

Abstract
Reliable real-time quantitative PCR assays to measure Epstein-Barr virus (EBV) DNA load (EBV) are useful for monitoring EBV-associated diseases. We evaluated a new commercial kit, EBV R-gene Quantification kit (Argene, Varilhes, France) to quantify EBV DNA load in whole blood. Assay performance was assessed with two PCR platforms (LightCycler 2.0 and SmartCycler 2.0) and three commercial DNA extraction methods. The assay was compared with our in-house real-time EBV PCR using samples from the Quality Control for Molecular Diagnostics 2006 EBV proficiency program and using 167 whole-blood specimens from individuals with infectious mononucleosis, from transplanted or HIV-infected patients, and from EBV-seropositive healthy carriers. The EBV R-gene assay was sensitive to 500 copies of EBV DNA per milliliter of whole blood with the two PCR platforms and the three extraction methods and was linear across 4 orders of magnitude. Intra- and interassay coefficients of variations were less than 20%. Nine of 10 samples tested with the EBV R-gene were in agreement with the expected qualitative results of the Quality Control for Molecular Diagnostics 2006 EBV proficiency program, and 7 of 10 samples were within ±0.5 log units of the expected quantitative values, with discrepant results mostly observed for low viral load (ie, <1000 copies/ml). In the clinical specimens, the correlation between the R-gene assay and the in-house PCR was high (r = 0.92). In conclusion, the EBV R-gene assay accurately assesses the EBV DNA load in whole blood of patients with various forms of EBV infections.
The Epstein-Barr virus (EBV), a ubiquitous human γ-herpesvirus, is the etiological agent of infectious mononucleosis (IM), a self-limited lymphoproliferative disease, and is also associated with lymphoid or epithelial malignancies both in immunocompetent and immunosuppressed individuals.1,2,3 The reliable measurement of the EBV DNA load in the blood using real-time quantitative PCR assays is a useful marker in the clinical management of the EBV-associated posttransplantation lymphoproliferative diseases (PTLDs).2,4 More recently, EBV DNA load measurement in blood also appears to be a potentially helpful tool for monitoring other EBV-associated cancers such as nasopharyngeal carcinoma, AIDS-associated lymphoma, and Hodgkin's disease.5,6,7,8 EBV DNA load measurements may also provide characterization of the natural history of IM.9,10 However, an appropriate standardization of EBV DNA load measurement is still needed to accurately establish the predictive value of EBV DNA load in specific clinical situations.
The use of unfractionated whole blood in diagnostic settings could be a first step toward standardization of EBV DNA load quantification. Indeed, this specimen combines all blood compartments that may harbor EBV, and it best reflects the absolute viral burden in the patient's circulation.11,12 The automation of the nucleic acid purification step before real-time PCR amplification could also simplify and improve the reproducibility of EBV DNA load measurement.11,13 Because the availability of a variety of in-house PCR assays using different real-time PCR platforms and software may challenge the standardization of EBV DNA load quantification, the development of a versatile commercial PCR amplification assay is another important step in providing clinical laboratories with reliable diagnostic tools.
The aim of this study was to evaluate the performance characteristics of a recently available commercial quantitative real-time PCR assay: the EBV R-gene Quantification kit (EBV R-gene assay; Argene SA, Varilhes, France) for EBV DNA load measurement in whole-blood samples. The sensitivity, specificity, and reproducibility of this assay were first assessed with different PCR platforms and with different DNA extraction methodologies. The performance characteristics of this assay were then compared with those of our in-house PCR14,15 using the Quality Control for Molecular Diagnostic 2006 EBV Proficiency panel (EBV QCMD 2006) and 167 clinical specimens from immunocompetent and immunosuppressed individuals with various forms of EBV infections.
Materials and Methods
Study Design
The sensitivity of the EBV R-gene assay was assessed with serial dilutions of an EDTA whole-blood sample tested as positive for EBV DNA with our in-house PCR and then diluted in EDTA whole blood tested as negative for EBV DNA and EBV antibodies (ie, negative for anti-Epstein-Barr nuclear antigen and anti-viral capsid antigen IgG antibodies). After manual DNA extraction with the QIAamp DNA Blood mini kit (Qiagen, Hilden, Germany), each dilution was tested six times on two different PCR platforms [SmartCycler 2.0 (Cepheid, Sunnyvale, CA), and LightCycler 2.0 (Roche Diagnostics, Mannheim, Germany)]. The specificity of the EBV R-gene assay was tested with two PCR platforms after manual DNA extractions from American Type Culture Collection (Rockville, MD) or wild strains of human herpesviruses (HSV1, HSV2, VZV, CMV, HHV6, HHV7, and HHV8), human polyomaviruses (JC virus and BK virus), and adenovirus 12.
The second step in evaluating the EBV R-gene assay assessed the intra- and interassay variations using the SmartCycler 2.0 PCR platform after manual or automated DNA extraction. Five EBV DNA-positive whole-blood samples ranging from 500 to more than 100,000 EBV DNA copies/ml (as determined by our in-house PCR assay) were tested after manual (QIAamp DNA Blood mini kit) or automated extraction [Qiagen BioRobot EZ1 System (Qiagen) or the MagNAPure compact instrument (Roche Molecular Biochemicals, Indianapolis, IN)]. Each sample was tested in six replicates from extraction to amplification in the same experiment, and this was repeated in six independent experiments.
The last part of the validation process compared the performance of the EBV R-gene assay after MagNaPureLC-extraction and with the SmartCycler 2.0 to our in-house quantitative real-time PCR based on MagNAPureLC extraction coupled with the LightCycler 2.0. For this purpose, methodologies were applied to the EBV QCMD 2006, which is sponsored by the European Society for Clinical Virology and the European Society for Clinical Microbiology and Infectious Diseases (the 10 panel samples were constructed by diluting electron microscope-counted viral particles in plasma). Among the 150 whole-blood clinical specimens randomly selected (these specimens were routinely sent to our institution for EBV DNA load measurement), 42 were obtained from patients with allogenic stem cell transplantation, 78 from solid organ transplant recipients (37 kidney, 24 liver, 14 lung, and 3 heart transplants), 6 samples from patients with clinically and serologically proven IM (ie, presence of IgM and IgG anti-viral capsid antigen antibodies and lack of anti-Epstein-Barr nuclear antigen antibodies), 17 samples from HIV-infected patients tested as EBV seropositive but without detectable EBV-associated disease, and finally 7 samples from voluntary EBV-seropositive healthy laboratory workers. Seventeen additional samples were obtained during the follow-up of two allogenic stem cell transplant recipients treated with anti-CD20 monoclonal antibodies, one for a biopsy-proven PTLD and the other for a rapid increase in EBV DNA load after graft-versus-host disease treated with anti-thymocyte globulins. Whole-blood samples were stored at −80°C until used for DNA isolation; two aliquots of whole blood were made to avoid further freezing and thawing. The local ethics committee approved the study.
DNA Isolation
DNA from 200 μl (except for EZ1 system, 350 μl) of whole blood was purified by one manual extraction using the QIAamp DNA Blood mini kit and by two robotic workstations: using the MagNAPure Compact instrument (MagNAPure Compact Nucleic Acid Isolation kit I) following the DNA isolation high-performance protocol and using the Qiagen BioRobot EZ1 System (following EZ1 DNA blood 350 μl protocol), according to the manufacturer's instructions. Two hundred microliters of whole blood were used with the MagNAPure LC instrument (MagNAPure LC Isolation kit I). The extracted DNA was eluted with 100 μl of elution buffer before the PCR reaction.
Quantitative Real-Time PCR
Blood specimens were run by the in-house quantitative real-time PCR assay and by the EBV R-gene assay. Both methods amplify a highly conserved region in the thymidine kinase (BXLF1) gene using similar primers, as previously described for the in-house method.14
EBV R-gene assay is based on the detection of the amplified product with a TaqMan probe. Each sample is amplified with both the amplification premix and the inhibition control premix. Amplification premix contains EBV primers and dual-labeled probe (5′-FAM/3′-TAMRA). Inhibition premix contains all necessary reagents to show potential inhibition of amplification: target DNA, primers, and dual-labeled probe (5′-FAM/3′-TAMRA). Each sample, and water as reference, was amplified with inhibition premix. After amplification, threshold cycle value of each sample was compared with reference value: If the difference was more than two cycles, the sample was considered as inhibited and had to be extracted again. PCR was performed in a 25-μl volume containing 15 μl of the amplification premix or inhibition premix and 10 μl of standard or sample DNA. A range of four quantification standards, sensitivity control, inhibition control, and negative control are supplied in the kit. The quantification standard is a composite plasmid containing a sequence that can be recognized by the primers, with thermodynamics (GC content, size, and melting temperature) similar to those of the amplified target.
The in-house PCR assay is based on detecting the amplified product with LightCycler hybridization probes (fluorescein, LC640). The PCR was performed in a LightCycler instrument (Roche Diagnostics) as described previously.14 EBV DNA was quantified using a serial 10-fold dilution of DNA extracted from Namalwa cells containing two integrated copies of EBV genome per cell. This method was able to detect 10 copies per reaction of EBV DNA. To monitor the whole process from isolation of nucleic acids to real-time detection, a universal internal control was used. This internal control sample consisted of a native DNA of coliphage PhiX174, which was added to the original clinical sample at a final concentration of 10,000 DNA copies/ml, equivalent to a threshold cycle value of approximately 30 in the real-time detection system used. A fragment of 298 bp of the PhiX174 was amplified using the primer set (position 4896 to 5194) upstream sequence IC1, 5′-CCCATCGCAGTTCGCT-3′/down-stream sequence IC2, 5′-AGCACTCCGTGGACAGA-3′ and the FRET hybridization probes 5′-ACTTCCCAAGAAGCTGTTCAGAATCAG-fluorescein-3′/5′-Red 705-ATGAGCCGCAACTTCGGGA-3′. The primers and probes were designed by LightCycler probe software (Roche Diagnostics).
Ten microliters of DNA sample were subjected to PCR with both methods, and the results are given in log copies per milliliter of whole blood.
Statistics
Data were analyzed using the SPSS 11.0 software for Windows (SPSS, Inc., Chicago, IL). The intra- and interassay variations of the EBV R-gene kit were evaluated with descriptive statistics. The correlation between EBV DNA loads obtained by the EBV R-gene assay and the in-house PCR assay was calculated with Spearman's correlation test.
Results
Sensitivity, Specificity, and Reproducibility of the EBV R-Gene Assay
Figure 1 shows that, after a manual DNA extraction, the EBV R-gene assay used with both PCR platforms detected all six replicates of specimen containing 10 copies of EBV DNA per reaction. This result corresponds to a limit of detection of 500 copies EBV DNA/ml whole blood (when using 200 μl of input sample volume and 10 μl of the 100-μl elution volume for the PCR). In this experiment, combining the EBV R-gene assay with the SmartCycler or with the LightCycler PCR platforms detected the replicates containing five copies per reaction (ie, 250 copies/ml) in four and two of the six cases, respectively (Figure 1). Using the standards provided by the manufacturer, the R-gene assay was linear from 500 to 5 million copies/ml. No cross-reaction was observed on the two PCR platforms with the other herpesviruses or with JC virus and BK virus or adenovirus.
Figure 1.
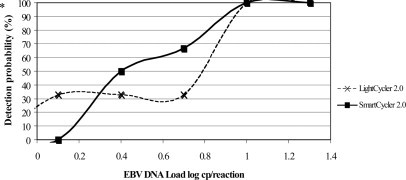
Analytic sensitivity of the EBV R-gene assay with two different PCR platforms. The EBV R-gene assay used with both PCR platforms, after manual extraction, detected all six replicates of specimen containing 10 copies of EBV DNA per reaction.
The intra-assay variability of the quantification kit plus the extraction step was evaluated using five EBV DNA-positive whole-blood samples ranging from less than 500 to more than 100,000 EBV DNA copies/ml. As shown in Table 1, depending on the EBV DNA load, the coefficients of variation (CV) ranged from 0.56 to 18.31% with the QIAamp DNA Blood mini kit extraction, from 2.13 to 19.29% with the MagNAPure compact instrument, and from 0.58 to 10.65% with the BioRobot EZ1 System. With the three DNA extraction methods, the highest variability was observed with the lowest template concentration. The interassay variability was determined by repeating the above experiment in six independent PCR runs. In this experiment, the CV varied from 0.60 to 11.77% with the QIAamp DNA Blood mini kit extraction, from 2 to 12.49% with the MagNAPure compact instrument, and from 0.66 to 13.82% with the BioRobot EZ1 System, again with the highest variability observed within the lowest template concentrations (Table 1).
Table 1.
Intra- and Interassay Reproducibility of the EBV R-Gene Quantification Kit in Whole-Blood Samples after Three Different Extraction Methods*
| Samples† | Manual DNA extraction‡ |
Automated DNA extraction 1 |
Automated DNA extraction 2 |
|||
|---|---|---|---|---|---|---|
| Mean of viral load (log copies/ml) | CV (%) | Mean of viral load (log copies/ml) | CV (%) | Mean of viral load (log copies/ml) | CV (%) | |
| Intra-assay reproducibility | ||||||
| 1 | 2.10 | 18.31 | 2.09 | 18.04 | 1.90 | 10.65 |
| 2 | 2.78 | 6.53 | 2.60 | 19.29 | 2.75 | 10.14 |
| 3 | 3.18 | 5.15 | 3.17 | 5.18 | 2.82 | 5.19 |
| 4 | 4.65 | 1.35 | 4.58 | 2.13 | 4.47 | 3.37 |
| 5 | 6.29 | 0.56 | 5.92 | 4.64 | 6.04 | 0.58 |
| Interassay reproducibility | ||||||
| 1 | 2.03 | 11.77 | 1.86 | 9.52 | 2.00 | 13.82 |
| 2 | 2.73 | 6.55 | 2.69 | 12.49 | 2.68 | 7.43 |
| 3 | 3.15 | 4.82 | 3.12 | 8.76 | 3.04 | 4.86 |
| 4 | 4.63 | 0.60 | 4.50 | 4.70 | 4.55 | 0.86 |
| 5 | 6.23 | 1.04 | 5.89 | 2.00 | 6.11 | 0.66 |
Experiments were conducted in six replicates and repeated in six independent PCR runs.
Manual DNA extraction, QIAamp DNA Blood mini kit; automated DNA extraction 1, the MagNAPure compact instrument; automated DNA extraction 2, Qiagen BioRobot EZ1 System.
Sample 1 < 500 copies/ml, 500 < sample 2 < 1000 copies/ml, 1000 < sample 3 < 10,000 copies/ml, 10,000 < sample 4 < 100,000 copies/ml, sample 5 > 100,000 copies/ml, as determined by in-house PCR.
Results of the EBV Quality Control for Molecular Diagnostic Proficiency Panel 2006 with the EBV R-Gene and the in-House EBV PCR Assays
Table 2 depicts the respective performance of the EBV R-gene assay and the in-house EBV PCR assay when tested with the EBV QCMD 2006 after automated DNA extraction. Nine of 10 and 10 of 10 were found in agreement with expected qualitative results with EBV R-gene and in-house PCR, respectively. The viral load of the discrepant sample is 250 copies/ml, below quantification range of both techniques. With the EBV R-Gene Quantification kit, 7 of 10 samples were found to be within ±0.5 log units of the expected results. The three remaining samples showed more than 1 log difference with the expected results: two samples, with expected results at 3 and 3.70 log copies/ml, were measured at 1.40 and 2.57 log copies/ml, and one sample, with an expected result at 2.40 log copies/ml, was found to be negative. When the in-house EBV PCR assay was used, 8 of 10 samples were found to be within ±0.5 log units of the expected result. The two remaining samples showed a difference lower than 1 log with the expected results.
Table 2.
Results of the QCMD 2006 Epstein-Barr Virus Proficiency Program
| Sample no. | Viral load (log copies/ml) |
||||
|---|---|---|---|---|---|
| QCMD expected results | R-gene assay | Delta log (R-gene − QCMD)* | In-house PCR | Delta log (in-house − QCMD)* | |
| EBV06-01 | 3.00 | 3.05 | 0.05 | 2.89 | −0.11 |
| EBV06-02 | 4.00 | 3.51 | −0.49 | 3.61 | −0.39 |
| EBV06-03 | 2.40 | Negative | ND† | (2.10)‡ | −0.30 |
| EBV06-04 | 2.00 | (1.58) | −0.42 | (1.81) | −0.19 |
| EBV06-05 | 3.40 | 2.93 | −0.47 | 3.09 | −0.31 |
| EBV06-06 | 3.00 | (1.40) | −1.60 | (2.33) | −0.67 |
| EBV06-07 | 2.40 | 2.51 | 0.11 | (1.69) | −0.71 |
| EBV06-08 | 3.70 | 2.57 | −1.13 | 3.58 | −0.12 |
| EBV06-09 | Negative | Negative | ND | Negative | ND |
| EBV06-10 | 2.70 | (2.48) | −0.22 | (2.39) | −0.31 |
Delta log, the log copies/ml difference between R-gene or in-house PCR assays and QCMD panels.
ND, not determined.
Values in parentheses are below the limit of quantification of both tests.
Comparison between the EBV R-Gene and the in-House PCR Assays in Clinical Specimens
Among the 150 samples tested, no PCR inhibitor was detected by the internal controls of both assays. We detected 127 (84.6%) and 128 (85.3%) specimens as positive with the EBV R-gene assay and the in-house PCR assay, respectively. Both assays showed the same percentages of EBV DNA detection in the allogenic stem cell transplant recipients, solid organ transplant recipients, HIV-infected individuals, and the patients with IM: 86, 91, 76, and 100%, respectively (Table 3). Only one of seven and two of seven of the EBV-seropositive healthy laboratory workers gave positive results with the EBV R-gene assay and the in-house PCR assay, respectively. Twenty samples were negative with both methods. Three samples were positive with our in-house PCR assay and negative with the EBV R-gene assay, whereas two other samples were positive with the EBV R-gene assay and negative with the in-house PCR assay. The five discrepant results corresponded to low EBV DNA loads (range, 2 to 2.88 log copies/ml; mean EBV DNA load of discrepant results, 2.69 log copies/ml). Overall, the logarithmic linear correlation between the two assays was high (r = 0.92; P < 0.0001) (Figure 2). Figure 3 presents EBV DNA monitoring in two allogenic stem cell transplant recipients treated with anti-CD20 monoclonal antibodies and confirms the good correlation between the two assays during the follow-up of these two patients.
Table 3.
Comparison between the EBV R-Gene Test and in-House EBV PCR in 150 Different Clinical Samples
| Group of patients | EBV R-gene quantification kit |
In-house real-time quantitative PCR |
||
|---|---|---|---|---|
| Number of positive samples (%) | Mean EBV load log copies/ml (range) | Number of positive samples (%) | Mean EBV load log copies/ml (range) | |
| Allogenic stem cell transplant recipients (n = 42) | 36 (86) | 3.78 (2.88 to 6.93) | 36 (86) | 3.77 (2.70 to 6.31) |
| Solid organ transplant recipients (n = 78) | 71 (91) | 3.42 (2.58 to 6.7) | 71 (91) | 3.53 (2.70 to 6.93) |
| Infectious mononucleosis patients (n = 6) | 6 (100) | 3.63 (2.96 to 4.55) | 6 (100) | 3.78 (3.08 to 4.59) |
| HIV-infected patients (n = 17) | 13 (76) | 3.66 (2.76 to 4.84) | 13 (76) | 3.79 (3.11 to 4.74) |
| Healthy laboratory workers (n = 7) | 1 (14) | BLQ* | 2 (28) | 2.94 (2.88 to 3.00) |
| Total positive samples/total samples | 127/150 | 128/150 | ||
BLQ, below the limit of quantification.
Figure 2.

Correlation of EBV DNA load quantified in EBV-positive whole-blood samples (n = 126) by the EBV R-gene quantification kit in the SmartCycler 2.0 and in-house PCR in the LightCycler 1.0, after automated extraction by the MagNAPure LC (r = 0.92; P < 0.0001).
Figure 3.
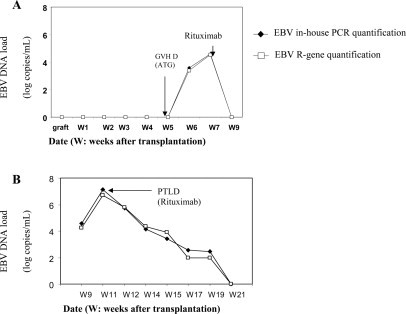
Correlation of longitudinal EBV DNA load dynamics measured by the EBV R-gene quantification kit and in-house PCR in two patients at risk for EBV-associated diseases (patients A and B). GVHD, graft-versus-host reaction disease; ATG, antithymocyte globulin.
Discussion
A wide variety of in-house quantitative real-time PCR assays has been described for the measurement of EBV DNA loads.16,17 Although these assays have been demonstrated individually to be of good diagnostic value, many clinical laboratories have no access to these “home brew” PCR assays. Moreover, the numerous in-house quantitative real-time PCR assays impair the direct comparison of EBV DNA load results from different laboratories. Therefore, the development of reliable, commercially available PCR assays may improve the accessibility and the feasibility of the EBV DNA load measurement.18,19,20,21
This study assessed the performance characteristics of the EBV R-gene Quantification kit for EBV DNA load measurement in whole-blood samples. Using whole-blood specimens, in all cases, the EBV R-gene assay detected 10 copies of EBV DNA per reaction, corresponding to a sensitivity of 500 copies EBV DNA/ml whole blood. This sensitivity of EBV R-gene assay was uniformly observed after manual or automated DNA extraction and with the two different PCR platforms. The detection of specimens containing fewer than 500 copies EBV DNA/ml whole blood was occasionally observed with both PCR platforms and considered as below the limit of quantification. Whatever the extraction method used, the reproducibility and repeatability of the R-gene assay were good with CVs less than 5% for high EBV DNA load (>4 log copies/ml) and less than 20% for lower EBV DNA load (<3 log copies/ml). This first set of data indicates that the R-gene assay for EBV DNA quantification in whole-blood specimens can be reliably performed after automated or manual extraction methods and with different PCR platforms. Since this report, we have also assessed the EBV R-gene assay for EBV DNA quantification in other biological fluids, and currently, we routinely use this assay for EBV DNA quantification in saliva, bronchoalveolar lavage, cerebrospinal fluid, and biopsies (data not shown).
In this validation study, we also compared the performance of the EBV R-gene assay after automated DNA extraction coupled with the SmartCycler PCR platform and our well validated in-house PCR using the same DNA extraction methodology but with the LightCycler 2.0 PCR platform.11,15 This comparison was made with the EBV QCMD 2006 Proficiency panel and with clinical specimens routinely sent to our institution for EBV DNA quantification. In clinical samples, the correlation for absolute values of EBV DNA load was very good (r = 0.92), and interestingly, this good agreement was also observed in the follow-up of two patients treated with anti-CD20 monoclonal antibodies. It should be noted that this good correlation was obtained although both assays used different quantification standards (Namalwa cells or composite plasmid) and different probe chemistry. From a medical point of view, the observed EBV DNA load results were in agreement with other studies that we and others have already published9,20: 100% of patients with acute uncomplicated IM showed an EBV DNA load in whole blood ranging between 2.9 and 4.5 log copies/ml in comparison with healthy EBV-seropositive individuals. The latter are less frequently EBV DNA positive in whole blood (one of seven and two of seven in the present study, depending on the test) and at a lower level: less than 3 log copies/ml. More than 85% of the patients undergoing transplantation were EBV DNA positive in whole blood, and they showed the highest absolute EBV DNA load values. However, to our knowledge, there is not yet a clear threshold value of EBV DNA load against which a pre-emptive strategy could be warranted to avoid PTLD (ie, reducing immunosuppression and anti-CD20 treatment).11,16,17 Furthermore, it should be noted here that the value of monitoring EBV DNA load in plasma rather than in whole blood in the transplantation setting is also a subject of controversy.22,23 In a previous study,11 we showed high viral loads in all blood compartments (plasma, peripheral blood mononuclear cells, and whole blood) in two patients with proven PTLD, whereas two recent studies24,25 demonstrated that whole blood was more sensitive for the quantification of EBV DNA in transplant patients than plasma. Moreover, Stevens et al12 described EBV-associated PTLDs without detection of EBV DNA in plasma, whereas the EBV DNA loads were high in whole blood. These discrepancies can perhaps be explained by the variable sensitivity of the EBV PCR in plasma where herpesvirus DNA may be highly fragmented, meaning that the length of expected amplicons is crucial for PCR sensitivity.26
Seventy-six percent of HIV-infected patients without EBV-associated diseases are EBV DNA positive in whole blood with, similarly to patients with IM, EBV DNA loads ranging from 2.7 to 4.8 log copies/ml. However, the usefulness of monitoring EBV DNA load in HIV-infected patients to predict a higher risk of AIDS-associated lymphomas is still a matter of debate.22,23 No significant differences were found between the mean and median of EBV DNA loads in the whole blood of patients with IM, HIV-infected patients, and transplant recipients.
The analysis of the EBV QCMD 2006 panel16 showed that the results of the in-house PCR assay were closer to the expected EBV DNA values than the EBV R-gene assay, even if 70% of the EBV DNA loads obtained with the EBV R-gene assay were within ± 0.5 log units compared with the expected values. One sample with an expected result lower than the limit of quantification of the R-gene assay was found to be negative, which is consistent with what we observed with the analytic sensitivity test. Two of 10 values showed discrepancies greater than 1 log copy/ml values in comparison with the expected EBV DNA levels. This problem of accurately quantifying low EBV DNA load (ie, between 2 and 3 log copies/ml) has already been reported for other commercial or in-house EBV DNA quantification assays tested with the previous EBV QCMD Proficiency panel 2002.16,20,21 Together, these results emphasize that evaluating new commercial EBV DNA quantification assays relies both on quality controls and clinical studies. They also confirm that it is more important to monitor EBV DNA load in follow-up samples and to correlate its dynamic decrease or increase with clinical events rather than to base the interpretation of viral load on absolute EBV levels only.
In conclusion, this study demonstrates that the EBV R-gene Quantification kit is able to accurately assess the EBV DNA load in whole blood after a manual or automated DNA extraction and with two different PCR platforms. Therefore, this assay is suitable for routine EBV-associated disease monitoring, particularly in clinical laboratories that cannot develop in-house methods or analyte-specific reagents. Overall, reliable commercial EBV DNA quantification assays combined with automated DNA extraction from a whole-blood sample should help standardize EBV DNA quantification.
Acknowledgements
We thank Linda Northrup for reviewing the English of this manuscript. Kits were provided by Argene.
Footnotes
Supported by grants from the Centre Hospitalier Universitaire, Grenoble.
S.F.-K., P.M., G.B., and J.-M.S. certify no affiliation with any organization or entity having a direct financial interest in the subject matter or materials discussed in the article. C.B., S.M., J.B., P.B., and M.J. are employed by Argene, whose product was studied in the present work.
References
- 1.Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343:481–492. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- 2.Thorley-Lawson DA, Gross A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med. 2004;350:1328–1337. doi: 10.1056/NEJMra032015. [DOI] [PubMed] [Google Scholar]
- 3.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 4.Gottschalk S, Rooney CM, Heslop HE. Post-transplant lymphoproliferative disorders. Annu Rev Med. 2005;56:29–44. doi: 10.1146/annurev.med.56.082103.104727. [DOI] [PubMed] [Google Scholar]
- 5.Fan H, Nicholls J, Chua D, Chan KH, Sham J, Lee S, Gulley ML. Laboratory markers of tumor burden in nasopharyngeal carcinoma: a comparison of viral load and serologic tests for Epstein-Barr virus. Int J Cancer. 2004;112:1036–1041. doi: 10.1002/ijc.20520. [DOI] [PubMed] [Google Scholar]
- 6.Keegan TH, Glaser SL, Clarke CA, Gulley ML, Craig FE, Digiuseppe JA, Dorfman RF, Mann RB, Ambinder RF. Epstein-Barr virus as a marker of survival after Hodgkin's lymphoma: a population-based study. J Clin Oncol. 2005;23:7604–7613. doi: 10.1200/JCO.2005.02.6310. [DOI] [PubMed] [Google Scholar]
- 7.Okano M, Kawa K, Kimura H, Yachie A, Wakiguchi H, Maeda A, Imai S, Ohga S, Kanegane H, Tsuchiya S, Morio T, Mori M, Yokota S, Imashuku S. Proposed guidelines for diagnosing chronic active Epstein-Barr virus infection. Am J Hematol. 2005;80:64–69. doi: 10.1002/ajh.20398. [DOI] [PubMed] [Google Scholar]
- 8.Young LS, Murray PG. Epstein-Barr virus and oncogenesis: from latent genes to tumours. Oncogene. 2003;22:5108–5121. doi: 10.1038/sj.onc.1206556. [DOI] [PubMed] [Google Scholar]
- 9.Fafi-Kremer S, Morand P, Brion JP, Pavese P, Baccard M, Germi R, Genoulaz O, Nicod S, Jolivet M, Ruigrok RW, Stahl JP, Seigneurin JM. Long-term shedding of infectious Epstein-Barr virus after infectious mononucleosis. J Infect Dis. 2005;191:985–989. doi: 10.1086/428097. [DOI] [PubMed] [Google Scholar]
- 10.van Laar JA, Buysse CM, Vossen AC, Hjalmarsson B, van Den Berg B, van Lom K, Deinum J. Epstein-Barr viral load assessment in immunocompetent patients with fulminant infectious mononucleosis. Arch Intern Med. 2002;162:837–839. doi: 10.1001/archinte.162.7.837. [DOI] [PubMed] [Google Scholar]
- 11.Fafi-Kremer S, Brengel-Pesce K, Bargues G, Bourgeat MJ, Genoulaz O, Seigneurin JM, Morand P. Assessment of automated DNA extraction coupled with real-time PCR for measuring Epstein-Barr virus load in whole blood, peripheral mononuclear cells and plasma. J Clin Virol. 2004;30:157–164. doi: 10.1016/j.jcv.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Stevens SJ, Pronk I, Middeldorp JM. Toward standardization of Epstein-Barr virus DNA load monitoring: unfractionated whole blood as preferred clinical specimen. J Clin Microbiol. 2001;39:1211–1216. doi: 10.1128/JCM.39.4.1211-1216.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beuselinck K, van Ranst M, van Eldere J. Automated extraction of viral-pathogen RNA and DNA for high-throughput quantitative real-time PCR. J Clin Microbiol. 2005;43:5541–5546. doi: 10.1128/JCM.43.11.5541-5546.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brengel-Pesce K, Morand P, Schmuck A, Bourgeat MJ, Buisson M, Bargues G, Bouzid M, Seigneurin JM. Routine use of real-time quantitative PCR for laboratory diagnosis of Epstein-Barr virus infections. J Med Virol. 2002;66:360–369. doi: 10.1002/jmv.2153. [DOI] [PubMed] [Google Scholar]
- 15.Choquet S, Leblond V, Herbrecht R, Socie G, Stoppa AM, Vandenberghe P, Fischer A, Morschhauser F, Salles G, Feremans W, Vilmer E, Peraldi MN, Lang P, Lebranchu Y, Oksenhendler E, Garnier JL, Lamy T, Jaccard A, Ferrant A, Offner F, Hermine O, Moreau A, Fafi-Kremer S, Morand P, Chatenoud L, Berriot-Varoqueaux N, Bergougnoux L, Milpied N. Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood. 2006;107:3053–3057. doi: 10.1182/blood-2005-01-0377. [DOI] [PubMed] [Google Scholar]
- 16.Niesters HG, van Esser J, Fries E, Wolthers KC, Cornelissen J, Osterhaus AD. Development of a real-time quantitative assay for detection of Epstein-Barr virus. J Clin Microbiol. 2000;38:712–715. doi: 10.1128/jcm.38.2.712-715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stevens SJ, Verkuijlen SA, Brule AJ, Middeldorp JM. Comparison of quantitative competitive PCR with LightCycler-based PCR for measuring Epstein-Barr virus DNA load in clinical specimens. J Clin Microbiol. 2002;40:3986–3992. doi: 10.1128/JCM.40.11.3986-3992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gulley ML, Fan H, Elmore SH. Validation of Roche LightCycler Epstein-Barr virus quantification reagents in a clinical laboratory setting. J Mol Diagn. 2006;8:589–597. doi: 10.2353/jmoldx.2006.050152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill CE, Harris SB, Culler EE, Zimring JC, Nolte FS, Caliendo AM. Performance characteristics of two real-time PCR assays for the quantification of Epstein-Barr virus DNA. Am J Clin Pathol. 2006;125:665–671. doi: 10.1309/ABEY-V2VK-E6DH-XAAA. [DOI] [PubMed] [Google Scholar]
- 20.Kozic S, Vince A, Bes JI, Rode OD, Lepej SZ, Poljak M, Bozic M, Kessler HH. Evaluation of a commercial real-time PCR assay for quantitation of Epstein-Barr virus DNA in different groups of patients. J Virol Methods. 2006;135:263–268. doi: 10.1016/j.jviromet.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Ruiz G, Pena P, de Ory F, Echevarria JE. Comparison of commercial real-time PCR assays for quantification of Epstein-Barr virus DNA. J Clin Microbiol. 2005;43:2053–2057. doi: 10.1128/JCM.43.5.2053-2057.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonnet F, Jouvencel AC, Parrens M, Leon MJ, Cotto E, Garrigue I, Morlat P, Beylot J, Fleury H, Lafon ME. A longitudinal and prospective study of Epstein-Barr virus load in AIDS-related non-Hodgkin lymphoma. J Clin Virol. 2006;36:258–263. doi: 10.1016/j.jcv.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Wagner HJ, Wessel M, Jabs W, Smets F, Fischer L, Offner G, Bucsky P. Patients at risk for development of posttransplant lymphoproliferative disorder: plasma versus peripheral blood mononuclear cells as material for quantification of Epstein-Barr viral load by using real-time quantitative polymerase chain reaction. Transplantation. 2001;72:1012–1019. doi: 10.1097/00007890-200109270-00006. [DOI] [PubMed] [Google Scholar]
- 24.Hakim H, Gibson C, Pan J, Srivastava K, Gu Z, Bankowski MJ, Hayden RT. Comparison of various blood compartments and reporting units for the detection and quantification of Epstein-Barr virus (EBV) in peripheral blood. J Clin Microbiol. 2007;45:2151–2155. doi: 10.1128/JCM.02308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wada K, Kubota N, Ito Y, Yagasaki H, Kato K, Yoshikawa T, Ono Y, Ando H, Fujimoto Y, Kiuchi T, Kojima S, Nishiyama Y, Kimura H. Simultaneous quantification of Epstein-Barr virus, cytomegalovirus, and human herpesvirus 6 DNA in samples from transplant recipients by multiplex real-time PCR assay. J Clin Microbiol. 2007;45:1426–1432. doi: 10.1128/JCM.01515-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boom R, Sol CJ, Schuurman T, Van Breda A, Weel JF, Beld M, Ten Berge IJ, Wertheim-Van Dillen PM, De Jong MD. Human cytomegalovirus DNA in plasma and serum specimens of renal transplant recipients is highly fragmented. J Clin Microbiol. 2002;40:4105–4113. doi: 10.1128/JCM.40.11.4105-4113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]