

Abstract
In ∼20 to 30% of patients with systemic mastocytosis (SM), an associated clonal hematological nonmast cell lineage disorder (AHNMD) is diagnosed. Although SM may be considered to be closely related to the myeloproliferative disorders (MPDs), it is unknown whether JAK2V617F+ MPD may occur as AHNMD in patients with SM. We here describe five patients with SM and co-existing chronic idiopathic myelofibrosis (SM-CIMF). In five of five patients, we detected the SM-related KIT mutation D816V, and in four of five patients, the MPD-related JAK2 mutation V617F. Surprisingly, JAK2V617F was found not only in the AHMMD component of the disease but also in microdissected mast cells in all four JAK2V617F-positive cases. Conversely, in two of the five patients, KITD816V was found not only in neoplastic mast cells but also in microdissected CD15+ neoplastic myeloid cells. Control experiments showed that 10 indolent SM patients without associated MPD did not carry the JAK2 mutation V617F and that 15 CIMF patients without SM did not carry the KIT mutation D816V. Altogether, these data suggest that KITD816V+ SM can co-exist with JAK2V617F+ CIMF and that, in some of these SM-CIMF cases, the two mutations are present in the neoplastic cells of both disease components.
Systemic mastocytosis (SM) is a myeloid neoplasm characterized by abnormal growth and accumulation of mast cells (MCs) in one or more extracutaneous organs.1,2 Based on clinical and histopathological features, SM now is considered to be closely related to the group of myeloproliferative disorders (MPDs).3,4 SM is a clonal disease exhibiting the transforming KIT mutation D816V in most patients.5,6 Clinically, SM presents as a hematological disease with a variable course and prognosis. The World Health Organization classification defines four distinct categories of SM: indolent SM (ISM), SM with an associated clonal hematological non-MC-lineage disease (SM-AHNMD), aggressive SM, and MC leukemia.7,8
In 20 to 30% of cases with SM, an AHNMD is detectable.9,10,11 In most patients, the AHNMD component of the disease is a myeloid neoplasm, such as acute myeloid leukemia or, more frequently, chronic myelomonocytic leukemia. In the latter, the KIT mutation D816V may be detected not only in neoplastic MCs but also in neoplastic monocytes.12,13,14 So far, only a few SM cases with an associated MPD have been reported.
Recently, the JAK2 mutation V617F has been introduced as a specific molecular marker for Ph-chromosome-negative variants of MPD.15,16,17,18 In the present study, we describe five SM-AHNMD patients in whom the AHNMD-component was classified as chronic idiopathic myelofibrosis (CIMF) based on clinical, histopathological, and molecular biological findings. To the best of our knowledge, this is the first report describing the coexistence of KITD816V+ SM with JAK2V617F+ MPD (SM-CIMF).
Materials and Methods
Patients
In a retrospective analysis, a group of five patients with SM and associated CIMF (SM-CIMF) was identified during a period of 6 years in a reference center of hematopathology. As control groups, 10 KITD816V+ patients with ISM lacking a co-existing MPD (pure ISM) and 15 cases of CIMF without co-existing SM (pure CIMF) were analyzed. Routinely processed bone marrow trephine biopsy specimens were used for immunohistochemical and molecular studies. Diagnoses were established according to World Health Organization criteria. Local ethic guidelines were followed for this study. The patient's characteristics are depicted in Table 1.
Table 1.
Patient Characteristics
| Diagnoses | Age | Gender | Splenomegaly | WBC count, ×109/L | Platelet count, ×109/L | Hgb concentration, g/dl | Skin lesions |
|---|---|---|---|---|---|---|---|
| SM-CIMF, no. 82 | 61 | M | Yes | 5.6 | 690 | 16.4 | No |
| SM-CIMF, no. 302 | 78 | M | NA | 7.1 | 1100 | 11.7 | No |
| SM-CIMF, no. 605 | 76 | F | No | 14.3 | 890 | 5.7 | No |
| SM-CIMF, no. 661 | 54 | M | Yes | 7.6 | 680 | 7.1 | No |
| SM-CIMF, no. 883 | 82 | F | NA | 11.3 | 650 | 14.8 | No |
| CIMF, no. 156 | 74 | F | NA | NA | NA | NA | No |
| CIMF, no. 287 | 64 | M | Yes | 1.3 | 46 | 7.9 | No |
| CIMF, no. 289 | 71 | M | NA | NA | NA | NA | No |
| CIMF, no. 315 | 65 | M | No | 4.4 | 110 | 10.1 | No |
| CIMF, no. 320 | 73 | F | NA | 25.0 | 141 | 13.7 | No |
| CIMF, no. 382 | 73 | M | NA | 31.0 | 1400 | 15.2 | No |
| CIMF, no. 562 | 83 | M | Yes | 21.0 | 28 | 8.7 | No |
| CIMF, no. 639 | 67 | M | Yes | 8.7 | 127 | 12.8 | No |
| CIMF, no. 643 | 63 | F | Yes | 17.3 | 540 | 15.6 | No |
| CIMF, no. 699 | 61 | M | Yes | 37.0 | 93 | 13.7 | No |
| CIMF, no. 1554 | 62 | M | NA | 11.3 | 740 | 13.5 | No |
| CIMF, no. 1593* | 57 | M | NA | 7.7 | 1200 | 13.4 | No |
| CIMF, no. 1691 | 38 | F | NA | 8.2 | 900 | 15.8 | No |
| CIMF, no. 1806 | 67 | F | NA | 5.6 | 790 | 14.0 | No |
| CIMF, no. 1821 | 70 | F | Yes | 13.6 | 250 | 10.9 | No |
| ISM, no. 20 | 27 | F | No | NA | NA | NA | Yes |
| ISM, no. 253 | 65 | F | No | NA | NA | NA | Yes |
| ISM, no. 263 | 51 | F | No | NA | NA | NA | Yes |
| ISM, no. 275 | 73 | F | No | NA | NA | NA | Yes |
| ISM, no. 374 | 59 | F | No | NA | NA | NA | Yes |
| ISM, no. 835 | 49 | M | No | NA | NA | NA | Yes |
| ISM, no. 1019 | 62 | F | No | NA | NA | NA | Yes |
| ISM, no. 1023 | 51 | F | No | NA | NA | NA | Yes |
| ISM, no. 1039 | 34 | F | No | NA | NA | NA | Yes |
| ISM, no. 1043 | 33 | M | No | NA | NA | NA | Yes |
WBC, white blood cell count; Hgb, hemoglobin; CM, cutaneous mastocytosis; M, male; F, female; NA, not assessable.
JAK2-wt.
Tissue Processing
All biopsies were fixed in 5% neutral buffered formalin and mildly decalcified overnight in ethylenediaminetetraacetic acid. The paraffin sections were cut at 5 μm and stained with hematoxylin and eosin, Giemsa, and naphthol AS-D chloroacetate esterase. Immunohistochemistry was performed using the avidin-biotin immunoperoxidase (ABC) staining technique19 with antibodies against CD15 (Biocarta, Hamburg, Germany), CD25 (Novocastra, New Castle, UK), CD34 (QBEND10; Biocare, Walnut Creek, CA), tryptase (AA-1; DAKO, Glostrup, Denmark), CD31 (DAKO Diagnostika, Hamburg, Germany), and chymase (DAKO Diagnostika). 3,3′-Diaminobenzidine (Zytomed, Berlin, Germany) was used as chromogen (Figure 1), and slides were counterstained with hemalaun (DAKO Diagnostika). For microdissection of immunostained single cells, AEC (3-amino 9 ethyl-carbazole, DAKO Diagnostika) was used as chromogen, and the counterstain step was omitted.
Figure 1.

SM with associated CIMF = SM-AHNMD/CIMF (case no. 302). A: Lower magnification clearly shows CIMF with extremely hypercellular BM, increased neutrophilic granulocytopoiesis (depicted in red), clustering of pleomorphic megakaryocytes, and a large (pale-appearing) infiltrate in the left central portion of the picture. A diagnosis of SM cannot be made without additional stainings. B: Higher magnification of this infiltrate exhibits an abundance of spindle-shaped cells with round nuclei and inconspicuous nucleoli. The overwhelming majority of spindle-shaped cells is not stained with CAE. C: Immunostaining clearly shows a strong cytoplasmic reactivity of these cells with an antibody against tryptase. Spindle-shaped tryptase-positive cells in the BM are atypical MCs allowing a diagnosis of SM to be established. D: To further emphasize the neoplastic character of the MCs, immunostaining with anti-CD25 should be performed, here leading to strong reactivity of MCs. The cytoplasmic staining of perifocal megakaryocytes can be used as an internal control. A and B: Naphthol AS-D chloroacetate esterase (CAE); C: ABC method; AA1 (anti-tryptase); D: ABC method; anti-CD25 (Novocastra, New Castle, UK).
Isolation of Total DNA for Mutational Screening Analysis
The presence of KITD816V and JAK2V617F mutations was investigated in formalin-fixed, paraffin-embedded tissue. Total genomic DNA was extracted from five 5-μm-thick paraffin sections according to a published protocol.20 In brief, the sections were dewaxed by vortexing in 1 ml of xylene (100%) for 1 minute and by centrifugation at 13,000 rpm for 5 minutes. The procedure was repeated three times. Then, the samples were washed twice in pure ethanol and were vacuum-dried. A proteinase K digestion step was performed at 55°C (2 hours) in a final volume of 200 μl buffer (50 mmol/L Tris, 1 mmol/L ethylenediaminetetraacetic acid, 0.5% Tween-20, pH 8.5) containing 0.2 mmol/L proteinase K. Total DNA was then extracted with phenol/chloroform/isoamyl alcohol (v/v/v, 25/24/1) and precipitated with ethanol (100%)/LiCl (8 mol/L) as described elsewhere.21 Samples of 100 ng of the total extracted DNA were used for polymerase chain reaction (PCR) amplification of the hot spot regions of the c-kit and the JAK2 genes.
Genomic DNA extracted from the MC leukemia cell line HMC-1, known to harbor the KIT mutation D816V heterozygously, served as a positive control for KITD816V and as a wild-type (wt) control for the JAK2 mutation V617F.22 DNA from the human erythroleukemia cell line HEL, carrying the JAK2 mutation V617F homozygously, was used to establish an assay for the detection of the JAK2 mutation and to determine the analytical sensitivity of this assay.
DNA Amplification and Mutation Detection in Total DNA
Screening investigations for KITD816V and JAK2V617F mutations were performed with the LightCycler using the DNA master hybridization probes kit as described by the manufacturer (Roche Molecular Systems, Mannheim, Germany). In addition to Taq polymerase, LightCycler buffer, and dNTPs as supplied in the kit, the PCR mixture used for c-kit mutational analyses contained 3 mmol/L MgCl2, 5 pmol (0.25 μmol/L) of primers c-kit 2F and c-kit 2B, 6 pmol (0.3 μmol/L) of both hybridization probes, and 15 pmol (0.75 μmol/L) of a peptide nucleic acid (PNA) molecule. In contrast, the PCR mixture for JAK2 mutational analyses contained 2 mmol/L MgCl2, 5 pmol (0.25 μmol/L) of primers JAK2 2F and JAK2 2B, 6 pmol (0.3 μmol/L) of both hybridization probes, and 12.5 pmol (0.63 μmol/L) of a wt-specific locked nucleic acid (LNA) molecule.23 The PNA was synthesized using reagents from Applied Biosystems (Weiterstadt, Germany). Sequences of primers and probes are listed in Table 2. All primers and probes, as well as the PNA and the LNA molecules were obtained from TIB Molbiol (Berlin, Germany).
Table 2.
Sequences of PCR Primers and Hybridization Probes
| Oligo | Sequence | Positions (nucleotides) | GenBank accession no. | |
|---|---|---|---|---|
| KIT primers | 1F | 5′-CACAGAGACTTGGCAGCCAG-3′ | 7097-7116 | L04143 |
| 1B | 5′-CAGGATTTACATTATGAAAGTCACGG-3′ | 7309-7284 | ||
| 2F | 5′-CAGCCAGAAATATCCTCCTTACT-3′ | 7110-7132 | ||
| 2B | 5′-TTGCAGGACTGTCAAGCAGAG-3′ | 7247-7227 | ||
| KIT probes | Sensor | 5′-AGCCAGAGTCATCAAGAATGATTCTA-3′(Fl)* | 7168-7193 | |
| Anchor | (LC)5′-ATGTGGTTAAAGGAAACGTGAGTACCCA(p)-3′† | 7197-7214 | ||
| PNA | 5′-GCCAGAGACATCAAGAATG-3′ | 7169-7187 | ||
| JAK2 primers | 1F | 5′-GGCAGAGAGAATTTTCTGAAC-3′ | 54,915-935 | AL161450 |
| 1B | 5′-CCTAGCTGTGATCCTGAAACTGA-3′ | 55,164-142 | ||
| 2F | 5′-AGCAGCAAGTATGATGAGC-3′ | 55,000-018 | ||
| 2B | 5′-AGCTGTGATCCTGAAACTGAATTTTCT-3′ | 55,161-135 | ||
| JAK2 probes | Sensor | 5′-GTCTCCACAGAAACATACTCCATAA-3′(Fl)* | 55,072-048 | |
| Anchor | (LC)5′-TAAAACCAAATGCTTGTGAGAAAGCTTGCT-3′(p)† | 55,045-016 | ||
| LNA | 5′-CCACAGACACATACTCCAp-3′ | 55,068-062 | ||
The sensor probes were 3′-fluorescein-labeled (Fl).
The anchor probes were 5′-terminal labeled with LightCycler Red 640 (LC) and 3′-terminal blocked with a phosphate group (p).
For both assays, samples were amplified by running 40 cycles of denaturation (10 seconds at 95°C), annealing of PNA/LNA (8 seconds at 68°C), annealing of primers (10 seconds at 56°C), and primer extension (10 seconds at 72°C). The first cycle was preceded by 10 minutes of denaturation at 95°C. Before melting point analysis the samples were denatured 5 minutes at 95°C. Melting curve analysis started at 40°C, increasing the temperature by 0.3°C/second up to 95°C.24
Microdissection of Immunostained Bone Marrow Cells
Single cells were microdissected by the technique of laser pressure catapulting (PALM, Bernried, Germany).25 Either MC chymase+ cells or CD15+ cells were pooled to a total of 50 to 100 cells per PCR tube. Microdissected cells were resuspended in 9 μl of proteinase K buffer (50 mmol/L Tris, 1 mmol/L ethylenediaminetetraacetic acid, 0.5% Tween-20, pH 8.5), centrifuged (5 minutes at 13.000 U/minute) and incubated at 95°C for 10 minutes. After a short centrifugation step, 3 μl of proteinase K (20 mg/ml) was added, and the mixture was incubated for 12 hours at 50°C. Proteinase K was then inactivated by heating at 95°C for 10 minutes. After centrifugation, 38 μl of PCR mixture was added to each PCR tube carrying the microdissected and pretreated cells.
Detection of KITD816V and JAK2V617F in Microdissected Cells
To increase the sensitivity, nested PCR amplification was performed with outer primers c-kit 1F and c-kit 1B and inner primers c-kit 2F and c-kit 2B. To optimize the specificity, the first round of amplification was performed as a time-release PCR in a final volume of 50 μl containing 50 mmol/L KCl, 10 mmol/L Tris-HCl, pH 8.3, 200 μmol/L of each dNTP, 1.5 mmol/L MgCl2, 1 U thermostable DNA polymerase (AmpliTaq Gold DNA polymerase; Perkin Elmer, Weiterstadt, Germany), and 15 pmol of each primer. Each of the 50 PCR cycles consisted of 30 seconds denaturation at 95°C, 30 seconds annealing at 56°C, and 45 seconds extension at 72°C. The first cycle was preceded by 45 seconds of denaturation at 95°C. The last cycle was extended by a 4-minute elongation at 72°C. First and second round amplifications were performed in a Perkin-Elmer GeneAmp 9700 thermocycler (PE Applied Biosystems, Weiterstadt, Germany). Two μl of the PCR product were used as template for nested PCR amplifications, which were performed as conventional hot start PCRs with an initial activation step of 10 minutes at 95°C and 40 cycles of amplification with identical cycling conditions as were used in the first round amplifications. Positive samples as identified by agarose gel electrophoresis of 10 μl of the nested PCR products were further analyzed. Another 10 μl of the nested PCR product was added to 10 μl of hybridization mixture containing 6 pmol (0.3 μmol/L) of either KIT or JAK2 hybridization probes in 3.0 mmol/L MgCl2. Subsequent melting point analysis was performed on the Light Cycler system as described before.24
Results
Clinical Presentation and Disease Classification
Clinical characteristics of the patients are shown in Table 1. In all patients with SM-CIMF, the SM component of the disease was classified as isolated bone marrow mastocytosis based on World Health Organization criteria (SM-CIMF), because signs of organomegaly or impairment of organ function attributable to MC infiltration could not be detected. Interestingly, none of the patients exhibited typical urticaria-pigmentosa-like skin lesions. Thus, with the exception of the skin lesions, the SM component in the five patients with SM-CIMF did not differ significantly from disease presentation in the control cases of patients with ISM in the absence of CIMF (Table 1).
Bone Marrow Histology and Immunohistochemistry
All patients with SM-CIMF and those patients with pure CIMF (control group) usually showed a marked hypercellular marrow with slight to moderate reticulin fibrosis corresponding to the cellular phase of the disease. Patients with subnormal platelet counts, however, exhibited the typical picture of advanced disease with marked fibrosis and patchy hypocellular areas. All cases exhibited striking clustering of pleomorphic megakaryocytes sometimes with bizarre nuclei. Neutrophilic cells were increased and granulopoiesis left-shifted. Erythropoiesis was hypoplastic in most cases. Immunohistochemically, the numbers of CD34+ progenitor/blast cells were slightly increased in most cases but did not exceed 5% of all nucleated bone marrow cells and did not aggregate. In patients with SM-CIMF and in all control cases with pure ISM, multifocal compact usually small MC infiltrates were easily detectable when tryptase/chymase and CD117 stainings were evaluated. The degree of MC infiltration in both disease groups did not exceed 5% of the section area. Cytomorphologically, there was a significant increase of spindle-shaped MCs with aberrant expression of CD25. In patients with pure CIMF, compact MC infiltrates were absent and atypical MCs were not detected, and even CD25-expressing cells were completely missing. A summary of the most important clinical features is shown in Figure 1, whereas the histomorphological and the immunohistochemical characteristics are depicted in Table 3.
Table 3.
Histomorphological and Immunohistochemical Characteristics
| Diagnoses | CD34+ cells, % | Megakaryocyte cluster | Fibrosis | Osteosclerosis | SM, degree of BM infiltration, % | CD25+ MC |
|---|---|---|---|---|---|---|
| SM-CIMF, no. 82 | 1 to 5 | Yes | + | + | <5 | Yes |
| SM-CIMF, no. 302 | <1 | Yes | +++ | ++ | 10 to 15 | Yes |
| SM-CIMF, no. 605 | <1 | Yes | + | + | <5 | Yes |
| SM-CIMF, no. 661 | 1 to 5 | Yes | +++ | ++ | 5 to 10 | Yes |
| SM-CIMF, no. 883 | 1 to 5 | Yes | + | + | <5 | Yes |
| CIMF, no. 156 | 1 to 5 | Yes | +++ | + | NA | No |
| CIMF, no. 287 | <1 | Yes | +++ | ++ | NA | No |
| CIMF, no. 289 | 1 to 5 | Yes | ++ | ++ | NA | No |
| CIMF, no. 315 | 1 to 5 | Yes | ++ | + | NA | No |
| CIMF, no. 320 | <1 | Yes | ++ | + | NA | No |
| CIMF, no. 382 | 1 to 5 | Yes | + | + | NA | No |
| CIMF, no. 562 | <1 | No | +++ | ++ | NA | No |
| CIMF, no. 639 | <1 | Yes | ++ | ++ | NA | No |
| CIMF, no. 643 | 1 to 5 | Yes | + | + | NA | No |
| CIMF, no. 699 | 1 to 5 | Yes | +++ | ++ | NA | No |
| CIMF, no. 1554 | <1 | No | + | + | NA | No |
| CIMF, no. 1593* | <1 | No | + | + | NA | No |
| CIMF, no. 1691 | 1 to 5 | Yes | ++ | + | NA | No |
| CIMF, no. 1806 | 1 to 5 | Yes | ++ | + | NA | No |
| CIMF, no. 1821 | 1 to 5 | Yes | ++ | ++ | NA | No |
| ISM, no. 20 | <1 | No | No | No | <5 | Yes |
| ISM, no. 253 | <1 | No | No | No | <5 | Yes |
| ISM, no. 263 | <1 | No | No | No | <5 | Yes |
| ISM, no. 275 | <1 | No | No | No | <5 | Yes |
| ISM, no. 374 | <1 | No | No | No | 5 to 10 | Yes |
| ISM, no. 835 | <1 | No | No | No | <5 | Yes |
| ISM, no. 1019 | <1 | No | No | No | 10 to 15 | Yes |
| ISM, no. 1023 | <1 | No | No | No | 5 to 10 | Yes |
| ISM, no. 1039 | <1 | No | No | No | <5 | Yes |
| ISM, no. 1043 | <1 | No | No | No | 5 to 10 | Yes |
Fibrosis and osteosclerosis: +, mild; ++, moderate; +++, marked.
SM, systemic mastocytosis; CIMF, chronic idiopathic myelofibrosis; NA, not assessed.
JAK2-wt.
Sensitivity of JAK2V617F LNA-PCR
Melting point analysis of amplified HEL DNA resulted in JAK2V617F-specific melting peaks at temperatures of ∼64.5°C, whereas the analysis of HMC-1 DNA gained melting temperatures of ∼60.0°C, specific for the JAK2 wt. Amplification of DNA from both cell lines in a single tube displayed both melting peaks (Figure 2A).
Figure 2.
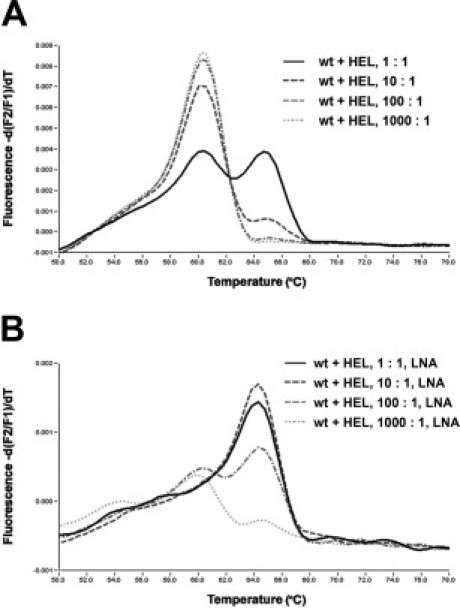
Effects of LNA-mediated PCR clamping. A: In melting curve analyses with Light Cycler hybridization probes wild-type (wt) JAK2 and mutation V617F can be discriminated through different melting peaks at ∼60°C (wt) and 65°C (V617F) (black continuous line). The method allows detection of the mutation in a 10:1 excess of normal versus mutated cells (dark gray broken line). B: LNA-mediated PCR-clamping results in suppression of the amplification of the JAK2 codon 617 wt allele (black continuous line and dark gray broken line) and in a 10-fold increase of sensitivity, yet allowing detection of the mutated allele in a 100-fold excess of wt background DNA (light gray broken line).
The analytical sensitivity of the melting analysis was determined in dilution experiments of varying amounts of HEL DNA, including 100 ng, 10 ng, 1 ng, and 100 pg, in constant amounts of 100 ng of HMC-1 DNA. The mutation-specific melting peaks were still detected in 10:1 mixtures, but the mutation was no longer detectable in a 100:1 mixture of 100 ng of HMC-1 DNA and 1 ng of HEL DNA (Figure 2A).
The LNA clamping probe was then used to suppress amplification of the wt target, thus, possibly increasing the analytical and clinical sensitivity of the assay in situations in which the tumor cells represent only a minor fraction of all cells present and there is thus a large amount of wt background DNA. A sufficient suppression of the amplification of the wt allele was achieved with 0.63 μmol/L LNA. Higher amounts of LNA resulted in a further suppression of the amplification of the mutated alleles (data not shown).
To estimate the relative detection limit of the LNA-mediated PCR-clamping assay, dilution series of 100 ng of HMC-1 DNA and various amounts of HEL DNA were amplified in the presence of a JAK2 wt-specific LNA molecule and again analyzed with JAK2V617F-specific hybridization probes. In repeated experiments, the mutation-specific melting peak at ∼64.5°C was reproducibly detected in a 100-fold excess of wt versus mutated cells. In contrast, detection of the JAK2 mutation failed in a 1000-fold excess of wt cells (Figure 2B).
Detection of KITD816V and JAK2V617F in Total Bone Marrow DNA
Screening analyses for the mutations KITD816V and JAK2V617F were performed on total extracted DNA from bone marrow biopsies of the five patients with SM-CIMF, of 15 patients with pure CIMF (without SM), and of 10 patients with typical ISM. Patients of the latter two groups were selected from our files.
Melting point analyses performed subsequently after PCR amplification revealed the presence of the KIT mutation D816V in all patients with ISM and in all patients with SM-CIMF but in none of the patients with pure CIMF. In every case amplification of total DNA was performed as conventional PCR, demonstrating the structural integrity of the extracted DNA and, by means of PNA-mediated PCR-clamping, preferably allowing amplification of the mutated alleles. KITD816V mutations were detected by melting point analysis after conventional PCR in 3 of 10 patients with ISM and in 1 of 5 patients with SM-CIMF. In the remaining cases, the KIT mutation was only detectable when PNA-mediated PCR-clamping of the WT alleles was performed before the melting point analysis. Presence or absence of this mutation resulted in a shift of the melting temperatures of ∼5°C (data not shown).
The JAK2 mutation V617F was present in four of five patients with SM-CIMF and in 14 of 15 patients with pure CIMF. The 10 patients with pure ISM, however, presented with the wt JAK2. In all CIMF patients examined (with or without SM), the JAK2 mutation V617F, if present, could be detected by melting point analysis after conventional amplification. Nevertheless, mutation-specific signals in melting point analyses were intensified when LNA-mediated PCR clamping was performed. Exclusively wt-specific melting curves were obtained when DNA of the patients with pure ISM was investigated for JAK2 V617F. All results are summarized in Table 4.
Table 4.
KIT Codon 816 and JAK2 Codon 617 Mutational Analyses
| Diagnoses | Total DNA |
Microdissected MC |
Microdissected CD15+ cells |
|||
|---|---|---|---|---|---|---|
| KIT D816V | JAK2 V617F | KIT D816V | JAK2 V617F | KIT D816V | JAK2 V617F | |
| SM-CIMF, no. 82 | + | + | + | + | + | + |
| SM-CIMF, no. 302 | + | − | + | − | + | − |
| SM-CIMF, no. 605 | + | + | + | + | − | + |
| SM-CIMF, no. 661 | + | + | + | + | − | + |
| SM-CIMF, no. 883 | + | + | + | + | − | + |
| CIMF (n = 15) | − | + (14/15) | NA | NA | NA | NA |
| ISM (n = 10) | + (10/10) | − | NA | NA | NA | NA |
MC, mast cells; NA, not assessed.
Detection of KITD816V and JAK2V617F in Microdissected Bone Marrow Cells
To investigate the clonal relationship of SM and CIMF in the five patients with ISM-CIMF, the two mutations KITD816V and JAK2V617F were used as markers for clonality. Chymase-immunostained MCs were microdissected as single cells and pooled to a total of 50 to 100 cells per PCR tube (Figure 3, A to D). Melting point analysis of nested PCR-amplified DNA revealed the presence of the KIT mutation D816V in MCs in all five cases with SM-CIMF. Moreover, this mutation was also found in DNA of pooled single microdissected CD15+ cells in two in the five SM-CIMF patients (Figures 3, E and F, and 4A). In one of these two patients, the mutation was also found in microdissected megakaryocytes (data not shown). Thus, the KIT mutation D816V could be detected in immunostained and microdissected neoplastic cells of both disease components, namely SM and CIMF, in a subset (two of five) of patients.
Figure 3.

Microdissection of bone marrow cells in SM-CIMF. A: Typical compact MC infiltrate with numerous spindle-shaped MCs before microdissection. B: The same detail as depicted in A after microdissection. Replaced single cells are indicated by white dots. C and D: Loosely scattered MC infiltrates before (C) and after (D) microdissection. Position of cells is indicated with circles. E and F: CD15+ myeloid cells before (E) and (F) after microdissection. Replaced single cells are indicated by white dots. (A–-F, ABC method/AEC; A-–D, anti chymase; E and F, anti-CD15).
Figure 4.
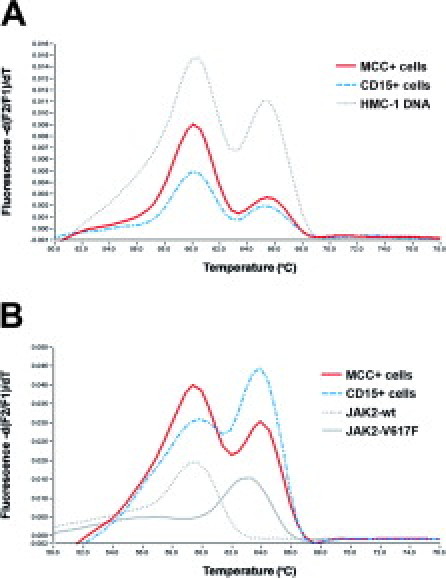
KIT and JAK2 mutation screening by melting point analysis of microdissected cells. A: Representative results obtained after melting point analysis of nested PCR-amplified, microdissected MCs (red continuous line) and CD15+ cells (blue broken line), both carrying the KIT mutation D816V (case 302). HMC-1 DNA served as control. The KIT codon 816 wt-specific melting peak is located at ∼60°C whereas the D816V-specific melting peak is located at ∼65°C. B: Detection of the JAK2 mutation V617F in microdissected CD15+ cells (blue broken line) and in MCs (red continuous line). The wt-specific melting peak is located at ∼60°C whereas the V617F-specific peak is located at ∼64°C (case 661). HMC-1 DNA served as the wt control (gray dotted line) and HEL cells as the V617F-specific control (gray continuous line).
Applying the same technique, the JAK2 mutation V617F was detected in microdissected CD15+ cells in four of five cases with SM-CIMF. The results perfectly matched with those obtained when total DNA was analyzed. Moreover, the mutation was also detectable in microdissected chymase+ MCs in each of the four JAK2V617F+ cases of SM-CIMF. Thus, this mutation was detected in microdissected cells of both disease components, SM and CIMF, in all four JAK2V617F+ cases (Figure 4B). All molecular biological results are summarized in Table 4.
Discussion
In a significant proportion of cases with SM, an associated clonal hematological non-MC lineage disease (AHNMD) is detected.9,10,11 We here describe five patients with SM, in whom a co-existing CIMF and the SM-related KIT mutation D816V was detected. The MPD-related JAK2 mutation V617F was detectable in four of five patients. Surprisingly, we also found that not only clonal cells of the AHNMD/MPD component but also microdissected MCs harbored the JAK2 mutation V617F. Moreover, in two of five patients, KITD816V was expressed not only in neoplastic MCs, but also in CD15+ myeloid cells. These data indicate that KITD816V+ SM may not only co-exist with JAK2V617F+ CIMF but that both molecular defects can be expressed in both components of the disease.
Although KITD816V is regarded as the molecular hallmark of SM, other molecular alterations may play a role in its pathogenesis, especially when an AHNMD is detected.22,26 It has thus been hypothesized that additional genetic hits may represent the molecular basis for the development of an AHNMD.27,28,29,30,31 On the other hand, it has also been demonstrated that KITD816V is expressed not only in neoplastic MCs but also by non-MC lineage cells including neoplastic myeloid cells in an AHNMD, especially in leukemic monocytes in SM chronic myelomonocytic leukemia.14 In the present study, we were able to show that KITD816V also occurs in neoplastic myeloid progenitor cells in SM-CIMF.
Recently, the JAK2 mutation V617F has been introduced as a specific molecular marker for the Ph-chromosome-negative MPDs.15,16,17,18 In the present study we used JAK2V617F as a marker of MPD and of clonality in SM-CIMF. Interestingly, JAK2V617F was detected in four of five (80%) patients with SM-CIMF, which seems to be a relatively high frequency compared to CIMF patients without SM. In fact, between 35% and 73% of patients with pure CIMF have been reported to display JAK2V617F.15,16,17,18,32 Notably, we detected this mutation in a series of consecutive cases with pure CIMF in approximately the same frequency (84%, 16 of 19) as in the SM-CIMF cases of this study. Because the mutation could be identified in all JAK2V617F-positive cases by PCR and conventional melting point analysis (without LNA-mediated PCR clamping), the high frequency of JAK2V617F in the present study is probably not a result of an increased analytical sensitivity of the technique applied but may best be explained by the fact that bone marrow biopsies were investigated instead of bone marrow aspirates or peripheral blood. Nevertheless, combining the advantages of PCR clamping and subsequent melting point analysis this technique overcomes the need for time-consuming analytical postamplification steps and generates reproducible and highly specific results within 1.5 hours of analysis.24,33,34
An important question is whether the two mutations, KITD816V and JAK2V617F, were indeed harbored by the same neoplastic cells. In fact, when pooled microdissected MCs and pooled microdissected CD15+ myeloid progenitors were analyzed, the KIT mutation was detected in both disease components in two of five patients, and the JAK2 mutation, notably, was detected in both disease components in all four JAK2V617F+ patients. In the former two patients, these observations argue for a co-expression of the two mutations in the same clone. In the latter group of patients, with KITD816V being present only in MCs but not in CD15+ cells, we believe that there is a JAK2-V617F-positive clone with a KIT-D816V-positive subclone. However, because these cell populations were not examined at a single-cell level, it cannot be excluded with certainty, that both defects were restricted to a subset of MCs and a subset of CD15+ cells.
The multihit theory of cancer development is well established.35,36 With regard to KITD816V and JAK2V617F, however, little is known about the critical co-expression of these oncogenic kinases. To ask whether the co-expression of KITD816V and JAK2V617F would cooperate as specific defects to cause transformation into a high-grade neoplasm, we compared clinical and histomorphological parameters of SM-CIMF patients with that of patients with either pure ISM or pure CIMF. However, clinically as well as morphologically, no significant differences were noted. Specifically, we were not able to find any signs of a more progressive disease in the SM-CIMF patients. These data would argue against a cooperative effect of KITD816V and JAK2V617F in patients with SM-CIMF.
In summary, our data show that KITD816V+ SM can co-exist with JAK2V617F+ MPD/CIMF and that in these patients both molecular alterations are expressed in neoplastic cells of both disease components. This observation may have implications for the pathogenesis of the disease as well as for the selection of targeted drugs, against which malignant cells may become resistant when certain mutations in distinct critical (signaling) kinases are expressed.
Acknowledgements
We thank Sema Colak for her excellent technical assistance.
Footnotes
Supported in part by the Fortüne Förderprogramm der Universität Tübingen (grant no. F1461141).
References
- 1.Lennert K, Parwaresch MR. Mast cells and mast cell neoplasia: a review. Histopathology. 1979;3:349–365. doi: 10.1111/j.1365-2559.1979.tb03017.x. [DOI] [PubMed] [Google Scholar]
- 2.Metcalfe DD. Classification and diagnosis of mastocytosis: current status. J Invest Dermatol. 1991;96:2S–4S. [PubMed] [Google Scholar]
- 3.Valent P, Akin C, Sperr WR, Horny HP, Arock M, Lechner K, Bennett JM, Metcalfe DD. Diagnosis and treatment of systemic mastocytosis: state of the art. Br J Haematol. 2003;122:695–717. doi: 10.1046/j.1365-2141.2003.04575.x. [DOI] [PubMed] [Google Scholar]
- 4.Tefferi A, Pardanani A. Clinical, genetic, and therapeutic insights into systemic mast cell disease. Curr Opin Hematol. 2004;11:58–64. doi: 10.1097/00062752-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 5.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Longley BJ, Jr, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA. 1999;96:1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nunez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, Parwaresch RM, Austen KF, Lennert K, Metcalfe DD, Vardiman JW, Bennett JM. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res. 2001;25:603–625. doi: 10.1016/s0145-2126(01)00038-8. [DOI] [PubMed] [Google Scholar]
- 8.Valent P, Horny H-P, Li CY, Longley JB, Metcalfe DD, Parwaresch RM, Bennett JM. Mastocytosis (mast cell disease): Tumours of haematopoietic and lymphoid tissues. In: Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and genetics, vol 1. World Health Organization (WHO) Classification of Tumours. World Health Organization; Geneva: 2001. pp. 291–302. [Google Scholar]
- 9.Travis WD, Li CY, Yam LT, Bergstralh EJ, Swee RG. Significance of systemic mast cell disease with associated hematologic disorders. Cancer. 1988;62:965–972. doi: 10.1002/1097-0142(19880901)62:5<965::aid-cncr2820620520>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 10.Sperr WR, Horny HP, Valent P. Spectrum of associated clonal hematologic non-mast cell lineage disorders occurring in patients with systemic mastocytosis. Int Arch Allergy Immunol. 2002;127:140–142. doi: 10.1159/000048186. [DOI] [PubMed] [Google Scholar]
- 11.Horny H-P, Sotlar K, Sperr WR, Valent P. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease (SM-AHNMD): a histopathological challenge. J Clin Pathol. 2004;57:604–608. doi: 10.1136/jcp.2003.014860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akin C, Kirshenbaum AS, Semere T, Worobec AS, Scott LM, Metcalfe DD. Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mastocytosis. Exp Hematol. 2000;28:140–147. doi: 10.1016/s0301-472x(99)00145-9. [DOI] [PubMed] [Google Scholar]
- 13.Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single-cell analysis of mutations in the c-kit gene. Blood. 2002;100:661–665. doi: 10.1182/blood-2002-01-0203. [DOI] [PubMed] [Google Scholar]
- 14.Sotlar K, Fridrich C, Mall A, Jaussi R, Bültmann B, Valent P, Horny H-P. Detection of c-kit point mutation Asp-816Val in microdissected pooled single mast cells and leukemic cells in a patient with systemic mastocytosis and concomitant chronic myelomonocytic leukemia. Leuk Res. 2002;26:979–984. doi: 10.1016/s0145-2126(02)00041-3. [DOI] [PubMed] [Google Scholar]
- 15.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Cancer Genome Project: acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 16.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 17.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 18.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D'Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland D. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 19.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- 20.Sotlar K, Horny H-P, Simonitsch I, Krokowski M, Aichberger KJ, Mayerhofer M, Printz D, Fritsch G, Valent P. CD25 indicates the neoplastic phenotype of mast cells: a novel immunohistochemical marker for the diagnosis of systemic mastocytosis (SM) on routinely processed bone marrow biopsy specimens. Am J Surg Pathol. 2004;28:1319–1325. doi: 10.1097/01.pas.0000138181.89743.7b. [DOI] [PubMed] [Google Scholar]
- 21.Sotlar K, Selinka HC, Menton M, Kandolf R, Bültmann B. Detection of human papillomavirus type 16 E6/E7 oncogene transcripts in dysplastic and nondysplastic cervical scrapes by nested RT-PCR. Gynecol Oncol. 1998;69:114–121. doi: 10.1006/gyno.1998.4994. [DOI] [PubMed] [Google Scholar]
- 22.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest. 1993;92:1736–1744. doi: 10.1172/JCI116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simeonov A, Nikiforov TT. Single nucleotide polymorphism genotyping using short, fluorescently labelled locked nucleic acid (LNA) probes and fluorescence polarization detection. Nucleic Acids Res. 2002;30:e91. doi: 10.1093/nar/gnf090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sotlar K, Escribano L, Landt O, Möhrle S, Herrero S, Torrelo A, Lass U, Horny HP, Bültmann B. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol. 2003;162:737–746. doi: 10.1016/S0002-9440(10)63870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schütze K, Lahr G. Identification of expressed genes by laser-mediated manipulation of single cells. Nat Biotechnol. 1998;16:737–742. doi: 10.1038/nbt0898-737. [DOI] [PubMed] [Google Scholar]
- 26.Féger F, Ribadeau Dumas A, Leriche L, Valent P, Arock M. Kit and c-kit mutations in mastocytosis: a short overview with special reference to novel molecular and diagnostic concepts. Int Arch Allergy Immunol. 2002;127:110–114. doi: 10.1159/000048179. [DOI] [PubMed] [Google Scholar]
- 27.Pullarkat VA, Pullarkat ST, Calverley DC, Brynes RK. Mast cell disease associated with acute myeloid leukemia: detection of a new c-kit mutation Asp816His. Am J Hematol. 2000;65:307–309. doi: 10.1002/1096-8652(200012)65:4<307::aid-ajh10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 28.Tefferi A, Lasho TL, Brockman SR, Elliott MA, Dispenzieri A, Pardanani A. FIP1L1-PDGFRA and c-kit D816V mutation-based clonality studies in systemic mast cell disease associated with eosinophilia. Haematologica. 2004;89:871–873. [PubMed] [Google Scholar]
- 29.Pardanani A, Ketterling RP, Brockman SR, Flynn HC, Paternoster SF, Shearer BM, Reeder TL, Li CY, Cross NC, Cools J, Gilliland DG, Dewald GW, Tefferi A. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood. 2003;102:3093–3096. doi: 10.1182/blood-2003-05-1627. [DOI] [PubMed] [Google Scholar]
- 30.Florian S, Esterbauer H, Binder T, Mullauer L, Haas OA, Sperr WR, Sillaber C, Valent P. Systemic mastocytosis (SM) associated with chronic eosinophilic leukaemia (SM-CEL): detection of FIP1L1/PDGFRalpha, classification by WHO criteria, and response to therapy with imatinib. Leuk Res. 2006;30:1201–1205. doi: 10.1016/j.leukres.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 31.Pullarkat V, Bedell V, Kim Y, Bhatia R, Nakamura R, Forman S, Sun J, Senitzer D, Slovak ML. Neoplastic mast cells in systemic mastocytosis associated with t(8;21) acute myeloid leukemia are derived from the leukemic clone. Leuk Res. 2007;31:261–265. doi: 10.1016/j.leukres.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 32.McClure R, Mai M, Lasho T. Validation of two clinically useful assays for evaluation of JAK2 V617F mutation in chronic myeloproliferative disorders. Leukemia. 2006;20:168–171. doi: 10.1038/sj.leu.2404007. [DOI] [PubMed] [Google Scholar]
- 33.Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques. 1997;22:176–181. doi: 10.2144/97221pf02. [DOI] [PubMed] [Google Scholar]
- 34.Orum H. PCR clamping. Curr Issues Mol Biol. 2000;2:27–30. [PubMed] [Google Scholar]
- 35.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 36.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]