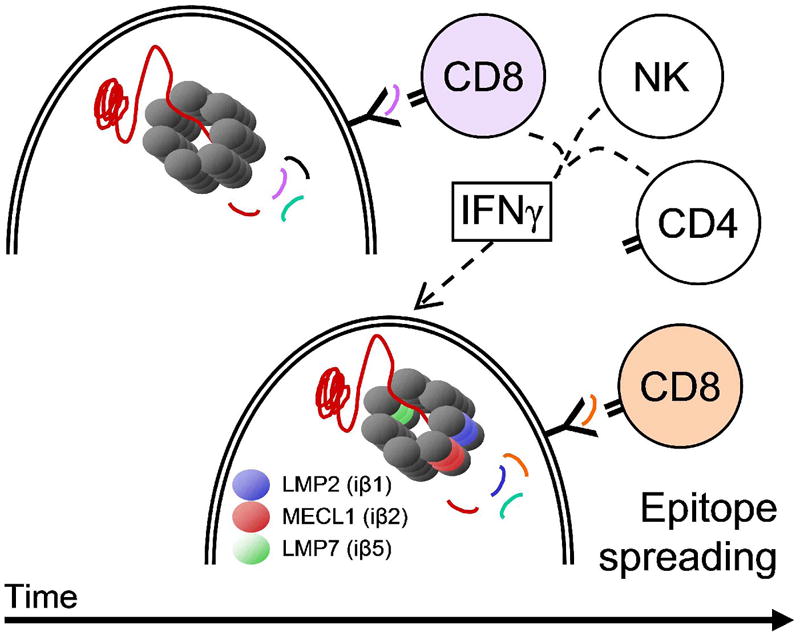
The hypothesis described in the text is presented in diagrammatic form. NK = natural killer. The double line represents the plasma membrane of the cell, enclosing the multimeric proteasome (upper cell) or immunoproteasome (lower cell); the “Y” motif represents an MHC class I molecule.
MOG-induced EAE in immunoproteasome deficient mice
LMP2 deficient C57BL/6 mice (LMP-2KO) and wildtype C57BL/6 mice (wt) were inoculated either with vehicle alone (complete freund’s adjuvant, CFA, n=6 per group; see Methods for details), or with vehicle plus one of the following 3 peptides: MOG35-55 (n=6 per group), MOG40-54 (n=3 per group), or MOG44-54 (n=6 per group). All mice were monitored daily for clinical signs of EAE. None of the mice treated with CFA developed any signs of EAE (Figure 2A). Mice immunized with MOG35-55 developed clinical signs of EAE at ~13 days post-inoculation, and clinical disease increased until it peaked at day 19. Most importantly, when considering the hypothesis outlined above (Figure 1), there was no statistically-significant difference between wt and LMP-2KO mice as measured by two criteria: the time of onset of EAE, and the kinetics of clinical disease progression after administration of MOG35-55 (Figure 2B). A similar conclusion can be drawn for wt and LMP-2KO mice that had been immunized with MOG40-54 peptide (Figure 2C); although the average score of clinical EAE among wt mice was slightly higher than that observed in LMP-2KO mice, the difference was not significantly different (P=0.457). In contrast to mice immunized with MOG35-55 or MOG40-54, neither wt nor LMP-2KO mice immunized with MOG44-54 developed any signs of clinical EAE over the same 19 day observation period (Figure 2D). Given this absence of EAE induced by MOG44-54, this peptide was not included in the subsequent analyses of histopathological changes and of T cell responses, described below.
Figure 2. Induction of EAE by MOG peptides, and the effect of immunoproteasome deficiency.
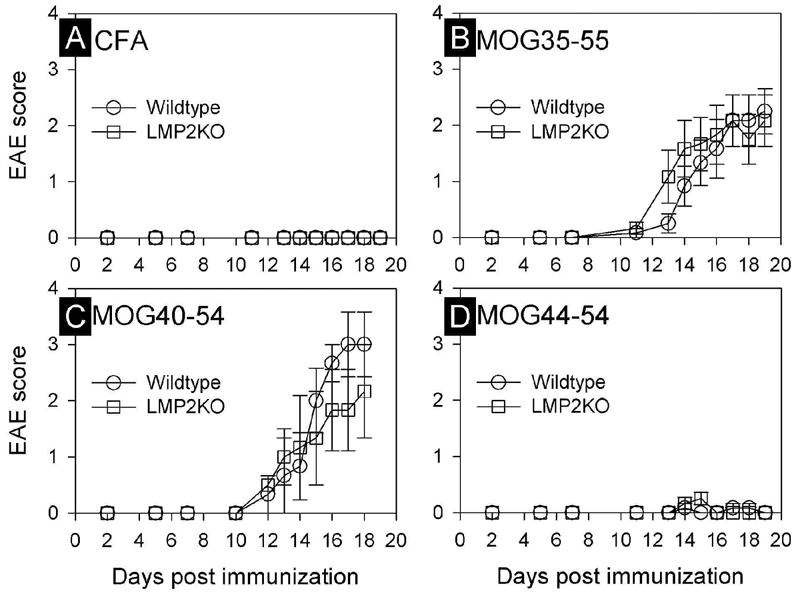
The development of clinical signs of EAE in wildtype C57B/6 or LMP-2KO/B6 mice immunized with either emulsified Complete Freund’s Adjuvant (CFA, panel A), or CFA with one of the three indicated myelin oligodendrocyte glycoprotein (MOG) peptides (panels B-D). Data points represent the EAE score (mean ± SEM) for each genotype and treatment group (n=3-6/group/time point).
Histopathological changes mirror the clinical picture
In order to evaluate the extent of demyelination following peptide inoculation, the brain stem and spinal cord were harvested on day 18 post-inoculation, and were stained by Luxol fast blue / periodic acid Schiff; inflammatory changes coupled with loss of myelin were observed in the CNS of mice that had received the MOG35-55 or MOG40-54 peptides, but there were no significant differences between wt and LMP-2KO strains (not shown). As a potentially more sensitive indicator of inflammatory changes within the CNS, the activation status, abundance and distribution of tissue macrophages / microglia were evaluated by histology and flow cytometry. Representative data from a mouse immunized 18 days previously with MOG35-55 are shown in Figure 3. Iba1, a marker of activated macrophages/microglia (Ito et al., 1998), was barely detectable in the brain stem/spinal cord of CFA-challenged Wt and LMP-2KO mice, but was much more readily detected in both mouse strains during EAE, indicative of widespread microglial activation during CNS demyelination (Figure 3A). However, this activation was accompanied by only a slight increase in abundance: flow cytometric analyses of cells extracted from the same CNS samples (Figure 3B) showed only a ~10% increase in the relative abundance of Mac-1+ cells during EAE, and this was true for both strains of mice.
Figure 3. Similar changes in microglia/macrophage activation and recruitment in wt and LMP-2KO mice.
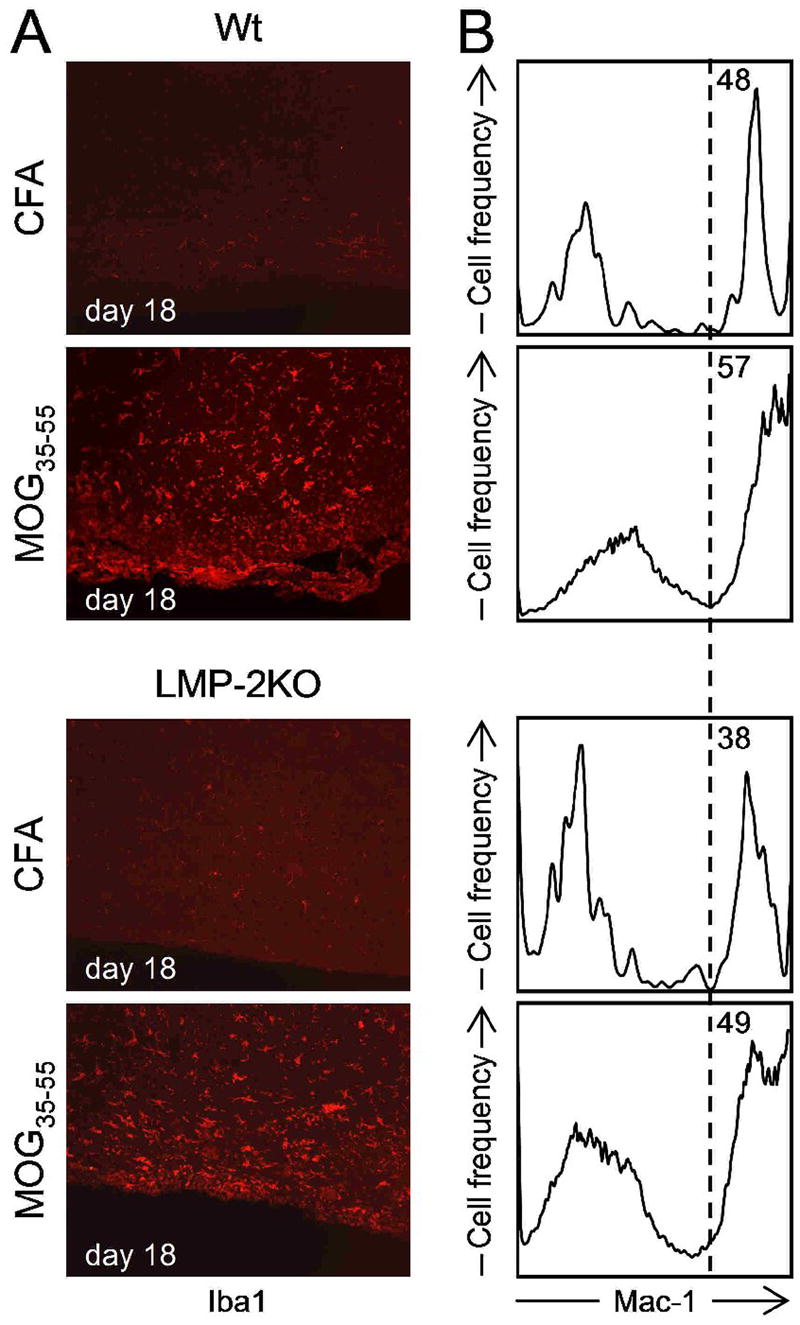
Wt and LMP-2KO mice were inoculated with either CFA alone, or CFA with MOG35-55 peptide and, 18 days later, were sacrificed, and their CNS was harvested. (A) Immunohistochemical detection of Iba-1, a marker of macrophage/microglial activation, revealed significant activation of these phagocytes during the acute phase of EAE. (B) Mononuclear cells were isolated from the CNS tissues, and the presence of Mac1+ cells was determined by flow cytometry. Mac1 is a marker for macrophages/microglia. The relative abundance of Mac1+ cells increases during the acute phase of EAE in both wt mice and LMP-2KO mice.
MOG-induced CD8+ T cell responses in wt and LMP-2KO mice
As noted above, accumulating evidence suggests that CD8+ T cells are involved in MS, and this study was initiated on the basis of (i) CD8+ T cells playing a role in peptide-induced MOG-EAE and (ii) the related hypothesis implicating the immunoproteasome, outlined in Figure 1 above. Therefore, we next evaluated the abundance and antigen-specific responsiveness of CD8+ T cells in the CNS (Figure 4). Mononuclear cells were isolated from the spinal cords of mice that been inoculated 18 days previously with CFA, or with MOG35-55 or MOG40-54. To determine CD8+ T cell abundance, the cells were stained without ex vivo peptide stimulation, and were enumerated by flow cytometry (Figure 4, rows labeled “No pep”). As expected, leukocytes were sparse in the CNS of mice that had received CFA alone (these mice had neither clinical EAE, nor any histological abnormality); the frequency of CD8+ T cells within the mononuclear population was variable (presumably due to the very low numbers of cells that were isolated), but low (around 2-6%). Mononuclear cells were more readily detected in mice with clinical EAE induced by inoculation of MOG peptide, but the relative abundance of CD8+ T cells was similar to, or only slightly higher than, that observed in CFA mice (MOG35-55 mice, 2-3%; MOG40-54 mice, 8-9%). There was no consistent difference observed between the wt and LMP-2KO mouse strains. The fact that the shorter of the two peptides induced more CD8+ T cells is intriguing, because MHC class I molecules usually present peptides of ~8-10 amino acids in length, and as peptide length increases beyond this, so peptide binding by MHC class I usually decreases. Next, to evaluate the antigen specificity of the infiltrating CD8+ T cells, the mononuclear cells were incubated for 6 hours ex vivo with cognate peptide followed by intracellular cytokine staining (ICCS). As shown in Figure 4 (rows labeled “Pep”) for both wt and LMP-2KO mice, very few of the infiltrating CD8+ T cells were peptide-specific (i.e., very few cells scored positive for IFNγ synthesis).
Figure 4. Very few peptide-specific CD8+ T cells in the spinal cords of mice inoculated with MOG peptides.
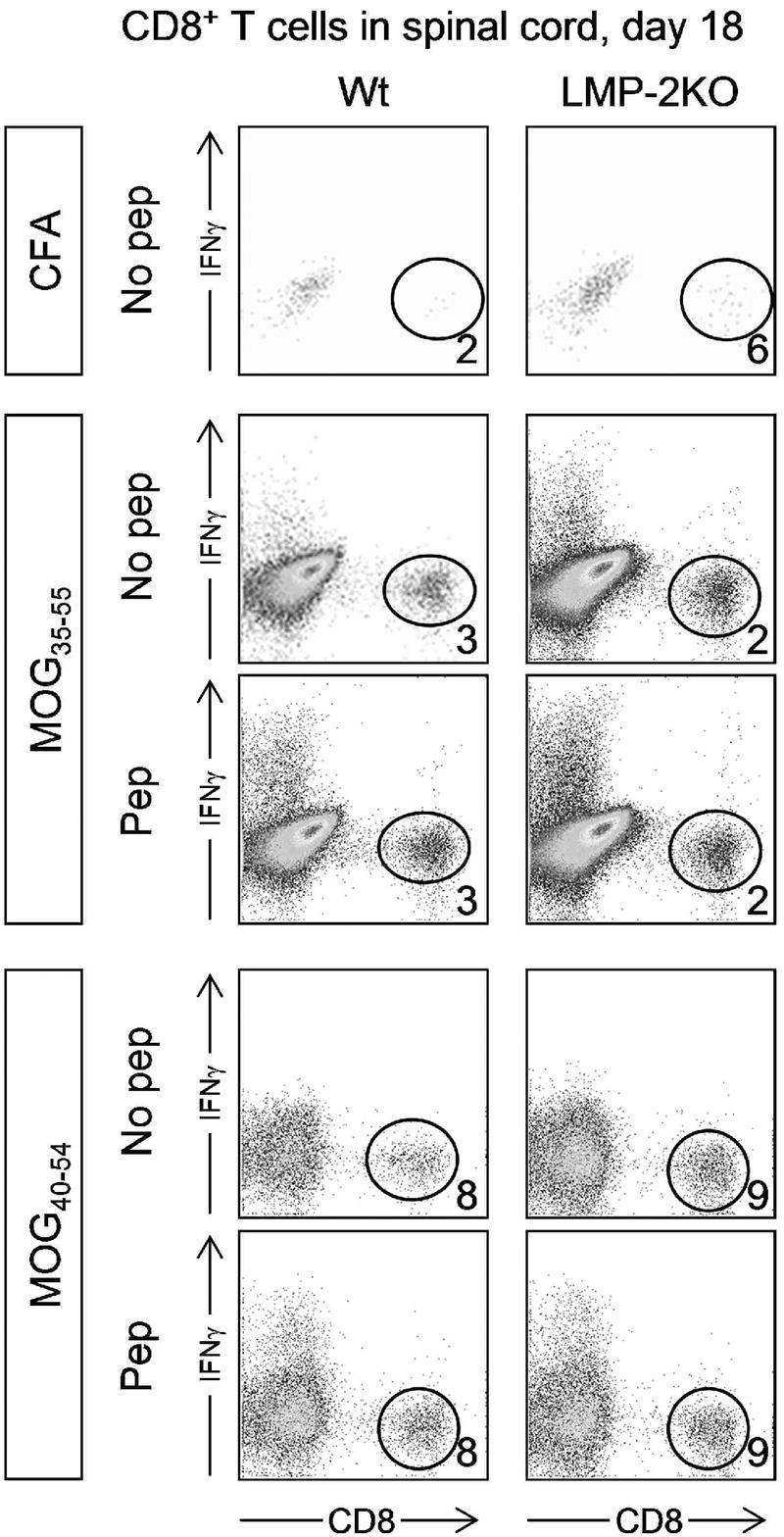
Wt C57BL/6 mice (left column) and LMP-2KO mice (right column) were immunized with either CFA, MOG35-55, or MOG40-54 (immunogen is indicated in boxes to the left of the dotplots). 18 days later, leukocytes were isolated from the spinal cords, and were analyzed for CD8+ T cell abundance (“No pep” rows) and peptide-responsiveness (“Pep” rows). Numbers represent the proportion of CD8+ T cells in each isolate (expressed as a percentage of all isolated leukocytes).
MOG-induced CD4+ T cell responses in wt and LMP-2KO mice
Although CD8+ T cells have been implicated in MOG peptide EAE, a substantial body of literature suggests that CD4+ T cells play a major role. Therefore, we assessed the abundance and antigen specificity of CD4+ T cells (Figure 5). In contrast to what was found for CD8+ T cells, immunization with MOG35-55 induced a robust influx of CD4+ T cells into the spinal cord; ~20% of cells were CD4+. An even more dramatic recruitment of CD4+ T cells was observed in mice that had received MOG40-54 peptide 18 days previously; in this case, ~45% of CNS leukocytes were CD4+. Mouse strain (wt or LMP-2KO) had no demonstrable influence on the proportion of CD4+ T cells in the infiltrates; this is unsurprising, because the immunoproteasome has no known effect on CD4+ T cell responses. Again in contrast to the CD8+ T cell response, a substantial proportion of the infiltrating CD4+ T cells were peptide-specific, as determined by the increase in IFNγ+ cells in response to ex vivo peptide stimulation (“Pep” rows in Figure 5). Again, and as expected, for each peptide the proportion of IFNγ-producing CD4+ T cells was similar in wt and LMP-2KO mice. However, it is interesting to note that many of the CNS-infiltrating CD4+ T cells were not peptide-responsive: in mice that had been immunized with MOG35-55, approximately ~70% of the CD4+ T cells failed to respond to cognate peptide; in mice that had received MOG40-54, the figure was even higher (~89%). In summary these data indicate that MOG35-55 or MOG40-54 peptides induce similar CD4+ T cell responses in both wt and LMP-2KO mice, that the CD4+ T cell infiltration correlated with the development of clinical EAE (Figure 2), but that many of the infiltrating cells are unresponsive to peptide.
Figure 5. Peptide-specific CD4+ T cells are readily detectable in the spinal cords of peptide-inoculated mice.
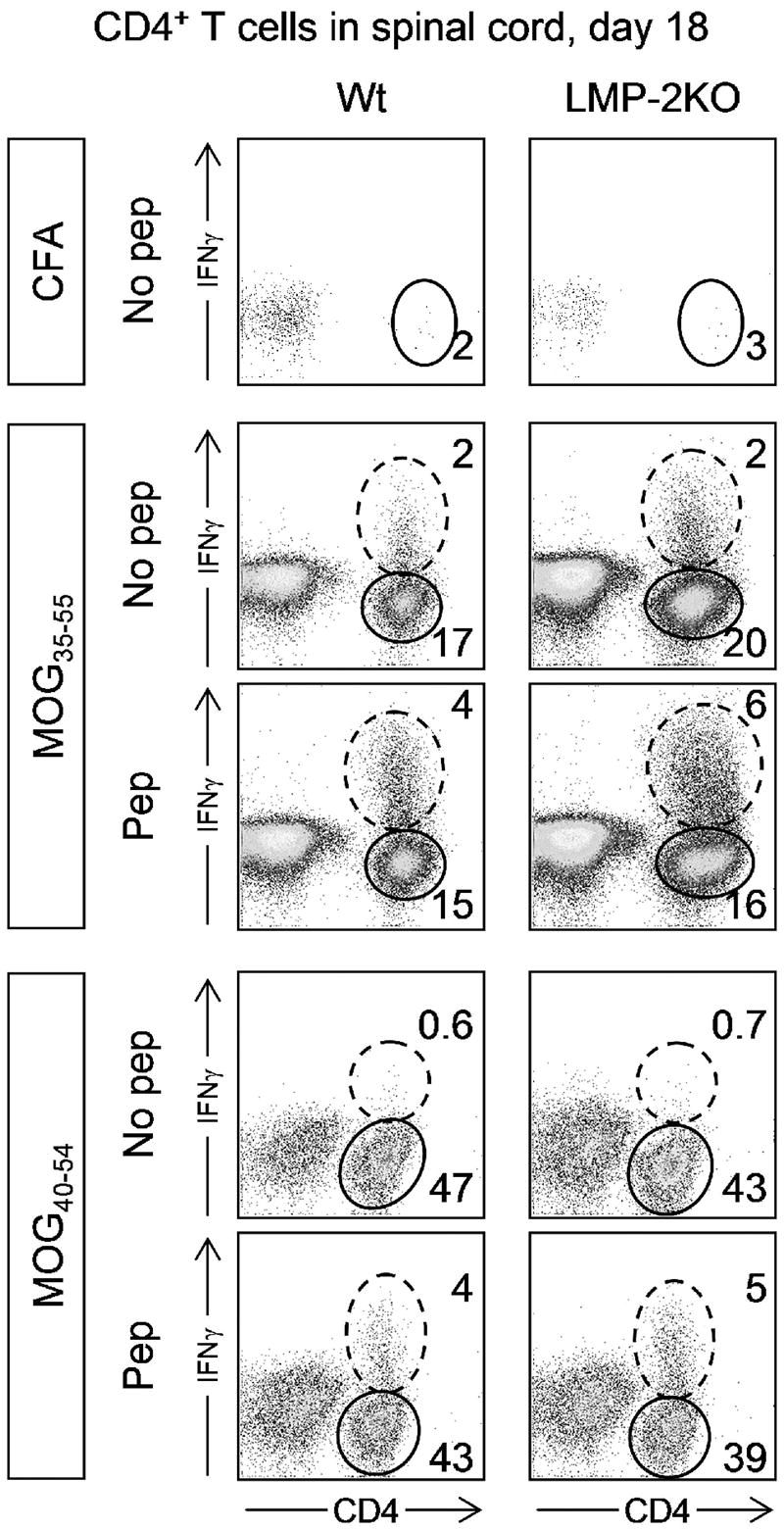
Wt C57BL/6 mice (left column) and LMP-2KO mice (right column) were immunized with either CFA, MOG35-55, or MOG40-54 (immunogen is indicated in boxes to the left of the dotplots). 18 days later, leukocytes were isolated from the spinal cords, and were analyzed for CD4+ T cell abundance (“No pep” rows) and peptide-responsiveness (“Pep” rows). Dashed ovals identify CD4+ T cells that are producing IFNγ, and solid ovals identify CD4+ T cells that do not produce this cytokine, even in response to the immunizing peptide. The numbers represent the proportion of CD4+ T cells in the adjacent oval (expressed as a percentage of all isolated leukocytes).
Identification of CD4+ and CD8+ T cells that are actively responding to in vivo antigen contact
Our laboratory recently has described a new technique to identify CD4+ and CD8+ T cells that are actively responding to in vivo antigen contact (Liu and Whitton, 2005; Foster et al., 2007). The method relies on the in vivo injection of brefeldin A (BFA), an antimetabolite that inhibits Golgi function and thus prevents the release of proteins that are normally destined for secretion. BFA is included in the incubation mixture for standard ICCS, because it causes peptide-stimulated cells to retain cytokines such as IFNγ, and thus renders the cells detectable by subsequent antibody staining. We have shown that the in vivo inoculation of BFA performs a similar function, and that cells harvested 6 hours post-injection from the spleens of such animals can be immediately stained (without any ex vivo peptide stimulation) allowing us to identify and enumerate T cells that have recently encountered antigen in vivo. Only cells that have recently encountered antigen in vivo will be identified using this approach, because T cells terminate IFNγ production immediately after they lose antigen contact (Slifka et al., 1999).
We applied this in vivo BFA approach to identify actively-responding T cells in the spleens wt mice that had received CFA alone, or MOG35-55 peptide. The results are shown in Figure 6. As expected, very few cells scored positive for IFNγ in the spleens of mice that had received CFA alone 19 days previously. In contrast, MOG35-55-immunized mice showed abundant IFNγ-producing cells at both 8 and 19 days post-peptide. At 8 days post-infection, some of the responding cells were CD4+ T cells; very few CD8+ T cells were actively synthesizing IFNγ. At day 19 – the peak of clinical EAE – IFNγ-positive CD4+ cells remained detectable, and CD8+ T cells too had been recruited. To our knowledge, this is the first demonstration that the in vivo BFA approach can be used to demonstrate active, ongoing, T cell responses in an autoimmune disease. Although informative, this in vivo BFA technique has two limitations: first, it does not reveal the antigen specificity of the responding cells; and, second, we have not yet been able to successfully apply the approach to non-lymphoid tissues (e.g., CNS), perhaps attributable to the longer time required for T cell isolation (Foster et al., 2007). However, the data in Figure 6 show that, despite these limitations, the BFA approach enhances our ability to detect CD4+ T cells in MOG-peptide immunized mice and, perhaps more significantly, it reveals – for the first time – quite abundant cytokine-producing CD8+ T cells in the spleens of primary peptide recipients.
Figure 6. At the height of clinical EAE, both CD4+ and CD8+ T cells are actively producing IFNγ, indicating recent in vivo antigen contact.
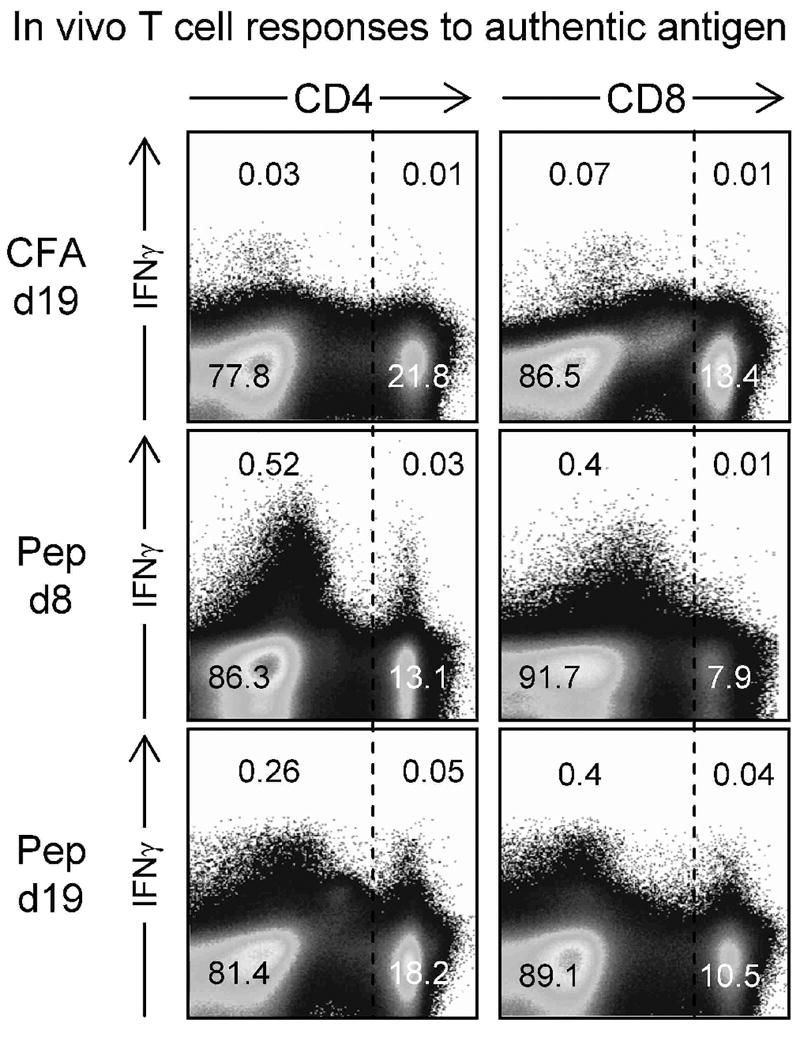
C57BL/6 mice received either CFA alone (CFA), or CFA with MOG35-55 peptide (rows labeled “Pep”). 8 or 19 days later (as indicated) the mice were injected with BFA (see Materials and Methods) and six hours later were sacrificed. The spleens were harvested, reduced to single cell suspension, and immediately stained (without ex vivo exposure to peptide) for CD4 or CD8, and for intracellular IFNγ. The numbers indicate the proportion of cells in each quadrant, as a percentage of total splenocytes.
DISCUSSION
The contribution of CD8+ T cells to CNS demyelination has been an under-investigated area of MS immunopathology (Gold et al., 2006). Herein we analyzed three separate MOG peptides for their propensity to induce EAE in wildtype mice and immunoproteasome-deficient mice, and we evaluated the ability of two of the peptides to induce CD4+ and CD8+ T cell responses. We determined that MOG peptides 35-55 and 40-54 induced robust CD4+ T cell responses in the CNS, and accompanying EAE, but peptide-specific CD8+ T cells were barely detectable.
The role of CD8+ T cells in EAE is controversial. Earlier studies on a myelin basic protein (MBP) model of EAE determined that administration of monoclonal antisera against CD8 did not alter initial EAE onset or clinical severity, but re-challenge with the MBP peptide resulted in an exacerbation of clinical disease, although with reduced mortality (Jiang et al., 1992). Similarly, analysis of MBP-induced EAE in CD8 deficient (CD8KO) mice resulted in a more chronic form of disease marked by increased frequency of spontaneous relapse (Koh et al., 1992). These findings have led to the suggestion that during immune-mediated demyelination CD8+ T cells may facilitate disease resolution or mediate adaptive protection against progressive disease, possibly through a CD8+ T cell-mediated lysis of myelinogenic CD4+ T cells (Sun et al., 1988; Jiang et al., 1992). These studies also point to a predominant regulatory role, rather than effector function, of CD8+ lymphocytes during autoimmune demyelination. Consistent with this idea, immunodepletion of CD8+ cells in C57B/6 mice immunized with MOG35-55 has been reported to result in a more severe clinical disease (Montero et al., 2004). Conversely, a study examining EAE induced by immunization with MOG1-125 in DBA/1- mice reported a diminished clinical severity and reduced myelin pathology in CD8KO mice (Abdul-Majid et al., 2003). However, a recent study of MOG-EAE in rats identified a possible confounding factor in CD8 depletion analyses: the majority of CD8+ cells were not T cells, but instead were activated macrophages/microglia (Schroeter et al., 2003).
Although there may be potentially diverse functions for CD8+ immune cells in EAE models, a direct pathogenic role for CD8+ T cells in CNS demyelination has recently been revealed. Adoptive transfer of MOG-reactive CD8+ T cells into naive syngeneic recipient mice (Sun et al., 2001; Ford and Evavold, 2005), or adoptive transfer of T cell lines derived from MBP knockout mice infected with a recombinant vaccinia virus expressing MBP (Huseby et al., 2001), can evoke a clinical syndrome reminiscent of EAE with inflammatory neuropathology that is distinct from CD4+ T cell mediated disease models (Huseby et al., 2001; Ji and Goverman, 2007). However, in some cases EAE is observed only if the recipient mice have been irradiated prior to cell transfer. Furthermore, the encephalitogenic effects of the CD8+ T cells have not, so far, been observed in the primary recipients (i.e., in the mice that received MOG peptide or recombinant MBP vaccinia virus); the demonstration of CD8+ T cell-mediated neuropathology requires that the T cells be harvested, purified, stimulated extensively in vitro (for 3-7 days in the presence of antigen ± IL2), then adoptively transferred into naïve mice. Nevertheless, these data support a direct pathogenic role for CD8+ effector T cells in CNS demyelination, and it was from this standpoint that we attempted to analyze the functional contribution of CD8 in MOG peptide-induced EAE, and compared wt mice with LMP-2KO mice. However, consistent with other studies, we observed that the injection of MOG peptides into C57BL/6 mice resulted in a predominance of CD4+ T cell responses in the CNS, with minimal CD8+ T infiltration. Thus the generation and precise function of CD8+ T cells in the context of EAE remains enigmatic. The results presented in this report confirm that neither MOG35-55 nor MOG40-54 induces a robust peptide-specific CD8+ T cell response in C57B/6 mice. The shorter of the two induces a more abundant CD8+ T cell response within the CNS (Figure 4) but, in both cases, very few of the infiltrating CD8+ cells were responsive to in vitro peptide stimulation. CD4+ T cell infiltration was more profound (Figure 5) but, again, relatively few of the cells responded to cognate peptide in vitro. What might be the antigen specificities of the non-responding CD4+ and CD8+ T cells? It is possible that some are specific for the inducing peptide, but are anergic / tolerized. However, it seems likely that some of the cells are specific for other self antigens, perhaps generated by epitope spreading as described above.
In conclusion, our findings suggest: that (i) the peptide-specific CD8+ T cells that develop during MOG-induced EAE in C57B/6 mice are not sufficiently abundant to allow their detection directly ex vivo using standard techniques; and (ii) the immunoproteasome is not required for the establishment of MOG-induced EAE, and may not be required for epitope spreading (although we cannot yet reject this possibility). Taken together, we conclude that this MOG peptide model may not be sufficiently robust to adequately address the functional contribution of CD8+ T cells to the pathogenesis of EAE and, by association, of MS. However, we do not interpret these findings as a repudiation of previous work; the published studies have convincingly demonstrated that peptide-induced and in vitro expanded CD8+ T cells can cause demyelinating disease following transfer into naive recipients. In this light, we have shown herein that MOG peptide injection leads to the activation of autoreactive CD4+ and CD8+ T cells that actively synthesize IFNγ as late as 19 days after peptide injection, indicative of recent in vivo antigen contact; it will be important to determine the antigen specificity of these cells, and to establish their role in EAE pathogenesis.
Acknowledgments
We are grateful to Annette Lord for excellent secretarial support. This work was supported by NIH grant R-01 AI027028 and P-01 AI058105, and by an advanced postdoctoral fellowship from the National Multiple Sclerosis Society (FA-1552-A-1 to SJC). This is manuscript number 18918 from the Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Majid KB, Wefer J, Stadelmann C, Stefferl A, Lassmann H, Olsson T, Harris RA. Comparing the pathogenesis of experimental autoimmune encephalomyelitis in CD4-/- and CD8-/-DBA/1 mice defines qualitative roles of different T cell subsets. J Neuroimmunol. 2003;141:10–19. doi: 10.1016/s0165-5728(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, Ravid R, Rajewsky K. Clonal expansions of CD8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett LA, Whitton JL, Wada Y, Fujinami RS. Enhancement of autoimmune disease using recombinant vaccinia virus encoding myelin proteolipid protein. J Neuroimmunol. 1993;44:15–25. doi: 10.1016/0165-5728(93)90263-x. [DOI] [PubMed] [Google Scholar]
- Barnett LA, Whitton JL, Wang LY, Fujinami RS. Virus encoding an encephalitogenic peptide protects mice from experimental allergic encephalomyelitis. J Neuroimmunol. 1996;64:163–173. doi: 10.1016/0165-5728(95)00165-4. [DOI] [PubMed] [Google Scholar]
- Barry MA, Lai WC, Johnston SA. Protection against mycoplasma infection using expression-library immunization. Nature. 1995;377:632–635. doi: 10.1038/377632a0. [DOI] [PubMed] [Google Scholar]
- Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. 1983;62:219–232. doi: 10.1016/0022-510x(83)90201-0. [DOI] [PubMed] [Google Scholar]
- Crocker SJ, Whitmire JK, Frausto RF, Chertboonmuang P, Soloway PD, Whitton JL, Campbell IL. Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am J Pathol. 2006;169:2104–2116. doi: 10.2353/ajpath.2006.060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado S, Sheremata WA. The role of CD4+ T-cells in the development of MS. Neurol Res. 2006;28:245–249. doi: 10.1179/016164106X98107. [DOI] [PubMed] [Google Scholar]
- Dressel A, Chin JL, Sette A, Gausling R, Hollsberg P, Hafler DA. Autoantigen recognition by human CD8 T cell clones: enhanced agonist response induced by altered peptide ligands. J Immunol. 1997;159:4943–4951. [PubMed] [Google Scholar]
- Driscoll J, Brown MG, Finley D, Monaco JJ. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature. 1993;365:262–264. doi: 10.1038/365262a0. [DOI] [PubMed] [Google Scholar]
- Feuer R, Mena I, Pagarigan RR, Harkins S, Hassett DE, Whitton JL. Coxsackievirus B3 and the neonatal CNS: the roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am J Pathol. 2003;163:1379–1393. doi: 10.1016/S0002-9440(10)63496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ML, Evavold BD. Specificity, magnitude, and kinetics of MOG-specific CD8+ T cell responses during experimental autoimmune encephalomyelitis. Eur J Immunol. 2005;35:76–85. doi: 10.1002/eji.200425660. [DOI] [PubMed] [Google Scholar]
- Ford ML, Evavold BD. Modulation of MOG 37-50-specific CD8+ T cell activation and expansion by CD43. Cell Immunol. 2006;240:53–61. doi: 10.1016/j.cellimm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Foster B, Prussin C, Liu F, Whitmire JK, Whitton JL. Detection of Intracellular Cytokines by Flow Cytometry. In: Coligan JE, Kruisbeek AM, Marguiles DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. John Wiley & Sons; 2007. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Aubert C, Burks JS, Kerr C, Lyon-Caen O, de TG, Brahic M. Analysis of human T-lymphotrophic virus sequences in multiple sclerosis tissue. Nature. 1986a;322:176–177. doi: 10.1038/322176a0. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986b;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- Hoftberger R, boul-Enein F, Brueck W, Lucchinetti C, Rodriguez M, Schmidbauer M, Jellinger K, Lassmann H. Expression of major histocompatibility complex class I molecules on the different cell types in multiple sclerosis lesions. Brain Pathol. 2004;14:43–50. doi: 10.1111/j.1750-3639.2004.tb00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlen C, Goverman J. A pathogenic role for myelin-specific CD8+ T cells in a model for multiple sclerosis. J Exp Med. 2001;194:669–676. doi: 10.1084/jem.194.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Ji Q, Goverman J. Experimental Autoimmune Encephalomyelitis Mediated by CD8+ T Cells. Ann N Y Acad Sci. 2007 doi: 10.1196/annals.1394.017. [DOI] [PubMed] [Google Scholar]
- Jiang H, Zhang SI, Pernis B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science. 1992;256:1213–1215. doi: 10.1126/science.256.5060.1213. [DOI] [PubMed] [Google Scholar]
- Jilek S, Schluep M, Rossetti AO, Guignard L, Le GG, Pantaleo G, Du Pasquier RA. CSF enrichment of highly differentiated CD8+ T cells in early multiple sclerosis. Clin Immunol. 2007;123:105–113. doi: 10.1016/j.clim.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Joly E, Mucke L, Oldstone MBA. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science. 1991;253:1283–1285. doi: 10.1126/science.1891717. [DOI] [PubMed] [Google Scholar]
- Joly E, Oldstone MBA. Neuronal cells are deficient in loading peptides onto MHC class I molecules. Neuron. 1992;8:1185–1190. doi: 10.1016/0896-6273(92)90138-4. [DOI] [PubMed] [Google Scholar]
- Jurewicz A, Biddison WE, Antel JP. MHC class I-restricted lysis of human oligodendrocytes by myelin basic protein peptide-specific CD8 T lymphocytes. J Immunol. 1998;160:3056–3059. [PubMed] [Google Scholar]
- Koh DR, Fung-Leung WP, Ho A, Gray D, cha-Orbea H, Mak TW. Less mortality but more relapses in experimental allergic encephalomyelitis in CD8-/- mice. Science. 1992;256:1210–1213. doi: 10.1126/science.256.5060.1210. [DOI] [PubMed] [Google Scholar]
- Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- Lehmann PV, Sercarz EE, Forsthuber T, Dayan CM, Gammon G. Determinant spreading and the dynamics of the autoimmune T-cell repertoire. Immunol Today. 1993;14:203–208. doi: 10.1016/0167-5699(93)90163-F. [DOI] [PubMed] [Google Scholar]
- Liblau R, van Endert PM, Sandberg-Wollheim M, Patel SD, Lopez MT, Land S, Fugger L, McDevitt HO. Antigen processing gene polymorphisms in HLA-DR2 multiple sclerosis. Neurol. 1993;43:1192–1197. doi: 10.1212/wnl.43.6.1192. [DOI] [PubMed] [Google Scholar]
- Lincoln MR, Montpetit A, Cader MZ, Saarela J, Dyment DA, Tiislar M, Ferretti V, Tienari PJ, Sadovnick AD, Peltonen L, Ebers GC, Hudson TJ. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet. 2005;37:1108–1112. doi: 10.1038/ng1647. [DOI] [PubMed] [Google Scholar]
- Lindsey JW, Hodgkinson S, Mehta R, Mitchell D, Enzmann D, Steinman L. Repeated treatment with chimeric anti-CD4 antibody in multiple sclerosis. Ann Neurol. 1994;36:183–189. doi: 10.1002/ana.410360210. [DOI] [PubMed] [Google Scholar]
- Liu F, Whitton JL. Cutting Edge: Re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J Immunol. 2005;174:5936–5940. doi: 10.4049/jimmunol.174.10.5936. [DOI] [PubMed] [Google Scholar]
- Maksymowych WP, Russell AS. Polymorphism in the LMP2 gene influences the relative risk for acute anterior uveitis in unselected patients with ankylosing spondylitis. Clin Invest Med. 1995;18:42–46. [PubMed] [Google Scholar]
- Maksymowych WP, Suarez-Almazor M, Chou CT, Russell AS. Polymorphism in the LMP2 gene influences susceptibility to extraspinal disease in HLA-B27 positive individuals with ankylosing spondylitis. Ann Rheum Dis. 1995;54:321–324. doi: 10.1136/ard.54.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksymowych WP, Tao S, Vaile J, Suarez-Almazor M, Ramos-Remus C, Russell AS. LMP2 polymorphism is associated with extraspinal disease in HLA-B27 negative Caucasian and Mexican Mestizo patients with ankylosing spondylitis. J Rheumatol. 2000;27:183–189. [PubMed] [Google Scholar]
- Maksymowych WP, Wessler A, Schmitt-Egenolf M, Suarez-Almazor M, Ritzel G, von Borstel RC, Pazderka F, Russell AS. Polymorphism in an HLA linked proteasome gene influences phenotypic expression of disease in HLA-B27 positive individuals. J Rheumatol. 1994;21:665–669. [PubMed] [Google Scholar]
- McCallum K, Esiri MM, Tourtellotte WW, Booss J. T cell subsets in multiple sclerosis. Gradients at plaque borders and differences in nonplaque regions. Brain. 1987;110(Pt 5):1297–1308. doi: 10.1093/brain/110.5.1297. [DOI] [PubMed] [Google Scholar]
- Medana I, Martinic MA, Wekerle H, Neumann H. Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol. 2001;159:809–815. doi: 10.1016/S0002-9440(10)61755-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SD, Katz-Levy Y, Neville KL, Vanderlugt CL. Virus-induced autoimmunity: epitope spreading to myelin autoepitopes in Theiler’s virus infection of the central nervous system. Adv Virus Res. 2001;56:199–217. doi: 10.1016/s0065-3527(01)56008-x. [DOI] [PubMed] [Google Scholar]
- Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, Katz-Levy Y, Carrizosa A, Kim BS. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- Monaco JJ. A molecular model of MHC class-I-restricted antigen processing. Immunol Today. 1992;13:173–179. doi: 10.1016/0167-5699(92)90122-N. [DOI] [PubMed] [Google Scholar]
- Montero E, Nussbaum G, Kaye JF, Perez R, Lage A, Ben-Nun A, Cohen IR. Regulation of experimental autoimmune encephalomyelitis by CD4(+), CD25(+) and CD8(+) T cells: analysis using depleting antibodies. J Autoimmun. 2004;23:1–7. doi: 10.1016/j.jaut.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science. 1995;269:549–552. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-γ and tumor necrosis factor (TNF)-α. J Exp Med. 1997;185:305–316. doi: 10.1084/jem.185.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedermann G. Immunological functions of the proteasome. Curr Top Microbiol Immunol. 2002;268:91–136. doi: 10.1007/978-3-642-59414-4_5. [DOI] [PubMed] [Google Scholar]
- Pryhuber KG, Murray KJ, Donnelly P, Passo MH, Maksymowych WP, Glass DN, Giannini EH, Colbert RA. Polymorphism in the LMP2 gene influences disease susceptibility and severity in HLA-B27 associated juvenile rheumatoid arthritis. J Rheumatol. 1996;23:747–752. [PubMed] [Google Scholar]
- Rodriguez F, An LL, Harkins S, Zhang J, Yokoyama M, Widera G, Fuller JT, Kincaid C, Campbell IL, Whitton JL. DNA immunization with minigenes: low frequency of memory cytotoxic T lymphocytes and inefficient antiviral protection are rectified by ubiquitination. J Virol. 1998;72:5174–5181. doi: 10.1128/jvi.72.6.5174-5181.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez F, Zhang J, Whitton JL. DNA immunization: ubiquitination of a viral protein enhances cytotoxic T-lymphocyte induction, and antiviral protection, but abrogates antibody induction. J Virol. 1997;71:8497–8503. doi: 10.1128/jvi.71.11.8497-8503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruijs TC, Louste K, Brown EA, Antel JP. Lysis of human glial cells by major histocompatibility complex-unrestricted CD4+ cytotoxic lymphocytes. J Neuroimmunol. 1993;42:105–111. doi: 10.1016/0165-5728(93)90217-m. [DOI] [PubMed] [Google Scholar]
- Sawcer S, Ban M, Maranian M, Yeo TW, Compston A, Kirby A, Daly MJ, De Jager PL, Walsh E, Lander ES, Rioux JD, Hafler DA, Ivinson A, Rimmler J, Gregory SG, Schmidt S, Pericak-Vance MA, Akesson E, Hillert J, Datta P, Oturai A, Ryder LP, Harbo HF, Spurkland A, Myhr KM, Laaksonen M, Booth D, Heard R, Stewart G, Lincoln R, Barcellos LF, Hauser SL, Oksenberg JR, Kenealy SJ, Haines JL. A high-density screen for linkage in multiple sclerosis. Am J Hum Genet. 2005;77:454–467. doi: 10.1086/444547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawcer S, Jones HB, Feakes R, Gray J, Smaldon N, Chataway J, Robertson N, Clayton D, Goodfellow PN, Compston A. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet. 1996;13:464–468. doi: 10.1038/ng0896-464. [DOI] [PubMed] [Google Scholar]
- Schroeter M, Stoll G, Weissert R, Hartung HP, Lassmann H, Jander S. CD8+ phagocyte recruitment in rat experimental autoimmune encephalomyelitis: association with inflammatory tissue destruction. Am J Pathol. 2003;163:1517–1524. doi: 10.1016/S0002-9440(10)63508-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Rodriguez F, Whitton JL. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature. 1999;401:76–79. doi: 10.1038/43454. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Steinman L. Myelin-specific CD8 T cells in the pathogenesis of experimental allergic encephalitis and multiple sclerosis. J Exp Med. 2001;194:F27–F30. doi: 10.1084/jem.194.5.f27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- Sun D, Qin Y, Chluba J, Epplen JT, Wekerle H. Suppression of experimentally induced autoimmune encephalomyelitis by cytolytic T-T cell interactions. Nature. 1988;332:843–845. doi: 10.1038/332843a0. [DOI] [PubMed] [Google Scholar]
- Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H, Raine CS. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol. 2001;166:7579–7587. doi: 10.4049/jimmunol.166.12.7579. [DOI] [PubMed] [Google Scholar]
- Sun D, Zhang Y, Wei B, Peiper SC, Shao H, Kaplan HJ. Encephalitogenic activity of truncated myelin oligodendrocyte glycoprotein (MOG) peptides and their recognition by CD8+ MOG-specific T cells on oligomeric MHC class I molecules. Int Immunol. 2003;15:261–268. doi: 10.1093/intimm/dxg023. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Kasahara M. The MHC class I ligand-generating system: roles of immunoproteasomes and the interferon-gamma-inducible proteasome activator PA28. Immunol Rev. 1998;163:161–176. doi: 10.1111/j.1600-065x.1998.tb01195.x. [DOI] [PubMed] [Google Scholar]
- The International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med. 2007 doi: 10.1056/NEJMoa073493. in press. [DOI] [PubMed] [Google Scholar]
- Theil DJ, Tsunoda I, Rodriguez F, Whitton JL, Fujinami RS. Viruses can silently prime for and trigger central nervous system autoimmune disease. J Neurovirol. 2001;7:1–8. doi: 10.1080/13550280152403263. [DOI] [PubMed] [Google Scholar]
- Tobery TW, Siliciano RF. Targeting of HIV-1 antigens for rapid intracellular degradation enhances cytotoxic T lymphocyte (CTL) recognition and the induction of de novo CTL responses in vivo after immunization. J Exp Med. 1997;185:909–920. doi: 10.1084/jem.185.5.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traugott U, Reinherz EL, Raine CS. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J Neuroimmunol. 1983;4:201–221. doi: 10.1016/0165-5728(83)90036-x. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A. 1994;91:10859–10863. doi: 10.1073/pnas.91.23.10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Endert PM, Liblau RS, Patel SD, Fugger L, Lopez T, Pociot F, Nerup J, McDevitt HO. Major histocompatibility complex-encoded antigen processing gene polymorphism in IDDM. Diabetes. 1994;43:110–117. doi: 10.2337/diab.43.1.110. [DOI] [PubMed] [Google Scholar]
- van Kaer L, Ashton-Rickardt PG, Eichelberger M, Gaczynska M, Nagashima K, Rock KL, Goldberg AL, Doherty PC, Tonegawa S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity. 1994;1:533–541. doi: 10.1016/1074-7613(94)90043-4. [DOI] [PubMed] [Google Scholar]
- van Oosten BW, Lai M, Hodgkinson S, Barkhof F, Miller DH, Moseley IF, Thompson AJ, Rudge P, McDougall A, McLeod JG, Ader HJ, Polman CH. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurol. 1997;49:351–357. doi: 10.1212/wnl.49.2.351. [DOI] [PubMed] [Google Scholar]
- Vanderlugt CL, Begolka WS, Neville KL, Katz-Levy Y, Howard LM, Eagar TN, Bluestone JA, Miller SD. The functional significance of epitope spreading and its regulation by co-stimulatory molecules. Immunol Rev. 1998;164:63–72. doi: 10.1111/j.1600-065x.1998.tb01208.x. [DOI] [PubMed] [Google Scholar]
- Wang LY, Theil DJ, Whitton JL, Fujinami RS. Infection with a recombinant vaccinia virus encoding myelin proteolipid protein causes suppression of chronic relapsing-remitting experimental allergic encephalomyelitis. J Neuroimmunol. 1999;96:148–157. doi: 10.1016/s0165-5728(99)00020-x. [DOI] [PubMed] [Google Scholar]
- Whitmire JK, Tan JT, Whitton JL. Interferon- acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201:1053–1059. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaouanq J, Semana G, Eichenbaum S, Quelvennec E, Roth MP, Clanet M, Edan G, Clerget-Darpoux F. Evidence for linkage disequilibrium between HLA-DRB1 gene and multiple sclerosis. The French Research Group on Genetic Susceptibility to MS. Science. 1997;276:664–665. doi: 10.1126/science.276.5313.661g. [DOI] [PubMed] [Google Scholar]
- Yeo TW, De Jager PL, Gregory SG, Barcellos LF, Walton A, Goris A, Fenoglio C, Ban M, Taylor CJ, Goodman RS, Walsh E, Wolfish CS, Horton R, Traherne J, Beck S, Trowsdale J, Caillier SJ, Ivinson AJ, Green T, Pobywajlo S, Lander ES, Pericak-Vance MA, Haines JL, Daly MJ, Oksenberg JR, Hauser SL, Compston A, Hafler DA, Rioux JD, Sawcer S. A second major histocompatibility complex susceptibility locus for multiple sclerosis. Ann Neurol. 2007;61:228–236. doi: 10.1002/ana.21063. [DOI] [PMC free article] [PubMed] [Google Scholar]