

Abstract
Fragile X syndrome, the most common inherited cause of intellectual impairment and the most common single gene associated with autism, generally occurs for fragile X mental retardation 1 (FMR1) alleles that exceed 200 CGG repeats (full-mutation range). Currently, there are no unbiased estimates of the number of full-mutation FMR1 alleles in the general population; a major obstacle is the lack of an effective screening tool for expanded FMR1 alleles in large populations. We have developed a rapid polymerase chain reaction (PCR)-based screening tool for expanded FMR1 alleles. The method utilizes a chimeric PCR primer that targets randomly within the expanded CGG region, such that the presence of a broad distribution of PCR products represents a positive result for an expanded allele. The method is applicable for screening both males and females and for allele sizes throughout the premutation (55 to 200 CGG repeats) and full-mutation ranges. Furthermore, the method is capable of rapid detection of expanded alleles using as little as 1% of the DNA from a single dried blood spot. The methodology presented in this work is suitable for screening large populations of newborn or those at high risk (eg, autism, premature ovarian failure, ataxia, dementia) for expanded FMR1 alleles. The test described herein costs less than $5 per sample for materials; with suitable scale-up and automation, the cost should approach $1 per sample.
Mutations of the fragile X mental retardation 1 (FMR1) gene (OMIM *309550) are responsible for several distinct forms of clinical involvement that together constitute a significant societal health burden across the life spectrum—for child health, reproductive fitness in women, and aging. Fragile X syndrome, mostly caused by large (noncoding) CGG repeat expansions (>200 repeats; full mutation) within the FMR1 gene, is the leading heritable form of cognitive impairment1 and the leading single-gene disorder associated with autism.2 Smaller expansions (55 to 200 CGG repeats; premutation) of the FMR1 gene give rise to the leading known cause of premature ovarian failure (menopause before age 40), with 4 to 14% of all cases of premature ovarian failure in the general population due to premutation alleles.3 Premutation FMR1 alleles are also responsible for a late-onset neurodegenerative disorder, fragile X-associated tremor/ataxia syndrome, with core features of progressive action tremor and gait ataxia and associated features of dementia, parkinsonism, and autonomic dysfunction.4
Prior estimates of frequencies of female carriers of premutation alleles, by direct population screening, have yielded values ranging from ∼1/100 from three Israeli studies5,6,7 to ∼1/260 from Israeli, Canadian, and Finnish studies.8,9,10 Frequency data for male premutation carriers, with premutation allele frequencies of ∼1/25011 and ∼1/800,12 are largely from two Canadian studies. In aggregate, these frequency estimates suggest that there may be between 1- and 2-million premutation carriers in the United States; however, there have been no screens of the general population within the United States to assess male and female carrier frequencies.
The situation is more difficult for the estimation of full-mutation alleles, those giving rise to fragile X syndrome, due to their much lower frequency. As a consequence, essentially all estimates to date have targeted populations with special education needs or mental retardation, with subsequent extrapolation to the general population. Earlier estimates of one full-mutation allele in 5000 to 9000 individuals13,14,15 are likely to be too low, because those estimates did not fully account for fragile X children without mental retardation. More recent estimates are generally higher, from one in 2500 to 5000 for the larger studies of special education needs children6,16,17,18,19,20; however, as for premutation alleles, there have been no large-scale unbiased (eg, newborn) screens for full-mutation FMR1 alleles in the general population, due in part to the absence of an effective screening tool.
Although newborn screening represents a logical approach to obtain unbiased estimates for expanded FMR1 alleles, there have been two main barriers to its implementation: the perceived lack of effective medical intervention for those identified with expanded (premutation or full mutation) alleles and the lack of a screening tool that is both rapid and cost-effective. The former issue has been discussed in detail elsewhere.21,22 However, despite the importance of continuing debate on newborn screening, it is clear that identification of expanded alleles during the newborn period would not only enable early intervention for learning delays but could also provide families the time needed to plan appropriately for subsequent pregnancies23 and provide important information about reproductive risks and reduce the stress and frustration of not knowing the basis of a child's developmental delay.
With respect to the absence of an effective screening tool, several previous PCR-based approaches have been developed to identify expanded FMR1 alleles.24,25,26,27,28,29,30 Unfortunately, none has been able to both rapidly and reliably identify full-mutation alleles due to the high GC content of the repeat expansion. This limitation of the PCR method does not represent a problem for diagnostic genotyping, in which a combination of PCR and Southern blot methods reliably detects all alleles and in which recent incorporation of capillary-based methods31 dramatically increases throughput. However, such approaches are neither time- nor cost-effective methods for screening large populations, particularly in situations such as newborn screening where blood spots are used. In addition, none of these methodologies has been shown to be applicable to blood spots. Although improved PCR methods using the osmolyte betaine are capable of detecting alleles as large as ∼300 CGG repeats,30 the betaine-based PCR approach is unable to distinguish between females who are homozygous for normal FMR1 alleles (single normal band following PCR; ∼40% of all females) and females with one normal allele and a second full-mutation allele that does not PCR-amplify (single normal band, apparent homozygote). This issue has been the greatest impediment to high-throughput screening.
In the current work, we describe a PCR test that effectively and reliably distinguishes between normal homozygous females and females with a very large (eg, full mutation) allele, which conventional PCR using primers that flank the CGG repeat fail to amplify. This approach, which utilizes a “chimeric” CGG-targeted primer in conjunction with betaine-based-PCR,30 allows rapid determination of the allele status of all males and females, including those females with a single band on standard PCR, regardless of the number of CGG repeats. Furthermore, the approach can be used to analyze allele status from blood spots, thus enabling newborn screening.
Materials and Methods
Samples
Blood samples were obtained from subjects seen at the M.I.N.D. Institute Clinic, following informed consent and according to an approved Institutional Review Board protocol. The DNA samples used for our in-house validation were isolated from peripheral blood leukocytes as described below, and only the code number was known to the technician who handled the samples.
For blood spots, a 1.5-mm punch or a ∼4 × 8 mm FTA card (Whatman Inc., Florham Park, NJ) was removed from the target sample and transferred to a PCR tube. Two hundred microliters of FTA purification reagent was added, followed by incubation for 5 minutes at room temperature. The reagent was then removed using a pipette, and the incubation of the disk was repeated twice (for a total of three times). Two hundred microliters of Tris sodium EDTA buffer (10 mmol/L Tris-HCl, pH 8, 0.1 mmol/L Na2EDTA) was added, followed by incubation for 5 minutes at room temperature. Buffer was removed, and the procedure was repeated once. The disk was then allowed to dry at room temperature for about 1 hour before starting the PCR reaction. PCR mix was added directly to the PCR tube containing the dry disk. For blood spots on 3MM paper (Whatman, Inc.), a 1.5-mm punch (or larger) was washed twice with 200 μl of dH20. One hundred microliters of dH20 was then added to the washed punch and boiled for 30 minutes. After a 2-minute spin, the eluted sample was used for PCR.
Southern Blot and PCR Analysis
Genomic DNA was isolated from peripheral blood leukocytes (3 to 5 ml of whole blood) using standard methods (Puregene Kit; Gentra Inc., Minneapolis, MN). For Southern blot analysis, 5 to 10 μg of isolated DNA was digested with EcoRI and NruI. Digested samples were separated on a 0.8% agarose/Tris acetate EDTA (TAE) gel, followed by partial depurination with HCl (0.4 N) for 15 minutes and denaturation in NaOH (0.5 N) for 30 minutes. DNA was transferred in 10× standard saline citrate (SSC) to a charged nylon membrane (Roche Diagnostics, Basel, Switzerland) using a vacuum transfer apparatus (Vacuum Blotter 785; Bio-Rad, Hercules, CA). A 1-kb DNA ladder (Invitrogen, Carlsbad, CA) was used as a size standard. The membranes were cross-linked (UV Cross linker; Fisher Scientific, Pittsburgh, PA) and were hybridized overnight at 42°C in roller bottles (Isotemp, Fisher Scientific) in Dig Easy Hybridization Buffer (Roche Diagnostics) with the FMR1 genomic probe StB12.3, labeled with Dig-11-dUTP by PCR (PCR Dig Synthesis Kit; Roche Diagnostics). After denaturation (boiling for 15 minutes), the probe was blocked with Cot1 DNA (Invitrogen) for 20 minutes at 65°C. Filters were washed twice for 5 minutes in 2× SSC/1% SDS and twice for 15 minutes in 1× SSC/0.1% SDS at 65°C. Filter blocking and FMR1 gene detection were performed using blocking solution and detection buffer according to the manufacture (Roche Diagnostics). Filters were exposed to X-ray film (Super RX; Fuji Medical X-Ray Film, Bedfordshire, UK) for 2 hours.
Genomic DNA was also amplified by PCR with primers c and f32 using the osmolite betaine according to Saluto et al.30 PCR fragments were visualized on a 2% agarose gel, ethidium bromide-stained. For a correct sizing, PCR products were separated on 6% denaturing polyacrylamide gels, followed by electroblot transfer (TE62; Amersham-Pharmacia Biotech, Pittsburgh, PA) to a nylon membrane (Roche Diagnostics) at 4 volts for 2 hours. Membranes were then cross-linked (UV Cross linker; Fisher Scientific). Dig-labeled DNA molecular weight Marker V (Roche Diagnostics) and a known size marker were used as size standards. Filters were hybridized overnight at 42°C in roller bottles (Isotemp; Fisher Scientific) in Dig Easy Hybridization Buffer (Roche Diagnostics) with a Dig-end-labeled oligonucleotide probe [(CGG)10] and Dig-labeled pBR322 DNA. Filters were washed at room temperature, twice for 5 minutes with 2× SSC/0.1% SDS (100 ml) and twice for 7 minutes in a larger volume (400 ml) with the same washing solution, followed by two washes of 25 minutes, each in 0.5× SSC/0.1 SDS at 45°C. Detection of the FMR1 PCR products was performed according to the manufacturer (Roche Diagnostics). Filters were exposed to X-ray film (Super RX; Fuji Medical X-Ray Film) at room temperature for approximately 30 minutes. Analysis of the repeat number for both Southern blot and PCR used an Alpha Innotech FluorChem 8800 Image Detection System (Alpha Innotech, San Leandro, CA).
PCR Amplification Using a CGG-Targeted Primer
We have further modified our PCR betaine protocol30 to consistently distinguish between females with a single PCR gel band, which arises either from comigrating equal CGG repeat number normal alleles (∼40% of females in the general population) or with a single (normal) allele and a second (nonamplifying) full-mutation allele. The principal feature of the current procedure is the use of a chimeric PCR primer, which anneals randomly within the CGG repeat expansion via a (CCG)n 3′ portion of the primer and has a remaining half-primer sequence that is unique, for the purpose of subsequent rounds of PCR amplification. Our PCR-based method is designed to be used for both purified DNA and blood spots; the latter being the preferred mode of collection of blood for newborn screening.
Standard primer PCR reactions were performed using the c and f primers (5′-GCTCAGCTCCGTTTCGGTTTCACTTCCGGT-3′; 5′-AGCCCCGCACTTCCACCACCAGCTCCTCCA-3′, respectively)32 and the Expand Long Template PCR System (Roche Diagnostics). Reaction mixtures included buffer 2 (Roche Diagnostics), 500 μmol/L dNTPs, 0.33 μM of each primer, and 100 to 500 ng of genomic DNA. The PCR buffer also included 2.2 M betaine (B0300; Sigma-Aldrich, St. Louis, MO). This higher concentration of betaine was based on a series of PCR optimization experiments using betaine concentrations from 1.3 to 2.2 M. Previous reports had recommended a concentration of 1.3 M,33 which we found to be too low for efficient expansion of the CGG element. PCR “hot-start” was performed as indicated by the manufacturer (Roche Diagnostics). The PCR cycling profile was as follows: denaturation at 98°C for 10 minutes, 10 cycles at 97°C for 35 seconds, 64°C for 35 seconds, and 68°C for 4 minutes; 25 cycles at 97°C for 35 seconds, 64°C for 35 seconds, 68°C for 4 minutes plus a 20-second increment for each cycle; and a final extension at 68°C for 10 minutes. The expected constant region of the PCR product (ie, excluding the CGG repeat region) was 221 bp. We recently modified this protocol by using the FastStart PCR kit (Roche Diagnostics), which allows us to eliminate the hot-start step and still be able to amplify FMR1 alleles in the upper premutation range. The elimination of the hot-start step allows us to amplify a much larger number of samples per PCR run, a necessary modification for a large-scale screening effort.
Samples that yielded standard primer PCR products with a single normal band for females (apparent homozygosity), or the absence of a normal band for males, were subjected to a second PCR screen with the c primer and the CCG-chimeric primer (5′-AGCGTCTACTGTCTCGGCACTTGC(CCG)4-3′; ie, targeting the CGG strand of the repeat expansion tract) in place of the f primer. Reaction mixture and PCR cycling conditions were as described above and in Saluto et al.30 Because the (CCG)4 portion of the primer anneals randomly within the CGG repeat region, PCR amplification using “c” and the chimeric primer will produce a smear on the gel that indicates the presence of expanded alleles (Figure 1); thus, the chimeric primer yields a “plus” for those cases of apparent homozygosity for which an expanded allele is present (approximately one in ∼2000 apparent homozygous females) in the general population. In the absence of an expanded allele, no large smear will be present on the analytical gel of the PCR reaction. As a screening tool, the current method is not designed to “size” high premutation or full-mutation alleles, only to flag their presence for subsequent workup that would include both Southern blot (sizing, methylation status, activation ratio) and further PCR sizing.
Figure 1.
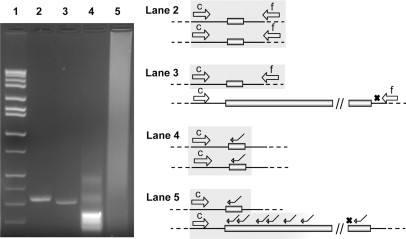
Schematic of the secondary PCR screening method to resolve apparent homozygosity in females. The approach uses a combination of betaine and a chimeric (CGG-targeted) PCR primer that primes randomly, but with a size bias for amplification of smaller expansions, within the CGG repeat. For a normal homozygous female (30,30 CGG repeats) (gel lane 2: c and f primers of Fu et al32; open arrows), only small PCR products are produced with the chimeric primer (gel lane 4: c, open arrow, and chimeric primer; line arrow). However, for a full-mutation female (28, 510, 615 CGG repeats) (lane 3: c and f primers, apparent homozygote), an extensive smear is produced with the chimeric primer (lane 5: c and chimeric primer), reflecting priming within the extended CGG repeat. For full-mutation alleles, priming by the downstream primer (primer f) does not occur. Representation of the PCR products for each lane is given to the right of the gel image (2% agarose). The chimeric primer comprises a 3′-(CCG)4 block for targeting and a 5′ unique N24 block for subsequent amplification [N24-(CCG)4]. Allele sizes of the normal and full-mutation females were obtained by both PCR (on acrylamide gel) and Southern blot analysis as described in Materials and Methods.
Results
A Rapid PCR Method to Distinguish Normal Homozygous Females from Full-Mutation Heterozygous Females: Secondary Screening Using the Chimeric CGG Repeat-Targeted Primer
The major difficulty encountered during the amplification of FMR1 alleles is the ambiguity associated with a single band (apparent homozygosity) in females, which represents ∼40 to 50% of female cases. In nearly all cases (∼99.9%), the single band indicates true homozygosity (ie, two alleles of the same size or alleles differing by 1 to 2 repeats). The problem is that it is not possible to deduce which of the 40 to 50% of female (apparent homozygous) cases that have a nonamplifying full-mutation or a very large premutation allele that may escape PCR detection. To eliminate this ambiguity, we have developed a novel PCR-based approach that involves a secondary PCR screening (of the apparent homozygous females) with a chimeric primer that randomly targets the CGG region itself. When used in combination with the standard primer c,32 the chimeric primer in the secondary PCR reaction produces an extended smear of amplified species only if an expanded allele is present (Figure 1). A smear will be visible on an agarose gel whether the FMR1 allele is in the high premutation range (data not shown) or in the full-mutation range (Figure 2A). Thus, the secondary screen returns a “yes” or “no” to the question of the presence of an expanded allele. This is also true for mosaic males, both for repeat size (presence of both premutation and full-mutation alleles) and/or for methylation (presence of partially methylated, full-mutation alleles), where expanded alleles are present and therefore visible as a smear on an agarose gel with secondary PCR screening (data not shown).
Figure 2.
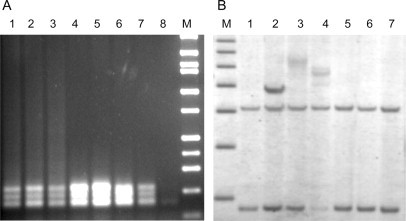
A: 2% agarose gel of PCR products using the chimeric primer. Lanes 1 to 3: DNA templates from three females with full-mutation alleles; lanes 4 to 7: DNA derived from four females with normal alleles; lane 8: control PCR without template DNA. B: Southern blot analysis of DNAs depicted in A. Lanes 2 to 4: full-mutation alleles corresponding to lanes 1 to 3 of A. Lanes 1, 5 to 7: normal alleles (2.8- and 5.2-kb bands) corresponding to lanes 4 to 7 of A. Different markers are used for each panel, and there is no direct size correspondence between gels.
As an initial test of this secondary screening method, we used the chimeric primer to analyze DNA samples from 15 normal females and 15 full-mutation females from our DNA archive, where the technician performing the PCR reactions and subsequent analysis was blinded to the allele status of the samples (in-house validation study). For all full-mutation alleles, the PCR reactions with the (CCG)4-containing primer consistently produced amplified products, visible as large smears on agarose gels (Figure 2A). The results were validated and confirmed by Southern blot and PCR/acrylamide gel analysis (Figure 2B).
We have further tested this approach by screening one hundred DNA samples sent to our laboratory in a blinded fashion by Dr. Annette Taylor (Kimball Genetics, Inc., Denver, CO). The samples had been stripped of all identifiers, retaining only a sample code and the gender of the sample donor. Although none of the investigators at University of California at Davis had any knowledge of the allele status of any sample, all samples had been characterized previously (by both PCR and Southern blot) in Dr. Taylor's laboratory in Denver. Results of the screening analysis were disclosed to Dr. Taylor, who was not blinded to sample genotypes, following completion of the screening process. In our screen, we identified 50 homozygous (normal) females and 50 full-mutation females, all correctly confirmed for genotype by Dr. Taylor.
Thus, the combination of two PCR approaches, one using standard c and f primers and the other using the c primer and a chimeric primer, allows us to detect expanded FMR1 alleles in females, regardless of the CGG repeat size, using a simple PCR-based approach.
Screening of Blood Spots Using the Chimeric Primer
Although our initial studies using the chimeric primer were performed on DNA samples isolated from peripheral blood leukocytes, the current PCR-based approach also performs well with DNA samples obtained directly from dried blood spots. Indeed, the sensitivity of the method allows us to detect FMR1 alleles using as little as 1/100 of the DNA sample extracted from the spot (Figure 3A). In addition, we have found that alleles can be scored, even if no prior processing (eg, extraction) has been performed on the spot itself (Figure 3B), thus further reducing the time required for sample processing. Furthermore, the method works with different types of blood spot paper (eg, FTA card, regular 3MM paper; Whatman, Inc.), reducing the likelihood that there will be differences in sample quality across laboratories. Of course, issues of spot storage integrity and age still need to be assessed. Finally, the method seems to be reliable whether blood spots are added directly to the PCR mixture or processed (washed twice with Tris-EDTA) (Figure 3B, lanes 3 and 4, respectively).
Figure 3.
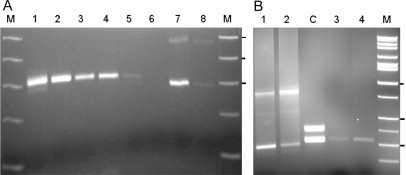
A: PCR product (normal allele of 28 CGG repeats) was detected from 10, 5, 2, 1, and 0.1% (where 1% or 1 μl contains between ∼1 and 5 ng of DNA) (lanes 1 to 5, respectively) of the eluted DNA from a single dried blood spot (∼4 × 8-mm FTA cards; Whatman, Inc.). Elution of DNA was according to manufacturer's instructions; almost no PCR product was detected from 0.01% of the dried spot (lane 6). Alleles from a premutation female (21 and 86 CGG repeats), amplified from 1 and 0.1% of eluted DNA, are shown in lanes 7 to 8. Five microliters of PCR product were loaded per lane. B: a mixture of control (21 CGG repeats) and premutation (∼200 CGG repeats) alleles were amplified using different proportions of DNAs from the control and carrier males. Both bands were visible in lane 1 (100 ng of DNA from each sample) and lane 2 (100 ng of premutation DNA and 50 ng of control DNA) of the 2% agarose gel. A female control (30 and 54 CGG repeats) is also shown (lane C). PCR products obtained from a small portion of a dried blood spot from a control male, with (lane 4) and without (lane 3) a washing step, added directly to the PCR reaction. M: marker (High-Low; Bionexus, Oakland, CA). Tick marks to the right of each figure indicate 300, 400, and 500 bp in A and 300, 500, and 750 bp in B.
To gauge the detection of an allele at the high end of the premutation range in a female carrier, we performed a PCR reaction on a mixture of DNAs from a normal (male) control and a carrier male, with an allele at the upper end of the premutation range (∼200 CGG repeats). As shown in Figure 3B, the larger allele is still visible in the presence of the normal allele, demonstrating that our method is capable of detecting all premutation alleles—for both males and females—at least to the upper end of the premutation range. However, even under conditions where the first PCR screen (c and f primers) would fail to detect a high premutation allele in a female, the presence of such an allele would be identified with the second PCR screen using the chimeric primer.
Discussion
One of the main impediments to the implementation of newborn screening for expanded alleles of the FMR1 gene has been the absence of a rapid, inexpensive screening tool, a tool that would be applicable to the screening of small amounts of biological material (eg, blood spots) and that would be capable of detecting all expanded alleles in both males and females. In the current work, we have described a novel PCR-based approach for FMR1 genotyping that combines modification of the betaine protocol30 with a use of a CGG-targeted (chimeric) primer, which would generate an extensive distribution of PCR products only in the presence of premutation or full-mutation alleles.
To underscore both the role and utility of this test, consider its application to a screen of 100,000 newborn bloodspots. For the ∼50,000 males in the population, the primary PCR screen (using primers c and f) would fail to yield a band in approximately 10 to 15 cases (assuming a frequency of full-mutation alleles of ∼1/3000 to 1/5000). These “null-allele” results would then be subjected to follow-up with the CGG-targeted primer (to distinguish expanded alleles from failure of the PCR reaction) and, for legitimate expanded alleles, more traditional Southern blots during newborn follow-up. However, for the females, single bands (apparent homozygosity) would be expected in approximately 20,000 cases (assuming 40% homozygosity) (Figure 4). In this instance, the use of the CGG-targeted primer should yield broad smears in another 10 to 15 cases (and perhaps a few additional cases with high-end premutation alleles), which would be subject to newborn follow-up and Southern blots for definitive genotyping. Although the current method is not designed to detect sexual chromosome aneuploidy (eg, XO, XXY, etc), it would flag those cases that present as double bands in males or more than two bands in females.
Figure 4.

Flow diagram for screening of DNA or blood spots from 50,000 females. First round PCR screening will size all normal and/or premutation bands and will pass through to a second PCR screen (using c and CGG-repeat targeted chimeric primers), all samples displaying a single band (∼20,000). Nearly all of these samples (∼19,985) will be homozygous normal alleles, with only ∼15 samples passed onto Southern blot analysis, based on the presence of a large smear on the analytical gel. This example is based on an assumed 15 full-mutation alleles per 50,000 samples in the general population. The analysis for males is much simpler; only those samples with no band on the first round are passed to the second PCR screening assay and/or directly to the Southern blot analysis.
We have recently applied the betaine PCR method (with primers c and f) to a high-risk screen of 903 adult males with a diagnosis of Parkinson's disease.34 A similar study of ∼600 adult females, using the CGG targeting method, has also been conducted (K. Amiri, J. Kraff, H.-T. Tang, R. Pan, S. Goldworm, J.P. Hagerman, F. Tassone, unpublished results), with no false-positive or negative genotypes to date. Furthermore, in the validation study performed in conjunction with Kimball Genetics, Inc., we were able to correctly identify all fifty females with full-mutation alleles (apparent homozygosity by standard PCR); this number corresponds to the expected number of full mutations in a general population of ∼150,000 females. Therefore, both in terms of effectiveness and cost ($2 to $5 in reagents and supplies per sample), the current screening test should enable large-scale newborn and/or high-risk screening for expanded alleles of the FMR1 gene. This capability will, in turn, facilitate investigation of the benefits and potential pitfalls of newborn screening and early intervention.21,22,23
However, to date, there is no published technology that allows the screening of blood spots for all mutation classes of the fragile X gene in both males and females. Neither Southern blot nor capillary electrophoresis methodologies are practical for large-scale screening, mainly due to the costs associated with such methods but also because such methods have not been validated for blood spots where very little DNA is available. Our procedure, which allows the detection of alleles in the normal, premutation, and full-mutation ranges in both males and females, will facilitate such large-scale screening studies since the identification of large alleles in females is not limited by either allele size for the chimeric primer or the small amounts of DNA recovered from portions of a single blood spot. Finally, although we have demonstrated the validity and feasibility of this procedure in terms of a manual gel-based method, extensions to include automated detection methods, such as the use of fluorescent primers and capillary- or gel-based automated scanning procedures, can be explored.
Note Added in Proof
Since the publication of this manuscript we have become aware that a similar approach has been reported for the amplification of large CAG repeat expansion.35 Fluorescent primers were used to amplify alleles with more than 100 CAG repeats because a standard PCR was not reliable in that range.
Acknowledgements
This work is dedicated to the memory of Matteo.
Footnotes
Supported by National Institutes of Child Health and Development Grants HD02274 (to F.T.) and HD040661 (to P.J.H.) and the Boory family.
References
- 1.Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome: Diagnosis, Treatment, and Research. 3rd Ed. The Johns Hopkins University Press; Baltimore: 2002. [Google Scholar]
- 2.Schaefer GB, Lutz RE. Diagnostic yield in the clinical genetic evaluation of autism spectrum disorders. Genet Med. 2006;8:549–556. doi: 10.1097/01.gim.0000237789.98842.f1. [DOI] [PubMed] [Google Scholar]
- 3.Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, Corrigan EC, Simpson JL, Nelson LM. The FMR1 premutation and reproduction. Fertil Steril. 2007;87:456–465. doi: 10.1016/j.fertnstert.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Jacquemont S, Hagerman RJ, Hagerman PJ, Leehey MA. Fragile-X syndrome and fragile X-associated tremor/ataxia syndrome: two faces of FMR1. Lancet Neurol. 2007;6:45–55. doi: 10.1016/S1474-4422(06)70676-7. [DOI] [PubMed] [Google Scholar]
- 5.Geva E, Yaron Y, Shomrat R, Ben-Yehuda A, Zabari S, Peretz H, Naiman T, Yeger H, Orr-Urtreger A. The risk of fragile X premutation expansion is lower in carriers detected by general prenatal screening than in carriers from known fragile X families. Genet Test. 2000;4:289–292. doi: 10.1089/10906570050501524. [DOI] [PubMed] [Google Scholar]
- 6.Pesso R, Berkenstadt M, Cuckle H, Gak E, Peleg L, Frydman M, Barkai G. Screening for fragile X syndrome in women of reproductive age. Prenatal Diag. 2000;20:611–614. doi: 10.1002/1097-0223(200008)20:8<611::aid-pd881>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 7.Toledano-Alhadef H, Basel-Vanagaite L, Magal N, Davidov B, Ehrlich S, Drasinover V, Taub E, Halpern GJ, Ginott N, Shohat M. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet. 2001;69:351–360. doi: 10.1086/321974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drasinover V, Ehrlich S, Magal N, Taub E, Libman V, Shohat T, Halpern GJ, Shohat M. Increased transmission of intermediate alleles of the FMR1 gene compared with normal alleles among female heterozygotes. Am J Med Genet. 2000;93:155–157. doi: 10.1002/1096-8628(20000717)93:2<155::aid-ajmg13>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 9.Rousseau F, Rouillard P, Morel ML, Khandjian EW, Morgan K. Prevalence of carriers of premutation-size alleles of the FMRI gene–and implications for the population genetics of the fragile X syndrome. Am J Hum Genet. 1995;57:1006–1018. [PMC free article] [PubMed] [Google Scholar]
- 10.Ryynanen M, Heinonen S, Makkonen M, Kajanoja E, Mannermaa A, Pertti K. Feasibility and acceptance of screening for fragile X mutations in low-risk pregnancies. Eur J Hum Genet. 1999;7:212–216. doi: 10.1038/sj.ejhg.5200285. [DOI] [PubMed] [Google Scholar]
- 11.Dawson AJ, Chodirker BN, Chudley AE. Frequency of FMR1 premutations in a consecutive newborn population by PCR screening of Guthrie blood spots. Biochem Mol Med. 1995;56:63–69. doi: 10.1006/bmme.1995.1057. [DOI] [PubMed] [Google Scholar]
- 12.Dombrowski C, Levesque S, Morel ML, Rouillard P, Morgan K, Rousseau F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11:371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- 13.de Vries BB, van den Ouweland AM, Mohkamsing S, Duivenvoorden H, Mol E, Gelsema K, van Rijn M, Halley DJ, Sandkuijl LA, Oostra BA, Tibben A, Niermeijer MF. Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Collaborative Fragile X Study Group. Am J Hum Genet. 1997;61:660–667. doi: 10.1086/515496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobs PA, Bullman H, Macpherson J, Youings S, Rooney V, Watson A, Dennis NR. Population studies of the fragile X: a molecular approach. J Med Genet. 1993;30:454–459. doi: 10.1136/jmg.30.6.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner G, Webb T, Wake S, Robinson H. Prevalence of fragile X syndrome. Am J Med Genet. 1996;64:196–197. doi: 10.1002/(SICI)1096-8628(19960712)64:1<196::AID-AJMG35>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 16.Crawford DC, Meadows KL, Newman JL, Taft LF, Scott E, Leslie M, Shubek L, Holmgreen P, Yeargin-Allsopp M, Boyle C, Sherman SL. Prevalence of the fragile X syndrome in African-Americans. Am J Med Genet. 2002;110:226–233. doi: 10.1002/ajmg.10427. [DOI] [PubMed] [Google Scholar]
- 17.Youings SA, Murray A, Dennis N, Ennis S, Lewis C, McKechnie N, Pound M, Sharrock A, Jacobs P. FRAXA and FRAXE: the results of a five year survey. J Med Genet. 2000;37:415–421. doi: 10.1136/jmg.37.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3:359–371. doi: 10.1097/00125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman S. Epidemiology. In: Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome: Diagnosis, Treatment and Research. 3rd Ed. The Johns Hopkins University Press; Baltimore: 2002. [Google Scholar]
- 20.Song FJ, Barton P, Sleightholme V, Yao GL, Fry-Smith A. Screening for fragile X syndrome: a literature review and modelling study. Health Technol Assess. 2003;7:1–106. doi: 10.3310/hta7160. [DOI] [PubMed] [Google Scholar]
- 21.Bailey DB, Jr, Beskow LM, Davis AM, Skinner D. Changing perspectives on the benefits of newborn screening. Ment Retard Dev Disabil Res Rev. 2006;12:270–279. doi: 10.1002/mrdd.20119. [DOI] [PubMed] [Google Scholar]
- 22.Bailey DB, Jr, Skinner D, Warren SF. Newborn screening for developmental disabilities: reframing presumptive benefit. Am J Public Health. 2005;95:1889–1893. doi: 10.2105/AJPH.2004.051110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey DB., Jr Newborn screening for fragil6e X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10:3–10. doi: 10.1002/mrdd.20002. [DOI] [PubMed] [Google Scholar]
- 24.Brown WT, Nolin S, Houck G, Jr, Ding X, Glicksman A, Li SY, Stark-Houck S, Brophy P, Duncan C, Dobkin C, Jenkins E. Prenatal diagnosis and carrier screening for fragile X by PCR. Am J Med Genet. 1996;64:191–195. doi: 10.1002/(SICI)1096-8628(19960712)64:1<191::AID-AJMG34>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 25.Haddad LA, Mingroni-Netto RC, Vianna-Morgante AM, Pena SD. A PCR-based test suitable for screening for fragile X syndrome among mentally retarded males. Hum Genet. 1996;97:808–812. doi: 10.1007/BF02346194. [DOI] [PubMed] [Google Scholar]
- 26.Hamdan H, Tynan JA, Fenwick RA, Leon JA. Automated detection of trinucleotide repeats in fragile X syndrome. Mol Diagn. 1997;2:259–269. doi: 10.1054/MODI00200259. [DOI] [PubMed] [Google Scholar]
- 27.Hecimovic S, Vlasic J, Barisic L, Markovic D, Culic V, Pavelic K. A simple and rapid analysis of triplet repeat diseases by expand long PCR. Clin Chem Lab Med. 2001;39:1259–1262. doi: 10.1515/CCLM.2001.202. [DOI] [PubMed] [Google Scholar]
- 28.Houdayer C, Lemonnier A, Gerard M, Chauve C, Tredano M, de Villemeur TB, Aymard P, Bonnefont JP, Feldmann D. Improved fluorescent PCR-based assay for sizing CGG repeats at the FRAXA locus. Clin Chem Lab Med. 1999;37:397–402. doi: 10.1515/CCLM.1999.065. [DOI] [PubMed] [Google Scholar]
- 29.O'Connell CD, Atha DH, Jakupciak JP, Amos JA, Richie K. Standardization of PCR amplification for fragile X trinucleotide repeat measurements. Clin Genet. 2002;61:13–20. doi: 10.1034/j.1399-0004.2002.610103.x. [DOI] [PubMed] [Google Scholar]
- 30.Saluto A, Brussino A, Tassone F, Arduino C, Cagnoli C, Pappi P, Hagerman P, Migone N, Brusco A. An enhanced polymerase chain reaction assay to detect pre- and full mutation alleles of the fragile X mental retardation 1 gene. J Mol Diagn. 2005;7:605–612. doi: 10.1016/S1525-1578(10)60594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strom CM, Huang D, Li Y, Hantash FM, Rooke J, Potts SJ, Sun W. Development of a novel, accurate, automated, rapid, high-throughput technique suitable for population-based carrier screening for fragile X syndrome. Genet Med. 2007;9:199–207. doi: 10.1097/gim.0b013e31803d3ac9. [DOI] [PubMed] [Google Scholar]
- 32.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST, Oostra BA, Nelson DL, Caskey CT. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 33.Baskaran N, Kandpal RP, Bhargava AK, Glynn MW, Bale A, Weissman SM. Uniform amplification of a mixture of deoxyribonucleic acids with varying GC content. Genome Res. 1996;6:633–638. doi: 10.1101/gr.6.7.633. [DOI] [PubMed] [Google Scholar]
- 34.Kraff J, Tang H-T, Cilia R, Canesi M, Pezzoli G, Goldwurm S, Hagerman PJ, Tassone F. Screen for excess FMR1 premutation alleles among males with parkinsonism. Arch Neurol. 2007;64:1002–1006. doi: 10.1001/archneur.64.7.1002. [DOI] [PubMed] [Google Scholar]
- 35.Warmer JP, Barron LH, Goudie D, Kelly K, Dow D, Fitzpatrick DR, Brock DJH. A general method for the detection of large CAG repeat expansion by fluorescent PCR. J Med Genet. 1996;33:1022–1026. doi: 10.1136/jmg.33.12.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]