

Abstract
Molecular classification of colorectal cancer is evolving. As our understanding of colorectal carcinogenesis improves, we are incorporating new knowledge into the classification system. In particular, global genomic status [microsatellite instability (MSI) status and chromosomal instability (CIN) status] and epigenomic status [CpG island methylator phenotype (CIMP) status] play a significant role in determining clinical, pathological and biological characteristics of colorectal cancer. In this review, we discuss molecular classification and molecular correlates based on MSI status and CIMP status in colorectal cancer. Studying molecular correlates is important in cancer research because it can 1) provide clues to pathogenesis, 2) propose or support the existence of a new molecular subtype, 3) alert investigators to be aware of potential confounding factors in association studies, and 4) suggest surrogate markers in clinical or research settings.
Why Molecular Classification?
Colorectal cancer (CRC) is not a single disease. Rather, CRC encompasses a heterogeneous complex of diseases, and each CRC patient has a unique disease that has been caused by distinctive genetic/epigenetic background. Theoretically, every CRC arises and behaves in a unique fashion that is unlikely to be exactly recapitulated by any other CRC. Nonetheless, we believe that tumors with similar characteristics most likely arise or behave in a similar way. The purpose of tumor classification is to find similar characteristics among individual tumors, to predict empirically the pathogenesis and biological behavior of a particular tumor.
Tumor classification is historically based on various clinical (eg, proximal versus distal), pathological (eg, mucinous versus nonmucinous; well-moderate versus poorly differentiated), and/or molecular features [eg, microsatellite instability status (MSI)-high versus microsatellite stable (MSS)].1,2,3 Molecular classification is important because it reflects underlying mechanisms of carcinogenesis. In other cancers, for example, molecular classification of leukemias and lymphomas has considerably advanced the field over the past couple of decades.4,5,6,7,8 Although clinical and pathological classifications are largely phenotypic, clinicopathological features are nonetheless very important because some of the features are associated with particular underlying molecular defects and are thus useful in estimating the likelihood of a particular molecular subtype.
There are different ways of determining molecular classification. One can theoretically divide tumors into different groups by the presence or absence of any molecular event(s). However, as a primary discriminator in classification, emphasis should be put on molecular classification based on global cellular events [such as chromosomal instability (CIN), MSI, and CpG island methylator phenotype (CIMP)]. Nonetheless, single molecular events are also useful classifiers, in particular, for predicting response to targeted therapies against those molecules.
Why Molecular Correlates?
CRCs arise through a multistep carcinogenic process in which genetic and epigenetic alterations accumulate in a sequential manner.9,10 Although these molecular alterations may occur in a stochastic fashion in many different cells, these alterations accumulate in a nonrandom fashion in a tumor, probably caused by selection advantages or disadvantages of many of these alterations. This nonrandom accumulation of molecular alterations creates an association between one alteration and another in tumors. Therefore, studying molecular correlates in tumor helps decipher nonrandomness in the multistep carcinogenic process. Molecular correlates in synchronous colorectal neoplasias support a nonrandom process of epigenetic alterations during carcinogenesis.11
The ultimate goal of studying molecular correlates is to identify clinically useful biomarkers, which correlate with patient survival or treatment response or help in treatment decision making or genetic counseling. Molecular correlates also advance further research; simple reports of some molecular correlates may give clues to other investigators.
The purposes of discovering molecular correlates in cancer research are multitude, and four major purposes are as follows.
Molecular Correlates Can Provide Clues to Pathogenesis
Positive correlations can support direct cause-effect relationship, and negative correlations can support the existence of mutually exclusive pathways of carcinogenesis. For example, an inverse association between CIN and MSI can propose the hypothesis that CIN and MSI represent mutually exclusive pathways of tumorigenesis. A mutually exclusive relationship between KRAS and BRAF activating mutations in CRC can confirm the theory that KRAS and BRAF are present in the same signal transduction pathway (see Table 1 for a full list of gene names and abbreviations). In contrast, a PIK3CA mutation in colorectal cancer tends to coexist with KRAS or BRAF mutation,12 supporting the parallel pathways of RAS-RAF-MAPK and PI3K-AKT and also suggesting synergistic effects of both pathways on the downstream mTOR (FRAP1) pathway. As another example, a strong correlation between CIN and Aurora kinase A amplification supports the hypothesis that Aurora kinase A amplification is one of the causes of CIN.13
Table 1.
Abbreviations and HUGO Gene Nomenclature Committee (HGNC)-Approved Official Gene Symbols
| APC | adenomatous polyposis coli |
| AURKA | Aurora kinase A (STK15/BTAK) |
| CACNA1G | calcium channel, voltage-dependent, T type alpha-1G subunit |
| CDKN1A | cyclin-dependent kinase inhibitor 1A (p21/CIP1) |
| CDKN1B | cyclin-dependent kinase inhibitor 1A (p27/KIP1) |
| CDKN2A | cyclin-dependent kinase inhibitor 1A (p16/INK4A) |
| CGH | comparative genomic hybridization |
| CHFR | checkpoint with forkhead and ring finger domains |
| CIMP | CpG island methylator phenotype |
| CIN | chromosomal instability |
| CRABP1 | cellular retinoic acid binding protein 1 |
| CRC | colorectal cancer |
| CTNNB1 | catenin, beta 1 (β-catenin) |
| FASN | fatty acid synthase |
| FBXW7 | F-box and WD repeat domain containing 7 (CDC4) |
| FISH | fluorescence in situ hybridization |
| 5-FU | 5-fluorouracil |
| HNPCC | hereditary nonpolyposis colorectal cancer |
| HR | hazard ratio |
| IGF2 | insulin-like growth factor 2 |
| LOH | loss of heterozygosity |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| MINT | methylated in tumor |
| MSI | microsatellite instability |
| MSI-H | microsatellite instability-high |
| MSI-L | microsatellite instability-low |
| MSS | microsatellite stable |
| NEUROG1 | neurogenin 1 |
| PTGS2 | prostaglandin-endoperoxide synthase 2 (COX-2, cyclooxygenase-2) |
| RUNX3 | runt-related transcription factor 3 |
| SNP | single nucleotide polymorphism |
| SOCS1 | suppressor of cytokine signaling 1 |
| TP53 | tumor protein p53 |
| WNT | wingless-type MMTV integration site |
Molecular Correlates Can Propose or Support the Existence of a New Molecular Subtype
The existence of a new molecular subtype can be supported by the presence of unique molecular correlates. For example, an association between CIMP-high and BRAF mutation supports the existence of CIMP-high as a distinct phenotype in CRC. An association between CIMP-low and KRAS mutation can propose CIMP-low as a new molecular subtype in CRC.14
Molecular Correlates Can Alert Investigators to Be Aware of Potential Confounding Factors in Association Studies
The presence of potential confounding factors is always a concern in any association study. For example, an investigator has found that MLH1 methylation is associated with resistance to 5-fluorouracil (5-FU)-based chemotherapy and may conclude that MLH1 methylation confers chemoresistance.15 However, MLH1 methylation is associated with MSI-high, and if chemoresistance is caused by MSI-high, but not by MLH1 methylation per se, anyone may be able to show the association between MLH1 methylation and chemoresistance. If this investigator knew the association between MLH1 methylation and MSI-high, this investigator could avoid a wrong conclusion.
As another example, investigators have found that an infiltrate of a subset of lymphocytes in CRC is associated with patient survival and concluded that a lymphocytic infiltrate kills tumor cells and helps patients live longer.16 However, in this study, longer survival may be due to the effect of MSI, which is associated with lymphocytic infiltrate. Alternatively, the known association between MSI and longer survival may be due to lymphocytic infiltrate. Unless these investigators perform multivariate analysis, including MSI data and lymphocytic infiltrate, results cannot support a causal link between lymphocytes and longer survival.
Molecular Correlates Can Suggest Surrogate Markers in Clinical or Research Settings
Surrogate markers are useful when it is difficult or impossible to test exactly what molecular alterations are present in an individual patient. There are many examples of good surrogate markers.
p53 immunohistochemistry is performed as a surrogate marker for the presence of p53 (TP53) mutation, because p53 positivity by immunohistochemistry correlates well with the presence of TP53 mutation and functional loss.17 Because mutations are distributed diversely in the TP53 gene, one may need to sequence the entire TP53 gene to achieve high sensitivity for mutation detection; however, it is often not practical to sequence the entire TP53 gene in a large-scale clinical study or clinical setting.
MSI markers (whether the 5 markers recommended by National Cancer Institute (NCI) or an expanded panel containing 10 or more markers) are good surrogate markers for global microsatellite instability level and underlying mismatch repair defect. Although it is not possible at this time to test all microsatellites in the human genome, testing 5 to 10 markers can certainly predict a defect of mismatch repair system and global microsatellite instability in CRC.
Molecular Classification of CRC
Global Molecular Classifiers: CIN, MSI, and CIMP
To study correlates with molecular events in CRC or patient outcomes, it is important to classify CRCs according to global genomic or epigenomic status. We herein discuss global molecular classifiers: CIN, MSI, and CIMP. Nonetheless, single molecular events are also useful classifiers, in particular for predicting response to targeted therapies against those molecules.
CIN
CIN appears to be a distinct phenotype in colorectal cancer, and tumors with CIN show frequent karyotypic abnormalities and chromosomal gains and losses.18 Allelic losses are quite common in CRC,19 and CIN is considered to promote carcinogenesis through loss of tumor suppressors and copy number gains of oncogenes. Although the occurrences of chromosomal abnormalities may be more stochastic than nonrandom, selection process can make a nonrandom pattern of chromosomal aberrations in tumor cells. CIN and MSI tend to be mutually exclusive in CRC.20
CIN has been commonly assessed by DNA ploidy analysis or loss of heterozygosity (LOH) analyses of microsatellite markers. For LOH analyses, markers in the 18q region have been shown to be generally more sensitive, compared with markers in other chromosomal regions such as 1p, 2p, 3p, 5q, 8p, and 17p.21,22,23,24,25 However, markers and criteria for CIN have not been standardized. In addition, LOH analyses are prone to have false-positive results (due to PCR bias or allele dropout), false-negative results (due to PCR bias or contamination of non-neoplastic cells), and uninformative results (due to homozygosity in a particular marker or unavailability of normal germline DNA).
Recently, array-based comparative genomic hybridization (array-CGH) and single nucleotide polymorphism (SNP) arrays have been used to study copy number gains/losses and LOH. Compared with conventional CGH, both techniques have higher resolution in genome-wide analysis of DNA copy number gains and losses. Major issues are assay cost and a requirement of high quality DNA. Although technically challenging, application to paraffin-embedded tissue is possible.
CIN may represent a heterogeneous phenomenon. CRC can have multiple reciprocal translocations with little changes in allele copy numbers or DNA content.26 Such a CRC would be misclassified as CIN negative by copy number variation assays including LOH or array-CGH. The existence of different mechanisms of CIN (whole chromosomal LOH, mitotic recombination, and mitotic gene conversion) is also suggested by a comprehensive study using array-CGH, SNP arrays, and multicolor fluorescence in situ hybridization (FISH).24
Causes of CIN are also likely heterogeneous. Mutations in genes encoding mitotic checkpoint proteins such as BUB1 and BUB1B (BUBR1) may cause CIN in a subset of CRCs.27 In addition, abnormal centrosome number and function have been a candidate mechanism for CIN. Amplification of AURKA (Aurora kinase A, STK15/BTAK), a centrosome-associated serine threonine kinase, has been found in a colon cancer cell line28 and is correlated with CIN in colon cancer.13 Overexpression of Aurora kinase A can induce aneuploidy in various cell lines.29 Other candidate causes of CIN include APC,30,31 TP53,32 FBXW7 (CDC4 ubiquitin ligase),33 CHFR34,35 (for controversial view, see Ref.36), and JC virus37,38 (for controversial view, see Ref.39).
MSI
MSI refers to altered lengths (“instability”) of short nucleotide repeat sequences (“microsatellites”) in tumor DNA compared with normal DNA.40 It has also been referred to as RER (replication error), mutator phenotype, and microsatellite instability (MIN); however, MSI has become the most commonly used term. MSI has been suggested as a carcinogenic mechanism alternative to the CIN pathway.40 Mutations of coding mononucleotide repeats in tumor suppressor genes such as the transforming growth factor (TGF)-β receptor type 2 (TGFBR2) and BAX have been shown to be important in carcinogenesis.41,42 A high degree of MSI (MSI-H) has been shown to be due to defects in the DNA mismatch repair system. Functional loss of MLH1 due to promoter methylation and gene silencing is the most common cause of MSI, particularly in sporadic MSI-H cancer. In contrast, in the setting of hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome, mutations in any of the mismatch repair genes, MSH2, MLH1, MSH6 and PMS2, can cause MSI.43 MSI-H is present as a distinct phenotype in approximately 15% of CRCs.18 A number of pathological features have previously been linked with MSI-H, such as mucinous differentiation, signet ring cell morphology, Crohn's-like lymphoid reaction, abundant tumor-infiltrating lymphocytes, tumor necrosis, and poor differentiation.2,44,45,46,47
MSI is typically assessed by analyzing five microsatellite markers (D2S123, D5S346, D17S250, BAT25, and BAT26) referred to as the NCI consensus panel,48 but additional microsatellite markers are commonly tested to increase the accuracy of classification. In clinical settings, MSI testing has been performed as a screening test for the identification of HNPCC/Lynch syndrome and sometimes as a prognostic marker (generally MSI-H implying a better prognosis) or a marker for predicting efficacy of chemotherapy (generally MSI-H implying resistance) (see Molecular Classification and Patient Outcomes).
MSI-Low versus MSS
Whether MSI-low (MSI-L) exists as a distinct phenotype from MSS has been controversial.49,50 A study has shown that virtually all CRCs show some degree of microsatellite instability when a large number of markers are tested.49 The study concluded that a difference between MSI-L and MSS is merely quantitative and that it is unlikely that there are qualitatively different genetic pathways to MSI-L tumors and MSS tumors.49 This study demonstrates that microsatellites may not be the best markers to identify MSI-L because there is no discrete phenotype or genotype associated with MSI-L determined by microsatellite markers.49 Thus, a newer MSI marker panel has been designed to separate a substantial number of MSI-L tumors into MSS and MSI-H.51,52
In contrast, there is evidence that supports the existence of MSI-L. MSI-L has been associated with shorter survival in Stage C colon cancer, compared with MSS tumors.53 A cDNA microarray expression study has also supported MSI-L as a distinct phenotype from MSS and MSI-H.54 MSI-L has been associated with MGMT methylation and loss.55 The association between MSI-L and MGMT methylation/loss is particularly strong among CIMP-low tumors.56 Among CIMP-low tumors, frequency of MGMT methylation and loss is much higher in MSI-L tumors than in MSI-H and MSS tumors; thus, MSI-L cannot be a mixture of MSI-H and MSS tumors.56 These data collectively support differences between MSI-L and MSS in colorectal cancer, although more studies are necessary to establish definitively the existence of MSI-L as a distinct phenotype. If MSI-L exists, additional studies are necessary to find underlining molecular defects for MSI-L and better biomarkers for MSI-L (maybe markers other than microsatellites).
CIMP
Transcriptional inactivation by cytosine methylation at promoter CpG islands of tumor suppressor genes is an important mechanism in human carcinogenesis, and a number of tumor suppressor genes have been shown to be silenced by promoter methylation in CRC.57,58,59,60,61,62 In fact, a subset of CRCs have been shown to exhibit widespread promoter CpG island methylation, which is referred to as the CIMP.57,63,64 CIMP has been established as a unique epigenetic phenotype in colorectal cancer, and CIMP-high colorectal tumors have a distinct clinical, pathological, and molecular profile, such as associations with proximal tumor location, female sex, poor differentiation, MSI, and high BRAF and low TP53 mutation rates.65,66,67,68,69,70,71,72,73 Even within MSI-H tumors and within MSI-L/MSS tumors, CIMP-high has been associated with proximal location,14 poor differentiation,46 BRAF mutation,71,72 and loss of nuclear p27 [cyclin-dependent kinase inhibitor 1A (CDKN1B)]74 and inversely associated with TP53 aberrations,75 loss of p21 (CDKN1A),75 overexpression of cyclooxygenase-2 (PTGS2),76 and cytoplasmic mislocalization of p27.77 Within MSI-H tumors, CIMP-high has been associated with TGFBR2 mononucleotide mutation.78 Using analyses on a large number of CpG island methylation markers, CIMP-high tumors form a distinct group by an unsupervised cluster analysis.73 These data collectively contradict the claims that CIMP does not represent a distinct phenotype in CRC and that characteristics of CIMP merely reflect those of MSI-H tumors.79,80 The serrated pathway of tumorigenesis has been suggested in the development of CIMP-high colorectal cancer,81,82,83,84 whereas flat-type adenomas do not appear to show frequent promoter methylation.85
At the present time, the panel of methylation markers and the method of assessment of CIMP is not standardized, although recent studies have found a fairly sensitive and specific identification of CIMP-high using MethyLight technology and evaluation of new panels of four to eight CpG islands (CACNA1G, CDKN2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3, and SOCS1).73,86 Any of these panels is a useful tool at this time to examine CRCs to diagnose CIMP-high versus non-CIMP-high.
Another question that has yet to be resolved is whether there are any sporadic MSI-H tumors that exhibit neither MLH1 methylation nor the CIMP phenomenon. Again, a recent study suggested that all sporadic MSI-H tumors were explainable by CIMP and MLH1 methylation,73 whereas other studies have suggested that there may be a subset of sporadic MSI-H non-CIMP-high tumors.71,86 The frequency of HNPCC/Lynch syndrome in the general population is estimated to be 1 to 3%.87 A large population-based study has suggested that the frequency of MSI-H non-CIMP-high tumors is ∼5%86; thus, it is likely that nearly one-half or more of MSI-H non-CIMP-high tumors do not arise through either HNPCC/Lynch syndrome or the CIMP-high pathway. Thus, the absence of CIMP-high in MSI-H tumors does not necessarily indicate HNPCC/Lynch syndrome, although it increases the likelihood of HNPCC/Lynch syndrome.
CIMP-High versus CIMP-Low versus CIMP-0
The original report of CIMP in 1999 used type C (cancer specific) methylated in tumor (MINT) clones as markers for CIMP, and methylation in MINT markers is correlated with CDKN2A (p16) methylation.63 The subsequent study demonstrated that CIMP determined by MINT markers exhibited distinct genetic features, including associations with KRAS mutation and wild-type TP53.65 Studies by other investigators basically used a similar panel of markers including MINT1, MINT2, and MINT31.66,67,68,69,71 In these studies, although the inverse association between CIMP and TP53 mutation appears to be consistent, the association between CIMP and KRAS mutation is not consistent; some studies have shown a positive correlation,65,71 whereas another study showed a negative correlation.66,88
Since BRAF mutation in CRC was first discovered, BRAF mutation has consistently been associated with CIMP or CIMP-high.69,70,71,72,73 It has been shown that MINT markers are not highly specific for CIMP-high tumors with BRAF mutation.73 Thus, the existence of CIMP-low that is separate from CIMP-high and CIMP-negative has recently been hypothesized.72 Using a panel of methylation markers that are specific for BRAF-mutated CIMP-high tumors, we have shown that CIMP-low is associated with KRAS mutation and male sex, whereas CIMP-high is associated with BRAF mutation and female sex, and CIMP-0 (CIMP-negative) is associated with wild-type BRAF/KRAS.14 Because the frequency of KRAS mutation in CIMP-low tumors is higher than in CIMP-high and CIMP-0 tumors, CIMP-low cannot be a mixture of misdiagnosed CIMP-high and CIMP-0.14 These findings also explain why previous studies using MINT markers and lower cutoffs for CIMP showed the positive correlation between CIMP and KRAS mutation. CIMP-positive tumors determined by MINT markers probably represent a heterogeneous group of tumors including substantial numbers of CIMP-low tumors with a high frequency of KRAS mutation.
A difference between CIMP-low and CIMP-0 is not as clear-cut as that between CIMP-low and CIMP-high, probably because methylation markers (CACNA1G, CDKN2A, CRABP1, IGF2, MLH, NEUROG1, RUNX3, and SOCS1) are specific for CIMP-high but are not ideal for the identification of CIMP-low.14,86 In future studies, markers that are sensitive and specific for CIMP-low need to be determined for the identification of CIMP-low, if CIMP-low really exists. Nonetheless, a difference between CIMP-low and CIMP-0 is more striking in terms of MGMT methylation and loss of expression.56 Among CIMP-low tumors, MSI-L tumors showed a significantly higher frequency of MGMT methylation/loss than MSI-H and MSS tumors, but no such relationship was observed in CIMP-0 tumors.56 Figure 1 represents current knowledge on different CIMP subtypes in CRC. The term “CIMP-0” is used to avoid confusion; “CIMP-negative” has been used for either “CIMP-low/0” or “CIMP-0”.
Figure 1.

CIMP classification in colorectal cancer and associations with clinical and molecular features.
MSI/CIMP Subtypes of CRC
Recently, Jass47 proposed a very comprehensive molecular classification system in which five groups of CRC are defined according to MSI and CIMP status in conjunction with clinical and pathological features. Here, we propose an updated classification system incorporating recent new data in this field. We still need to accumulate more data to further validate our proposed classification system. There are some differences from the Jass classification, in particular groups 2 and 4. It is theoretically possible to have nine groups (three MSI status × three CIMP status). However, some of the nine groups have similar features, so we combined some of the nine groups to make six groups as shown in Figure 2. Because the differences between MSI-L and MSS are subtle and because the differences between CIMP-low and CIMP-0 are also subtle, CRCs can also be classified into four major subtypes [two MSI status (MSI-H versus MSI-L/MSS) × two CIMP status (CIMP-high versus CIMP-low/0)] (Figure 3). In other words, groups 4, 5, and 6 can be combined into one major subtype, MSI-L/MSS CIMP-low/0, because they share similar clinical, pathological and molecular features.
Figure 2.

The six groups of colorectal cancer according to MSI and CIMP status.
Figure 3.
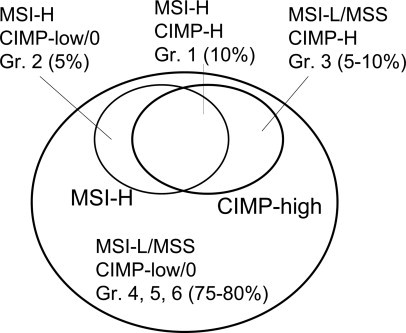
The four major subtypes of colorectal cancer according to MSI and CIMP status.
Note that MSI and CIMP are not the only molecular classifiers in CRC; however, MSI and CIMP status reflect global genomic/epigenomic status, and compared with CIN, MSI and CIMP are still relatively well defined. The frequency of each subtype among CRCs in the general population is an approximate number, depending on markers and criteria, in particular for CIMP status.
Group 1: MSI-H CIMP-High (10%86)
This group of tumors commonly shows MLH1 methylation, BRAF mutation, CIN negative, wild-type TP53, intact p21 (CDKN1A) expression,75 loss of nuclear p27 (CDKN1B),74 poor differentiation, lymphocytic reactions, and mucinous and/or signet ring cell features. Clinically, this is generally known as sporadic MSI-H and is associated with good prognosis, elderly female, and proximal colon.
Group 2: MSI-H CIMP-Low/0 (5%86)
This group of tumors includes HNPCC/Lynch syndrome (1 to 3%). However, because the frequency of HNPCC/Lynch syndrome is estimated to be 1 to 3% of CRCs in the general population,87 nearly one-half or even a majority of MSI-H CIMP-low/0 tumors are sporadic and unrelated to HNPCC/Lynch syndrome. Recently identified CIMP-high-specific markers have a high power of separating CIMP-low/0 from CIMP-high in MSI-H tumors with virtually no overlap between CIMP-high and CIMP-low/0.86 Thus, the 5% estimated frequency based on almost 900 tumors is quite accurate.86 This group (MSI-H CIMP-low/0) of tumors are associated with KRAS mutation, wild-type TP53, CIN negative, fatty acid synthase overexpression,89 proximal colon (compared with MSI-L/MSS CIMP-low/0),86 lymphocytic reactions, and mucinous features, but not with poor differentiation or signet ring cell features.46 Therefore, the presence of poor differentiation or signet ring cells perhaps by itself does not increase the likelihood of HNPCC/Lynch syndrome. Prognostic significance of sporadic MSI-H CIMP-low/0 needs further investigation.
Group 3: MSI-L/MSS CIMP-High (5 to 10%86)
This group of tumors commonly shows BRAF mutation,71,72 wild-type TP53,71,75 CIN negative,90 poor differentiation, and signet ring cell features46 and is associated with poor prognosis,91 elderly female, and right colon.14,71
Group 4: MSI-L CIMP-Low (∼5%56)
This group of tumors is associated with high frequencies of MGMT methylation and KRAS mutation.56 Previous studies have shown an association between MSI-L and MGMT methylation,53,55 which is due to the association between MSI-L and MGMT methylation among CIMP-low tumors.56
Group 5: MSS CIMP-Low (30 to 35%56)
This group of tumors is associated with KRAS mutation,14 CIN negative,92 and male sex.14
Group 6: MSI-L/MSS CIMP-0 (∼40%56)
This group of tumors are associated with CIN,90,92 wild-type KRAS/BRAF,14 and distal colon14 and show no sex predilection.14
Molecular Correlates in CRC
General Approach to Molecular Correlates Based on MSI/CIMP Classification
As mentioned previously, molecular classification based on global genomic/epigenomic aberrations, including CIN, MSI, and CIMP, is important. However, classification based on CIN is a challenge for several reasons. Currently, the methods/markers and criteria for CIN are far from uniform. LOH analysis has been known to have false positive and false negative results and substantial uninformative results. Thus, we illustrate how MSI/CIMP classification can decipher correlates with other molecular alterations.
According to MSI status, CRCs can be classified into two categories, MSI-H and MSI-L/MSS, because a distinction between MSI-L and MSS is subtle. According to CIMP status, CRCs can also be classified into two categories, CIMP-high and CIMP-low/0, because a distinction between CIMP-low and CIMP-0 is subtle. Considering the status of both MSI and CIMP, CRC can be classified into four major groups: MSI-H CIMP-high (10% of all CRCs; group 1), MSI-H CIMP-low/0 (5% of all CRCs; group 2), MSI-L/MSS CIMP-high (5 to 10% of all CRCs; group 3), and MSI-L/MSS CIMP-low/0 (75 to 80% of all CRCs; groups 4, 5, and 6) (Figure 3). Because each of MSI-H CIMP-low/0 and MSI-L/MSS CIMP-high constitutes only 5 to 10% of all CRCs, a large number of samples are required to properly dissect molecular correlates using the combined MSI and CIMP classification system.
The classification system based on combined MSI and CIMP status is useful in analyzing molecular correlates. For example, one can examine BRAF mutation frequencies in these four subtypes of colorectal cancer as in Figure 4A (data in Ref.86). This figure shows much higher BRAF mutation frequencies in MSI-H CIMP-high and MSI-L/MSS CIMP-high tumors than in MSI-H CIMP-low/0 and MSI-L/MSS CIMP-low/0 tumors. It is evident that MSI status has no effect on BRAF mutation frequencies. These results indicate that BRAF mutation is positively correlated with CIMP-high, independent of MSI status. As another example, in Figure 4B (data in Ref.76), both MSI-H and CIMP-high exhibit synergistic effect of lowering the frequency of p53 positivity (by immunohistochemistry); ie, both MSI-H and CIMP-high are inversely associated with p53 positivity. Because p21 (CDKN1A) is one of the major downstream effectors of p53, the interrelationship between p53, p21, and MSI (or CIMP) is examined in Figure 4C (data in Ref.75). After CRCs are stratified by p53 and p21 status, p53 status exhibits very little effect on the frequencies of CIMP-high and MSI-H. In contrast, p21 loss is inversely correlated with CIMP-high and MSI-H regardless of p53 status.
Figure 4.
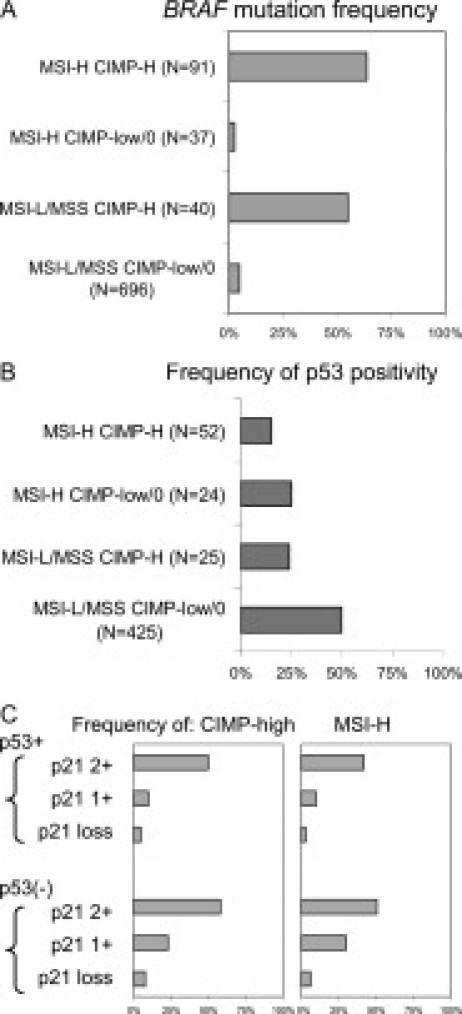
A: Frequency of BRAF mutation in the four major subtypes of colorectal cancer (data in Ref.86). B: Frequency of p53 positivity (by immunohistochemistry) in the four major subtypes of colorectal cancer (data in Ref.76). C: Frequencies of CIMP-high and MSI-H in colorectal cancer according to combined p53 and p21 (CDKN1A) status (data in Ref.75).
Molecular Correlates
Figure 5A illustrates our current knowledge of molecular correlates in CRC. When CIMP and BRAF mutation were unknown, our understanding was something simpler, as in Figure 5B. It is likely that we will have a better (although more complicated) figure than Figure 5A in the future with advancement of science. Table 2 lists findings on molecular correlates mainly based on the MSI/CIMP (and CIN) classification. It does not list findings by studies that only analyze either MSI or CIMP classifiers.
Figure 5.
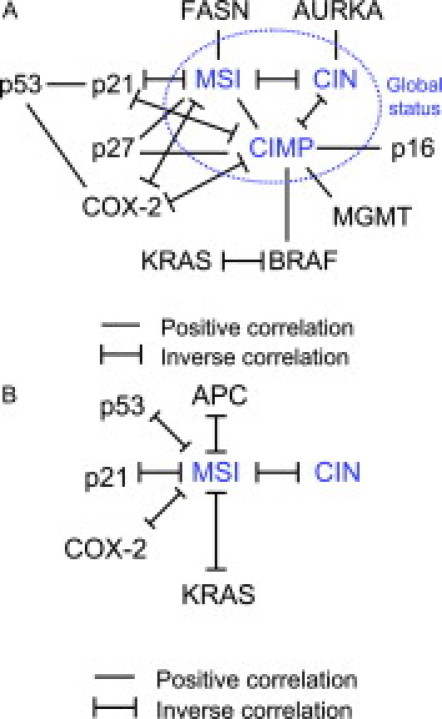
A: Molecular correlates in colorectal cancer. Note that MSI and CIMP (in the blue circle) are present centrally in this figure of the molecular correlates, implying their influence on various molecular alterations (indicated as “p53,” “p21,” etc) and a phenotype in colorectal cancer. B: Molecular correlates in colorectal cancer in the past (particularly without our current knowledge on CIMP and BRAF mutation). This figure is much simpler than A.
Table 2.
Molecular Correlates with MSI and CIMP in Colorectal Cancer
| Molecular event | Molecular subtypes | Positive or inverse association | Note | Reference |
|---|---|---|---|---|
| AURKA amplification | CIN | Positive | AURKA amplification is not correlated with MSI or CIMP status | 13 |
| BRAF mutation | CIMP-high | Positive | BRAF mutation is not correlated with MSI-H independent of CIMP status | 71,72 |
| β-Catenin (CTNNB1) nuclear localization (activation) | CIMP-high | Inverse | Inverse correlation between β-catenin nuclear localization (or activation) and CIMP-high is independent of MSI status; the β-catenin activation score system is based on the system described by Jass et al21 | 93 |
| CIN | CIMP | Inverse | 90 | |
| Cyclooxygenase-2 (PTGS2) overexpression | MSI-H and CIMP-high | Inverse | COX-2 and p53 expressions synergistically decrease the frequencies of both MSI-H and CIMP- high in CRC | 76 |
| Fatty acid synthase (FASN) overexpression | MSI-H | Positive | Positive correlation between FASN overexpression and CIMP-high is mediated by MSI | 89 |
| JC virus T antigen | CIMP and CIN | Positive | JC virus T antigen is not correlated with MSI status | 94 |
| KRAS mutation | CIMP-low | Positive | Inverse relationship between KRAS mutation and CIMP-0 is not observed in MSI-H tumors because of the small number of MSI-H CIMP-0 tumors | 14 |
| KRAS mutation | CIMP-high | Inverse | Inverse relationship between KRAS mutation and CIMP-high is particularly strong among MSI-H tumors | 86 |
| 18q loss of heterozygosity | CIMP-high and CIMP-low | Inverse | MSI-H tumors are excluded | 92 |
| 18q loss of heterozygosity | MSI-L | Positive | MSI-H tumors are excluded | 92 |
| p21 (CDKN1A) loss | MSI-H and CIMP-high | Inverse | Inverse relationship between p21 loss and BRAF mutation is mediated by CIMP | 75 |
| p53 (TP53) positivity (expression) | MSI-H and CIMP-high | Inverse | Inverse relationship between p53 positivity and MSI/CIMP is mediated by p2175; p21 loss and p53 positivity are positively correlated75 | 68,75 |
| p27 (CDKN1B) loss (nuclear) | MSI-H and CIMP-high | Positive | Relationship between p27 loss and BRAF mutation is mediated by CIMP; relationship between p27 and MSI/CIMP appears to be limited to p53-negative CRC | 74 |
| p27 (CDKN1B) cytoplasmic expression | MSI-H and CIMP-high | Inverse | p27 cytoplasmic expression is inversely correlated with nuclear p27 loss; relationship between cytoplasmic p27 and MSI/CIMP appears to be limited to p53-negative CRC | 77 |
| TGFBR2 mononucleotide mutation | CIMP-high | Positive | Only in MSI-H tumors | 78 |
We limit studies to only those that analyzed molecular correlates with combined MSI and CIMP status because analyzing both MSI and CIMP would be necessary to delineate molecular correlates around MSI and CIMP (Figure 5A). For example, one can find a correlation between MSI and BRAF mutation without analyzing CIMP. However, the relationship between MSI and BRAF mutation is actually mediated by CIMP (Figure 4A).
The relationship of APC mutation and β-catenin (CTNNB1) nuclear localization with MSI/CIMP warrants discussion. APC mutations are common causes of WNT/CTNNB1 activation in CRC. Although there are conflicting data regarding correlations of APC mutation or CTNNB1 nuclear localization with MSI status,21,95,96,97,98,99,100,101,102 CTNNB1 mutation is associated specifically with HNPCC MSI-H tumors.103 Recent reports have shown an inverse correlation between APC mutation and BRAF mutation104 and an inverse correlation between CTNNB1 nuclear localization and CIMP-high independent of MSI status.93 In addition, APC methylation may be inversely associated with CIMP-high.88
Pathological Features and MSI/CIMP
Various pathological features have been associated with MSI-H, but few studies with a large sample size have examined the relationship between pathological features and MSI/CIMP.46,71,86 Pathological features have been used to assess risks of HNPCC/Lynch syndrome. It is widely thought that poor tumor differentiation increases the likelihood of HNPCC/Lynch syndrome because of the association of poor differentiation with MSI-H. However, recent data indicate that poor differentiation is associated with CIMP-high tumors, but not MSI-H CIMP-low/0 tumors (which include most HNPCCs).86 Thus, it is unlikely that poor differentiation alone increases HNPCC risk. On the other hand, mucinous features (≥50% mucinous), lymphocytic reactions, and proximal tumor location increase HNPCC risk because these features are associated with both MSI-H CIMP-high and MSI-H CIMP-low/0.46,86
Molecular Classification and Patient Outcomes
There are numerous studies on individual molecular alterations (such as KRAS mutation, TP53 mutation, etc) and patient outcomes. Discussion in this article focuses on global genomic and epigenomic status and patient outcomes. A correlation of a single molecular event (such as TP53 mutation, BRAF mutation, etc) with patient outcomes needs to be interpreted with caution; the molecule examined might be associated with global genomic or epigenomic aberrations and improved or adverse outcomes might be caused by aberrations in other molecules.
With regard to MSI status and CRC patient survival, a systematic review of 32 studies that reported survival data on a total of 7642 colorectal cancer patients, including 1277 with MSI-H tumors, showed that MSI-H tumors were associated with better prognosis compared with MSS tumors [the combined hazard ratio estimate for overall survival associated with MSI-H was 0.65 (95% confidence interval, 0.59 to 0.71)].105 In a meta-analysis of six studies106,107,108,109,110,111 that investigated overall survival stratified by MSI status in patients who received adjuvant 5-FU, patients with MSI-H tumors had a better prognosis (hazard ratio, 0.76; 95% confidence interval, 0.65 to 0.88).105 In addition, MSI-H tumors showed no benefit from adjuvant 5-FU.106,109,112 However, other studies have shown no predictive value of MSI status on overall survival of CRC patients treated with adjuvant 5-FU-based chemotherapy or on survival benefit from adjuvant 5-FU-based chemotherapy.113,114,115 Although evidence has been accumulating for MSI-H as a good prognostic indicator, additional investigation is needed to understand the mechanisms by which MSI influences colorectal cancer survival.
MSI-H tumors frequently show mutations in TGFBR2.116,117,118 TGFBR2 mediates signaling from TGF-β to its signal transducers, such as SMADs and further downstream targets, and functions as a tumor suppressor.18 Mutations in TGFBR2 observed in MSI-H cancers truncate and inactivate the TGFBR2 protein, abolishing its growth-regulating function.41 Among MSI-H cancers, TGFBR2 mutations have been associated with a significantly improved survival in one population of patients with stage III colon cancer.107 In a separate analysis, the survival benefit of TGFBR2 mutations in MSI-H tumors appeared to be particularly strong in the presence of coexistent BAX mutations.119 However, other studies have failed to confirm these data with one study finding worse survival associated with TGFBR2 mutations among 16 MSI-H tumors118 and another analysis finding no influence of TGFBR2 mutations on survival among 174 MSI-H tumors.117 Additional studies are necessary to assess whether survival benefit of MSI-H status is influenced by the presence of TGFBR2 or BAX mutation.
Why MSI-H tumors show better prognosis is currently unknown. A study has shown that MSI-H is not an independent predictor of survival in a multivariate model including DNA ploidy status.120 Although DNA ploidy may be a crude measure of CIN status, this study suggests that the association between MSI-H and better survival may be due to the confounding effect of CIN, which is a predictor of worse survival independent of MSI status.120 Another study has also shown that CIN is an independent predictor of worse survival, but survival is not correlated with MSI-H when CIN-positive tumors are excluded.121
With regard to the influence of CIMP on CRC patient survival, previous studies have been conflicting. Although some studies have found no significant relationship,68,122 one study did demonstrate poor prognosis associated with CIMP in non-MSI-H tumors but not in MSI-H tumors,91 and another study has also shown that CIMP in advanced MSS tumors treated with chemotherapy predicts worse survival.123 BRAF mutations are associated with poor prognosis in non-MSI-H tumors (although no effect was seen on the good prognosis of MSI-H tumors).122 Because non-MSI-H tumors with BRAF mutations are most likely CIMP-high,71,72 it is possible that the relationship between prognosis and CIMP-high is actually due to the relationship between prognosis and BRAF mutation.
It is also controversial whether CIMP confers survival benefit from chemotherapy in CRC. In one study of stage III colorectal cancer treated with surgery and adjuvant chemotherapy,124 patients with CIMP-positive tumors experienced a significant survival benefit from chemotherapy in contrast to those with CIMP-negative tumors, and this effect was independent of MSI and p53 mutation status; however, this study was not designed to randomly assign treatment groups. Therefore, an unidentified confounding factor cannot be excluded. Randomized trials are necessary to definitively assess treatment efficacy.
Limitations in Molecular Classification and Correlates
Lack of Gold Standard and Uniform Methods, Definition, and Criteria
Gold standard methods to assess global genomic and epigenomic changes are important in various clinical studies. Gold standard methods are also important to evaluate performance characteristics of any potential surrogate markers. However, defining such standard is not a trivial task, particularly when one needs to analyze global genomic and epigenomic status in the tumor cell. The use of a limited set of markers (such as MSI, CIMP, and LOH marker panels) can serve as a gold standard with careful validation and correlative studies; however, it is less than ideal. The use of microarray technology may be a solution, but validation and cost of implementing microarrays in clinical laboratory settings are currently difficult issues.
Lack of uniform markers, criteria, and definition in the literature is also a problem. A validation study of markers and criteria requires a large number of markers and samples, thus making such a study difficult to perform. With regard to CIN and CIMP, for example, there are no uniform methods, criteria, and definition in the literature, which makes a comparison between studies challenging. With regard to MSI, the implementation of the NCI marker panel48 has been successful, and a comparison between various studies has become easier.
False Positives and False Negatives
False-positive and false-negative results may obscure true associations. It is important to design a large enough study to have adequate statistical power, even with predictable frequencies of false-positive and false-negative results. False results may also lead to a false association if assay errors are systematic and nonrandom. For example, KRAS mutation and MSI have been shown to be inversely associated,125 but the inverse relation could be caused by abundant reactive lymphocytes commonly seen in MSI-H tumors, which can cause false negative results in KRAS sequencing assays. The inverse association has been confirmed by a more sensitive KRAS pyrosequencing assay,126 particularly in CIMP-high tumors.14
As another example, poor quality paraffin tissue samples, which fail to react with a specific antibody (ie, false negatives), tend to fail to react with another antibody. Thus, negativity of one protein tends to coincide with negativity of another protein even with the absence of any true association. In other words, one should be always be cautious when obtaining a positive correlation between overexpression in two proteins by immunohistochemistry. The presence of internal control in immunohistochemically stained slides may solve this problem to some extent.
Sampling Bias
Sampling bias is an inevitable problem when one can analyze only a finite number of cases. Thus, one should make the best effort to eliminate any source of bias. Compared with a single-hospital-based study, certainly a population-based study or multicenter study is designed to decrease the degree of sampling bias, but it is not always an option to most investigators. Academic hospitals may have more advanced or complicated cases than community hospitals. Racial or geographic bias can be present in any hospital setting. As an example of sampling bias, it is well known that the frequency of HNPCC/Lynch syndrome has been reported to be 3 to 5% in many studies; however, in the general population, the frequency of HNPCC/Lynch syndrome is estimated to be 1 to 3% among all CRCs87; the reported higher frequency of 3 to 5% is likely due to a combination of geographic and/or referral bias in a setting of academic hospitals.87
Small Sample Size
A small sample size is the cause of a number of problems. Studies with a small number of samples are more susceptible to the adverse effects of sampling bias and false positives/negatives, which may lead to erroneous conclusion. Even if the total number of cases is not small, a number of a particular subtype (especially a rare subtype such as group 2, 3, or 4 in Figure 2) may become small. If the total number of CRCs in a study is 100, for example, the number of cases in group 2, 3, or 4 is at most 5 to 10 without significant sampling bias. A study has shown an inverse association between MGMT methylation and MLH1 methylation using 110 single-hospital-based CRCs (with only 13 cases showing MLH1 methylation).127 In contrast, a much larger study has shown a positive association between MGMT and MLH1 methylation using 920 population-based CRCs (with 115 cases showing MLH1 methylation).56
Issues in MSI Classification
Despite the presence of the recommended panel of markers (the NCI panel),48 markers used for studies on MSI are still not uniform. This is partly because the NCI panel has only five markers, and an increase in the number of markers increases the accuracy of classification, and because the five NCI markers may not be the best markers for the identification of MSI-H.
Another issue is MSI-L. It is still controversial whether MSI-L represents a distinct phenotype from MSI-H and MSS. We discussed the rationale for the existence of MSI-L (see above, MSI-L versus MSS). Yet, it is very clear that we need better markers for the identification of MSI-L if it really exists. Ideal MSI-L markers, if any, may not be microsatellite markers.
Issues in CIMP Classification
Definition of CIMP and markers and criteria for CIMP diagnosis are still controversial issues. It has been repeatedly shown that BRAF mutation can be a good surrogate marker for CIMP-high independent of MSI status, and originally described MINT markers may not be ideal for identification of CIMP-high tumors with a high BRAF mutation rate.
In addition to methylation markers and criteria, methods of methylation detection are not uniform. Most previous studies on molecular correlates with CIMP in CRC used nonquantitative methylation-specific PCR, which may show positivity in tumors with biologically insignificant low-level methylation and overestimate the frequency of methylation in any markers as well as the frequency of CIMP. In contrast, a quantitative methods such as quantitative methylation-specific PCR (MethyLight) are robust and can reproducibly differentiate high-level from low-level methylation in paraffin-embedded tissue, and low-level methylation is likely due to biological noise because it is not associated with gene silencing.128
The concept of CIMP-low is no less problematic than that of MSI-L. We discussed the rationale for the existence of CIMP-low (see above, CIMP-high versus CIMP-low versus CIMP-0). Yet, it is very clear that we need better markers for the identification of CIMP-low, if it really exists.
Issues in CIN Classification
Lack of standardized definition of CIN is a substantial issue. CIN has been evaluated by many different methods, including metaphase karyotyping, flow cytometric DNA ploidy study, microsatellite LOH analysis, fluorescence in situ hybridization, CGH, array-based CGH, SNP array, and spectral karyotyping. Thus, data in the literature have been based on a diverse array of methods, which makes cross-comparison between studies very difficult. Moreover, markers (chromosomal loci) used for evaluation are also diverse. Each method has advantages and disadvantages. In particular, microsatellite LOH analysis has been in widespread use because of its low cost and easy applicability to paraffin-embedded archival tissue samples. However, it requires normal DNA for comparison with tumor DNA, microsatellite markers are frequently uninformative because of homozygosity, and there are false positives (because of PCR bias and allele drop-out) and false negatives (because of PCR bias and normal cell contamination).
Interpretation of Molecular Correlates or Association Studies
When one has found a positive correlation between two molecules, it is often erroneously concluded that two molecules are pathogenetically linked. However, one should be cautious in interpreting molecular correlates. A significant positive correlation is not synonymous to a causal or pathogenetic link. Unidentified potential confounding factors should also be kept in mind when interpreting molecular correlates.
Future Directions
It should always be kept in mind that molecular classification and correlates in colorectal cancer are still evolving and that the current knowledge only represents our best understanding at the present. Advances in technology in the field of cancer research are rapid and very promising. High-throughput technology for the detection of molecular alterations in tumors will enable us to evaluate molecular correlates in CRC in a much more comprehensive way. However, challenges still exist, including cost issues in newer technologies, inevitable sampling bias, and time required to assess patient outcomes. The latter two will persist even with the state-of-the-art laboratory technologies. We can make great efforts to decrease the degree of sampling bias by organizing multicenter studies and population-based studies. Data obtained from molecular correlate studies will help further understanding of carcinogenesis and help implement cost-effective laboratory tests for various purposes: prognostication, treatment decision making, and genetic risk assessment for family members.
Conclusions
Molecular classification and molecular correlates will continue to be very important in colorectal cancer research. Studying molecular correlates can 1) provide clues to pathogenesis, 2) propose or support the existence of a new molecular subtype, 3) alert investigators to potential confounding factors in association studies, and 4) suggest surrogate markers in clinical or research settings. In molecular classification, global genomic or epigenomic classifiers such as MSI, CIN, and CIMP are increasingly important, and investigators should always try to analyze such global cellular status in colorectal cancer whenever feasible.
Notes Added in Proof
Recently, Shen et al (Ref.129) have described “CIMP2” associated with KRAS mutation, separate from CIMP1 and CIMP-negative in colorectal cancer. This CIMP2 appears to have overlapping features with CIMP-low.14,56,86,92
We have recently shown that density of methylation in CIMP-high-specific promoters (CACNA1G, CDNK2A, CRABP1, IGF2, MLH1, NEUROG1, RUNX3, and SOCS1) is generally lower in CIMP-low tumors than in CIMP-high tumors on average, even after adjusting for the number of methylated loci, suggesting a possibility of different underlying methylation defects in these two types of tumors.130
Acknowledgements
We thank all of the investigators who contributed to this field. We truly regret that we could not cite all of the published papers in this field. S.O. thanks Charles Fuchs, Massimo Loda, Walter Willett, Sue Hankinson, Edward Giovannucci, Gregory Kirkner, Takako Kawasaki, and other laboratory members for their support and assistance in various aspects of this work.
Footnotes
S.O. was supported by National Institutes of Health grants P01 CA87969 and P01 CA55075 and in part by the Bennett Family Fund for Targeted Therapies Research and by the Entertainment Industry Foundation through the Entertainment Industry Foundation National Colorectal Cancer Research Alliance.
References
- 1.Hamilton SR, Vogelstein B, Kudo S. In: Carcinoma of the Colon and Rectum. Aaltonen LA, Hamilton SR, editors. IARC Press; Lyon, France: 2000. [Google Scholar]
- 2.Redston M. Carcinogenesis in the GI tract: from morphology to genetics and back again. Mod Pathol. 2001;14:236–245. doi: 10.1038/modpathol.3880292. [DOI] [PubMed] [Google Scholar]
- 3.Compton CC. Colorectal carcinoma: diagnostic, prognostic, and molecular features. Mod Pathol. 2003;16:376–388. doi: 10.1097/01.MP.0000062859.46942.93. [DOI] [PubMed] [Google Scholar]
- 4.Bagg A, Kallakury BV. Molecular pathology of leukemia and lymphoma. Am J Clin Pathol. 1999;112:S76–S92. [PubMed] [Google Scholar]
- 5.Arber DA. Molecular diagnostic approach to non-Hodgkin's lymphoma. J Mol Diagn. 2000;2:178–190. doi: 10.1016/S1525-1578(10)60636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventura RA, Martin-Subero JI, Jones M, McParland J, Gesk S, Mason DY, Siebert R. FISH analysis for the detection of lymphoma-associated chromosomal abnormalities in routine paraffin-embedded tissue. J Mol Diagn. 2006;8:141–151. doi: 10.2353/jmoldx.2006.050083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akin C. Molecular diagnosis of mast cell disorders: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2006;8:412–419. doi: 10.2353/jmoldx.2006.060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steensma DP. JAK2 V617F in myeloid disorders: molecular diagnostic techniques and their clinical utility: a paper from the 2005 William Beaumont Hospital Symposium on Molecular Pathology. J Mol Diagn. 2006;8:397–411. doi: 10.2353/jmoldx.2006.060007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 10.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 11.Ogino S, Brahmandam M, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. Epigenetic profiling of synchronous colorectal neoplasias by quantitative DNA methylation analysis. Mod Pathol. 2006;19:1083–1090. doi: 10.1038/modpathol.3800618. [DOI] [PubMed] [Google Scholar]
- 12.Velho S, Oliveira C, Ferreira A, Ferreira AC, Suriano G, Schwartz S, Jr, Duval A, Carneiro F, Machado JC, Hamelin R, Seruca R. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41:1649–1654. doi: 10.1016/j.ejca.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 13.Nishida N, Nagasaka T, Kashiwagi K, Boland CR, Goel A. High copy amplification of the Aurora-A gene is associated with chromosomal instability phenotype in human colorectal cancers. Cancer Biol Ther. 2007;6:525–533. doi: 10.4161/cbt.6.4.3817. [DOI] [PubMed] [Google Scholar]
- 14.Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn. 2006;8:582–588. doi: 10.2353/jmoldx.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- 16.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 17.Hall PA, McCluggage WG. Assessing p53 in clinical contexts: unlearned lessons and new perspectives. J Pathol. 2006;208:1–6. doi: 10.1002/path.1913. [DOI] [PubMed] [Google Scholar]
- 18.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 19.Vogelstein B, Fearon ER, Kern SE, Hamilton SR, Preisinger AC, Nakamura Y, White R. Allelotype of colorectal carcinomas. Science. 1989;244:207–211. doi: 10.1126/science.2565047. [DOI] [PubMed] [Google Scholar]
- 20.Eshleman JR, Casey G, Kochera ME, Sedwick WD, Swinler SE, Veigl ML, Willson JK, Schwartz S, Markowitz SD. Chromosome number and structure both are markedly stable in RER colorectal cancers and are not destabilized by mutation of p53. Oncogene. 1998;17:719–725. doi: 10.1038/sj.onc.1201986. [DOI] [PubMed] [Google Scholar]
- 21.Jass JR, Biden KG, Cummings MC, Simms LA, Walsh M, Schoch E, Meltzer SJ, Wright C, Searle J, Young J, Leggett BA. Characterisation of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J Clin Pathol. 1999;52:455–460. doi: 10.1136/jcp.52.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goel A, Arnold CN, Niedzwiecki D, Chang DK, Ricciardiello L, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR. Characterization of sporadic colon cancer by patterns of genomic instability. Cancer Res. 2003;63:1608–1614. [PubMed] [Google Scholar]
- 23.Matsuzaki K, Deng G, Tanaka H, Kakar S, Miura S, Kim YS. The relationship between global methylation level, loss of heterozygosity, and microsatellite instability in sporadic colorectal cancer. Clin Cancer Res. 2005;11:8564–8569. doi: 10.1158/1078-0432.CCR-05-0859. [DOI] [PubMed] [Google Scholar]
- 24.Rowan A, Halford S, Gaasenbeek M, Kemp Z, Sieber O, Volikos E, Douglas E, Fiegler H, Carter N, Talbot I, Silver A, Tomlinson I. Refining molecular analysis in the pathways of colorectal carcinogenesis. Clin Gastroenterol Hepatol. 2005;3:1115–1123. doi: 10.1016/s1542-3565(05)00618-x. [DOI] [PubMed] [Google Scholar]
- 25.Sugai T, Habano W, Jiao YF, Tsukahara M, Takeda Y, Otsuka K, Nakamura S. Analysis of molecular alterations in left- and right-sided colorectal carcinomas reveals distinct pathways of carcinogenesis: proposal for new molecular profile of colorectal carcinomas. J Mol Diagn. 2006;8:193–201. doi: 10.2353/jmoldx.2006.050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdel-Rahman WM, Katsura K, Rens W, Gorman PA, Sheer D, Bicknell D, Bodmer WF, Arends MJ, Wyllie AH, Edwards PA. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc Natl Acad Sci USA. 2001;98:2538–2543. doi: 10.1073/pnas.041603298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 28.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iacopetta BJ, Welch J, Soong R, House AK, Zhou XP, Hamelin R. Mutation of the transforming growth factor-beta type II receptor gene in right-sided colorectal cancer: relationship to clinicopathological features and genetic alterations. J Pathol. 1998;184:390–395. doi: 10.1002/(SICI)1096-9896(199804)184:4<390::AID-PATH1230>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 30.Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 31.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 32.Gualberto A, Aldape K, Kozakiewicz K, Tlsty TD. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc Natl Acad Sci USA. 1998;95:5166–5171. doi: 10.1073/pnas.95.9.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: rAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 34.Scolnick DM, Halazonetis TD. Chfr defines a mitotic stress checkpoint that delays entry into metaphase. Nature. 2000;406:430–435. doi: 10.1038/35019108. [DOI] [PubMed] [Google Scholar]
- 35.Corn PG, Summers MK, Fogt F, Virmani AK, Gazdar AF, Halazonetis TD, El-Deiry WS. Frequent hypermethylation of the 5′ CpG island of the mitotic stress checkpoint gene Chfr in colorectal and non-small cell lung cancer. Carcinogenesis. 2003;24:47–51. doi: 10.1093/carcin/24.1.47. [DOI] [PubMed] [Google Scholar]
- 36.Bertholon J, Wang Q, Falette N, Verny C, Auclair J, Chassot C, Navarro C, Saurin JC, Puisieux A. Chfr inactivation is not associated to chromosomal instability in colon cancers. Oncogene. 2003;22:8956–8960. doi: 10.1038/sj.onc.1207078. [DOI] [PubMed] [Google Scholar]
- 37.Ricciardiello L, Baglioni M, Giovannini C, Pariali M, Cenacchi G, Ripalti A, Landini MP, Sawa H, Nagashima K, Frisque RJ, Goel A, Boland CR, Tognon M, Roda E, Bazzoli F. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003;63:7256–7262. [PubMed] [Google Scholar]
- 38.Niv Y, Goel A, Boland CR. JC virus and colorectal cancer: a possible trigger in the chromosomal instability pathways. Curr Opin Gastroenterol. 2005;21:85–89. [PubMed] [Google Scholar]
- 39.Newcomb PA, Bush AC, Stoner GL, Lampe JW, Potter JD, Bigler J. No evidence of an association of JC virus and colon neoplasia. Cancer Epidemiol Biomarkers Prev. 2004;13:662–666. [PubMed] [Google Scholar]
- 40.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 41.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 42.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 43.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. [DOI] [PubMed] [Google Scholar]
- 44.Shia J, Ellis NA, Paty PB, Nash GM, Qin J, Offit K, Zhang XM, Markowitz AJ, Nafa K, Guillem JG, Wong WD, Gerald WL, Klimstra DS. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am J Surg Pathol. 2003;27:1407–1417. doi: 10.1097/00000478-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Alexander J, Watanabe T, Wu TT, Rashid A, Li S, Hamilton SR. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158:527–535. doi: 10.1016/S0002-9440(10)63994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogino S, Odze RD, Kawasaki T, Brahmandam M, Kirkner GJ, Laird PW, Loda M, Fuchs CS. Correlation of pathologic features with CpG island methylator phenotype (CIMP) by quantitative DNA methylation analysis in colorectal carcinoma. Am J Surg Pathol. 2006;30:1175–1183. doi: 10.1097/01.pas.0000213266.84725.d0. [DOI] [PubMed] [Google Scholar]
- 47.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 48.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 49.Tomlinson I, Halford S, Aaltonen L, Hawkins N, Ward R. Does MSI-low exist? J Pathol. 2002;197:6–13. doi: 10.1002/path.1071. [DOI] [PubMed] [Google Scholar]
- 50.Jass JR. Re: Tomlinson et al. Does MSI-low exist. J Pathol 2002; 197:6–13. J Pathol. 2003;199:267–269. doi: 10.1002/path.1266. author reply 269–270. [DOI] [PubMed] [Google Scholar]
- 51.Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, Megid WM, Thibodeau SN. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Dis Markers. 2004;20:237–250. doi: 10.1155/2004/136734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, Berg KD, Eshleman JR. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn. 2006;8:305–311. doi: 10.2353/jmoldx.2006.050092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohonen-Corish MR, Daniel JJ, Chan C, Lin BP, Kwun SY, Dent OF, Dhillon VS, Trent RJ, Chapuis PH, Bokey EL. Low microsatellite instability is associated with poor prognosis in stage C colon cancer. J Clin Oncol. 2005;23:2318–2324. doi: 10.1200/JCO.2005.00.109. [DOI] [PubMed] [Google Scholar]
- 54.Mori Y, Selaru FM, Sato F, Yin J, Simms LA, Xu Y, Olaru A, Deacu E, Wang S, Taylor JM, Young J, Leggett B, Jass JR, Abraham JM, Shibata D, Meltzer SJ. The impact of microsatellite instability on the molecular phenotype of colorectal tumors. Cancer Res. 2003;63:4577–4582. [PubMed] [Google Scholar]
- 55.Whitehall VL, Walsh MD, Young J, Leggett BA, Jass JR. Methylation of O-6-methylguanine DNA methyltransferase characterizes a subset of colorectal cancer with low-level DNA microsatellite instability. Cancer Res. 2001;61:827–830. [PubMed] [Google Scholar]
- 56.Ogino S, Kawasaki T, Kirkner GJ, Suemoto Y, Meyerhardt JA, Fuchs CS. Molecular correlates with MGMT promoter methylation and silencing support CpG island methylator phenotype-low (CIMP-low) in colorectal cancer. Gut. 2007;56:1564–1571. doi: 10.1136/gut.2007.119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 58.Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 59.Laird PW. Cancer epigenetics. Hum Mol Genet. 2005;14:R65–R76. doi: 10.1093/hmg/ddi113. [DOI] [PubMed] [Google Scholar]
- 60.Rashid A, Issa JP. CpG island methylation in gastroenterologic neoplasia: a maturing field. Gastroenterology. 2004;127:1578–1588. doi: 10.1053/j.gastro.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 61.Wong JJ, Hawkins NJ, Ward RL. Colorectal cancer: a model for epigenetic tumorigenesis. Gut. 2007;56:140–148. doi: 10.1136/gut.2005.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Agrawal A, Murphy RF, Agrawal DK. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20:711–721. doi: 10.1038/modpathol.3800822. [DOI] [PubMed] [Google Scholar]
- 63.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samowitz W. The CpG island methylator phenotype in colorectal cancer. J Mol Diagn. 2007;9:281–283. doi: 10.2353/jmoldx.2007.070031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Rijnsoever M, Grieu F, Elsaleh H, Joseph D, Iacopetta B. Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut. 2002;51:797–802. doi: 10.1136/gut.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whitehall VL, Wynter CV, Walsh MD, Simms LA, Purdie D, Pandeya N, Young J, Meltzer SJ, Leggett BA, Jass JR. Morphological and molecular heterogeneity within nonmicrosatellite instability-high colorectal cancer. Cancer Res. 2002;62:6011–6014. [PubMed] [Google Scholar]
- 68.Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, O'Connor T, Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–1387. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 69.Kambara T, Simms LA, Whitehall VLJ, Spring KJ, Wynter CVA, Walsh MD, Barker MA, Arnold S, McGivern A, Matsubara N, Tanaka N, Higuchi T, Young J, Jass JR, Leggett BA. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut. 2004;53:1137–1144. doi: 10.1136/gut.2003.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagasaka T, Sasamoto H, Notohara K, Cullings HM, Takeda M, Kimura K, Kambara T, MacPhee DG, Young J, Leggett BA, Jass JR, Tanaka N, Matsubara N. Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol. 2004;22:4584–4594. doi: 10.1200/JCO.2004.02.154. [DOI] [PubMed] [Google Scholar]
- 71.Samowitz W, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 72.Ogino S, Cantor M, Kawasaki T, Brahmandam M, Kirkner G, Weisenberger DJ, Campan M, Laird PW, Loda M, Fuchs CS. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–1006. doi: 10.1136/gut.2005.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 74.Ogino S, Kawasaki T, Kirkner GJ, Yamaji T, Loda M, Fuchs CS. Loss of nuclear p27 (CDKN1B/KIP1) in colorectal cancer is correlated with microsatellite instability and CIMP. Mod Pathol. 2007;20:15–22. doi: 10.1038/modpathol.3800709. [DOI] [PubMed] [Google Scholar]
- 75.Ogino S, Kawasaki T, Kirkner GJ, Ogawa A, Dorfman I, Loda M, Fuchs CS. Down-regulation of p21 (CDKN1A/CIP1) is inversely associated with microsatellite instability and CpG island methylator phenotype (CIMP) in colorectal cancer. J Pathol. 2006;210:147–154. doi: 10.1002/path.2030. [DOI] [PubMed] [Google Scholar]
- 76.Ogino S, Brahmandam M, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. Combined analysis of COX-2 and p53 expressions reveals synergistic inverse correlations with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Neoplasia. 2006;8:458–464. doi: 10.1593/neo.06247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ogino S, Kawasaki T, Ogawa A, Kirkner GJ, Loda M, Fuchs CS. Cytoplasmic localization of p27 (cyclin-dependent kinase inhibitor 1B/KIP1) in colorectal cancer: inverse correlations with nuclear p27 loss, microsatellite instability, and CpG island methylator phenotype. Hum Pathol. 2007;38:585–592. doi: 10.1016/j.humpath.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ogino S, Kawasaki T, Ogawa A, Kirkner GJ, Loda M, Fuchs CS. TGFBR2 mutation is correlated with CpG island methylator phenotype in microsatellite instability-high colorectal cancer. Hum Pathol. 2007;38:614–620. doi: 10.1016/j.humpath.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 79.Anacleto C, Leopoldino AM, Rossi B, Soares FA, Lopes A, Rocha JC, Caballero O, Camargo AA, Simpson AJ, Pena SD. Colorectal cancer “methylator phenotype”: fact or artifact? Neoplasia. 2005;7:331–335. doi: 10.1593/neo.04502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell. 2003;4:121–131. doi: 10.1016/s1535-6108(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 81.Jass JR. Serrated adenoma of the colorectum: a lesion with teeth. Am J Pathol. 2003;162:705–708. doi: 10.1016/S0002-9440(10)63865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jass JR. Serrated adenoma of the colorectum and the DNA-methylator phenotype. Nat Clin Pract Oncol. 2005;2:398–405. doi: 10.1038/ncponc0248. [DOI] [PubMed] [Google Scholar]
- 83.Minoo P, Jass J. Senescence and serration: a new twist to an old tale. J Pathol. 2006;210:137–140. doi: 10.1002/path.2047. [DOI] [PubMed] [Google Scholar]
- 84.Dong SM, Lee EJ, Jeon ES, Park CK, Kim KM. Progressive methylation during the serrated neoplasia pathway of the colorectum. Mod Pathol. 2005;18:170–178. doi: 10.1038/modpathol.3800261. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi T, Nosho K, Yamamoto H, Mikami M, Taniguchi H, Miyamoto N, Adachi Y, Itoh F, Imai K, Shinomura Y. Flat-type colorectal advanced adenomas (laterally spreading tumors) have different genetic and epigenetic alterations from protruded-type advanced adenomas. Mod Pathol. 2007;20:139–147. doi: 10.1038/modpathol.3800722. [DOI] [PubMed] [Google Scholar]
- 86.Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, Fuchs CS. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn. 2007;9:305–314. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer. 2005;4:233–237. doi: 10.1007/s10689-004-5811-3. [DOI] [PubMed] [Google Scholar]
- 88.Iacopetta B, Grieu F, Li W, Ruszkiewicz A, Caruso M, Moore J, Watanabe G, Kawakami K. APC gene methylation is inversely correlated with features of the CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2006;119:2272–2278. doi: 10.1002/ijc.22237. [DOI] [PubMed] [Google Scholar]
- 89.Ogino S, Kawasaki T, Ogawa A, Kirkner GJ, Loda M, Fuchs CS. Fatty acid synthase overexpression in colorectal cancer is associated with microsatellite instability, independent of CpG island methylator phenotype. Hum Pathol. 2007;38:842–849. doi: 10.1016/j.humpath.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 90.Goel A, Nagasaka T, Arnold CN, Inoue T, Hamilton C, Niedzwiecki D, Compton C, Mayer RJ, Goldberg R, Bertagnolli MM, Boland CR. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology. 2007;132:127–138. doi: 10.1053/j.gastro.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 91.Ward RL, Cheong K, Ku SL, Meagher A, O'Connor T, Hawkins NJ. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J Clin Oncol. 2003;21:3729–3736. doi: 10.1200/JCO.2003.03.123. [DOI] [PubMed] [Google Scholar]
- 92.Ogino S, Kawasaki T, Kirkner GJ, Ohnishi M, Fuchs CS. 18q loss of heterozygosity in microsatellite stable colorectal cancer is correlated with CpG island methylator phenotype-negative (CIMP-0) and inversely with CIMP-low and CIMP-high. BMC Cancer. 2007;7:72. doi: 10.1186/1471-2407-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Meyerhardt JA, Fuchs CS, Ogino S. Correlation of beta-catenin localization with cyclooxygenase-2 expression and CpG island methylator phenotype (CIMP) in colorectal cancer. Neoplasia. 2007;9:569–577. doi: 10.1593/neo.07334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goel A, Li MS, Nagasaka T, Shin SK, Fuerst F, Ricciardiello L, Wasserman L, Boland CR. Association of JC virus T-antigen expression with the methylator phenotype in sporadic colorectal cancers. Gastroenterology. 2006;130:1950–1961. doi: 10.1053/j.gastro.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 95.Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, Kinzler KW, Vogelstein B, Dunlop MG. APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA. 1996;93:9049–9054. doi: 10.1073/pnas.93.17.9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Olschwang S, Hamelin R, Laurent-Puig P, Thuille B, De Rycke Y, Li YJ, Muzeau F, Girodet J, Salmon RJ, Thomas G. Alternative genetic pathways in colorectal carcinogenesis. Proc Natl Acad Sci USA. 1997;94:12122–12127. doi: 10.1073/pnas.94.22.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim IJ, Kang HC, Park JH, Shin Y, Ku JL, Lim SB, Park SY, Jung SY, Kim HK, Park JG. Development and applications of a beta-catenin oligonucleotide microarray: beta-catenin mutations are dominantly found in the proximal colon cancers with microsatellite instability. Clin Cancer Res. 2003;9:2920–2925. [PubMed] [Google Scholar]
- 98.Løvig T, Meling GI, Diep CB, Thorstensen L, Norheim Andersen S, Lothe RA, Rognum TO. APC and CTNNB1 mutations in a large series of sporadic colorectal carcinomas stratified by the microsatellite instability status. Scand J Gastroenterol. 2002;37:1184–1193. doi: 10.1080/003655202760373407. [DOI] [PubMed] [Google Scholar]
- 99.Ricciardiello L, Ceccarelli C, Angiolini G, Pariali M, Chieco P, Paterini P, Biasco G, Martinelli GN, Roda E, Bazzoli F. High thymidylate synthase expression in colorectal cancer with microsatellite instability: implications for chemotherapeutic strategies. Clin Cancer Res. 2005;11:4234–4240. doi: 10.1158/1078-0432.CCR-05-0141. [DOI] [PubMed] [Google Scholar]
- 100.Shitoh K, Furukawa T, Kojima M, Konishi F, Miyaki M, Tsukamoto T, Nagai H. Frequent activation of the beta-catenin-Tcf signaling pathway in nonfamilial colorectal carcinomas with microsatellite instability. Genes Chromosomes Cancer. 2001;30:32–37. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1065>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 101.Thorstensen L, Lind GE, Lovig T, Diep CB, Meling GI, Rognum TO, Lothe RA. Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia. 2005;7:99–108. doi: 10.1593/neo.04448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wong NA, Morris RG, McCondochie A, Bader S, Jodrell DI, Harrison DJ. Cyclin D1 overexpression in colorectal carcinoma in vivo is dependent on beta-catenin protein dysregulation, but not k-ras mutation. J Pathol. 2002;197:128–135. doi: 10.1002/path.1113. [DOI] [PubMed] [Google Scholar]
- 103.Johnson V, Volikos E, Halford SE, Eftekhar Sadat ET, Popat S, Talbot I, Truninger K, Martin J, Jass J, Houlston R, Atkin W, Tomlinson IP, Silver AR. Exon 3 beta-catenin mutations are specifically associated with colorectal carcinomas in hereditary non-polyposis colorectal cancer syndrome. Gut. 2005;54:264–267. doi: 10.1136/gut.2004.048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Samowitz WS, Slattery ML, Sweeney C, Herrick J, Wolff RK, Albertsen H. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007;5:165–170. doi: 10.1158/1541-7786.MCR-06-0398. [DOI] [PubMed] [Google Scholar]
- 105.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 106.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, III, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liang JT, Chang KJ, Chen JC, Lee CC, Cheng YM, Hsu HC, Chien CT, Wang SM. Clinicopathologic and carcinogenetic appraisal of DNA replication error in sporadic T3N0M0 stage colorectal cancer after curative resection. Hepatogastroenterology. 1999;46:883–890. [PubMed] [Google Scholar]
- 109.Barratt PL, Seymour MT, Stenning SP, Georgiades I, Walker C, Birbeck K, Quirke P. DNA markers predicting benefit from adjuvant fluorouracil in patients with colon cancer: a molecular study. Lancet. 2002;360:1381–1391. doi: 10.1016/s0140-6736(02)11402-4. [DOI] [PubMed] [Google Scholar]
- 110.Colombino M, Cossu A, Manca A, Dedola MF, Giordano M, Scintu F, Curci A, Avallone A, Comella G, Amoruso M, Margari A, Bonomo GM, Castriota M, Tanda F, Palmieri G. Prevalence and prognostic role of microsatellite instability in patients with rectal carcinoma. Ann Oncol. 2002;13:1447–1453. doi: 10.1093/annonc/mdf240. [DOI] [PubMed] [Google Scholar]
- 111.Hemminki A, Mecklin JP, Jarvinen H, Aaltonen LA, Joensuu H. Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology. 2000;119:921–928. doi: 10.1053/gast.2000.18161. [DOI] [PubMed] [Google Scholar]
- 112.Percesepe A, Borghi F, Menigatti M, Losi L, Foroni M, Di Gregorio C, Rossi G, Pedroni M, Sala E, Vaccina F, Roncucci L, Benatti P, Viel A, Genuardi M, Marra G, Kristo P, Peltomaki P, Ponz de Leon M. Molecular screening for hereditary nonpolyposis colorectal cancer: a prospective, population-based study. J Clin Oncol. 2001;19:3944–3950. doi: 10.1200/JCO.2001.19.19.3944. [DOI] [PubMed] [Google Scholar]
- 113.Westra JL, Schaapveld M, Hollema H, de Boer JP, Kraak MM, de Jong D, ter Elst A, Mulder NH, Buys CH, Hofstra RM, Plukker JT. Determination of TP53 mutation is more relevant than microsatellite instability status for the prediction of disease-free survival in adjuvant-treated stage III colon cancer patients. J Clin Oncol. 2005;23:5635–5643. doi: 10.1200/JCO.2005.04.096. [DOI] [PubMed] [Google Scholar]
- 114.Kim GP, Colangelo LH, Wieand HS, Paik S, Kirsch IR, Wolmark N, Allegra CJ. Prognostic and predictive roles of high-degree microsatellite instability in colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J Clin Oncol. 2007;25:767–772. doi: 10.1200/JCO.2006.05.8172. [DOI] [PubMed] [Google Scholar]
- 115.Lamberti C, Lundin S, Bogdanow M, Pagenstecher C, Friedrichs N, Buttner R, Sauerbruch T. Microsatellite instability did not predict individual survival of unselected patients with colorectal cancer. Int J Colorectal Dis. 2007;22:145–152. doi: 10.1007/s00384-006-0131-8. [DOI] [PubMed] [Google Scholar]
- 116.Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55:5548–5550. [PubMed] [Google Scholar]
- 117.Samowitz WS, Curtin K, Neuhausen S, Schaffer D, Slattery ML. Prognostic implications of BAX and TGFBRII mutations in colon cancers with microsatellite instability. Genes Chromosomes Cancer. 2002;35:368–371. doi: 10.1002/gcc.10125. [DOI] [PubMed] [Google Scholar]
- 118.Fernández-Peralta AM, Nejda N, Oliart S, Medina V, Azcoita MM, Gonzalez-Aguilera JJ. Significance of mutations in TGFBR2 and BAX in neoplastic progression and patient outcome in sporadic colorectal tumors with high-frequency microsatellite instability. Cancer Genet Cytogenet. 2005;157:18–24. doi: 10.1016/j.cancergencyto.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 119.Jung B, Smith EJ, Doctolero RT, Gervaz P, Alonso JC, Miyai K, Keku T, Sandler RS, Carethers JM. Influence of target gene mutations on survival, stage and histology in sporadic microsatellite unstable colon cancers. Int J Cancer. 2006;118:2509–2513. doi: 10.1002/ijc.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sinicrope FA, Rego RL, Halling KC, Foster N, Sargent DJ, La Plant B, French AJ, Laurie JA, Goldberg RM, Thibodeau SN, Witzig TE. Prognostic impact of microsatellite instability and DNA ploidy in human colon carcinoma patients. Gastroenterology. 2006;131:729–737. doi: 10.1053/j.gastro.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 121.Choi SW, Lee KJ, Bae YA, Min KO, Kwon MS, Kim KM, Rhyu MG. Genetic classification of colorectal cancer based on chromosomal loss and microsatellite instability predicts survival. Clin Cancer Res. 2002;8:2311–2322. [PubMed] [Google Scholar]
- 122.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 123.Ogino S, Meyerhardt JA, Kawasaki T, Clark JW, Ryan DP, Kulke MH, Enzinger PC, Wolpin BM, Loda M, Fuchs CS. CpG island methylation, response to combination chemotherapy, and patient survival in advanced microsatellite stable colorectal carcinoma. Virchows Arch. 2007;450:529–537. doi: 10.1007/s00428-007-0398-3. [DOI] [PubMed] [Google Scholar]
- 124.Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–2903. [PubMed] [Google Scholar]
- 125.Samowitz WS, Holden JA, Curtin K, Edwards SL, Walker AR, Lin HA, Robertson MA, Nichols MF, Gruenthal KM, Lynch BJ, Leppert MF, Slattery ML. Inverse relationship between microsatellite instability and K-ras and p53 gene alterations in colon cancer. Am J Pathol. 2001;158:1517–1524. doi: 10.1016/S0002-9440(10)64102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, Mino-Kenudson M, Lauwers GY, Loda M, Fuchs CS. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–421. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fox EJ, Leahy DT, Geraghty R, Mulcahy HE, Fennelly D, Hyland JM, O'Donoghue DP, Sheahan K. Mutually exclusive promoter hypermethylation patterns of hMLH1 and O6-methylguanine DNA methyltransferase in colorectal cancer. J Mol Diagn. 2006;8:68–75. doi: 10.2353/jmoldx.2006.050084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–217. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, Hamilton SR, Issa JP. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kawasaki T, Ohnishi M, Nosho K, Suemoto Y, Kirkner GJ, Meyerhardt JA, Fuchs CS, Ogino S. CpG island methylator phenotype (CIMP)-low colorectal cancer shows not only fewer methylated CIMP-high-specific CpG island, but also low-level methylation in individual loci. Mod Pathol. 2008 doi: 10.1038/modpathol.3800982. (in press) [DOI] [PubMed] [Google Scholar]