

Abstract
Inflammation results in CNS damage in multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), an animal model of MS. It is uncertain how much repair of injured myelin and axons can occur following highly selective anti-inflammatory therapy in EAE and MS. In this study, SJL/J mice with established EAE were treated successfully with an antigen-specific recombinant T cell receptor ligand (RTL), RTL401, a mouse I-As/PLP-139–151 construct, after the peak of EAE. To define the mechanisms by which late application of RTL401 inhibits EAE, we evaluated mice at different time points to assess the levels of neuroinflammation and myelin and axon damage in their spinal cords. Our results showed that RTL401 administered after the peak of acute EAE induced a marked reduction in inflammation in the CNS, associated with a significant reduction of demyelination, axonal loss and ongoing damage. Electron microscopy showed that RTL-treated mice had reduced pathology compared with mice treated with vehicle and mice at the peak of disease, as demonstrated by a decrease in continued degeneration, increase in remyelinating axons and the presence of an increased number of small, presumably regenerative axonal sprouts. These findings indicate that RTL therapy targeting encephalitogenic T cells may promote CNS neuro-regenerative processes.
Keywords: axonal loss, demyelination, multiple sclerosis, T lymphocytes
The early inflammatory phase of multiple sclerosis (MS) is believed to involve an autoimmune process similar to that induced in experimental autoimmune encephalomyelitis (EAE) upon immunization with myelin antigens in adjuvants. In both instances it is apparent that encephalitogenic T cells penetrate the blood–brain barrier and cause neuroinflammation, demyelination and axonal damage in the CNS (Steinman 1996). Clinically, MS has a very diverse clinical presentation and course, with a majority of MS patients experiencing a relapsing-remitting phase (RRMS) that may evolve into a secondary progressive phase (SPMS) characterized by persistent and advancing neurological impairments. In primary progressive MS (PPMS), the symptoms are progressive from onset without remissions. Collective evidence suggests axonal loss occurs at all stages of MS and is responsible for the accrual of disability in progressive MS.
Development of effective treatments aimed at preventing or restoring CNS myelin and axonal damage is crucial for successful management of MS. Current therapies such as glatiramer acetate and recombinant, B-interferons can reduce MRI enhancing lesions and slow clinical progression of MS, but little is known about the effects of these therapies on myelin and axonal injury. Moreover, although blockade of immune cell trafficking into the CNS with anti-VLA-4 antibodies (Natalizumab) appears to effectively reduce CNS inflammation, this therapeutic approach can cause life-threatening complications, such as progressive multifocal leucoencephalopathy (Lublin 2005), and may inhibit the protective role of neuroinflammation (Moalem et al. 1999; Kerschensteiner et al. 2003; Schwartz and Kipnis 2005). On the other hand, the development of direct neuroregenerative or neuroprotective therapy in MS is hampered by a limited understanding of the mechanism of neurodegeneration in MS, and by lack of clinically proven agents with unequivocal neuroprotective effects.
The immune attack in MS and EAE may be initiated by neuroantigen-specific CD4+ Th1 cells, which home to the CNS where autoantigens are present, and after local activation, selectively produce inflammatory mediators that recruit and activate macrophages and monocytes that ultimately destroy myelin and axons. Antigen specific regulation of encephalitogenic Th1 cells has long been considered an appealing strategy for therapy due to its selectivity and limited effects on protective immune defense mechanisms. We showed previously that the function of Ag-specific CD4+ T cells can be selectively regulated using recombinant TCR ligands (RTLs) that contain soluble MHC domains linked to specific antigenic peptides (Burrows et al. 2000; Burrows et al. 2001; Vandenbark et al. 2003; Wang et al. 2003; Huan et al. 2004). When administered to mice at the onset of EAE, RTL401, a mouse I-As/PLP-139–151 construct, significantly reduced the clinical severity and prevented disease relapse (Huan et al. 2004). However, whether this molecule is capable of ameliorating CNS damage in established EAE has not been investigated. We found in this study that administration of RTL401 after the peak of EAE, when substantial CNS damage had already occurred, prevented further relapses and induced a marked reduction in inflammation in the CNS, associated with a significant reduction of demyelination, axonal loss and ongoing damage. These findings suggest that selective immune modulation of encephalitogenic T cells may allow emergence of natural repair mechanisms that promote remyelination and recovery from axonal injury.
Materials and methods
Animals
Female SJL/J mice were obtained from The Jackson Laboratory at 7–8 week of age. The mice were housed at the animal facility at Portland Veterans Affairs Medical Center in accordance with institutional guidelines.
RTL construction and production
General methods for the design, cloning, and expression of RTLs have been described previously (Burrows et al. 2000; Chang et al. 2001). In brief, mRNA was isolated from the splenocytes of SJL/J mice using an Oligotex Direct mRNA mini kit (Qiagen, Valencia, CA, USA). cDNA of the Ag binding/TCR recognition domain (β1 and α1) of murine I-As MHC class II was derived from mRNA using two pairs of PCR primers. The two chains were sequentially linked by a 5-aa linker (GGQDD) in a two-step PCR with NcoI and XhoI restriction sites being added to the N terminus of the β1 chain and to the C terminus of the α1 chain. The PLP-139–151 peptide with a linker (GGGGSLVPRGSGGGG) was covalently attached to the 5′ end of the α1 domain to form RTL401. The murine I-As β1α1 insert was then ligated into pET21d+ vector and transformed into Nova blue Escherichia coli host (Novagen, San Diego, CA) for positive colony selection and sequence verification. RTL401 plasmid constructs were then transformed into E. coli strain BL21(DE3) expression host (Novagen, Madison, WI, USA). The purification of proteins has been described previously (Chang et al. 2001). The final yield of purified protein varied between 15 and 30 mg/L bacterial culture.
Induction and scoring of EAE
SJL/J mice were inoculated s.c. in the flanks with 0.2 mL of an emulsion containing 150 μg of PLP-139–151 peptide and an equal volume of CFA containing 150 μg of heat-killed Mycobacterium tuberculosis H37RA (M.Tb.; Difco, Detroit, MI, USA). After immunization, the mice were assessed daily for signs of EAE according to the following scale: 0 = normal; 1 = limp tail or mild hind limb weakness; 2 = moderate hind limb weakness or mild ataxia; 3 = moderately severe hind limb weakness; 4 = severe hind limb weakness or mild forelimb weakness or moderate ataxia; 5 = paraplegia with no more than moderate forelimb weakness; and 6 = paraplegia with severe forelimb weakness or severe ataxia or moribund condition.
Evaluation of CNS damage and RTL401 treatment
After immunization, mice with EAE were killed at different time-points to monitor the development of CNS damage in EAE and investigate the impact of RTL401 treatment. On day 11 (11 days after immunization), 8 mice with disease score 1.5, designated as the ‘Onset’ group, were killed as described below. From day 15 to day 20, a total of 8 mice, designated as the ‘Peak’ group, were killed individually when their disease score reached 4.5. On day 20, additional mice (16) with matched disease scores were divided into two groups, ‘RTL401’ and ‘Vehicle’, with 8 mice in each group. The RTL401 group received 5 consecutive daily i.v. injections of 100 μg of RTL401 from day 20–24, and 3 daily s.c. injection of 100 μg of RTL401 from day 32–34. An independent experiment evaluating different administration strategies for RTL in DR2-transgenic mice showed that this tandem administration strategy is highly efficacious and carries low toxicity (Link et al. submitted to Clinical Immunology). The vehicle group received saline on the same treatment schedule as the RTL401 group. Both groups were euthanized on day 60 at the conclusion of the experiment.
From each group, half of the mice (4, randomly chosen) were sedated and killed by perfusion with 4% paraformaldehyde (PFA) plus 5% glutaraldehyde for histopathology and electron microscopy study (see Histopathology and Electron Microscopy section, Figs 2, 6 and 7). The other half of the mice (4) were killed by perfusion with ice-cold PBS. The thoracic spinal cords of PBS-perfused mice were used for immunohistochemistry (see Immunohistochemistry section, Figs 3 and 4) and their lumbar spinal cords were used for western blots (see Western blot section and Fig. 5).
Fig. 2.
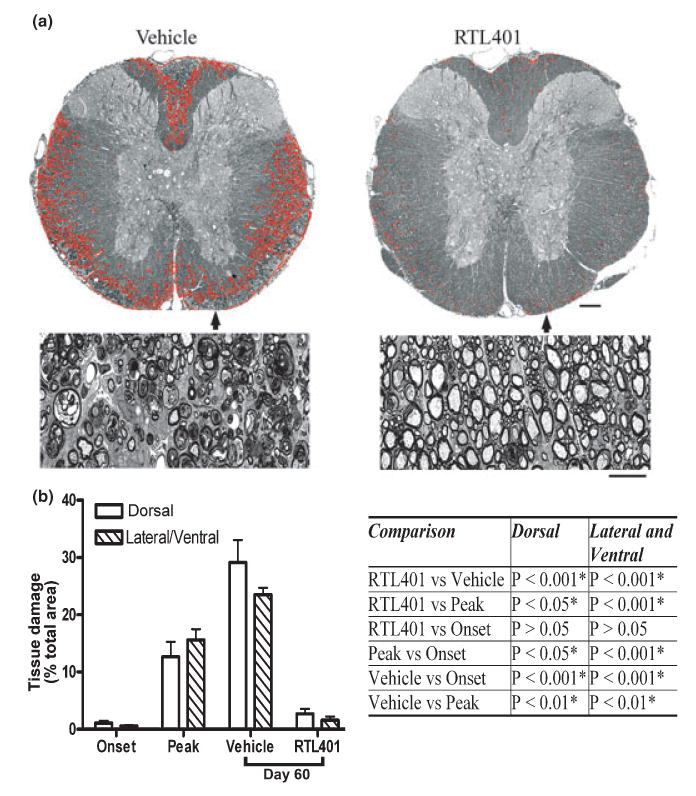
RTL treatment ameliorated tissue (myelin) damage in EAE. (a) Representative thoracic spinal cord sections from vehicle (left panel) or RTL401 (right panel)-treated mice stained with toluidine blue for myelin on day 60 of EAE. (b) Morphometric analysis of the development of myelin damage in the white matter of thoracic spinal cords of EAE treated with vehicle or RTL401. The cords were dissected from paraformaldehyde (PFA) plus glutaraldehyde-perfused EAE mice (4 mice randomly chosen from a group of 8, the rest were euthanized by perfusion with PBS, see Figs 3,4 and 5) euthanized at disease onset (day 11, disease score 1.5), peak (between day 15 and day 20 when the disease score for each mouse reached 4.5), or at the termination of the experiment (day 60 of EAE, after RTL401 or vehicle treatment). Tissue sections were stained with toluidine blue and images were captured with a compound microscope equipped with a digital camera. Areas with tissue damage were measured and analyzed using BIOQUANT classic 95 software. Scale bars = 25 μM (low power views) or 100 μM (high power views). Inserted Table: RTL401-treated mice show a significant reduction in spinal cord white matter tissue damage, demonstrated by one-way ANOVA followed by Newman-Kuels multiple comparisons test. *Comparison statistically significant.
Fig. 6.
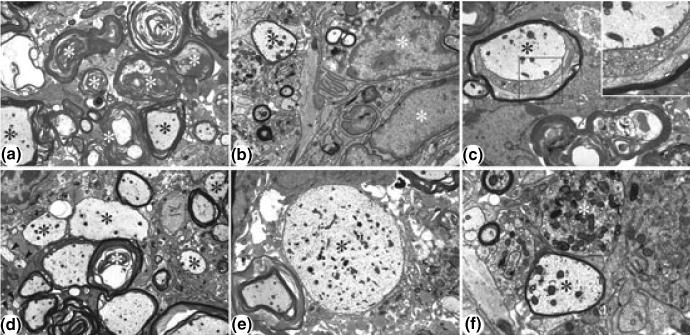
Representative electron micrographs showing lesion areas in spinal cords from EAE mice at the peak of the disease. (a) Low power view of typical lesion area. Wallerian-like axonal degeneration (white asterisks) is the most prominent feature. Active demyelination (black asterisks) is also present. Magnification × 4000. (b) Inflammatory cells (white asterisks) are also present, enlarging the lesion area. Note remyelinating axon (black asterisk). Magnification × 8000. (c) Active demyelination (black asterisk), as revealed by the loosening of the myelin sheath. Inset: Higher power view of boxed region showing active demyelination. Magnification × 6700; × 14 000 (inset). (d) Low power view showing active demyelination (white asterisk), medium to large sized remyelinating axons (black asterisk) and several very small axons (arrowheads), presumably representing regenerating axonal sprouts. Magnification × 5000. (e) Low power view of a large, demyelinated axon (black asterisk). Magnification × 5000. (f) Higher power view of large remyelinating axon (black asterisk) and an end bulb of a degenerating axon (dystrophic axon) (white asterisk). Magnification × 14 000.
Fig. 7.
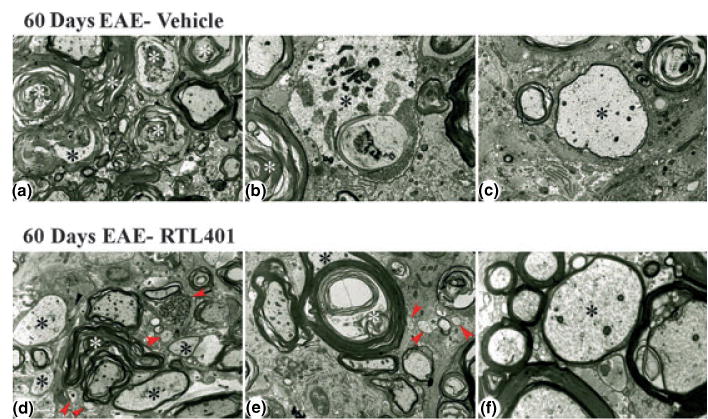
Representative electron micrographs showing lesion areas in spinal cords from mice with EAE on day 60 after treatment with vehicle (panels a–c) or RTL401 (panels d–f). (a) Low power view of typical lesion area showing marked continued Wallerian-like axonal degeneration (white asterisks) and demyelination (black asterisk). Note paucity of infiltrating cells and lack of small, regenerating axonal sprouts. Magnification × 4000. (b) Higher power view showing Wallerian-like axonal degeneration (white asterisk) and active demyelination (black asterisk). Magnification × 8000. (c) Higher power view of a large, remyelinating axon as shown by the relatively thin myelinated sheath (black asterisk). Magnification × 6700. (d) Low power view of typical lesion area showing continued Wallerian-like axonal degeneration (white asterisk), including a dystrophic axon (black arrow), and demyelinated and remyelinating axons (black asterisks). However, there are also prominent remyelinating axons and several small axonal sprouts (red arrowheads). Note paucity of infiltrating cells. Magnification × 4000. (e) Low power view of a large fiber (black asterisk) undergoing active demyelination (white asterisk). Note also three very small axons/regenerating sprouts (arrowheads). Magnification × 5000. (f) Higher power view of a medium-sized, remyelinating axon as shown by the relatively thin myelinated sheath (black asterisk). Magnification × 14 000.
Fig. 3.
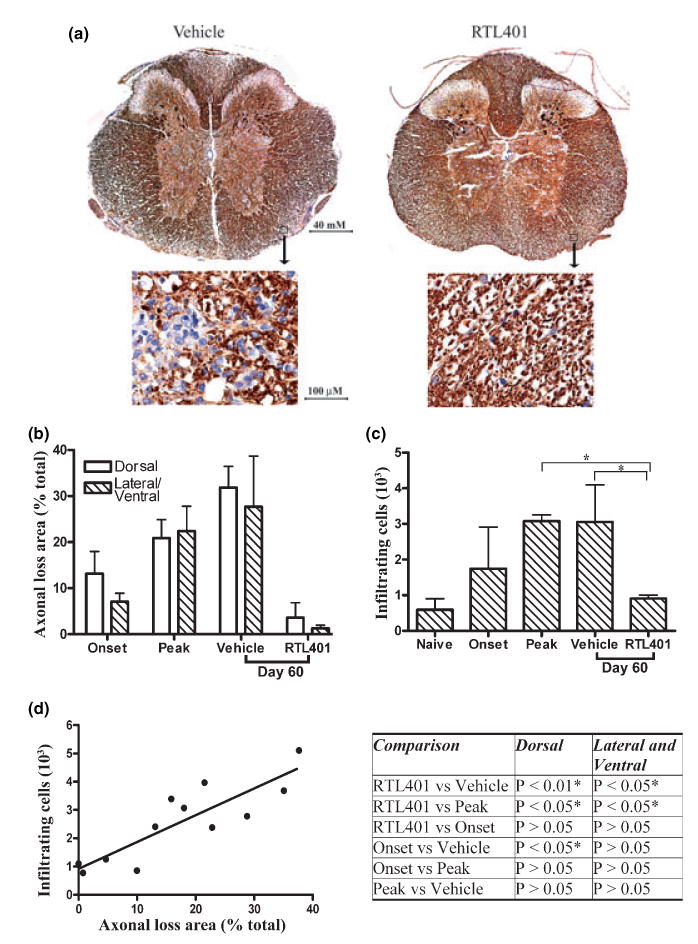
RTL401-treatment decreased axonal loss and inflammation in the spinal cord in EAE. (a) Representative phosphorylated neuro-filament staining of axons in thoracic spinal cord sections from EAE mice treated with vehicle (left panel) or RTL401 (right panel) 60 days after disease induction. Tissue sections were dissected from PBS-perfused mice (4 available mice from a group of eight after 4 randomly chosen mice were perfused by PFA, see Fig. 2), fixed and stained with SMI312, an antibody cocktail for neurofilaments (brown). The nuclei of inflammatory mononuclear cells were visualized by hematoxylin (blue). Images were captured with a compound microscope equipped with a digital camera. (b) Morphometric analysis of the development of axonal loss in EAE mice treated with vehicle or RTL401. EAE mice were euthanized at disease onset (day 11, disease score 1.5), peak (between day 15 and day 20, when disease scores reached 4.5), or at the termination of the experiment (day 60 of EAE, after RTL401 or vehicle treatment). Digitally acquired images were analyzed with BIOQUANT software. Areas with loss of phosphorylated NF staining of axons were circled by hand and traced by BIOQUANT. The percentage of axonal loss area was calculated by dividing total axonal loss areas by the total area of dorsal or lateral/ventral white matter. Data represent mean ± SD (n = 4). (c) RTL401-treatment reduced inflammatory mononuclear cells in the CNS of EAE mice. Total numbers of inflammatory mononuclear cells (stained blue with hematoxylin) in whole thoracic spinal cord sections were counted manually. Data represent mean ± SD (n = 4). *Comparison statistically significant as demonstrated by one-way ANOVA followed by Newman-Kuels multiple comparisons test, n = 4. (d) Correlation of axonal loss to number of inflammatory cells in individual mice. Pearson’s correlation analysis showed a significant correlation, r = 0.8636, p (two-tailed) = 0.0003. Inserted Table: RTL401-treated mice show a significant reduction in axonal loss, demonstrated by one-way ANOVA followed by Newman-Kuels multiple comparisons test. *Comparison statistically significant.
Fig. 4.
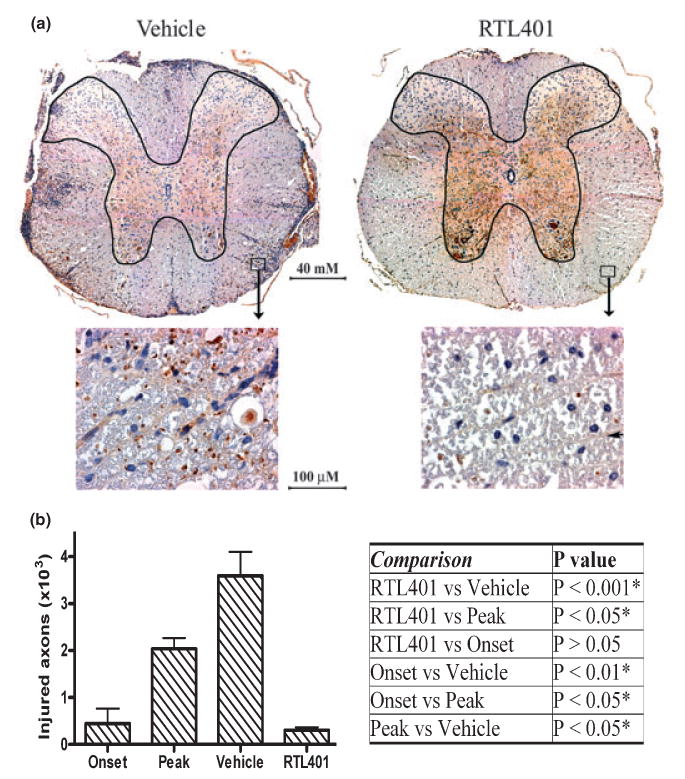
RTL401-treatment reduced axonal injury in the spinal cord of EAE mice. (a) Representative non-phosphorylated neurofilaments (NPNFL), which are abnormally expressed in injured axons, of the thoracic spinal cord from EAE mice treated with vehicle (left panel) or RTL401 (right panel) 60 days after disease induction. Tissue sections were dissected from 4 mice per group, fixed and stained with antibody SMI32 for NPNFL (brown). The nuclei of inflammatory mononuclear cells were stained with hematoxylin (blue). Images were captured with a compound microscope equipped with a digital camera. (b) Morphometric analysis of the total number of injured (NPNFL-positive) axons in the whole thoracic spinal cord white matter in EAE mice. EAE mice were euthanized by perfusion with PBS at disease onset (day 11, disease score 1.5), peak (between day 15 and day 20, when disease scores reached 4.5), or at the termination of the experiment (day 60 of EAE, after RTL401 or vehicle treatment). The numbers of injured axons were counted manually by an investigator with no knowledge of treatment conditions. Data = mean ± SD (n = 4). Inserted Table: RTL401-treated mice show a significant reduction in the number of injured axons, demonstrated by one-way ANOVA followed by Newman-Kuels multiple comparisons test. *Comparison statistically significant.
Fig. 5.
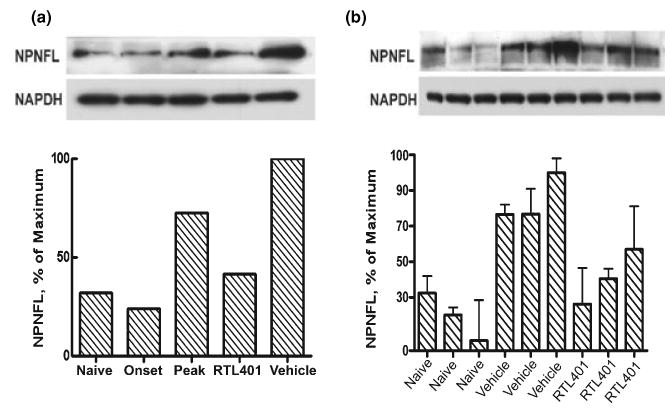
RTL treatment ongoing axonal injury in the lumbar spinal cord of EAE mice. Left panel: Immunoblotting result showing that RTL401 treatment reversed the development of axonal injury as indicated by abnormal expression of non-phosphorylated neurofilaments in lumbar spinal cords from mice with EAE. EAE mice were euthanized by perfusion with PBS at disease onset (day 11, disease score 1.5), peak (between day 15 and day 20, when disease scores reached 4.5), or at the termination of the experiment (day 60 of EAE, after RTL401 or vehicle treatment). The lysates of whole lumbar spinal cords from each group (4 PBS-perfused mice from a group of 8) were pooled and the amount of NPNFL was detected after immunoblotting. Each column represents pooled samples from 4 mice from a single group. Right panel: Immunoblotting results showed the amount of NPNFL in the lumbar spinal cord samples from three randomly chosen mice (3 out of the 4 samples in the pool) from naïve, vehicle-treated (day 60 of EAE) or RTL401-treated (day 60 of EAE) groups. Each band represents a lumbar spinal cord sample from an individual mouse. The experiment was repeated two times.
Histopathology and electron microscopy
The method for histopathology and electron microscopy has been published before (Gold et al. 2004). After deep anesthesia with inhaled isofluorane, each mouse was fixed by perfusion of ice-cold 4% PFA in 0.1 M sodium phosphate buffer (pH 7.4) for 10 s, followed by perfusion of 100 mL 5% glutaraldehyde in 0.1 M sodium phosphate buffer. The mice were left at 4°C for 24 h before their spinal cords were dissected out and cut into sections 1–2 mm in length. Tissues were placed in 0.1 M sodium phosphate buffer (pH. 7.4), post-fixed with 1% osmium tetroxide (in 0.1 M phosphate buffer) for 2.5 h, dehydrated in ethanol and embedded in plastic. Semi-thin sections (0.5 μm) were stained with toluidine blue and photographed at 25× magnification. Thin sections (80–90 nm) were stained with uranyl acetate and lead citrate and examined using a JOEL 100CX electron microscope.
Immunohistochemistry
PBS-perfused thoracic spinal cord was fixed in 4% paraformaldehyde dissolved in 0.1 M sodium phosphate buffer (pH 7.4) at 4°C for at least 48 h. The spinal cords were dissected from the spinal columns, cut into sections 1–2 mm in length from the sampled thoracic cords, re-fixed briefly in 10% Zn-buffered formalin, dehydrated and embedded in paraffin blocks. Then, 10 μM thick sections were cut from paraffin blocks and mounted onto pre-cleaned Superfrost®/Plus microscope slides (Fisher Scientific, Pittsburgh, PA, USA). PloyBath regent (American MasterTech, Lodi, CA, USA) was applied to enhance attachment of sections. The sections were dewaxed and rehydrated sequentially by xylene (2 min), gradient ethanol (100%, 95%, 85%, 2 min each) and PBS (5 min), and then cooked (120°C) in antigen unmasking agent Trilogy® (Cell Marque, Hot Springs, AR, USA) for 10min in a pressure steamer. The endogenous peroxidase activity was blocked with 3% hydrogen peroxide in tap water for 5 min. The sections were incubated 1 h in working solution of Mouse Ig Blocking Reagent from the VECTOR® M.O.M.™ Immunodetection peroxidase kit (Vector Laboratories, Burlingame, CA, USA), and then incubated sequentially with primary antibody (SMI312 1 : 3000 or SMI32 1 : 1000 diluted in M.O.M.™ diluent, 30min), M.O.M.™ biotinylated anti-Mouse IgG reagent (10 min), VECTASTAIN® ABC reagent (5min), and DakoCytomation liquid DAB substrate (DakoCytomation, Carpinteria, CA, USA). The slides were counterstained with VECTOR Hematoxylin QS for 30–60 s to visualize nuclei, dehydrated, and mounted with Cytoseal™ XYL mounting medium (Richard-Allan Scientific, Kalamazoo, MI, USA).
CNS morphometric analysis
Tissue sections were analyzed by an investigator blinded to treatment status. The percentage of the spinal cord showing tissue damage was determined in the mid-thoracic cord. Regions in the (1) dorsal columns and (2) the lateral/ventral white matter tracts containing tissue damage, including disrupted compact myelin, demyelinated axons and degenerating axons, were circumscribed on photomontages (final magnification × 100) of the entire spinal cord. Damaged areas were measured using a SummaSketch III (Summa-graphics, Seymour, CT, USA) digitizing tablet and BIOQUANT Classic 95 software (R & M Biometrics, Nashville, TN, USA). Measurements were also made of the total area (damaged and intact) of (1) the dorsal columns and (2) the lateral/ventral columns. Cumulative percent lesion areas were calculated for each region (dorsal column and lateral/ventral columns) and for the combined total damage from the two regions. Electron microscopy of lesioned areas was also conducted to determine whether these regions contained demyelinated, remyelinating and possibly regenerating (small caliber sprouts) axons.
Western blot (immunoblotting)
The procedure was modified from that reported by Pitt et al. (Pitt et al. 2000). Briefly, the mice were sedated and perfused with cold PBS and their spinal cords were dissected out and cut into thoracic and lumbar cords. The lumbar cords were homogenized in ice-cold RIPA+ buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Non-idet P-40, 0.5% deoxycholate, 0.1% SDS, 1 mM NaCO3, with protease and phosphatase inhibitors) and incubated for 15 min with shaking. After centrifugation (14 000 × g at 4°C for 15 min), the supernatant was collected and the protein concentration was measured and adjusted using extra RIPA+ buffer. Samples were denatured in SDS-sampling buffer for 10min at 70°C, then separated by 10% SDS–PAGE and blotted onto a PVDF membrane. After transfer, the membrane was blocked for 1 h in Tris-buffered saline (TBS) with 3% BSA (Sigma, St. Louis, MO). Immunodetection was accomplished by incubation overnight at 4°C with primary antibody SMI 32 (1/5000 dilution in 3% BSA and 0.05% Tween 20; purchased from Sternberger Monoclonals) specific for non-phosphorylated neurofilaments (NPNFL). After being washed, the blots were incubated with HRP-labeled goat antibody against mouse IgG (1/5000 dilution in 3% BSA and 0.05% Tween® 20) for 1 h and then washed. Blots were developed with a SuperSignal West Pico Chemiluminescent kit (Pierce). To precisely control the amounts of protein loaded, the membranes were stripped with the Restore Western Blot Stripping Buffer (Pierce) and detected again with an antibody for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) purchased from Chemicon International. After being developed, the films were scanned and quantified with ImageQuant software (Amersham Biosciences).
Results
We investigated whether established clinical EAE is treatable by RTL401 in this study. After immunization with PLP-139–151 peptide/CFA, SJL/J mice developed a typically relapsing EAE disease course, with onset of the initial episode of acute disease occurring on or after day 11, and peak clinical scores developing between day 15–20, followed by a short clinical improvement in length varying from mouse to mouse. After two groups of mice (‘Onset’ and ‘Peak’, 8 per group) were euthanized on disease onset and peak, respectively, additional mice were divided into two groups on day 20 according to matched collective disease scores (2.8 ± 0.7 for vehicle-treated mice and 2.6 ± 0.5 for RTL401-treated mice) and treated with RTL401 or vehicle. As shown in Fig. 1, 5 daily i.v. plus 3 sc. injections of RTL401 administered after the peak of disease steadily lowered the disease scores of EAE. Compared to mice in the vehicle-treated group, RTL401-treated mice did not show further disease progression after the first relapse (Fig. 1). At the end of the experiment on day 60, the mean EAE score of RTL401-treated mice was 2.0 ± 0.4, compared to 3.7 ± 0.7 for vehicle-treated mice. RTL401 treatment also significantly lowered the cumulative disease index (CDI) of EAE mice (Inserted table in Fig. 1). Our results show that established EAE can be treated by delayed administration of RTL401.
Fig. 1.
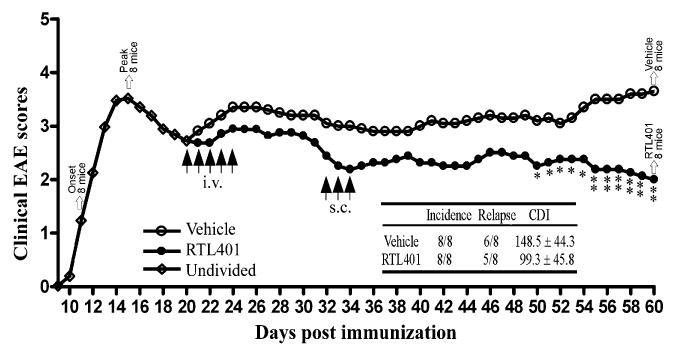
RTL401 treatment of SJL mice with established clinical EAE. Active EAE was induced in 32 female SJL mice by immunization with PLP-139–151 peptide/CFA. Eight mice (Onset) were euthanized at disease onset and another 8 (Peak) were euthanized at the peak of disease. On day 20, just after the peak of the first episode of EAE, the remaining mice were divided into two groups with matched disease scores (2.8 ± 0.7 for vehicle-treated mice vs 2.6 ± 0.5 for RTL401 treated mice) and treated daily for 5 days i.v. with RTL401 (100 μg/mouse) or vehicle. On day 32, the two groups of mice were boosted with 3 daily s.c. injections of RTL401 or vehicle and monitored for change of disease scores until the conclusion of the experiment (day 60). Inserted table: The effect of RTL401 treatment on cumulative disease index (CDI) and number of relapses of SJL mice with EAE. Data represent averages of summed daily disease scores for all available mice. Statistically significant difference in daily or collective EAE scores between RTL401- vs. vehicle-treated mice is demonstrated by Student t-test, p < 0.05* or 0.01**.
We assessed the impact of RTL401-treatment on tissue injury, including areas in which there was loosening of the myelin sheaths, demyelinated axons and degenerating axons, in established EAE by morphometric quantification of the area of tissue injury in mice during different disease phases, with or without RTL401 treatment. Demyelination was demonstrated by quantification of toluidine blue stained plastic sections of spinal cords, a method used previously by other investigators to show or quantify demyelination and remyelination (Kondo et al. 2005; Papadopoulos et al. 2006). At the onset of the disease (disease score 1.5 for all mice), tissue damage in the dorsal and ventral/lateral white matter was mild, but progressively worsened by peak (between day 15 and day 20, disease score 4.5 for all mice) and day 60 (disease score 3.7 ± 0.7 for vehicle-treated mice, Fig. 2). Mice treated with RTL401 (disease score 2.0 ± 0.4) but not with vehicle showed a marked reduction in the total damaged areas in dorsal and lateral/ventral thoracic spinal cords (Fig. 2a). The damaged areas in the dorsal and lateral/ventral white matter of RTL401-treated mice were 2% and 1%, respectively, compared to 29% and 24%, respectively, in vehicle-treated mice (Fig. 2b). In addition, the degree of myelin damage in RTL401-treated mice on day 60 was significantly lower than those in mice prior to treatment on day 20 and in vehicle-treated mice on day 60 (Fig. 2b), suggesting that RTL401 treatment promoted recovery from myelin injury in EAE.
Additionally, we determined the level of axonal loss in the thoracic spinal cord of 4 mice from each group. Existing axons can be visualized by immunohistochemistry staining with SMI312, an antibody cocktail for neurofilaments. As depicted in Fig. 3(a), axons were stained dark brown with SMI312 and inflammatory cells were stained bright blue with hematoxylin. Without therapeutic intervention, axonal staining was markedly reduced in the presence of inflammatory mononuclear cells, resulting in severe loss of SMI312 staining in the outer region of white matter, where most neuroinflammation occurred. Axons in the spinal cord of RTL401-treated mice, to the contrary, were well preserved. Hematoxylin blue stained immune cells were much less frequent in the spinal cords of RTL-treated mice (Fig. 3a and c). The areas of axonal loss in the dorsal and lateral/ventral spinal cords of vehicle vs. RTL401-treated mice were 31.8% and 27.6% vs. 3.5% and 1.3%, respectively. Statistical analyses indicated that RTL401-treatment significantly reversed the trend of progressive development of both axonal injury and neuroinflammation (Fig. 3b, c). As shown in Fig. 3(d), the degree of axonal damage correlated significantly with neuroinflammation, as demonstrated by Pearson’s correlation analysis (r = 0.8636, p (two-tailed) = 0.0003). This observation suggests that RTL401 may reduce CNS damage by reducing infiltration of immune cells into the spinal cord.
The degree of ongoing damage in EAE mice was also investigated by detecting the number of injured axons with SMI32 staining. Different from SMI312, the SMI32 antibody specifically stains non-phosphorylated neurofilaments (NPNFL) that are present only in injured and demyelinated axons (Trapp et al. 1998; Pitt et al. 2000). This staining thus demonstrates the degree of ongoing damage rather than a reduction in axonal staining. As is shown in Fig. 4, RTL401-treated mice showed much less axonal injury and secondary demyelination in the white matter of the thoracic spinal cord. Similar to reduced axonal staining, the degree of ongoing axonal injury and demyelination appeared to be associated closely with inflammation. Additionally, immunoblotting for NPNFL with SMI32 demonstrated that axonal injury in both lumbar and thoracic spinal cord tissue from EAE mice was reduced on day 60 after RTL401-treatment compared to samples from mice at the peak of EAE or in vehicle-treated mice evaluated on day 60 (Fig. 5).
Electron microscopy of spinal cord sections revealed considerable ongoing Wallerian-like axonal degeneration and large numbers of inflammatory cells at the peak of disease (15–20 days, Fig. 6). However, normal recovery processes were apparently able to compensate for the degree of damage, as evidenced by the presence of remyelinating axons and very small axons, most likely representing regenerating sprouts (Fig. 6). Vehicle treated control mice showed continued worsening of the disease process by 60 days, as shown by the increase in Wallerian-like axonal degeneration, continued axonal demyelination and the lack of axonal sprouts (Fig. 7, upper row). In contrast, the RTL401-treated animals on day 60 demonstrated reduced pathology compared to the peak of disease, as demonstrated by the decrease in continued degeneration, increase in remyelinating axons and the presence of an increased number of axonal sprouts (Fig. 7, lower row).
Discussion
Our previous studies showed that RTL401, a highly effective antigen-specific immunomodulatory agent, could ameliorate EAE when administered at disease onset. This is clearly a therapeutic effect on disease progression, but differs from the situation in MS where therapy is initiated some time after onset of the initial symptoms. In recent years, neuroaxonal damage in both white and gray matter tissue has been recognized as a central feature in the pathogenesis of progressive MS (Trapp et al. 1998). Therefore, it is crucial to assess the effect of RTL401 in treating established EAE and associated myelin and axonal damage before testing its human version in MS. In this study, we induced active EAE in SJL/J mice with PLP-139–151 peptide/CFA and waited until after the peak of the initial clinical episode (day 20) to start the RTL401 treatment. Although the effect was not as pronounced as earlier intervention (Huan et al. 2004), delayed treatment with RTL401 still ameliorated the disease course, thus indicating a therapeutic effect during the relapsing phase of disease. Administration of RTL401 displayed its therapeutic effect slowly and gradually compared to vehicle-treated controls, only significantly lowering the disease scores beginning on day 50. Our clinical data is consistent with the notion that it is more difficult and time consuming to impact or reverse axonal damage, which steadily accumulates during EAE, than neuroinflammation, using RTL therapy directed at myelin-antigen-specific T cells.
It is not totally surprising that RTL401 treatment reversed the progression of demyelination in the CNS of EAE mice. It has been suggested that up to 40% of MS plaques in the CNS show signs of remyelination (Barkhof et al. 2003; Bruck 2005). Nonetheless, remyelination is generally incomplete in patients and is characterized by thinner myelin sheaths and shorter internodal lengths than in normal myelin. One of the possible factors that may impede this otherwise biologically spontaneous process is the low level inflammation that may exist even in relatively inactive lesions. We show here that RTL401 treatment apparently enables a nearly complete reversal of demyelination. A recent report suggested that accumulation of hyaluronic acid (HA) in the white matter may contribute directly to the inhibition of remyelination (Back et al. 2005), and our future studies in collaboration with these investigators will address whether our antigen-specific therapy can prevent HA from accumulating in the CNS.
Of importance to the neurodegenerative process, we showed in the current study that RTL401 treatment reversed the progression of axonal loss and ongoing injury in EAE. This finding has potential clinical significance since axonal injury appears to be the main factor responsible for the accrual of neurological deficits manifested in progressive MS (Waxman 1998; Rieckmann and Smith 2001; Rammohan 2003). In contrast to demyelination, injured or transected adult CNS axons do not regenerate spontaneously in brain trauma or traditional neurodegenerative diseases. Evidence suggests that this failure of regeneration is not due to an intrinsic inability of axons to regenerate, but to unfavorable CNS environments, such as inhibition from myelin-associated inhibitors (Grados-Munro and Fournier 2003). Specifically, blockade of the signaling pathway employed by myelin-associated inhibitors promoted regeneration of CNS axons both in vitro and in vivo (Koprivica et al. 2005). Thus, reversal of axonal damage might only necessitate overcoming the inhibitory signals. In MS and EAE, axons may have lost their immediate contact with myelin-associated inhibitors due to demyelination, thus allowing axonal repair to occur once inflammation has ceased. We found that CNS inflammatory cells were significantly reduced in RTL401-treated mice, which was correlated with the enhanced axonal staining. Therefore, RTL401 might facilitate the natural axonal repair process through antigen-specific inhibition of neuroinflammation.
There is no direct evidence from our study that RTL401 can stimulate neuroregeneration by acting directly on neurons. However, we cannot exclude the possibility that RTL401 may provide neuroprotection by inducing secretion of anti-inflammatory Th2 cytokines. While excessive production of Th1 proinflammatory cytokines has been shown to cause neuronal cell injury and death, the presence of Th2 anti-inflammatory cytokines, including IL-4 and IL-10, tends to promote neuronal protection and survival in the CNS (D’Souza et al. 1995; Szelényi 2001). Interestingly, we showed before that RTL401 can induce a Th1 to Th2 cytokine switch in encephalitogenic T cells (Burrows et al. 2001; Huan et al. 2004).
Our electron microscopic observations also indicate that RTL treatment prevents continued inflammation, thereby reducing the degree of damage from the peak of disease and, consequently, enabling the normal processes of recovery (i.e. remyelination and axonal regeneration) intrinsic to these animals to occur. Remyelination and axonal sprouting were also observed previously in SJL/J mice given FK506 (at either an immunosuppressant or non-immunosuppressant dose) or a non-immunosuppressant FK506 derivative (FK1706) (Gold et al. 2004), indicating that these are common features of these models regardless of the underlying process (reduced inflammation or axonal protection) leading to recovery from damage.
It is worthy to note that functional integrity of whole axons may not be restored completely on day 60, despite a nearly complete morphological CNS recovery. Compared to vehicle-treated mice, RTL401-treated ones showed significant and consistent improvement during the late stage of treatment (mean EAE score 2.0 ± 0.4, compared to 3.7 ± 0.7 for vehicle-treated mice, Fig. 1). However, signs of mild EAE were still detectable in the majority of these mice, indicating an incomplete functional recovery. Electrophysiological dysfunction in the axons of both vehicle- and RTL401-treated mice will be investigated in our future studies.
Although it is generally accepted that demyelination in MS is caused by immune attack, whether axonal damage is directly associated with neuroinflammation is still controversial. First, axonal injury tends to be more severe in active inflammatory MS lesions than inactive lesions (Ferguson et al. 1997; Trapp et al. 1998), suggesting neuroinflammation might contribute to axonal damage in MS. Indeed, excess production of proinflammatory cytokines, including IL-1, TNF-α, IFN-α, and IL-12, has been shown to cause neuronal cell injury and apoptosis in the CNS (D’Souza et al. 1995; Szelényi 2001). The expression of MHC class I molecules in neurons, which can be up-regulated by TNF-α and IFN-α, and the presence of clonally expanded CD8+ T cells in MS lesions implied cytotoxic lymphocyte mediated axonal damage. Recently, a study using two-photon microscopy showed that encephalitogenic CD4+ T cells could also attack neurons directly (Nitsch et al. 2004). Finally, we showed that passively transferred myelin-reactive T cells can induce axonal injury in recipient mice (Offner et al. 2005). On the other hand, there is separate evidence indicating axonal damage may develop independent of neuroinflammation. For instance, while inflammatory activities are mostly restricted to focal lesions, neuroaxonal damage is more diffuse in both lesioned and normal-appearing white and grey matter (Arnold et al. 1992; Fu et al. 1998). In MS patients who received autologous hematopoietic cell transplantation, autopsy and MRI studies showed that axonal damage and brain atrophy continued, even though inflammation in the CNS appeared to cease (Mancardi et al. 2001; Fassas et al. 2002; Inglese et al. 2004). Additionally, neuroinflammation may be neuroprotective by releasing neurotrophic factors and removing excitotoxic glutamic acid (Kerschensteiner et al. 2003; Moalem et al. 1999; Szelényi 2001). Here, we found that axonal injury was correlated with neuroinflammation spatially, temporally and in degree. Reduction of inflammatory cells by RTL401-treatment reversed the progression of EAE and axonal damage. These results support a direct etiological relationship between neuroaxonal damage and neuroinflammation in EAE. Better understanding of the etiological relationship between neuroaxonal damage and neuroinflammation is important for drug development in MS.
Taken together, we showed that RTL401, when administered after the peak of relapsing EAE, not only prevented disease relapses, but also markedly reduced demyelination, axonal damage and ongoing injury in the CNS. This study provides the necessary foundation for the clinical application of RTLs in MS patients to prevent or treat myelin and axonal damage. Our findings also support a direct etiological relationship between neuroaxonal damage and neuroinflammation in EAE.
Acknowledgments
The authors wish to thank Ms. Eva Niehaus for assistance in preparing the manuscript. This work was supported by NIH Grants NS47661, AI43960, NS41965, and NS46877; National Multiple Sclerosis Society Grants RG3468A, RG3794A and PP1104; Artielle Immuno-Therapeutics, Inc.; The Nancy Davis MS Center without Walls; and the Biomedical Laboratory R & D Service, Department of Veterans Affairs.
Abbreviations used
- EAE
experimental autoimmune encephalomyelitis
- MS
multiple sclerosis
- PPMS
primary progressive MS
- RRMS
relapsing-remitting phase
- RTL
recombinant T cell receptor ligand
- SPMS
secondary progressive MS
References
- Arnold DL, Matthews PM, Francis GS, O’Connor J, Antel JP. Proton magnetic resonance spectroscopic imaging for metabolic characterization of demyelinating plaques. Ann Neurol. 1992;31:235–241. doi: 10.1002/ana.410310302. [DOI] [PubMed] [Google Scholar]
- Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Jr, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Barkhof F, Bruck W, De Groot CJ, Bergers E, Hulshof S, Geurts J, van Polman CH, van Polman dV. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch Neurol. 2003;60:1073–1081. doi: 10.1001/archneur.60.8.1073. [DOI] [PubMed] [Google Scholar]
- Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol. 2005;252:v3–v9. doi: 10.1007/s00415-005-5002-7. [DOI] [PubMed] [Google Scholar]
- Burrows GG, Adlard KL, Bebo BF, Jr, Chang JW, Tenditnyy K, Vandenbark AA, Offner H. Regulation of encephalitogenic T cells with recombinant TCR ligands. J Immunol. 2000;164:6366–6371. doi: 10.4049/jimmunol.164.12.6366. [DOI] [PubMed] [Google Scholar]
- Burrows GG, Chou YK, Wang C, Chang JW, Finn TP, Culbertson NE, Kim J, Bourdette DN, Lewinsohn DA, Lewinsohn DM, Ikeda M, Yoshioka T, Allen CN, Offner H, Vandenbark AA. Rudimentary TCR signaling triggers default IL-10 secretion by human Th1 cells. J Immunol. 2001;167:4386–4395. doi: 10.4049/jimmunol.167.8.4386. [DOI] [PubMed] [Google Scholar]
- Chang JW, Mechling DE, Bachinger HP, Burrows GG. Design, engineering, and production of human recombinant T cell receptor ligands derived from human leukocyte antigen DR2. J Biol Chem. 2001;276:24 170–24 176. doi: 10.1074/jbc.M101808200. [DOI] [PubMed] [Google Scholar]
- D’Souza S, Alinauskas K, McCrea E, Goodyer C, Antel JP. Differential susceptibility of human CNS-derived cell populations to TNF-dependent and independent immune-mediated injury. J Neurosci. 1995;15:7293–7300. doi: 10.1523/JNEUROSCI.15-11-07293.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassas A, Passweg JR, Anagnostopoulos A, et al. Hematopoietic stem cell transplantation for multiple sclerosis. A retrospective multicenter study. J Neurol. 2002;249:1088–1097. doi: 10.1007/s00415-002-0800-7. [DOI] [PubMed] [Google Scholar]
- Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- Fu L, Matthews PM, De Stefano N, Worsley KJ, Narayanan S, Francis GS, Antel JP, Wolfson C, Arnold DL. Imaging axonal damage of normal-appearing white matter in multiple sclerosis. Brain. 1998;121:103–113. doi: 10.1093/brain/121.1.103. [DOI] [PubMed] [Google Scholar]
- Gold BG, Voda J, Yu X, Mckeon G, Bourdette DN. FK506 and a nonimmunosuppressant derivative reduce axonal and myelin damage in experimental autoimmune encephalomyelitis: neuroimmunophilin ligand-mediated neuroprotection in a model of multiple sclerosis. J Neurosci Res. 2004;77:367–377. doi: 10.1002/jnr.20165. [DOI] [PubMed] [Google Scholar]
- Grados-Munro EM, Fournier AE. Myelin-associated inhibitors of axon regeneration. J Neurosci Res. 2003;74:479–485. doi: 10.1002/jnr.10803. [DOI] [PubMed] [Google Scholar]
- Huan J, Subramanian S, Jones R, Rich C, Link J, Mooney J, Bourdette DN, Vandenbark AA, Burrows GG, Offner H. Monomeric recombinant TCR ligand reduces relapse rate and severity of experimental autoimmune encephalomyelitis in SJL/J mice through cytokine switch. J Immunol. 2004;172:4556–4566. doi: 10.4049/jimmunol.172.7.4556. [DOI] [PubMed] [Google Scholar]
- Inglese M, Mancardi GL, Pagani E, Rocca MA, Murialdo A, Saccardi R, Comi G, Filippi M. Brain tissue loss occurs after suppression of enhancement in patients with multiple sclerosis treated with autologous haematopoietic stem cell transplantation. J Neurol Neurosurg Psychiatry. 2004;75:643–644. [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Stadelmann C, Dechant G, Wekerle H, Hohlfeld R. Neurotrophic cross-talk between the nervous and immune systems: implications for neurological diseases. Ann Neurol. 2003;53:292–304. doi: 10.1002/ana.10446. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Wenger DA, Gallo V, Duncan ID. Galacto-cerebrosidase-deficient oligodendrocytes maintain stable central myelin by exogenous replacement of the missing enzyme in mice. Proc Natl Acad Sci USA. 2005;102:18 670–18 675. doi: 10.1073/pnas.0506473102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprivica V, Cho KS, Park JB, Yiu G, Atwal J, Gore B, Kim JA, Lin E, Tessier-Lavigne M, Chen DF, He Z. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310:106–110. doi: 10.1126/science.1115462. [DOI] [PubMed] [Google Scholar]
- Link JM, Rich CM, Korat M, Burrows GG, Offner H, Vandenbark AA. Preclinical studies demonstrate rapid and potent modulation of experimental encephalomyelitis in humanized DR2+ transgenic mice treated with a soluble recombinant TCR ligand containing DR2/MOG-35-55 peptide. Clin Immunol. 2006 submitted. [Google Scholar]
- Lublin F. Multiple sclerosis trial designs for the 21 (st) century: Building on recent lessons. J Neurol. 2005;252:v46–v53. doi: 10.1007/s00415-005-5008-1. [DOI] [PubMed] [Google Scholar]
- Mancardi GL, Saccardi R, Filippi M, et al. Autologous hematopoietic stem cell transplantation suppresses Gd-enhanced MRI activity in MS. Neurology. 2001;57:62–68. doi: 10.1212/wnl.57.1.62. [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- Nitsch R, Pohl EE, Smorodchenko A, Infante-Duarte C, Aktas O, Zipp F. Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J Neurosci. 2004;24:2458–2464. doi: 10.1523/JNEUROSCI.4703-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Wang C, Afentoulis M, Vandenbark AA, Huan J, Burrows GG. Treatment of passive experimental autoimmune encephalomyelitis in SJL mice with a recombinant TCR ligand induces IL-13 and prevents axonal injury. J Immunol. 2005;175:4103–4111. doi: 10.4049/jimmunol.175.6.4103. [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol. 2006;197:373–385. doi: 10.1016/j.expneurol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Rammohan KW. Axonal injury in multiple sclerosis. Curr Neurol Neurosci Rep. 2003;3:231–237. doi: 10.1007/s11910-003-0083-0. [DOI] [PubMed] [Google Scholar]
- Rieckmann P, Smith KJ. Multiple sclerosis: more than inflammation and demyelination. Trends Neurosci. 2001;24:435–437. doi: 10.1016/s0166-2236(00)01860-9. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J. Protective autoimmunity and neuro-protection in inflammatory and noninflammatory neurodegenerative diseases. J Neurol Sci. 2005;233:163–166. doi: 10.1016/j.jns.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Steinman L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell. 1996;85:299–302. doi: 10.1016/s0092-8674(00)81107-1. [DOI] [PubMed] [Google Scholar]
- Szelényi J. Cytokines and the central nervous system. Brain Res Bull. 2001;54:329–338. doi: 10.1016/s0361-9230(01)00428-2. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Vandenbark AA, Rich C, Mooney J, Zamora A, Wang C, Huan J, Fugger L, Offner H, Jones R, Burrows GG. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35–55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J Immunol. 2003;171:127–133. doi: 10.4049/jimmunol.171.1.127. [DOI] [PubMed] [Google Scholar]
- Wang C, Mooney JL, Meza-Romero R, Chou YK, Huan J, Vandenbark AA, Offner H, Burrows GG. Recombinant TCR ligand induces early TCR signaling and a unique pattern of downstream activation. J Immunol. 2003;171:1934–1940. doi: 10.4049/jimmunol.171.4.1934. [DOI] [PubMed] [Google Scholar]
- Waxman SG. Demyelinating diseases – new pathological insights, new therapeutic targets. N Engl J Med. 1998;338:323–325. doi: 10.1056/NEJM199801293380610. [DOI] [PubMed] [Google Scholar]