

Abstract
Multiple osteochondromas (MO) is an autosomal-dominant skeletal disorder characterized by the formation of multiple cartilage-capped protuberances. MO is genetically heterogeneous and is associated with mutations in the EXT1 and EXT2 genes. In this study we describe extensive mutation screening in a set of 63 patients with clinical and radiographical diagnosis of MO. Denaturing high-performance liquid chromatography analysis revealed mutations in 43 patients. Additional deletion analysis by fluorescence in situ hybridization and a newly developed multiplex ligation-dependent probe amplification probe set identified one patient with an intragenic EXT1 translocation, three patients with a partial EXT1 deletion, and one patient with a partial EXT2 deletion. Thirty-six patients harbored an EXT1 mutation (57%), and 12 had an EXT2 mutation (19%). We show that our optimized denaturing high-performance liquid chromatography/sequencing/multiplex ligation-dependent probe amplification protocol represents a reliable and highly sensitive diagnostic strategy for mutation screening in MO patients. Clinical analysis showed no clear genotype-phenotype correlation in our cohort of MO patients.
Multiple osteochondromas (MO) is an autosomal-dominant skeletal disorder characterized by the formation of multiple cartilage-capped protuberances, or osteochondromas. Osteochondromas are the result of excessive chondrocyte proliferation and bone growth at the juxta-epiphyseal regions of long tubular bones.1 In theory, they can arise in every bone with an endochondral origin, but they mainly occur in distal femur, proximal humerus, and proximal tibia. The great variability in size and number of osteochondromas reflects the clinical heterogeneity and variable severity of MO.2 This disorder has an estimated prevalence of ∼1/50,000, making it one of the most frequent skeletal dysplasias.3 Osteochondromas are rarely present at birth, but in more than 80% of the patients they develop gradually during the first decade of life and increase in size until closure of the growth plates at the end of puberty.1
Although osteochondromas are benign, they can cause several secondary complications. By exerting pressure on neighboring tissues, osteochondromas cause pain, nerve compression, and disturbance of the blood circulation as a result of blood vessel compression. Additionally, complications of abnormal skeletal growth are observed in MO patients with shortening of the long bones, restricted range of joint movement, limb length inequalities, and short stature. Especially deformities of the forearm are characteristic. Surgery may be required to correct the most severe deformities. The most serious complication of MO, however, is malignant transformation of osteochondromas resulting in peripheral secondary chondrosarcomas, which occurs in 0.5 to 2% of cases.2,3,4
MO is genetically heterogeneous and is associated with mutations in the EXT15 and EXT2 genes.6,7 In ∼10 to 20% of the patients no EXT1 or EXT2 mutation can be detected. Both EXT genes belong to the larger EXT gene family, which also comprises three homologues EXT-like genes (EXTL1, EXTL2, and EXTL3). EXT1 and EXT2 are ubiquitously expressed and encode proteins that function as glycosyltransferases in the biosynthesis of heparan sulfate. Both proteins interact in the Golgi apparatus to form a hetero-oligomeric complex that catalyzes the transfer of N-acetyl-glucosamine (Glcnac) and d-glucuronic acid (GlcA) to the elongating heparan sulfate glycosaminoglycans chains.8,9
Both EXT1 and EXT2 are presumed to be tumor suppressor genes based on mutation and loss of heterozygosity studies. Indeed, loss of heterozygosity has been demonstrated in the EXT1 8q24 region in both sporadic and hereditary osteochondromas10,11,12,13 and chondrosarcomas.10,14 DNA copy number aberrations of this region have also been detected in non-MO related tumors such as colorectal carcinoma.15 Moreover, somatic homozygous EXT1 deletions have been found in nonhereditary osteochondromas.16 Loss of heterozygosity of the EXT2 11p11 region has been described previously in chondrosarcomas.14
In this study we describe extensive mutation screening in a large set of MO patients. To improve the identification of intragenic EXT1 and EXT2 deletions, a new multiplex ligation-dependent probe amplification (MLPA) probe set was designed and validated.
Materials and Methods
Patients
In this study we investigated patients from 63 families with MO. Diagnosis was based on the presence of MOs confirmed by radiographical examination (X-rays). Ethylenediaminetetraacetic acid or heparin blood samples were obtained from patients and available relatives for DNA mutation screening of the EXT1 and EXT2 genes. Genomic DNA was isolated from peripheral blood according to standard procedures. Eleven additional MO patients, in whom no point mutation was found in previous mutation screening studies, were also included for MLPA analysis.
DHPLC, Sequencing Analysis, and Fluorescence in Situ Hybridization (FISH) Analysis
Polymerase chain reaction (PCR) amplification of the EXT1 and EXT2 coding exons and DHPLC analysis on a WAVE-3500HT fragment analysis system (Transgenomic, Crewe, UK) were performed as previously described.17 If a fragment showed an aberrant chromatograph in DHPLC analysis it was PCR reamplified and the sequence was determined using ABI v1.1 chemistry with sequencing analysis on an ABI3130xl genetic analyzer (Applied Biosystems, Foster City, CA). DNA mutation and nucleotide numbering for EXT1 and EXT2 were based on the cDNA reference sequences (GenBank accession numbers NT_023811.12 and NT_009237.13, respectively) with base one corresponding to the first base of the initiation codon. FISH analysis was performed with EXT1 probes 46F10 and 65G55 and EXT2 probes A1151 and D0694.7
MLPA
For MLPA analysis a new MLPA probe set with 13 EXT1 and 16 EXT2 probes was developed (Table 1). All probes were designed in such a way that they are located in the EXT1 and EXT2 exon sequences. For each exon one probe was developed, with exception of the large EXT1 exons 1 and 11 for which two probes were designed. Also probes against EXT2 noncoding exons 1, 1a, and 1b were included. Additionally, 15 probes located outside the EXT1 and EXT2 regions were included as reference probes (Table 1). For these reference probes we selected regions that have not been reported to be implicated in osteochondroma or chondrosarcoma development. The MLPA reactions were basically performed as described by Schouten and colleagues18 with an annealing temperature for all exons of 60°C. Fragment data were quantitatively analyzed using capillary electrophoresis on an ABI3130xl genetic analyzer (Applied Biosystems). Thresholds for deletion and duplication were set at 0.80 and 1.35, respectively.
Table 1.
Overview of MLPA-P215 Probes
| Amplicon size (bp) | Upstream hybridizing sequence | Downstream hybridizing sequence | |
|---|---|---|---|
| EXT probes | |||
| EXT1-ex 1_1 | 292 | 5′-GGACACATGCAGGCCAAAAAACGCTA-3′ | 5′-TTTCATCCTGCTCTCAGCTGGCTCTTGTCTCGCCCTTTTGTTTTCATG-3′ |
| EXT1-ex 1_2 | 328 | 5′-CCAAAGCCAGCATCAGTACTGAAAACT-3′ | 5′-TCCGACCCAACTTTGATGTTTCTATTCCCCTCTTTTCTAAGGACATG-3′ |
| EXT1-ex 2 | 247 | 5′-GGAAATGCTGCACAATGCCACTT-3′ | 5′-TCTGTCTGGTTCCTCGTGGTCGCAGGCTTGGGTCCTTCAGACATG-3′ |
| EXT1-ex 3 | 310 | 5′-CTATGACGGCAGCTTGGTTCCAATTA-3′ | 5′-ATCACTTCAGAGAATGGCAACTCCCATCCATTGCTGAGCATCACCATG-3′ |
| EXT1-ex 4 | 382 | 5′-CCTAGCACTTAGACAGCAGACACAATTCT-3′ | 5′-TGTGGGAGGCTTATTTTTCTTCAGTTGAGAAGATTGTATTAACTACACATG-3′ |
| EXT1-ex 5 | 208 | 5′-CCTGGAGGATTGTTCGTACTACCACAGTAT-3′ | 5′-TCATCTTATCTGGGAGATTTTCCTTACTACTATGCTAATTTAGGTAAGTCATG-3′ |
| EXT1-ex 6 | 148 | 5′-GTGGTCTCTCAGTCCCAGCCAGTGT-3′ | 5′-TGAAGCTTCTCGTGGCTGCAGCCAAGTCCCAGTACTGTGCCATG-3′ |
| EXT1-ex 7 | 160 | 5′-GTTCTATGGAATTGTGACAAGCCCCT-3′ | 5′-ACCAGCCAAACACCGCTGGCCTGCCACTGCTGTGCCATG-3′ |
| EXT1-ex 8 | 178 | 5′-TGAGCAGCCGTTTTCTGCCCTACGACA-3′ | 5′-ACATCATCACAGACGCCGTGCTCAGCCTTGACGAGGACACCATG-3′ |
| EXT1-ex 9 | 214 | 5′-GAGGAGCGGTGGGGATACACATCAA-3′ | 5′-AGTGGACGAACGACTACTCCATGGTGTTGACAGGAGCTGCTATTTACCATG-3′ |
| EXT1-ex 10 | 355 | 5′-CAGCCTGAAGAACATGGTGGACCAAT-3′ | 5′-TGGCCAATTGTGAGGACATTCTCATGAACTTCCTGGTGTCTCATG-3′ |
| EXT1-ex 11_1 | 265 | 5′-TCAGCGACAGAGCTGCATGAATACGTT-3′ | 5′-TGCCAGCTGGTTTGGCTACATGCCGCTGATCCACTCTCAGACATG-3′ |
| EXT1-ex 11_2 | 346 | 5′-GCTGCTCTCTCTTCCCAGTGCAGA-3′ | 5′-TCCACTCATCAGCAGAGCCAGATTGTGCCAACTATCCCATG-3′ |
| EXT2-ex 1 | 154 | 5′-TGAGCGCGCCTGCCTGGGAAA-3′ | 5′-ACACTGCAGCGGTGCTCGGACTCCTCCTGTCCAGCAGCATG-3′ |
| EXT2-ex 1a | 220 | 5′-GAAGGGGGATGTCCTGCGCCTCAG-3′ | 5′-GGTCCGGTGGTGGCCTGCGGCATCCCTTGCGCATG-3′ |
| EXT2-ex 1b | 283 | 5′-GAGCTACTCAGAGTTGCTGTTTCTCCT-3′ | 5′-TGAGATGCTTTTGGTAAGTATATTTTAAAATAATTTTTCCATGTTATCTGAGCATG-3′ |
| EXT2-ex 2 | 247 | 5′-GGCCCCATTCTATCGAGTCCTCA-3′ | 5′-AATGACTGGAATGTAGAGAAGCGCAGCATCCGTGATGTGCCGGTCATG-3′ |
| EXT2-ex 3 | 184 | 5′-TGGGATCGAGGTACGAATCACCTGTTGTT-3′ | 5′-CAACATGTTGCCTGGAGGTCCCCCAGATTATAACACAGCCCCATG-3′ |
| EXT2-ex 4 | 337 | 5′-CTGTTGGCTGGTGGCGGCTTTTCT-3′ | 5′-ACGTGGACTTACCGGCAAGGCTACGATGTCAGCACATG-3′ |
| EXT2-ex 5 | 409 | 5′-CCAACCTCTCAGAGGGTGTCCTT-3′ | 5′-TCTGTCCGTAAGCGCTGCCACAAGCACCAGGTCTTCGATTACCCCATG-3′ |
| EXT2-ex 6 | 202 | 5′-TCTACTTTCTGTGTGGTTCTTCGTGGA-3′ | 5′-GCTCGGCTGGGCCAGGCAGTATTGAGCGATGTGTTACAAGCCATG-3′ |
| EXT2-ex 7 | 190 | 5′-CTGTTGTCTCCAGAGCATCTGTGGTT-3′ | 5′-GTACCAGAAGAAAAGATGTCAGATGTGTACAGTATTTTGCACATG-3′ |
| EXT2-ex 8 | 166 | 5′-TGCAGATTATCAATGACCGGATCTATCCAT-3′ | 5′-ATGCTGCCATCTCCTATGAAGAATGGAATGACCCTCCTGCTGTCATG-3′ |
| EXT2-ex 9 | 418 | 5′-CGCTGATCCCACCACAGTCTCAA-3′ | 5′-GGGTTCACCGCCATAGTCCTCACCTACGACCGAGTAGAGAGCCTCATG-3′ |
| EXT2-ex 10 | 364 | 5′-CTGGCCCAAAATCCGGGTTCCATTAA-3′ | 5′-AAGTTGTGAGGACTGCTGAAAACAAGTTAAGTAACCGTTTCTTCCCTCATG-3′ |
| EXT2-ex 11 | 136 | 5′-TGGCGGGAATTTCCTGACCGGTT-3′ | 5′-GGTGGGTTACCCGGGTCGTCTGCATCTCTGGGACCATGCATG-3′ |
| EXT2-ex 12 | 142 | 5′-TGGGTAGATGCTCATATGAACTGTGAAGA-3′ | 5′-TATTGCCATGAACTTCCTGGTGGCCAACGTCACGGGAAAAGCAGCATG-3′ |
| EXT2-ex 13 | 229 | 5′-GAAATTCAAGTGTCCTGAGTGCACAGCCAT-3′ | 5′-AGATGGGCTTTCACTAGACCAAACACACATGGTGGAGAGGCATG-3′ |
| EXT2-ex 14 | 436 | 5′-CAGAGTGCATCAACAAGTTTGCTTCAGTCT-3′ | 5′-TCGGGACCATGCCTCTCAAGGTGGTGGAACACCGAGCTGACATG-3′ |
| Reference probes | |||
| SLC7A1 | 130 | 5′-GAGACGCGGCTGTCTCGCTGCCTGAACACT-3′ | 5′-TTTGATCTGGTGGCCCTCGGGGTGGGCAGCA-3′ |
| VIPR2 | 172 | 5′-CGCATTCACCCAGAATGCCGATTTCAT-3′ | 5′-CTGGAAATACAGGAGGAAGAAACAAAATGTGCAGAGCTTCT-3′ |
| KIF21A | 196 | 5′-TCTCAGGTCTTCCTAGGGAAAGATAAGGCT-3′ | 5′-TTTACTTTTGACTATGTATTTGACATTGACTCCCAGCAAGAGC-3′ |
| KCNH2 | 238 | 5′-GGACCTGCTCACCGCCCTGTACT-3′ | 5′-TCATCTCCCGGGGCTCCATCGAGATCCTGCGG-3′ |
| IL1A | 256 | 5′-GATGCCTGAGATACCCAAAACCAT-3′ | 5′-CACAGGTAGTGAGACCAACCTCCTCTTCTTC-3′ |
| GJB6 | 301 | 5′-GGATTGGGGGACGCTGCACACTT-3′ | 5′-TCATCGGGGGTGTCAACAAACACTCCACCAGCATC-3′ |
| GBAS | 321 | 5′-GGTGTTGCCAAAGATTCACGAAGATA-3′ | 5′-AACACTACCCTTGTACTTTGGTGGGGACTTGGAACACGTGGT-3′ |
| LRRK2 | 373 | 5′-GCTGAACAATGTCCAGGAAGGAAAACAGAT-3′ | 5′-AGAAACGCTGGTCCAAATCCTGGAGGATCTGCTGGTG-3′ |
| NIPBL | 391 | 5′-GGTGATGATGATGAAATTCCTCAGGAACT-3′ | 5′-GCTCTTAGGAAAACATCAGCTTAATGAACTTGGCAGTGAATCTG-3′ |
| STCH | 441 | 5′-CCAGAAAATACTGGTACCCATTCAGCAAGTA-3′ | 5′-TTGAAAGAAGGCCACCTGGAAAAGACTGAGATTGATGAGGT-3′ |
| GNAS | 454 | 5′-CTGTGAACACCCCACGTGTCTTTCTTT-3′ | 5′-TTCTCCCAGCTTCCTGGACAAGATCGACGTGATC-3′ |
| PKP2 | 463 | 5′-CCGACATCAGTGGCTCAGACAGT-3′ | 5′-TGTCCAGAAGGAAAGTGGCCTGCAGCACACCC-3′ |
| MCCC1 | 472 | 5′-CCAGGAAGAAACAGTGACAGGCA-3′ | 5′-ACCAAGAGGGGGAGGATGGAGAAAACCCTTGCCTTAGTCAGAGCAGG-3′ |
| CASR | 481 | 5′-CCAGTGCCTGTAACAAGTGCCCAGATGACT-3′ | 5′-TCTGGTCCAATGAGAACCACACCTCCTGCATTGCCAAGGA-3′ |
RNA Analysis
RNA was isolated from EBV lymphoblastoid cell lines using QIAamp RNA blood mini kit (Qiagen, Hilden, Germany). cDNA was subsequently prepared with SuperScript III first-strand synthesis system for RT-PCR (Invitrogen, Carlsbad, CA).
Long-Range PCR
Long-range PCR for the characterization of the EXT2 exon 2 deletion in family 150 was performed with primers 5′-TCAGAGTTGCTGTTTCTCCTTGAG-3′ and 5′-AACCCATCATAAGGACAGCCC-3′ located in exon 1b and exon 3, respectively.
Results
DHPLC, Sequencing Analysis, and FISH Analysis
Genomic DNA from 63 families, clinically diagnosed with MO, was analyzed for mutations in the EXT1 and EXT2 genes by DHPLC analysis of all coding exons from both genes. This point mutation analysis resulted in the identification of a mutation in 43 families (Table 2). In total, 32 families harbored an EXT1 mutation and in 11 families an EXT2 mutation was identified. Additionally, FISH analysis revealed a partial deletion of EXT1 with probe 46F10 deleted in two families and a translocation between chromosomes 4 and 8 involving EXT1 in one family. No mutation was detected in 17 families.
Table 2.
Overview of Families in Which an EXT1 or EXT2 Mutation Was Identified
| Gene | Exon/intron | Mutation at DNA level | Protein change | Mutation type | First reference | Detection technique | Family number |
|---|---|---|---|---|---|---|---|
| EXT1 | |||||||
| Exon 1 | c.247dupC | Frame shift | This study | DHPLC | 155 | ||
| Exon 1 | c.538-539delAG | Frame shift | This study | DHPLC | 183 | ||
| Exon 1 | c.560-562delGTAinsC | Frame shift | This study | DHPLC | 188 | ||
| Exon 1 | c.644-664del | Frame shift | This study | DHPLC | 179 | ||
| Exon 1 | c.786C>G | p.Y262X | Nonsense | This study | DHPLC | 158 | |
| Exon 1 | c.793delG | Frame shift | This study | DHPLC | 164 | ||
| Intron 1 | c.962-2G>A | Splice site | This study | DHPLC | 199 | ||
| Exon 2 | c.992C>A | p.A331D | Missense | This study | DHPLC | 204 | |
| Exon 2 | c.992C>A | p.A331D | Missense | This study | DHPLC | 220 | |
| Exon 2 | c.1019G>T | p.R340L | Missense | 20 | DHPLC | 166 | |
| Exon 2 | c.1019G>T | p.R340L | Missense | 20 | DHPLC | 171 | |
| Exon 2 | c.1019G>T | p.R340L | Missense | 20 | DHPLC | 197 | |
| Intron 2 | c.1056 + 1G>A | Splice site | 21 | DHPLC | 187 | ||
| Intron 4 | c.1284 + 1G>T | Splice site | 22 | DHPLC | 145 | ||
| Exon 5 | c.1362dupA | Frame shift | This study | DHPLC | 144 | ||
| Exon 5 | c.1374T>G | p.Y458X | Nonsense | This study | DHPLC | 207 | |
| Intron 5 | c.1417 + 1G>A | Splice site | 23 | DHPLC | 181 | ||
| Exon 6 | c.1431delC | Frame shift | This study | DHPLC | 161 | ||
| Exon 6 | c.1468dupC | Frame shift | This study | DHPLC | 184 | ||
| Exon 6 | c.1469delT | Frame shift | 5 | DHPLC | 160 | ||
| Exon 6 | c.1469delT | Frame shift | 5 | DHPLC | 180 | ||
| Exon 6 | c.1477C>T | p.Q493X | Nonsense | This study | DHPLC | 176 | |
| Intron 6 | c.1536-1G>C | Splice site | This study | DHPLC | 165 | ||
| Exon 7 | c.1557T>A | p.C519X | Nonsense | This study | DHPLC | 174 | |
| Intron 7 | c.1632-2A>G | Splice site | 22 | DHPLC | 146 | ||
| Exon 8 | c.1659C>A | p.Y553X | Nonsense | This study | DHPLC | 186 | |
| Exon 8 | c.1685T>C | p.L562P | Missense | This study | DHPLC | 191 | |
| Exon 8 | c.1685T>C | p.L562P | Missense | This study | DHPLC | 178 | |
| Exon 8 | Deletion exon 8 | Deletion | This study | MLPA | 200 | ||
| Intron 8 | c.1722 + 1G>T | Splice site | This study | DHPLC | 152 | ||
| Intron 8 | c.1722-2A>C | Splice site | This study | DHPLC | 159 | ||
| Exon 9 | c.1810G>T | p.E604X | Nonsense | This study | DHPLC | 194 | |
| Exon 10 | c.2051delG | Frame shift | This study | DHPLC | 182 | ||
| Deletion exon 2-11 | Deletion | This study | FISH/MLPA | 147 | |||
| Deletion exon 2-11 | Deletion | This study | FISH/MLPA | 163 | |||
| t(4;8)(q25;q24.1) | Translocation | This study | FISH | 156 | |||
| Exon 3 | c.1065C>T | Polymorphism | |||||
| Exon 6 | c.1431C>T | Polymorphism | |||||
| Exon 9 | c.1761G>A | Polymorphism | |||||
| EXT2 | |||||||
| Exon 2 | c.178A>T | p.K60X | Nonsense | This study | DHPLC | 170 | |
| Exon 2 | c.269-271GTT>AAA | p.C90X | Nonsense | This study | DHPLC | 193 | |
| Exon 2 | c.451-461dup | Frame shift | This study | DHPLC | 185 | ||
| Exon 2 | c.484delC | Frame shift | This study | DHPLC | 177 | ||
| Exon 2 | Deletion EXT2 exon 2 | Deletion | This study | MLPA | 150 | ||
| Exon 4 | c.668G>C | p.R223P | Missense | 24 | DHPLC | 202 | |
| Intron 4 | c.743 + 1G>C | Splice site | This study | DHPLC | 169 | ||
| Exon 7 | c.1139dupT | Frame shift | This study | DHPLC | 172 | ||
| Exon 7 | c.1151-1154delAGAT | Frame shift | This study | DHPLC | 203 | ||
| Intron 7 | c.1173 + 1G>T | Splice site | 25 | DHPLC | 151 | ||
| Exon 8 | c.1191delA | Frame shift | This study | DHPLC | 175 | ||
| Exon 8 | c.1250G>C | p.R417P | Missense | This study | DHPLC | 198 | |
| Intron 6 | c.1080-18T>A | Polymorphism | |||||
| Intron 7 | c.1174-18G>T | Polymorphism | |||||
| Intron 11 | c.1807-51C>T | Polymorphism | |||||
MLPA Assay
To create a new MLPA assay, 13 EXT1 and 16 EXT2 MLPA probes covering all exons of EXT1 and EXT2, respectively, were designed, and 15 reference probes were included. This MLPA probe set was validated using 10 EXT-negative (Figure 1) and 22 EXT-positive samples, consisting of 13 positive controls for mutations in EXT1 and 9 for EXT2 (Table 3). All control patients harbored a (partial) EXT1 or EXT2 deletion or duplication. The mutations in these positive control samples were previously identified using FISH with intragenic EXT1 and EXT2 probes, two-color MLPA,19 real-time PCR or DHPLC, and sequencing analysis. No false-positive results were obtained for EXT-negative samples, and all mutations in EXT1- or EXT2-positive control samples were identified accurately. The MLPA probe set will be referred to as MLPA-P215 hereafter.
Figure 1.
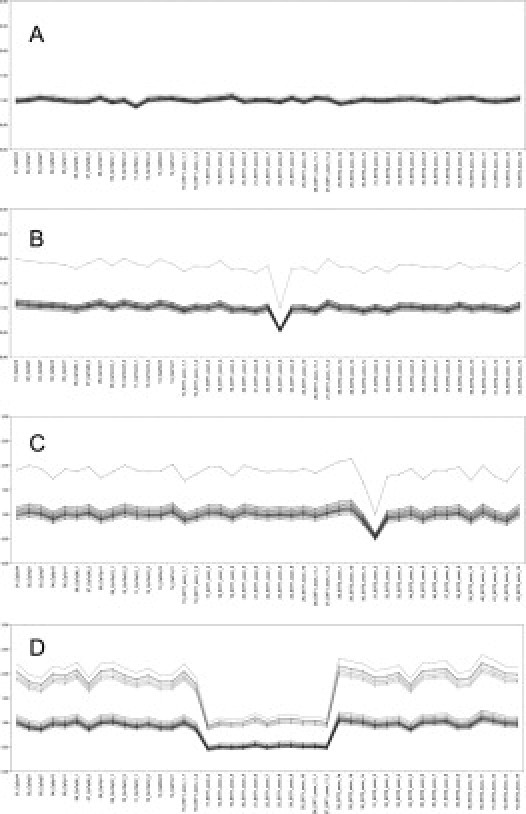
Example of the MLPA curve of EXT deletion-negative sample (A), EXT1 exon 8 deletion (family number 200) (B), EXT2 exon 2 deletion (family number 150) (C), and EXT1 exon 2-11 deletion (family number 147) (D). Lines represent the series of dosage coefficients (DQ) for each probe (DQamp n/amp n+ 1 = [sample (peak area amplicon n/peak area amplicon n + 1)]/[control (peak area amplicon n/peak area amplicon n + 1)].
Table 3.
Positive Control Samples Used for the Validation of MLPA-P215
| Family number | Mutation at DNA level | Detection technique | Reference |
|---|---|---|---|
| 87 | Deletion EXT1 | FISH | Unpublished results |
| Italy_3 | Deletion EXT1 | Real-time PCR | Unpublished results |
| 99 | Deletion EXT1, exon 1 | FISH | Unpublished results |
| 132 | Deletion EXT1, exon 1 | 2-color MLPA | Unpublished results |
| Italy_6 | Deletion EXT1, exon 1 | Real-time PCR | Unpublished results |
| Italy_5 | Deletion EXT1, exon 8 | DHPLC and sequencing analysis | Unpublished results |
| Italy_7 | Deletion EXT1, exon 8 | DHPLC and sequencing analysis | Unpublished results |
| Italy_4 | Deletion EXT1, exon 2-5 | Real-time PCR | Unpublished results |
| 89 | Deletion EXT1, exon 2-11 | FISH | Unpublished results |
| 128 | Deletion EXT1, exon 2-11 | 2-color MLPA | Unpublished results |
| 147 | Deletion EXT1, exon 2-11 | FISH | Unpublished results |
| Italy_1 | Duplication EXT1, exon 4 | Real-time PCR | Unpublished results |
| Italy_2 | Duplication EXT1, exon 4 | Real-time PCR | Unpublished results |
| 8 | Deletion EXT2 | FISH | 26 |
| Defect 11_1 | Deletion EXT2 | FISH | 27 |
| Defect 11_2 | Deletion EXT2 | FISH | 27 |
| Defect 11_3 | Deletion EXT2 | FISH | 27 |
| Defect 11_4 | Deletion EXT2 | FISH | 27 |
| Italy_8 | Deletion EXT2 | Real-time PCR | Unpublished results |
| Italy_9 | Deletion EXT2 exon 1-5 | Real-time PCR | Unpublished results |
| Italy_10 | Deletion EXT2 exon 1-5 | Real-time PCR | Unpublished results |
| 125 | Deletion EXT2 exon 1-10 | 2-color MLPA | Unpublished results |
After validation, MLPA-P215 was used to screen 17 samples from the set of 63 families that failed to show a variant chromatograph profile with DHPLC analysis and in which no genetic aberration could be found using FISH, as well as 11 samples from former mutation screenings failing to identify a pathogenic mutation. In the set of 17 families, family 200 harbored an EXT1 exon 8 deletion, and in family 150 an EXT2 exon 2 deletion was found (Figure 1, Table 2). The presence of the EXT1 exon 8 deletion was confirmed by RNA analysis showing an aberrant mRNA lacking the complete exon 8 in the proband of this family. The EXT2 exon 2 deletion was confirmed by long-range PCR with primers located in the exon 2 flanking exons. All affected patients in this family showed a secondary amplification product of ∼3 kb, in addition to the expected 5.6-kb PCR amplification product. Sequencing analysis revealed that the deletion actually comprises 2748 bp, with the deletion breakpoints located 1470 bp upstream of the ATG start codon in exon 2 and 742 bp downstream of exon 2. In the set of 11 families from the former screening, one patient with a deletion from EXT1 exon 2-11 was identified, as well as one patient with an EXT2 exon 8 deletion. The presence of the latter single exon deletion was confirmed by RNA analysis showing an aberrant EXT2 mRNA lacking only the complete exon 8 sequence.
Clinical Analysis
Detailed clinical data were available for 28 of the EXT1 and 6 of the EXT2 patients, for 5 patients without a mutation identified as well as for 6 relatives from EXT1 patients, in whom the same mutation was identified as in the index patient of their family. The number of affected sites was found to be more than 10 in 73% of the EXT1 patients, 80% of the patients with an EXT2 mutation, and 75% in the mutation-negative patients. The average number of sites affected was 6.8 sites in EXT1 patients, 9.0 for EXT2 patients, and 7.4 in mutation-negative patients. Deformities were present in 80% of both EXT1 and EXT2 patients and in all mutation-negative patients. Twenty-seven percent of the EXT1 patients reported to have secondary complications, whereas in EXT2 patients this group accounted for 67%. No complications were reported in the mutation-negative group. Stature evaluation showed that 85% of the EXT1 patients fell below the 50th percentile with 61% of these smaller than the 10th percentile. Eighty percent of the EXT2 patients were below the 50th percentile, with 75% below the 10th percentile. For the mutation-negative group, stature data were available for only two patients, with both below the 50th percentile and one of them below the 10th percentile.
Discussion
In the past, mutation screening of MO patients was based on direct sequencing or less-sensitive screening methods such as single-strand conformation polymorphism. To facilitate and reduce the cost of this mutation screening, optimization of a DHPLC-based protocol for all EXT1 and EXT2 coding exons has been described.17,20 After all, most MO patients have point mutations or small deletions or insertions of a few bp in one of both genes and the EXT1 and EXT2 coding regions contain very little polymorphisms, making them very suitable for DHPLC analysis. However, because mutation screening was performed almost exclusively at the sequence level, quantitative (deletions and duplications) and positional (inversions and translocations) changes were not detected by this technique. To complement DHPLC screening, MLPA and/or FISH16 and RNA analysis can be performed. MLPA is a quick and simple technique for quantitative analysis. It is based on the ligation of two probes that hybridize to adjacent sites of the target sequence. All ligated probes have common end sequences, permitting simultaneous PCR amplification of all target sequences using only one primer pair. The resulting PCR products can be separated according to size and be quantified.18 In this study, a new MLPA assay (MLPA-P215) was validated and used for screening of MO patients that did not show an aberrant DHPLC profile. We found this new MLPA probe set to produce more reliable and reproducible results compared to a previously developed two-color MLPA for the EXT1 and EXT2 genes.19 This latter MLPA is composed of chemically synthesized oligonucleotide probes that are restricted in length. As a consequence, the various probes differ only minimally in size, complicating the analysis. MLPA-P215, however, was designed with cloned probes, allowing the construction of larger probes and more size difference between the individual probes, resulting in improved results. When MLPA-P215 was validated using 10 negative and 22 positive control samples, no false-positive results were obtained for negative control samples, and all mutations in positive control samples were sensitively identified. These observations lead to a theoretical sensitivity and specificity of both 100% in our validation.
Mutation screening for alterations in EXT1 and EXT2 was performed on a set of 63 MO families, resulting in the identification of 48 disease-causing mutations (Figure 2). Forty-three mutations were identified using DHPLC (90%); four deletions and one translocation were detected with FISH/MLPA (10%). No disease-causing genetic alteration was detected in 15 families (24%). This gave an overall mutation detection rate of 76% in 63 families, in accordance with previous studies.21,22,23 Thirty-six families (57%) harbored an EXT1 mutation, whereas only 12 families (19%) had an EXT2 mutation, giving a mutation frequency ratio of 75% for EXT1 versus 25% for EXT2. The observed higher frequency of EXT1 mutations is in agreement with previous mutation studies performed in western populations.20,21,24,25 For EXT1 we found a frame shift in 11 families, a nonsense mutation in 6 families, and a splice site mutation in 8 families. In seven families a missense mutation was found. Next to these small mutations, three deletions were identified as well as one translocation. Frameshift mutation c.1469delT was found in two families as well as missense mutations c.992C>A (p.A331D) and c.1685T>C (p.L562P). Three families were diagnosed with the missense mutation c.1019G>T (p.R340L). The c.992C>A (p.A331D) and c.1685T>C (p.L562P) mutations have not been described before but there are several arguments in favor of a pathogenic effect. They were found both absent in our control population (>100 chromosomes). Alanine 331 is conserved between the EXT1-EXT2-EXTL1 proteins and between several species (human-mouse-Xenopus). Theoretical prediction programs SIFT and Polyphen predict this variant to be pathogenic. Unfortunately only the proband of families 204 and 220 were available for analysis. Also leucine 562 is highly conserved in all members of the EXT/EXTL protein family and between human and Drosophila. SIFT and Polyphen also predict the c.1685T>C (p.L562P) variant to be pathogenic. In family 178 both proband and affected father showed the c.1685T>C (p.L562P), which was absent in the nonaffected sibling. In family 191 both parents of the proband patient were reported unaffected, but the c.1685T>C (p.L562P) was also found in the father of the patient. Detailed clinical examination needs to be performed for further clarification and evaluation. The c.1019G>T (p.R340L) missense mutation has previously been shown to result in impaired EXT1 function.26 In the EXT2 families five frame shifts, two nonsense, two splice site, and two missense mutations were found. In one family an EXT2 exon 2 deletion was present. The two missense mutations identified were c.668G>C (p.R223P, family 202) and c.1250G>C (p.R417P, family 198). Both were absent in our control population (>100 chromosomes) and are conserved between EXT1 and EXT2 and between several species from human to Drosophila. SIFT and Polyphen predict both variants to be pathogenic. c.668G>C (p.R223P) has been described before in MO patients27 and was also found to segregate with MO in family 202 (two affected and three unaffected individuals). For family 198, only the proband was available for analysis. In total, ∼70% of all identified genetic aberrations were truncating mutations, whereas missense mutations represented ∼20% and deletions and translocations were responsible for MO in ∼10% of cases. Additionally, our finding of four deletions in a cohort of 63 families (6%) confirmed the suggestion of White and colleagues19 that in a series of MO patients one can expect to find a deletion of one or more exons in ∼5 to 8% of cases. We were able to characterize the deletion breakpoint in detail for one family harboring an EXT2 exon 2 deletion. Analysis of the deletion breakpoint regions did not show the presence of low copy repeats nor did these two regions share homology. The mechanism behind the deletion event therefore remains unknown.
Figure 2.
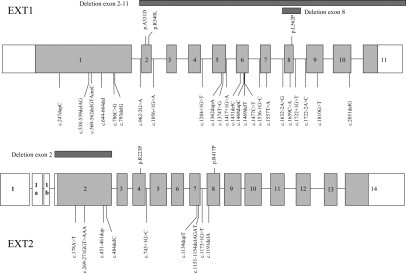
Distribution of mutations detected in the EXT1 gene (top) and EXT2 gene (bottom) in this study.
Of the 48 mutations identified, 34 mutations (71%) were found only once in our cohort of MO families and had never been reported before. The identification of several novel private mutations confirms the strong allelic heterogeneity of the EXT1 and EXT2 genes in MO patients. In addition to the private mutations, we identified some mutations located in previously reported mutation hot spots that might represent functional sites in the EXT1 and EXT2 genes. Mutation c.668G>C (p.R223P), found in one family, is located in the EXT2 region between amino acids 211 and 230, which has been reported to be a MO mutation hot spot.23,27,28,29 Two families were diagnosed with deletion 1469delT in exon 6 from EXT1, which was already mentioned to be located in a mutation hot spot by Francannet and colleagues.25 Finally, missense mutation c.1019G>T (p.R340L) in EXT1 exon 2 was found in three families and is known to be a recurrent missense mutation in a region that harbors key elements for EXT1 function.8,23,26,28,30
In 24% of cases no mutation could be identified with the performed multistep mutation screening. Several plausible reasons exist as to why disease-causing mutations could not be detected, including mutations that may occur in introns, regulatory regions, or promoter regions of EXT1 and EXT2. A recent study however, suggests that the latter is not a frequent cause of MO because no promoter mutations were found in any of the EXT1- and EXT2-negative MO patients of a large British-Caucasian cohort.21 Also, the existence of causative mutations located in another MO-causing gene cannot be excluded. To explain the relatively high percentage of patients without an identified mutation, one has to consider that it is also uncertain whether in all cases the phenotype of the patients fully matches the clinical picture of MO. MO can for example be confused with other skeletal disorders affecting multiple bones, such as metachondromatosis.22 Multidisciplinary re-evaluation of the radiographical and histological material by bone tumor experts is advised in these cases to exclude misdiagnosis. Interestingly, for the majority of mutation-negative patients (11 of the 15) there was no familial history reported. It is possible that these patients harbor MO causing somatic mutations, which cannot be detected in peripheral blood.
To study the genotype-phenotype correlation, the phenotype was reviewed for all 63 families in whom a mutation was identified. Detailed clinical descriptions were available for 34 EXT1, 6 EXT2 patients, and 5 mutation-negative patients. Previous studies suggested a more severe phenotype associated with EXT1 mutations,17,25,31 although this could not always be confirmed.32 We also could not confirm this observation in our cohort with none of the differences in evaluated parameters (number of osteochondromas, P = 1.000; deformities, P = 1.000; presence of secondary complications, P = 0.149; and stature ≤ P10, P = 1.000) being statistically significant. Also when making an evaluation between EXT1 patients, EXT2 patients and patients without an identified mutation, no significant differences could be found for the same parameters (number of osteochondromas,; P = 1.000; deformities, P = 0.813; presence of secondary complications, P = 0.067; and stature ≤ 10th percentile, P = 1.000). Possibly, the population of especially EXT2 patients with a clinical overview was too small to make relevant conclusions about the genotype-phenotype correlation. The same parameters were evaluated for patients with truncating mutations versus patients with missense mutations. No significant difference could be identified (number of osteochondromas, P = 1.000; deformities, P = 0.068; presence of secondary complications, P = 0197; and stature ≤ P10, P = 1.000). Most of the patients in our study were index patients, and only a small number of relatives were included in the clinical study. This may have created a bias toward more severely affected patients in all groups because they tend to go for genetic testing more rapidly. We are aiming to expand our clinical data by including more relatives and pooling larger cohorts of patients to evaluate this further. In conclusion we provided a sensitive molecular screening strategy with improved deletion analysis for the EXT1 and EXT2 genes applicable for MO mutation analysis.
Footnotes
Supported by the Multiple Hereditary Exostoses Coalition/Multiple Hereditary Exostoses Research Foundation (grant to W.W.).
The Department of Medical Genetics of Antwerp, MRC-Holland, and the Rizzoli Orthopaedic Institute of Bologna are partners of the EuroBoNeT consortium, a European Commission granted Network of Excellence for studying the pathology and genetics of bone tumors.
References
- 1.Solomon L. Hereditary multiple exostosis. J Bone Joint Surg Br. 1963;45:292–304. [Google Scholar]
- 2.Hennekam RCM. Hereditary multiple exostoses. J Med Genet. 1991;28:262–266. doi: 10.1136/jmg.28.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmale GA, Conrad EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg Br. 1994;76:986–992. doi: 10.2106/00004623-199407000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Wicklund CL, Pauli RM, Johnston D, Hecht JT. Natural history study of hereditary multiple exostoses. Am J Med Genet. 1995;55:43–46. doi: 10.1002/ajmg.1320550113. [DOI] [PubMed] [Google Scholar]
- 5.Ahn J, Lüdecke H, Lindow S, Horton WA, Lee B, Wagner MJ, Horsthemke B, Wells DE. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1) Nat Genet. 1995;11:137–143. doi: 10.1038/ng1095-137. [DOI] [PubMed] [Google Scholar]
- 6.Stickens D, Clines G, Burbee D, Ramos P, Thomas S, Hogue D, Hecht JT, Lovett M, Evans GA. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat Genet. 1996;14:25–32. doi: 10.1038/ng0996-25. [DOI] [PubMed] [Google Scholar]
- 7.Wuyts W, Van Hul W, Wauters J, Nemtsova M, Reyniers E, Van Hul E, De Boulle K, de Vries BBA, Hendrickx J, Herrygers I, Bossuyt P, Balemans W, Fransen E, Vits L, Coucke P, Nowak NJ, Shows TB, Mallet L, van den Ouweland AMW, McGaughran J, Halley DJJ, Willems PJ. Positional cloning of a gene involved in hereditary multiple exostoses. Hum Mol Genet. 1996;5:1547–1557. doi: 10.1093/hmg/5.10.1547. [DOI] [PubMed] [Google Scholar]
- 8.McCormick C, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, Tufaro F. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat Genet. 1998;19:158–161. doi: 10.1038/514. [DOI] [PubMed] [Google Scholar]
- 9.Lind T, Tufaro F, McCormick C, Lindahl U, Lidholt K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J Biol Chem. 1998;273:26265–26268. doi: 10.1074/jbc.273.41.26265. [DOI] [PubMed] [Google Scholar]
- 10.Hecht JT, Hogue D, Strong LC, Hansen MF, Blanton SH, Wagner M. Hereditary multiple exostosis and chondrosarcoma: linkage to chromosome 11 and loss of heterozygosity for EXT-linked markers on chromosomes 11 and 8. Am J Hum Genet. 1995;56:1125–1131. [PMC free article] [PubMed] [Google Scholar]
- 11.Bridge JA, Nelson M, Orndal C, Bhatia P, Neff JR. Clonal karyotypic abnormalities of the hereditary multiple exostoses chromosomal loci 8q24.1 (EXT1) and 11p11-12 (EXT2) in patients with sporadic and hereditary osteochondromas. Cancer. 1998;82:1657–1663. doi: 10.1002/(sici)1097-0142(19980501)82:9<1657::aid-cncr10>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 12.Bovée J, Cleton-Jansen A, Wuyts W, Caethoven G, Taminiau A, Bakker E, Van Hul W, Cornelisse P, Hogendoorn P. EXT mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am J Hum Genet. 1999;65:689–698. doi: 10.1086/302532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feely MG, Boehm AK, Bridge RS, Krallman PA, Neff JR, Nelson M, Bridge JA. Cytogenetic and molecular cytogenetic evidence of recurrent 8q24.1 loss in osteochondroma. Cancer Genet Cytogenet. 2002;137:102–107. doi: 10.1016/s0165-4608(02)00557-5. [DOI] [PubMed] [Google Scholar]
- 14.Raskind WH, Conrad EU, Chansky H, Matsushita M. Loss of heterozygosity in chondrosarcomas for markers linked to hereditary multiple exostoses loci on chromosomes 8 and 11. Am J Hum Genet. 1995;56:1132–1139. [PMC free article] [PubMed] [Google Scholar]
- 15.Lassmann S, Weis R, Makowiec F, Roth J, Danciu M, Hopt U, Werner M. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med. 2007;85:289–300. doi: 10.1007/s00109-006-0126-5. [DOI] [PubMed] [Google Scholar]
- 16.Hameetman L, Szuhai K, Yavas A, Knijnenburg J, van Duin M, van Dekken H, Taminiau AH, Cleton-Jansen AM, Bovee JV, Hogendoorn PC. The role of EXT1 in nonhereditary osteochondroma: identification of homozygous deletions. J Natl Cancer Inst. 2007;99:396–406. doi: 10.1093/jnci/djk067. [DOI] [PubMed] [Google Scholar]
- 17.Wuyts W, Radersma R, Storm K, Vits L. An optimized DHPLC protocol for molecular testing of the EXT1 and EXT2 genes in hereditary multiple osteochondromas. Clin Genet. 2005;68:542–547. doi: 10.1111/j.1399-0004.2005.00538.x. [DOI] [PubMed] [Google Scholar]
- 18.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White SJ, Vink GR, Kriek M, Wuyts W, Schouten J, Bakker B, Breuning MH, den Dunnen JT. Two-color multiplex ligation-dependent probe amplification: detecting genomic rearrangements in hereditary multiple exostoses. Hum Mutat. 2004;24:86–92. doi: 10.1002/humu.20054. [DOI] [PubMed] [Google Scholar]
- 20.Pedrini E, De Luca A, Valente EM, Maini V, Capponcelli S, Mordenti M, Mingarelli R, Sangiorgi L, Dallapiccola B. Novel EXT1 and EXT2 mutations identified by DHPLC in Italian patients with multiple osteochondromas. Hum Mutat. 2005;26:280. doi: 10.1002/humu.9359. [DOI] [PubMed] [Google Scholar]
- 21.Lonie L, Porter DE, Fraser M, Cole T, Wise C, Yates L, Wakeling E, Blair E, Morava E, Monaco AP, Ragoussis J. Determination of the mutation spectrum of the EXT1/EXT2 genes in British Caucasian patients with multiple osteochondromas, and exclusion of six candidate genes in EXT negative cases. Hum Mutat. 2006;27:1160. doi: 10.1002/humu.9467. [DOI] [PubMed] [Google Scholar]
- 22.Vink GR, White SJ, Gabelic S, Hogendoorn PCW, Breuning MH, Bakker E. Mutation screening of EXT1 and EXT2 by direct sequence analysis and MLPA in patients with multiple osteochondromas: splice site mutations and exonic deletions account for more than half of the mutations. Eur J Hum Genet. 2005;13:470–474. doi: 10.1038/sj.ejhg.5201343. [DOI] [PubMed] [Google Scholar]
- 23.Wuyts W, Van Hul W, De Boulle K, Hendrickx J, Bakker E, Vanhoenacker F, Mollica F, Lndecke HJ, Sayli BS, Pazzaglia UE, Mortier G, Hamel B, Conrad EU, Matsushita M, Raskind WH, Willems PJ. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am J Hum Genet. 1998;62:346–354. doi: 10.1086/301726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wuyts W, Van Hul W. Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum Mutat. 2000;15:220–227. doi: 10.1002/(SICI)1098-1004(200003)15:3<220::AID-HUMU2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 25.Francannet C, Cohen-Tanugi A, Le MM, Munnich A, Bonaventure J, Legeai-Mallet L. Genotype-phenotype correlation in hereditary multiple exostoses. J Med Genet. 2001;38:430–434. doi: 10.1136/jmg.38.7.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung UI, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest. 2001;107:295–304. doi: 10.1172/JCI11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi YR, Wu JY, Hsu YA, Lee CC, Tsai CH, Tsai FJ. Mutation screening of the EXT genes in patients with hereditary multiple exostoses in Taiwan. Genet Test. 2002;6:237–243. doi: 10.1089/109065702761403441. [DOI] [PubMed] [Google Scholar]
- 28.Philippe C, Porter DE, Emerton ME, Wells DE, Hamish A, Simpson RW, Monaco AP. Mutation screening of EXT1 and EXT2 genes in patients with hereditary multiple exostoses. Am J Hum Genet. 1997;61:520–528. doi: 10.1086/515505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Xia J, Jiang H, Zhou J, Li H, Wang D, Pan Q, Long Z, Fan C, Deng HX. Mutation analysis of hereditary multiple exostoses in the Chinese. Hum Genet. 1999;105:45–50. doi: 10.1007/s004399900058. [DOI] [PubMed] [Google Scholar]
- 30.Raskind WH, Conrad EU, Matsushita M, Wijsman EM, Wells DE, Chapman N, Sandell LJ, Wagner M, Houck J. Evaluation of locus heterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum Mutat. 1998;11:231–239. doi: 10.1002/(SICI)1098-1004(1998)11:3<231::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 31.Porter DE, Lonie L, Fraser M, Dobson-Stone C, Porter JR, Monaco AP, Simpson AH. Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study. J Bone Joint Surg Br. 2004;86:1041–1046. doi: 10.1302/0301-620x.86b7.14815. [DOI] [PubMed] [Google Scholar]
- 32.Signori E, Massi E, Matera MG, Poscente M, Gravina C, Falcone G, Rosa MA, Rinaldi M, Wuyts W, Seripa D, Dallapiccola B, Fazio VM. A combined analytical approach reveals novel EXT1/2 gene mutations in a large cohort of Italian multiple osteochondromas patients. Genes Chromosom Cancer. 2007;46:470–477. doi: 10.1002/gcc.20429. [DOI] [PubMed] [Google Scholar]
Uncited references
- 33.Hecht JT, Hogue D, Wang Y, Blanton SH, Wagner M, Strong LC, Raskind W, Hansen MF, Wells D. Hereditary multiple exostoses (EXT): mutational studies of familial EXT1 cases and EXT-associated malignancies. Am J Hum Genet. 1997;60:80–86. [PMC free article] [PubMed] [Google Scholar]
- 34.Wells DE, Hill A, Lin X, Ahn J, Brown N, Wagner MJ. Identification of novel mutations in the human EXT1 tumor suppressor gene. Hum Genet. 1997;99:612–615. doi: 10.1007/s004390050415. [DOI] [PubMed] [Google Scholar]
- 35.Bartsch O, Wuyts W, Van Hul W, Hecht JT, Meinecke P, Hogue D, Werner W, Zabel B, Hinkel GK, Powell CM, Shaffer LG, Willems PJ. Delineation of a contiguous gene syndrome with multiple exostoses, enlarged parietal foramina, craniofacial dysostosis and mental retardation, caused by deletions on the short arm of chromosome 11. Am J Hum Genet. 1996;58:734–742. [PMC free article] [PubMed] [Google Scholar]
- 36.Wuyts W, Di Gennaro G, Bianco F, Wauters J, Morocutti C, Pierelli F, Bossuyt P, Van Hul W, Casali C. Molecular and clinical examination of an Italian DEFECT 11 family. Eur J Hum Genet. 1999;7:579–584. doi: 10.1038/sj.ejhg.5200339. [DOI] [PubMed] [Google Scholar]