

Abstract
Clinical workup of metastatic malignancies of unknown origin is often arduous and expensive and is reported to be unsuccessful in 30 to 60% of cases. Accurate classification of uncertain primary cancers may improve with microarray-based gene expression testing. We evaluated the analytical performance characteristics of the Pathwork tissue of origin test, which uses expression signals from 1668 probe sets in a gene expression microarray, to quantify the similarity of tumor specimens to 15 known tissues of origin. Sixty archived tissue specimens from poorly and undifferentiated tumors (metastatic and primary) were analyzed at four laboratories representing a wide range of preanalytical conditions (eg, personnel, reagents, instrumentation, and protocols). Cross-laboratory comparisons showed highly reproducible results between laboratories, with correlation coefficients between 0.95 to 0.97 for measurements of similarity scores, and an average 93.8% overall concordance between laboratories in terms of final tissue calls. Bland-Altman plots (mean coefficients of reproducibility of 32.48 ± 3.97) and κ statistics (κ > 0.86) also indicated a high level of agreement between laboratories. We conclude that the Pathwork tissue of origin test is a robust assay that produces consistent results in diverse laboratory conditions reflecting the preanalytical variations found in the everyday clinical practice of molecular diagnostics laboratories.
In the initial pathological evaluation of tumors with an uncertain primary origin, especially those found in unexpected or multiple locations or with poorly differentiated morphologies, the tissue of origin (TOO) can remain hard to identify. These malignancies often require extensive clinical workup. Recently, diagnostic algorithms to aid clinicians in their management of the most challenging patients with uncertain primary cancers have been developed (National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. Occult Primary (Version 2.2007). 2007. Available at: http://www.nccn.org/professionals/physician_gls/PDF/occult.pdf. Accessed March 1, 2007).1,2,3 However, even with guideline-directed use of immunohistochemistry, electron microscopy, and advanced imaging procedures, the primary tumor is ultimately identified in only ∼20 to 25% of living patients with metastatic tumors for which the primary site is not apparent after the initial workup.4,5 Overall, patients are given the diagnosis of exclusion—tumor of unknown origin or carcinoma of unknown primary—in ∼2 to 5% of all diagnosed cancers.5,6,7 The recent guidelines of the National Comprehensive Cancer Network (NCCN) emphasize the importance of identifying the TOO in carcinoma of unknown primary patients so that cancer-specific treatment recommendations can be followed. Patients in whom the primary cancer is diagnosed have been shown in a prospective clinical study to have a longer survival compared to patients in whom the TOO remains unknown.8
Recently, newer diagnostic techniques have been evaluated to improve classification of clinicopathologically ambiguous tumors and thereby allow more tissue-directed therapy. Advanced immunohistochemistry panels, proteomic expression tests, and gene expression-based analyses using quantitative real-time polymerase chain reaction (qRT-PCR) have shown some promising preliminary results in identifying the TOO, but these tests have generally been limited to categorization of seven or fewer tissue types.9,10,11,12,13 One qRT-PCR-based assay recently demonstrated an overall 87% accuracy in classifying tumor specimens into 28 tissue types, covering 32 different tumor classes, but that accuracy dropped to 71% when classifying high-grade specimens (ie, poorly differentiated or undifferentiated), and assay reproducibility in diverse laboratory settings was not reported.14
Gene expression microarrays, which capture data from tens of thousands of expressed genes in a single test, have the potential to allow a more accurate classification of tumors of unknown primary, including those with high histological grade. In recent years, researchers have used microarray platforms to address several of the most challenging diagnostic and prognostic situations in oncology. These include, for example, identification of gene-expression-based models that proved to have high rates of concordance in their outcome predictions for breast cancer patients15 and creation of gene expression profiles that predicted the risk of recurrence in non-small-cell lung cancer.16
Several studies have demonstrated the feasibility of using genome-wide gene expression profiling to classify uncertain tumors according to TOO and have envisioned a future application in the clinical setting.11,14,17,18 Until now, using high-density microarray platforms in the clinical environment has proved challenging for two main reasons. First is the inherent multivariate aspect of testing using this technology, which is related to the complexity of accurately interrogating and interpreting expression signals from the thousands of genes that might help distinguish the dozen or more tissue types of highest interest. The fundamental challenges are both logistical and computational, eg, the need for a 1000 or more well-characterized tissue specimens in the training set, and the tendency to overfit an algorithm to a specific training set, as well as the risk of uncorrected artifacts related to signal saturation or sequence error that reduce sensitivity. Although microarrays are theoretically suited to these challenges, reliability in accurately classifying several hundred clinical specimens into more than a dozen potential tissue types has not been demonstrated.
The second major challenge that microarray technology has faced in becoming a routine diagnostic tool is related to interlaboratory variability. Preanalytical variability can be introduced at any stage of the multistep procedure of microarray analysis, including collecting and storing tissue specimens and then extracting, isolating, and preparing labeled RNA. Differences in specimen handling,19,20 total RNA isolation,21,22 and RNA amplification and labeling,23,24,25,26,27 have been identified as prime sources of analytical variability. Importantly, microarray design, manufacturing, and quality control have matured to the point of providing excellent technical performance,28 and thus, implementation of robust standard operating procedures for gene expression analysis in high-complexity molecular diagnostics laboratories might be able to overcome this challenge.29
The Pathwork TOO test (Pathwork Diagnostics, Sunnyvale, CA) is a microarray-based gene expression diagnostic test for determining the similarity of tumors of unknown origin to cancers from 1 of 15 known TOOs. The test uses proprietary normalization and classification algorithms and a companion high-density oligonucleotide microarray (Pathchip, manufactured for Pathwork Diagnostics by Affymetrix, Inc., Santa Clara, CA) to measure the expression of 1668 gene probe sets or markers. The molecular similarity of each tumor specimen's expression pattern is compared to 15 distinctive patterns from the different tissue types covered by this test. For each specimen, the algorithm reduces the highly complex expression data into 15 separate similarity scores (SSs), one score for each potential tissue type. These continuous scores (scale 0 to 100) are reported to the pathologist, who then establishes whether or not a particular tissue type is present in the specimen. As of the date of submission of this manuscript, the test was under Food and Drug Administration review and could not yet be used for diagnostic procedures.
In this study, we evaluated the analytical performance characteristics of this microarray-based test for tumor TOO. By comparing test results from replicate specimens analyzed in four laboratories representing a wide range of preanalytical and analytical conditions (eg, protocols, personnel, reagents, and timing), the study primarily addresses the test's reproducibility across multiple sites and, thereby, gauges the test's potential usefulness in actual clinical environments.
Materials and Methods
Overall Study Design
This study was designed to evaluate the performance of the Pathwork TOO test when performed at multiple laboratories using archived frozen clinical specimens. Three academic laboratories and one commercial laboratory were enrolled to ensure that the variability found during clinical testing in molecular diagnostics laboratories was adequately represented in this study (Figure 1). Sixty frozen tissue specimens from metastatic and primary tumors were procured and processed for microarray-based gene expression analysis at these four sites. The resulting microarray data files were sent electronically in blinded manner to Pathwork Diagnostics for processing and generation of scores for each TOO type. One-page reports with the quantitative scores were sent back to pathologists (blinded) for their use in generating an interpretation based on predetermined cutoff levels of the physician-guided conclusion (PGC), a categorical parameter that established the TOO call for each sample. Data were compared between laboratories to determine reproducibility of the assay. The PGC results were also compared in blinded manner to the clinical truth of the tissue type as originally established in the surgical pathology report.
Figure 1.

Study design. Sixty frozen tissue specimens from metastatic and primary tumors were assayed at four sites, three academic laboratories and one commercial laboratory, to adequately represent the variability found during routine clinical testing in molecular diagnostics laboratories. Microarray data files for samples passing quality control criteria only were analyzed by Pathwork Diagnostics, and reports were evaluated by pathologists (blinded). Data were compared between laboratories to determine reproducibility of the assay.
Tumor Specimens
Fifty-seven archived tissue specimens were obtained from the Health Sciences Tissue Bank at the University of Pittsburgh, Pittsburgh, PA. In addition, three metastatic gastric specimens were acquired from a commercial tissue repository (ProteoGenex, Inc., Culver City, CA). All 15 tissue types included in the Pathwork TOO test were represented in the 60 specimens (Supplemental Table 1, see http://jmd.amjpathol.org/). Five tumor tissue types (breast, colorectal, non-small-cell lung, non-Hodgkin's lymphoma, and pancreas) were represented by six specimens each, and all other tissue types (bladder, gastric, germ cell, hepatocellular, kidney, melanoma, ovarian, prostate, soft tissue sarcoma, and thyroid) were represented by three specimens each. Forty (74%) of these specimens were metastatic in origin, and 14 (26%) were primary tumors classified as poorly differentiated or undifferentiated (not including the six lymphomas). A copy of the deidentified surgical pathology report was obtained for each sample to establish the origin of each tissue, referred to in this article as the clinical truth; however, this information was blinded from all personnel performing the gene expression assays and the pathologists issuing the PGC (J.R.-P. and J.L.Z.). Each of the 57 specimens was collected during routine pathological examination under an institutional review board-approved banking program. For all tissues from the University of Pittsburgh Tissue Bank, tissues were embedded in Tissue-Tek OCT Compound (Sakura Finetek USA, Inc., Torrance, CA), snap-frozen, and stored at −80°C within 60 minutes of removal from the patient. Before inclusion in this study, a frozen section was prepared, stained with hematoxylin and eosin (H&E), and examined by a pathologist (F.A.M.) to record tumor content and necrosis. The inclusion criterion for this study was >25% viable tumor tissue in the sample. This criterion was selected before the assay manufacturer definitions for excessive necrosis (>20%) and insufficient tumor (<60%) were issued. Three replicate portions of frozen tumor, each at least 0.1 g, were scraped from the original frozen tumor tissue block using RNAlater (Ambion, Inc., Austin, TX) at room temperature to thaw a superficial layer of tissue, and were distributed among sites 1, 3, and 4. Site 2 received the remaining portion of the frozen tissue block from which serial frozen sections were prepared for RNA extraction. ProteoGenex specimens (three metastatic gastric specimens) were processed with a freezer/mill (SPEX CertiPrep, Inc., Metuchen, NJ). All tissue samples were shipped overnight on dry ice.
Laboratories and Protocols
Three university-based pathology laboratories and one commercial microarray facility prepared and analyzed the replicates in this study: University of Pittsburgh (site 1); Virginia Commonwealth University, Richmond, VA (site 2); Cogenics, Inc., Morrisville, NC (site 3); and Stanford University, Stanford, CA (site 4). Sites 1 to 3 were laboratories with years of experience in running microarrays and in developing standard operating procedures and quality control practices typically used in routine microarray-based gene expression analysis with the Affymetrix platform. The fourth site belongs to a major center for microarray research, but the clinical molecular diagnostics laboratory and personnel had not been previously engaged in microarray-based gene expression testing.
RNA Extraction
Each site performed RNA extraction using the following protocols: site 1 used the manufacturer-recommended protocols for total RNA extraction with the miRVana (Ambion, Inc.) for approximately half of the samples, and RNeasy mini and midi kits (Qiagen Inc., Valencia, CA) for the rest. Sites 3 and 4 used the manufacturer-recommended protocol for RNeasy mini and midi kits. Site 2 used the following protocol for tissue handling and RNA extraction: 1) multiple 10-μm frozen sections of the tissue samples were prepared; 2) the first and last frozen sections were stained with H&E, and a pathologist documented histopathological scoring of standard features (tumor content, stromal contribution, and presence of necrosis); 3) the in-between sections were placed directly in TRIzol reagent (Invitrogen Corp., Carlsbad, CA) for RNA extraction, with a subsequent cleanup process with the RNeasy mini kit. Total RNA concentration was assessed by spectrophotometry (OD 260 nm), and purity was judged by the ratio of absorbance at 260 nm to 280 nm (A260/A280).
Gene Expression Assays
A detailed table of protocol variations and instrumentation is shown in Supplemental Table 2 (see http://jmd.amjpathol.org/). The gene expression protocols at all sites were primarily based on the standard protocol for GeneChip expression assays from Affymetrix, as described by the manufacturer and elsewhere.26 These protocols met the broad specifications for recommended clinical laboratory protocol set by the TOO test developer. Variations in the protocols at each site included the use of laboratory-prepared reagents for array washing and staining (sites 2 and 4) or commercially available reagent kits (Affymetrix, Inc.) (sites 1 and 3).
Samples were hybridized to Affymetrix U133A or Pathwork Pathchip microarrays (sites 1, 2, and 3) or Pathwork Pathchip arrays only (site 4). Pathwork Pathchip arrays are manufactured by Affymetrix and are based on the U133A array design. Pathwork designed the Pathchip microarray to be functionally equivalent to the U133A microarray for the 1668 genes used in the TOO test. Only the Pathchip may be used for the Pathwork TOO test, which at the time of submission of this article, is investigational and not for clinical use in the United States. All sites used the Affymetrix GeneChip instrumentation (fluidics station and scanner) and the GeneChip operating software to generate gene expression data (.CEL files). Further details on instrumentation used are provided in Supplemental Table 2 (see http://jmd.amjpathol.org/).
Microarray-Based Gene Expression Test
The Pathwork TOO test is an in vitro diagnostic test for evaluating the TOO in poorly differentiated or undifferentiated tumors. This microarray-based gene expression test quantifies the similarity of tumor specimens to 15 known TOOs. These tissues are bladder, breast, colorectal, gastric, germ cell, hepatocellular, kidney, non-small-cell lung, non-Hodgkin's lymphoma, melanoma, ovarian, pancreatic, prostate, soft tissue sarcoma, and thyroid.
Gene expression data (.CEL files) were standardized on the basis of 121 endogenous mRNA markers that were found to be relatively stable in their expression patterns and were used to correct for variations expected to exist between clinical laboratory settings. The standardization model, which was developed before the development of the tissue classifier, was based on a proprietary standardization algorithm and gene expression signals from 5539 human tissue specimens processed by 11 laboratories.30 The resulting standardized expression (SE) values underwent a data verification algorithm that addresses RNA quality, inadequate amplification, insufficient quantity of labeled RNA, as well as inadequate hybridization time or temperature.
After data verification, the SE values are analyzed using a tissue classification model that uses 1550 markers chosen by gene ranking. The SE values for the optimal markers are used in the proprietary machine learning algorithm trained on 2039 well-characterized tumor specimens, acquired from 14 laboratories. The tissues and number of specimens used in algorithm training are shown in Supplemental Table 3 (see http://jmd.amjpathol.org/). The tissue classification analysis yields a SS ranging from 0 (very low similarity) to 100 (very high similarity) for each of the 15 potential TOOs.
The SSs are reported in the form of an electronic one-page graphical report (Figure 2), which is meant to be interpreted by a pathologist who uses predetermined cutoffs to draw a conclusion about the tissue's origin (ie, the TOO call, also referred to as the PGC). Pathwork established that for an SS greater than or equal to 30 there is a >95% probability that the tissue indicated is present in the specimen, as either the tumor or the biopsy site. If, on the same report, two SSs are greater than or equal to 30 and one of these SSs indicates the biopsy site, then the other SS indicates the tumor with the probability described above. If neither SS indicates the biopsy site, then the result is indeterminate and no TOO is indicated. If the maximum SS is less than 30, then the result is indeterminate and no TOO is indicated. An SS less than 5 allows that tissue to be excluded as a TOO with a probability of >95%.
Figure 2.
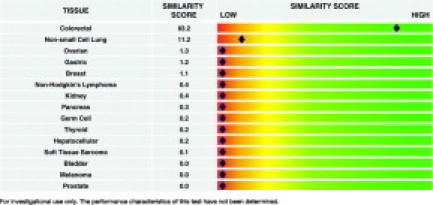
Example of Pathwork TOO report. Results shown are for study specimen 06-0360 from site 1. A SS ≥30 is the cutoff value to indicate a positive call, and an SS <5 confirms a negative call for each TOO.
Statistical Analysis and Data Exclusions
Reproducibility was analyzed by crosswise comparisons of all four laboratories for all three categories of results (ie, SE, SS, and PGC). For the continuous variables SE and SS, linear regression analysis was used for cross-laboratory comparisons to generate correlation coefficients. For a relative measure of the value of the test's normalization algorithm, raw expression values from all replicates were also standardized using the established MAS5 algorithm.31 In addition, Bland and Altman32 plots of the difference between the SS for the true TOO versus the average of the SS for each site compared to all of the other sites were prepared to test for possible systematic bias between laboratories and to measure the agreement and reproducibility of the TOO test among all laboratories. Thus, agreement according to the method of Bland and Altman32 was evaluated by the percentage of values outside the ± 1.96 SD range for every comparison; if the latter was ≤10%, agreement was considered to be good. The coefficient of repeatability (CR; 1.96 × SD of the mean difference between the intersite values), which defines the 95% confidence intervals of such values, was also examined. For the categorical variable PGC, contingency tables were created in which the TOO calls, including indeterminate calls, for each site were compared to all of the other sites. Interlaboratory agreement for the PGC was evaluated by use of the κ statistic. The κ statistic measures agreement between two raters that is beyond chance, with chance being a value of 0 and 1.00 being complete agreement.33 Values of κ along with a confidence interval (CI) of 95% were calculated and ranked as poor (κ < 0.20), fair (κ = 0.21 to 0.40), moderate (κ = 0.41 to 0.60), good (κ = 0.61 to 0.80), and very good (κ = 0.81 to 1.00). In assessing reproducibility, all specimens with reportable raw expression values in all four sites were included in the analyses, even those specimens that fell outside the recommended criteria of the test manufacturer for excessive necrosis (>20%) and insufficient tumor (<60%). Specimens with a failed data verification flag were not included in the correlation analyses.
Results
RNA Samples Quality Control
Replicate samples from 60 individual tumors were distributed among the four laboratories for a total of 237 tissue samples (site 4 did not receive samples from gastric tumors because of insufficient tumor volume.) Thus, samples from 57 tumors were processed in all four laboratories and samples for three tumors in three laboratories. RNA extraction yields ranged from 0.25 to 107.2 μg. All extraction methods used produced RNA with A260/A280 ratios greater than 1.8 in all but three samples, which were from the same tumor (06-460). After RNA extraction, seven additional samples failed to yield sufficient RNA for the gene expression assay (minimum of 1 μg of total RNA required). Comparison of two extraction methods at site 1 indicated that higher RNA yields were generally obtainable with the Ambion miRVana extraction reagents (44.07 ± 26.46, n = 29) in comparison with the Qiagen RNeasy kit (18.95 ± 19.11, n = 31; P = 7.33E-05 with Student's t-test).
Sites 1, 2, and 3 performed RNA quality control analysis with capillary electrophoresis on the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA). Agilent RNA integrity numbers (RINs) ranged from 3 to 10, with a median RIN of 8.9 for all three sites (7.6, 9.7, and 9.1 for sites 1, 2, and 3, respectively). There was no statistical difference in RNA quality metrics between Ambion miRVana and Qiagen RNeasy kits at site 1 (t-test, P = 0.61540). Site 4 evaluated RNA quality by agarose gel electrophoresis.
Performance of Gene Expression Assays
All 227 samples with adequate RNA quantity and quality produced sufficient labeled cRNA for hybridization to Affymetrix HG-U133A or Pathchip arrays (15 μg of fragmented, labeled cRNA; 10 μg applied to each array). Thirty-one samples required more than one labeling reaction (26 because of three separate batch failures and 5 because of individual sample underperformance). In three samples, cRNA from two in vitro transcription (IVT) reactions was combined to obtain sufficient material for hybridization. Thus, gene expression assay result files on all 227 samples were submitted to Pathwork Diagnostics for analysis with the TOO test.
A total of 218 gene expression data files passed the data verification step performed by the TOO test algorithm (see Materials and Methods). Only nine samples returned a failed data verification result. It is noteworthy that all failed data files were produced by samples with evidence of RNA degradation, as judged by a low Agilent RIN (RIN < 5.5) or degraded RNA by agarose gel electrophoresis (site 4). However, nine samples with evidence of RNA degradation (RIN < 5.5) still produced expression data that passed the data quality step. Thus, 50% of samples with low-quality RNA as judged by Agilent RINs produced gene expression data of sufficient quality for the TOO test.
Reproducibility across Four Laboratories
Cross-laboratory comparisons of gene expression values from the 1668 genes used in the TOO test algorithm were performed after normalization with the Affymetrix MAS5 algorithm and with the TOO test algorithm. Results were correlated across laboratories and Pearson correlation coefficients were calculated. MAS5 normalized values showed Pearson correlation coefficients between 0.65 and 0.82 in one-to-one laboratory comparisons (Figure 3). Data standardization with the Pathwork TOO test algorithm was performed, and the SE showed a significant improvement in the reproducibility of results between laboratories, with Pearson correlation coefficients between 0.81 and 0.87 for each individual comparison between two laboratories (Wilcoxon one-sided paired test, P value = 0.01563). With the SS, the calculated results level of the TOO test, there was further improvement of the correlations, with all comparisons between independent laboratories showing Pearson correlation coefficients greater than 0.95.
Figure 3.

Comparison of reproducibility of gene expression measurements and SS between test sites, as measured by the Pearson correlation coefficient. Data for 1668 genes normalized with the MAS5 and Pathwork TOO test algorithms show that correlation coefficients are significantly higher for the Pathwork SE value (Wilcoxon one-sided paired test, P = 0.01563) than those achieved with MAS5 normalization. Reproducibility across laboratories is further improved by the calculation of the SS from the SE data.
When assessing for systematic bias, using Bland-Altman plots and allowing a constant 95% limit of agreement, the SS showed no direct dependence of the differences on the average between sites. As depicted in Figure 4, the plots of differences versus averages show no evidence of spreading of the differences with increasing or decreasing magnitude of average SS. Moreover, the variation in these plots decreases slightly with SS > 80. A high level of agreement among all four sites was observed, with coefficients of reproducibility (CR) of 32.48 ± 3.97, and an overall percentage of outliers of <10%. Interestingly, although the Bland-Altman analysis identified 19 outlier samples, only 10 tumors accounted for these outliers. Five of these tumors produced outliers in multiple site-to-site comparisons. Three of the ten tumors with outlier samples corresponded to tumor samples that failed one or more of the TOO test manufacturer's tissue quality control parameters (ie, tumor and/or necrosis content).
Figure 4.
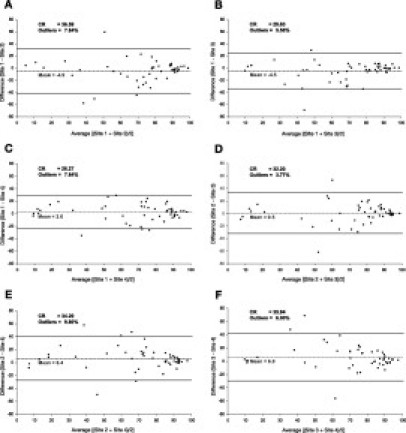
Bland-Altman plots graph the difference between the SS for the true TOO between sites (y axis) versus the average of the SS (x axis) of any given pair of sites. The mean and ± 1.96 SD intervals are shown by dotted and plain horizontal lines, respectively. The coefficients of reproducibility (CR) and percentage of outliers are indicated for each comparison. The number of samples included in each comparison were n = 51 (A), n = 54 (B), n = 51 (C), n = 53 (D), n = 51 (E), and n = 50 (F).
At the reported result level, the PGCs showed an overall level of concordance between laboratories of 89.4% (range, 87.0 to 92.5%) in individual site comparisons (Table 1). Moreover, the κ analysis indicates very good agreement (κ > 0.86) between laboratories in the final TOO call (Table 2). Also, in terms of the reported results for all replicate specimens, the overall percent agreement of PGC with the known TOO (ie, clinical truth) was 86.7% (range, 84.9 to 89.3%) (Table 3) for all laboratories, with 4.6% discordant and 8.7% indeterminate results. Results of the TOO test for all samples are shown in Supplemental Table 4 (see http://jmd.amjpathol.org/).
Table 1.
Concordance of Test Results for Final Call for Tissue of Origin (Physician-Guided Conclusion) between Sites
| Site | n | % Concordance | % Discordance |
|---|---|---|---|
| 1 versus 2 | 51 | 88.2 | 11.8 |
| 1 versus 3 | 54 | 87.0 | 13.0 |
| 1 versus 4 | 51 | 88.2 | 11.8 |
| 2 versus 3 | 53 | 92.5 | 7.5 |
| 2 versus 4 | 51 | 88.2 | 11.8 |
| 3 versus 4 | 50 | 92.0 | 8.0 |
Table 2.
Interlaboratory Agreement in Final Call for Tissue of Origin (Physician-Guided Conclusion): Statistic κ with 95% CI
| Site | Site 1 |
Site 2 |
Site 3 |
Site 4 |
||||
|---|---|---|---|---|---|---|---|---|
| κ | CI = 95% | κ | CI = 95% | κ | CI = 95% | κ | CI = 95% | |
| 1 | − | − | − | − | − | − | − | − |
| 2 | 0.872 | (0.775, 0.968) | − | − | − | − | − | − |
| 3 | 0.860 | (0.763, 0.957) | 0.918 | (0.840, 0.995) | − | − | − | − |
| 4 | 0.872 | (0.775, 0.968) | 0.872 | (0.775, 0.968) | 0.913 | (0.831, 0.995) | − | − |
CI, confidence interval.
Table 3.
Agreement between the Test Results for Final Call for Tissue of Origin (Physician-Guided Conclusion) and Clinical Truth per Site
| Site | n | % Agreement | % Nonagreement | % Indeterminate |
|---|---|---|---|---|
| 1 | 55 | 87.3 | 7.3 | 5.5 |
| 2 | 55 | 85.5 | 5.5 | 9.1 |
| 3 | 56 | 89.3 | 1.8 | 8.9 |
| 4 | 53 | 84.9 | 3.8 | 11.3 |
Interestingly, when the analysis was restricted to tissue specimens that met the sample criteria set by the TOO test manufacturer for tumor content (>60% tumor and <20% necrosis, n = 52), the average percent agreement between sites increased to 93.8% (range, 93.3 to 95.5%) (Table 4). Conversely, when the analysis was expanded to include samples with RNA degradation (n = 18), 50% of which failed the quality control (data verification step) of the Pathwork TOO test, the agreement between sites diminished. Of the nine samples failing data verification, five yielded an indeterminate result for the TOO test PGC, three gave a correct tissue identification, and one a discordant result (other replicates of this sample were also discordant). In contrast, from the remaining nine samples with RNA degradation that passed the data verification step, all but one showed agreement between the PGC and the clinical truth. The remaining sample gave an indeterminate result for all laboratory sites tested (regardless of RNA quality).
Table 4.
Concordance of Test Results for Final Call for Tissue of Origin (Physician-Guided Conclusion) between Sites, after Removing Samples that Failed the Test Manufacturer's Tissue Quality Control Parameters
| Site | n | % Concordance | % Discordance |
|---|---|---|---|
| 1 versus 2 | 45 | 93.3 | 6.7 |
| 1 versus 3 | 46 | 93.5 | 6.5 |
| 1 versus 4 | 45 | 93.3 | 6.7 |
| 2 versus 3 | 47 | 93.6 | 6.4 |
| 2 versus 4 | 45 | 93.3 | 6.7 |
| 3 versus 4 | 44 | 95.5 | 4.6 |
Discussion
For a diagnostic microarray to perform adequately within the clinical setting, the test's robustness should be assessed and is expected to account for variations in both laboratory technique and RNA input. Given the anticipated variations between laboratories in terms of personnel, reagents, equipment, and protocols,26,34,35,36 evidence of reliable results across sites is clearly a prerequisite for routine use of any microarray-based gene expression test in the clinical laboratory. Although early concerns about the analytical performance of microarray technology itself37,38,39 have been allayed by demonstrations of repeatability within sites, across sites, and across platforms,28,36,40,41 the impacts of analytical variability on specific tests must be validated anew for each test. Some existing gene expression tests avoid this issue of procedural variability by requiring that all tissue specimens be sent to a central national laboratory for processing and analysis (Genomic Health OncotypeDX Breast Cancer Assay, Redwood City, CA; MammaPrint, Amsterdam, The Netherlands).42,43 In contrast, the Pathwork TOO test is designed to be performed by local molecular diagnostics laboratories, with the bioinformatics analysis done at a centralized facility. In this study, we assessed the robustness and reproducibility of this test through a multicenter study representing diverse laboratory environments. The test was challenged with replicates of 60 cancer specimens analyzed at four separate laboratories.
The results of this study show reproducibility of the Pathwork TOO test, despite the differences between sites in terms of instruments, reagents, RNA extraction protocols, users, and days. Good reproducibility was observed in all three categories of evaluated results: the SE values, the SS, and the PGC, with correlation coefficients greater than 0.84 for SE and greater than 0.95 for SS in crosswise comparisons between laboratories. No evidence of a systematic bias was observed, and the agreement between laboratories was considered to be good by the Bland-Altman analysis (less than 10% outliers). It is important to note that analytical variations found in everyday clinical practice were part of this study; thus, the study reflects the environment where this test would be performed clinically and therefore markedly differs from other studies of reproducibility of microarrays in which total RNA samples, not tissues, were analyzed.27,28
At the level of the reported result (PGC), there was an overall concordance of 89.1% between laboratories (κ > 0.86), demonstrating that the test is robust and reproducible. When we excluded the samples that failed tissue quality parameters, an increase in reproducibility was observed, strongly suggesting that the TOO test is more reproducible when good quality tissue and RNA samples are analyzed. This result also underscores the need for adequate tissue quality in molecular testing. Although blending patient specimens to create uniform replicates can overcome potential tissue-related sources of variability, we opted to use direct sampling from frozen tissue blocks to replicate the most likely clinical scenario in which this test would be used. The overall high correlation between laboratories reported here for all of the tissue samples with greater than 60% tumor content and less than 20% necrosis implies that any actual heterogeneity in these replicates had virtually no impact on the TOO test results.
Because prompt collection and preservation steps are critical to maintaining the high quality of RNA,20,35,44 we preserved RNA integrity by collecting tissue samples at only one site. Interestingly, we found that only samples with low-quality RNA (RIN < 5.5, or evidence of degradation based on gel electrophoresis) showed failed data verification flags for the TOO test. Most importantly, samples with evidence of degradation that passed the data verification step yielded either correct tissue identifications (eight of nine) or indeterminate results (one of nine). Hence, none of the TOO results obtained from degraded RNA samples yielded the wrong tissue type for that sample. On the other hand, the fact that 50% of samples with low RNA quality failed to pass the built-in quality control criteria of the TOO algorithm strongly accentuates the need for an RNA extraction quality control program for successful implementation of this test in the clinical setting. Other studies may be needed to explore alternatives to both freezing and formalin fixation (eg, RNA stabilizers).19,20
The algorithm used by the TOO test normalizes all raw expression input from the patient specimen based on that specimen's aggregate expression levels for 121 mRNA markers that are expressed in a stable manner across cell types. When compared to the default Affymetrix standardization algorithm (MAS5) (correlation coefficients of 0.66 to 0.81) the test's normalization algorithm (SE) improved correlation coefficients to 0.84 to 0.90. The additional increase in correlation observed between the SE and SS levels indicates that the algorithm for calculating SS scores further contributes to the overall reproducibility of the assay.
The results of the TOO test are based on the overall pattern of expression detected by the 1668 probe sets or markers in which single genes not individually predictive may become predictive when evaluated in combination with other genes.45,46 Examining all of the potential gene-gene combinations to create such a highly multiplex class prediction method requires an extraordinarily large number of specimens in the original training set. Given the reproducibility of results in this study, this strategy may be more effective for these types of multivariate index assays than a reductionist approach in which a panel of individual genes first discovered with microarray analysis is selected for use on a nonmicroarray platform.14,42,47
The main goal of the present study was to address the analytical performance of the TOO test, and as such it was not designed to evaluate assay performance in identifying the correct site of origin. A much larger clinical validation study that will address this question is under way. Nonetheless, we found very good agreement with clinical truth that improved when only samples meeting the tissue quality criteria for the TOO test were included. Moreover, 14 of the 18 high-grade, poorly differentiated and undifferentiated tumor specimens were properly identified by the TOO test, in agreement with the clinical truth in at least three of the four laboratories, whereas the remaining four of those tumor specimens either yielded an indeterminate TOO result or failed to agree with the clinical truth in all four sites. These results are encouraging, especially given the tendency of previously reported PCR-based14 and microarray-based18 assays to produce inaccurate or low-confidence results in specimens that were poorly differentiated or undifferentiated—the very specimens requiring an improved diagnostic approach. This historic difficulty in classifying poorly differentiated tumors had bolstered the suggestion that these may be molecularly distinct entities,18,48 and therefore, perhaps unlikely to be classified using molecular stratification techniques. However, confirmation of accurate and consistent results with microarray-based gene expression tests in tumors across a range of differentiation stages would indicate that at least a portion of the TOO molecular signature remains intact as the tumor progresses. Such findings may have implications not only for the feasibility of molecular diagnosis, but also, in a broader context, for the resolution of long-standing questions about the relationship of metastatic cells to their TOO,49 and for the development of targeted treatment approaches for TOO.50
In summary, the Pathwork TOO test delivered reproducible results across a wide range of laboratory settings and therefore, at least from an analytical perspective, may be suitable for clinical application. Although the user guide for this microarray-based test gave general guidance in terms of tissue handling and target RNA preparation, in fact, variations in these protocols because of different operators, reagents, and instrumentation were actually used in the different laboratories. Thus, the study provides a realistic assessment of this microarray-based test's performance in the clinical setting. If the clinical performance of this test can be validated in large and appropriately designed multicenter trials, then this test may become a valuable alternative for those challenging malignancies for which the TOO remains uncertain after the initial pathological evaluation.
Acknowledgements
We thank Amelia Hensler and Aprel Delo from the University of Pittsburgh Health Sciences Tissue Bank for their assistance with tissue procurement and preparation of frozen sections and Paul Courter and Jane Seck for their editorial support.
Footnotes
Supported by a sponsored research agreement from Pathwork Diagnostics, Sunnyvale, CA (to F.A.M.).
C.T.R., L.J.B., and G.G.A. are employees and shareholders of Pathwork Diagnostics, which developed the tissue of origin test described in this article. F.A.M. has received honoraria for consultation with Pathwork Diagnostics regarding new test development unrelated to the tissue of origin test. There are no other potential conflicts of interest. C.I.D. and F.A.M. had full access to all the data in the study and have final responsibility for the results of the study reported in this article.
Supplemental material for this article can be found on http://jmd.amjpathol.org/.
Supplementary data
References
- 1.Pavlidis N, Briasoulis E, Hainsworth J, Greco FA. Diagnostic and therapeutic management of cancer of an unknown primary. Eur J Cancer. 2003;39:1990–2005. doi: 10.1016/s0959-8049(03)00547-1. [DOI] [PubMed] [Google Scholar]
- 2.Bugat R, Bataillard A, Lesimple T, Voigt JJ, Culine S, Lortholary A, Merrouche Y, Ganem G, Kaminsky MC, Negrier S, Perol M, Laforet C, Bedossa P, Bertrand G, Coindre JM, Fizazi K. FNCLCC: summary of the standards, options and recommendations for the management of patients with carcinoma of unknown primary site (2002) Br J Cancer. 2003;89:S59–S66. doi: 10.1038/sj.bjc.6601085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeYoung BR, Wick MR. Immunohistologic evaluation of metastatic carcinomas of unknown origin: an algorithmic approach. Semin Diagn Pathol. 2000;17:184–193. [PubMed] [Google Scholar]
- 4.Hillen HF. Unknown primary tumours. Postgrad Med J. 2000;76:690–693. doi: 10.1136/pmj.76.901.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavlidis N, Merrouche Y. The importance of identifying CUP subsets. In: Fizazi K, editor. Carcinoma of Unknown Primary Site. Taylor & Francis Group; New York: 2006. pp. 37–48. [Google Scholar]
- 6.Van de Wouw AJ, Laplanche A. What we know about carcinomas of unknown primary site (CUP) almost for sure: incidence, survival, and necropsy data. In: Fizazi K, editor. Carcinoma of Unknown Primary Site. Taylor & Francis Group; New York: 2006. pp. 1–10. [Google Scholar]
- 7.Greco FA, Hainsworth JD. Cancer of unknown primary site. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. ed 7. Lippincott Williams & Wilkins; Philadelphia: 2005. pp. 2213–2236. [Google Scholar]
- 8.Abbruzzese JL, Abbruzzese MC, Lenzi R, Hess KR, Raber MN. Analysis of a diagnostic strategy for patients with suspected tumors of unknown origin. J Clin Oncol. 1995;13:2094–2103. doi: 10.1200/JCO.1995.13.8.2094. [DOI] [PubMed] [Google Scholar]
- 9.Dennis JL, Hvidsten TR, Wit EC, Komorowski J, Bell AK, Downie I, Mooney J, Verbeke C, Bellamy C, Keith WN, Oien KA. Markers of adenocarcinoma characteristic of the site of origin: development of a diagnostic algorithm. Clin Cancer Res. 2005;11:3766–3772. doi: 10.1158/1078-0432.CCR-04-2236. [DOI] [PubMed] [Google Scholar]
- 10.Bloom GC, Eschrich S, Zhou JX, Coppola D, Yeatman TJ. Elucidation of a protein signature discriminating six common types of adenocarcinoma. Int J Cancer. 2007;120:769–775. doi: 10.1002/ijc.22041. [DOI] [PubMed] [Google Scholar]
- 11.Tothill RW, Kowalczyk A, Rischin D, Bousioutas A, Haviv I, van Laar RK, Waring PM, Zalcberg J, Ward R, Biankin AV, Sutherland RL, Henshall SM, Fong K, Pollack JR, Bowtell DD, Holloway AJ. An expression-based site of origin diagnostic method designed for clinical application to cancer of unknown origin. Cancer Res. 2005;65:4031–4040. doi: 10.1158/0008-5472.CAN-04-3617. [DOI] [PubMed] [Google Scholar]
- 12.Buckhaults P, Zhang Z, Chen Y-C, Wang T-L, St Croix B, Saha S, Bardelli A, Morin PJ, Polyak K, Hruban RH, Velculescu VE, Shih I-M. Identifying tumor origin using a gene expression-based classification map. Cancer Res. 2003;63:4144–4149. [PubMed] [Google Scholar]
- 13.Talantov D, Baden J, Jatkoe T, Hahn K, Yu J, Rajpurohit Y, Jiang Y, Choi C, Ross JS, Atkins D, Wang Y, Mazumder A. A quantitative reverse transcriptase-polymerase chain reaction assay to identify metastatic carcinoma tissue of origin. J Mol Diagn. 2006;8:320–329. doi: 10.2353/jmoldx.2006.050136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma XJ, Patel R, Wang X, Salunga R, Murage J, Desai R, Tuggle JT, Wang W, Chu S, Stecker K, Raja R, Robin H, Moore M, Baunoch D, Sgroi D, Erlander M. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch Pathol Lab Med. 2006;130:465–473. doi: 10.5858/2006-130-465-MCOHCU. [DOI] [PubMed] [Google Scholar]
- 15.Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DSA, Nobel AB, van't Veer LJ, Perou CM. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 16.Potti A, Mukherjee S, Petersen R, Dressman HK, Bild A, Koontz J, Kratzke R, Watson MA, Kelley M, Ginsburg GS, West M, Harpole DH, Jr, Nevins JR. A genomic strategy to refine prognosis in early-stage non-small-cell lung cancer. N Engl J Med. 2006;355:570–580. doi: 10.1056/NEJMoa060467. [DOI] [PubMed] [Google Scholar]
- 17.Su AI, Welsh JB, Sapinoso LM, Kern SG, Dimitrov P, Lapp H, Schultz PG, Powell SM, Moskaluk CA, Frierson HF, Jr, Hampton GM. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61:7388–7393. [PubMed] [Google Scholar]
- 18.Ramaswamy S, Tamayo P, Rifkin R, Mukherjee S, Yeang CH, Angelo M, Ladd C, Reich M, Latulippe E, Mesirov JP, Poggio T, Gerald W, Loda M, Lander ES, Golub TR. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci USA. 2001;98:15149–15154. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mutter GL, Zahrieh D, Liu C, Neuberg D, Finkelstein D, Baker HE, Warrington JA. Comparison of frozen and RNALater solid tissue storage methods for use in RNA expression microarrays. BMC Genomics. 2004;5:88. doi: 10.1186/1471-2164-5-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang SS, Sherman ME, Rader JS, Carreon J, Schiffman M, Baker CC. Cervical tissue collection methods for RNA preservation: comparison of snap-frozen, ethanol-fixed, and RNAlater-fixation. Diagn Mol Pathol. 2006;15:144–148. doi: 10.1097/01.pdm.0000213460.53021.cd. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Robinson JF, Khan HM, Carter DE, McKinney J, Miskie BA, Hegele RA. Optimizing RNA extraction yield from whole blood for microarray gene expression analysis. Clin Biochem. 2004;37:741–744. doi: 10.1016/j.clinbiochem.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 22.Egyhazi S, Bjohle J, Skoog L, Huang F, Borg AL, Frostvik Stolt M, Hagerstrom T, Ringborg U, Bergh J. Proteinase K added to the extraction procedure markedly increases RNA yield from primary breast tumors for use in microarray studies. Clin Chem. 2004;50:975–976. doi: 10.1373/clinchem.2003.027102. [DOI] [PubMed] [Google Scholar]
- 23.Baugh LR, Hill AA, Brown EL, Hunter CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res. 2001;29:E29. doi: 10.1093/nar/29.5.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Puskás LG, Zvara A, Hackler L, Jr, Van Hummelen P. RNA amplification results in reproducible microarray data with slight ratio bias. Biotechniques. 2002;32:1330–1340. doi: 10.2144/02326mt04. [DOI] [PubMed] [Google Scholar]
- 25.Gold D, Coombes K, Medhane D, Ramaswamy A, Ju Z, Strong L, Koo JS, Kapoor M. A comparative analysis of data generated using two different target preparation methods for hybridization to high-density oligonucleotide microarrays. BMC Genomics. 2004;5:2. doi: 10.1186/1471-2164-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma C, Lyons-Weiler M, Liang W, LaFramboise W, Gilbertson JR, Becich MJ, Monzon FA. In vitro transcription amplification and labeling methods contribute to the variability of gene expression profiling with DNA microarrays. J Mol Diagn. 2006;8:183–192. doi: 10.2353/jmoldx.2006.050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ach RA, Floore A, Curry B, Lazar V, Glas AM, Pover R, Tsalenko A, Ripoche H, Cardoso F, Saghatchian d'Assignies M, Bruhn L, Van 't Veer LJ. Robust interlaboratory reproducibility of a gene expression signature measurement consistent with the needs of a new generation of diagnostic tools. BMC Genomics. 2007;8:148. doi: 10.1186/1471-2164-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Amur S, Bao W, Barbacioru CC, Lucas AB, Bertholet V, Boysen C, Bromley B, Brown D, Brunner A, Canales R, Cao XM, Cebula TA, Chen JJ, Cheng J, Chu TM, Chudin E, Corson J, Corton JC, Croner LJ, Davies C, Davison TS, Delenstarr G, Deng X, Dorris D, Eklund AC, Fan XH, Fang H, Fulmer-Smentek S, Fuscoe JC, Gallagher K, Ge W, Guo L, Guo X, Hager J, Haje PK, Han J, Han T, Harbottle HC, Harris SC, Hatchwell E, Hauser CA, Hester S, Hong H, Hurban P, Jackson SA, Ji H, Knight CR, Kuo WP, LeClerc JE, Levy S, Li QZ, Liu C, Liu Y, Lombardi MJ, Ma Y, Magnuson SR, Maqsodi B, McDaniel T, Mei N, Myklebost O, Ning B, Novoradovskaya N, Orr MS, Osborn TW, Papallo A, Patterson TA, Perkins RG, Peters EH, Peterson R, Philips KL, Pine PS, Pusztai L, Qian F, Ren H, Rosen M, Rosenzweig BA, Samaha RR, Schena M, Schroth GP, Shchegrova S, Smith DD, Staedtler F, Su Z, Sun H, Szallasi Z, Tezak Z, Thierry-Mieg D, Thompson KL, Tikhonova I, Turpaz Y, Vallanat B, Van C, Walker SJ, Wang SJ, Wang Y, Wolfinger R, Wong A, Wu J, Xiao C, Xie Q, Xu J, Yang W, Zhang L, Zhong S, Zong Y, Slikker W. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dumur CI, Nasim S, Best AM, Archer KJ, Ladd AC, Mas VR, Wilkinson DS, Garrett CT, Ferreira-Gonzalez A. Evaluation of quality-control criteria for microarray gene expression analysis. Clin Chem. 2004;50:1994–2002. doi: 10.1373/clinchem.2004.033225. [DOI] [PubMed] [Google Scholar]
- 30.Moraleda J, Grov N, Tran Q, Doan J, Hull J, Nguyen L, Pattin A, Anderson G. Gene expression data analytics with interlaboratory validation for identifying anatomical sites of origin of metastatic carcinomas. ASCO Annual Meeting Proceedings (post-meeting edition) 2004. J Clin Oncol. 2004;22:S9625. [Google Scholar]
- 31.Hubbell E, Liu W-M, Mei R. Robust estimators for expression analysis. Bioinformatics. 2002;18:1585–1592. doi: 10.1093/bioinformatics/18.12.1585. [DOI] [PubMed] [Google Scholar]
- 32.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1:307–310. [PubMed] [Google Scholar]
- 33.Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Measurement. 1960;20:37–46. [Google Scholar]
- 34.Dobbin KK, Beer DG, Meyerson M, Yeatman TJ, Gerald WL, Jacobson JW, Conley B, Buetow KH, Heiskanen M, Simon RM, Minna JD, Girard L, Misek DE, Taylor JM, Hanash S, Naoki K, Hayes DN, Ladd-Acosta C, Enkemann SA, Viale A, Giordano TJ. Interlaboratory comparability study of cancer gene expression analysis using oligonucleotide microarrays. Clin Cancer Res. 2005;11:565–572. [PubMed] [Google Scholar]
- 35.Tumor Analysis Best Practices Working Group, Hoffman EP, Awad T, Palma J, Webster T, Hubbell E, Warrington JA, Spira A, Wright G, Buckley J, Triche T, Davis R, Tibshirani R, Xiao W, Jones W, Tompkins R, West M. Expression profiling—best practices for data generation and interpretation in clinical trials. Nat Rev Genet. 2004;5:229–237. doi: 10.1038/nrg1297. [DOI] [PubMed] [Google Scholar]
- 36.Bammler T, Beyer RP, Bhattacharya S, Boorman GA, Boyles A, Bradford BU, Bumgarner RE, Bushel PR, Chaturvedi K, Choi D, Cunningham ML, Deng S, Dressman HK, Fannin RD, Farin FM, Freedman JH, Fry RC, Harper A, Humble MC, Hurban P, Kavanagh TJ, Kaufmann WK, Kerr KF, Jing L, Lapidus JA, Lasarev MR, Li J, Li YJ, Lobenhofer EK, Lu X, Malek RL, Milton S, Nagalla SR, O'Malley JP, Palmer VS, Pattee P, Paules RS, Perou CM, Phillips K, Qin LX, Qiu Y, Quigley SD, Rodland M, Rusyn I, Samson LD, Schwartz DA, Shi Y, Shin JL, Sieber SO, Slifer S, Speer MC, Spencer PS, Sproles DI, Swenberg JA, Suk WA, Sullivan RC, Tian R, Tennant RW, Todd SA, Tucker CJ, Van Houten B, Weis BK, Xuan S, Zarbl H. Members of the Toxicogenomics Research Consortium: standardizing global gene expression analysis between laboratories and across platforms. Nat Methods. 2005;2:351–356. doi: 10.1038/nmeth754. [DOI] [PubMed] [Google Scholar]
- 37.Naef F, Socci ND, Magnasco M. A study of accuracy and precision in oligonucleotide arrays: extracting more signal at large concentrations. Bioinformatics. 2003;19:178–184. doi: 10.1093/bioinformatics/19.2.178. [DOI] [PubMed] [Google Scholar]
- 38.Tan PK, Downey TJ, Spitznagel EL, Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC. Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res. 2003;31:5676–5684. doi: 10.1093/nar/gkg763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miklos GL, Maleszka R. Microarray reality checks in the context of a complex disease. Nat Biotechnol. 2004;22:615–621. doi: 10.1038/nbt965. [DOI] [PubMed] [Google Scholar]
- 40.Canales RD, Luo Y, Willey JC, Austermiller B, Barbacioru CC, Boysen C, Hunkapiller K, Jensen RV, Knight CR, Lee KY, Ma Y, Maqsodi B, Papallo A, Peters EH, Poulter K, Ruppel PL, Samaha RR, Shi L, Yang W, Zhang L, Goodsaid FM. Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol. 2006;24:1115–1122. doi: 10.1038/nbt1236. [DOI] [PubMed] [Google Scholar]
- 41.Shippy R, Fulmer-Smentek S, Jensen RV, Jones WD, Wolber PK, Johnson CD, Pine PS, Boysen C, Guo X, Chudin E, Sun YA, Willey JC, Thierry-Mieg J, Thierry-Mieg D, Setterquist RA, Wilson M, Lucas AB, Novoradovskaya N, Papallo A, Turpaz Y, Baker SC, Warrington JA, Shi L, Herman D. Using RNA sample titrations to assess microarray platform performance and normalization techniques. Nat Biotechnol. 2006;24:1123–1131. doi: 10.1038/nbt1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cronin M, Pho M, Dutta D, Stephans JC, Shak S, Kiefer MC, Esteban JM, Baker JB. Measurement of gene expression in archival paraffin-embedded tissues: development and performance of a 92-gene reverse transcriptase-polymerase chain reaction assay. Am J Pathol. 2004;164:35–42. doi: 10.1016/S0002-9440(10)63093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 44.Ramaswamy S, Golub TR. DNA microarrays in clinical oncology. J Clin Oncol. 2002;20:1932–1941. doi: 10.1200/JCO.2002.20.7.1932. [DOI] [PubMed] [Google Scholar]
- 45.Pusztai L, Hess KR. Clinical trial design for microarray predictive marker discovery and assessment. Ann Oncol. 2004;15:1731–1737. doi: 10.1093/annonc/mdh466. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Weinberg CR, Darden TA, Pedersen LG. Gene selection for sample classification based on gene expression data: study of sensitivity to choice of parameters of the GA/KNN method. Bioinformatics. 2001;17:1131–1142. doi: 10.1093/bioinformatics/17.12.1131. [DOI] [PubMed] [Google Scholar]
- 47.Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N Engl J Med. 2004;350:1828–1837. doi: 10.1056/NEJMoa032520. [DOI] [PubMed] [Google Scholar]
- 48.Busson P, Daya-Grosjean L, Pavlidis N, Va deWouw AJ. The biology of the unknown primary tumors: the little we know, the importance of learning more. In: Fizazi K, editor. Carcinoma of Unknown Primary Site. Taylor & Francis Group; New York: 2006. pp. 159–174. [Google Scholar]
- 49.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 50.Pentheroudakis G, Pavlidis N. Perspectives for targeted therapies in cancer of unknown primary site. Cancer Treat Rev. 2006;32:637–644. doi: 10.1016/j.ctrv.2006.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.