

Abstract
Inhibition of aminopeptidase N and neutral endopeptidase-24.11, two zinc metallopeptidases involved in the inactivation of the opioid peptides enkephalins, produces potent physiological analgesic responses, without major side-effects, in all animal models of pain in which morphine is active. Dual inhibitors of both enzymes could fill the gap between opioid analgesics and antalgics. Until now, attempts to find a compound with high affinity both for neutral endopeptidase and aminopeptidase N have failed. We report here the design of dual competitive inhibitors of both enzymes with KI values in the nanomolar range. These have been obtained by selecting R1, R2, and R3 determinants in aminophosphinic-containing inhibitors: NH2—CH(R1)P(O)—(OH)CH2—CH(R2)CONH—CH(R3)COOH, for optimal recognition of the two enkephalin inactivating enzymes, whose active site peculiarities, determined by site-directed mutagenesis, have been taken into account. The best inhibitors were 10× more potent than described dual inhibitors in alleviating acute and inflammatory nociceptive stimuli in mice, thus providing a basis for the development of a family of analgesics devoid of opioid side effects.
Acute and chronic pain are incapacitating diseases, and an improvement in their management is a high priority. Two classes of pain-alleviating substances currently are used in clinic. The first one is constituted by morphine and surrogates, which are the most potent and useful compounds to reduce severe pain, including pain associated with terminal issues. Antalgics including aspirin, paracetamol, and related substances provide the second group. These compounds inhibit the formation of hyperalgesic substances such as prostaglandins and are efficient in reducing inflammatory pain. However, there is a need for compounds capable of filling the gap between opioids and antalgics, which could be used for the treatment of postoperative, osteoarticular, and neuropathic pain as well as pain in children and in the elderly.
One of the most promising avenues in the search for such compounds is to improve the potency of the physiological system of pain control (1), constituted by the endogenous opioid peptides, enkephalins which interact with two specific binding sites, the μ and the δ receptors, strategically located at various levels of nociceptive pathways (2). This can be realized by inhibition of the membrane-bound zinc metallopeptidases involved in the rapid inactivation of the enkephalins. One of these enzymes is neutral endopeptidase-24.11 (NEP, neprilysin, EC 3.4.24.11) and other one is an exopeptidase, aminopeptidase N (APN, EC 3.4.11.2). Biological studies performed on rat brain and spinal cord slices (3, 4) have shown that thiorphan, a selective NEP inhibitor, or bestatin, an APN inhibitor, did not significantly prevent [3H]Met-enkephalin catabolism whereas their combination resulted in a clear reduction of the peptide degradation. This result has been confirmed in vivo. Thus, because of the complementary role of NEP and APN in enkephalin inactivation, selective inhibition of only one of these peptidases gives weak antinociceptive responses whereas strong analgesic effects can be obtained after complete inhibition of opioid peptide metabolism with combination of selective inhibitors (5). This leads us to propose the concept of “dual” inhibitors. Accordingly, compounds able to interact with the S′1 and S′2 subsites of NEP and APN, such as kelatorphan (5, 6) and RB 38A (7), were shown to produce strong analgesic responses caused by a large increase in the extracellular levels of enkephalins (4). However, these compounds (Fig. 1), which contain a hydroxamate as zinc chelating group, were unable to cross the blood–brain barrier, and their affinities for APN were ≈100× lower than those for NEP (KI ≈ 10−9 M). These problems were recently overcome by designing “mixed inhibitor prodrugs” made of potent and selective thiol-containing inhibitors of APN and NEP linked by a disulfide bond (8), which was shown to be cleaved in brain (9), where the targeted enzymes are localized (2). One of these compounds, RB 101, has been studied extensively. After i.p. administration, it has been shown by microdialysis in the nucleus accumbens of freely moving rats that RB 101 induced a long lasting increase in the extracellular level of Met-enkephalin-like material (10). Moreover, RB 101 has proved to be very potent after systemic administration in all animal models of pain without producing the side effects of morphine (11). RB 101 is now in preclinical studies. Given these promising results, it was of major interest to develop a single molecule capable of inhibiting both NEP and APN with nanomolar affinities as achieved for dual inhibitors of the two endopeptidases NEP and angiotensin-converting enzyme (12). Previous attempts with hydroxamate or thiol inhibitors were unsuccessful mainly because these compounds did not fit perfectly the active site of each enzyme.
Figure 1.

Structure of the three families of dual NEP/APN inhibitors.
It recently has been confirmed, by site directed mutagenesis experiments, that the positively charged amino group in APN substrates or inhibitors is stabilized within the active site of the peptidase by interacting with a glutamate residue (13). Taking these results into account, we have selected compounds containing a phosphinic moiety as a zinc coordinating ligand and a free N-terminal amino group for optimal APN binding. Numerous specific or multiple-phosphinic inhibitors of zinc metallopeptidases, particularly matrixins, have been described (14), but none of them contains a free amino group that was assumed to be detrimental for endopeptidase recognition. In this study, we show that aminophosphinic-containing compounds of the general formula NH2—CH(R1)P(O)—(OH)CH2—CH(R2)CONH—CH(R3)COOH, in which the side chains have been selected for optimal recognition of NEP and APN, are the first dual competitive inhibitors with nanomolar affinities for both enzymes. As expected, these compounds were more potent than described dual APN/NEP inhibitors in both severe and chronic animal models of pain, thus providing a basis for the development of new analgesics fulfilling the gap between antalgics and opioids.
MATERIALS AND METHODS
Chemicals.
Cbz-Phe-His-Leu (Cbz, carbobenzoxy) was from Bachem. Alanine p nitro-anilide was from Sigma, and Dansyl-Gly-(pNO2)Phe-βAla (DGNPA) was prepared as described (15). Details on the synthesis of the various inhibitors, summarized in Fig. 2, will be described elsewhere.
Figure 2.

Scheme for the synthesis of the phosphinic inhibitors.
Assay For in Vitro NEP and APN Inhibition.
NEP was purified to homogeneity from rabbit kidney (16). IC50 values were determined with DGNPA (Km = 37 μM) as substrate according to the procedure described (15). Aminopeptidase from hog kidney was purchased from Boehringer Mannheim, and inhibitory potencies were determined by using l-alanine-p-nitroanilide (Ala-pNA, Km = 400 μM) as substrate (17). Dixon plot analyses (Fig. 3 a and b) of the inhibition of both enzymes by compound 9B showed that this compound is a competitive inhibitor. The IC50 values of all the inhibitors tested were transformed into Ki values by the Cheng–Prussof relationship (Ki = IC50/1 + S/Km) (18).
Figure 3.

Dixon plot analysis of the inhibitory activity of compound 9B for NEP and APN. The experiments were performed at 25°C in 50 mM Tris⋅HCl buffer (pH 7.4). (a) The inhibitory potency of 9B for NEP was measured by using DGNPA as substrate at the concentrations indicated (□, 5μm; ▴, 10 μM; ○, 25 μM; ■, 50 μM; ▵, 100 μM). (b) The inhibitory potency of 9B for APN was measured by using Ala-PNA as substrate at the concentrations indicated (□, 25 μM; ▴, 50 μM; ○, 100 μM; ■, 200 μM). Each point of the Dixon plots represents the mean of triplicate determinations. These experiments were repeated five times, leading to a mean Ki value (± SEM) of 0.80 ± 0.05 nM for NEP and 2.5 ± 0.3 nM for APN.
In Vivo Inhibition of NEP.
The inhibition of cerebral NEP, induced by i.v. injection of 100 mg/kg compound 9B in mice, was evaluated as described (9). Fifteen minutes after injection, mice were anesthetized with chloral hydrate and were fixed by transcardial perfusion of paraformaldehyde, followed by phosphate buffer. Then, the brain was removed, was homogenized in cold, 50 mM Tris⋅HCl buffer, and was incubated with bestatin and captopril and with or without thiorphan. Then, [3H]-D.Ala2-Leu-enkephalin was added, and the amount of [3H]Tyr-D.Ala-Gly was evaluated. NEP inhibition was calculated as the difference of [3H]Tyr-D.Ala-Gly formation in the absence and in the presence of thiorphan. Controls corresponded to animals treated with saline.
Pharmacological Assays.
The inhibitors were dissolved in water and the pH of the solutions was adjusted to 7.0. Drugs and vehicles (controls) were administered intracerebroventricularly (ICV) to male Swiss mice (20–22 g, Depré, Fallaviers, France) 15 min before the test. Mice were housed and used strictly in accordance with European Community guidelines for the care and use of laboratory animals and after approval of the proposed experiments by the ethic committee of the Faculty of Pharmacy. Inhibitors or vehicle were slowly (15 sec) injected free hand into the left lateral ventricle of mice by using a modified Hamilton microliter syringe in a volume of 10 μl per mouse according to the method of Haley and McCormick (19).
Hot-Plate Test.
The test was based on that described by Eddy and Leimbach (20). A glass cylinder (16 cm high and 16 cm in diameter) was used to keep the mouse on the heated surface of the plate (53 ± 0.5°C). The latency of jump (cut-off time of 240 sec) was measured. Dose-response curves were established by expressing the data as a percentage of analgesia calculated by the equation: % analgesia = (test latency−control latency)/(cut-off time−control latency) × 100. Statistical analysis was carried out by ANOVA followed by Dunnett’s t test or Newman–Keuls test for multiple comparisons.
Writhing Test.
This test was derived from that of Koster et al. (21). Mice received i.p. 0.1 ml/10 g of body weight of a solution of 0.6% acetic acid, generating typical contractions of the abdominal musculature followed by extension of the hind limbs. The mice were placed in individual transparent containers, and the number of writhes per animal, in the 10-min period between 5 and 15 min after i.p. injection of acetic acid, was counted. Results were analyzed with ANOVA followed by the Dunnett’s t test.
Analysis of Data.
The ED50 values and their 95% confidence limits were calculated by log-probit analysis according to the method of Litchfield and Wilcoxon (22). The ED50 is defined as the dose required to elicit 50% analgesia.
RESULTS
Synthesis.
The synthesis of the various phosphinic inhibitors is summarized in Fig. 2 and will be described in detail elsewhere. In brief, the N-protected aminophosphinic acid 1 prepared following reported methods (23) was added to various 2-substituted ethyl acrylates. After alkaline hydrolysis of the ethyl esters, C-terminal amino acids were introduced. Compounds 3–9 were isolated, after final deprotection of the functional groups, as mixtures of four stereoisomers because of the presence of two unresolved asymmetric carbons. It was not possible to separate the four isomers by preparative HPLC because they had similar retention times, and two fractions were obtained, each containing a mixture of two isomers, A+B and C+D. No stereochemical assignment could be made at this step.
In Vitro Inhibitory Potency of Phosphinic Compounds on NEP and APN.
In a preliminary approach, the inhibitory potencies of several phosphinic compounds, containing benzyl groups in R2 and R3 positions and various substituents in position R1, were studied on NEP and APN activities (Table 1). For each compound, the two HPLC fractions A+B and C+D were tested separately on both enzymes, and, in all cases, the mixture A+B displayed a higher activity than C+D. Consequently, only the results on the mixture A+B will be discussed in detail.
Table 1.
Inhibitory potencies of phosphinic derivatives containing various R1 residues for NEP and APN
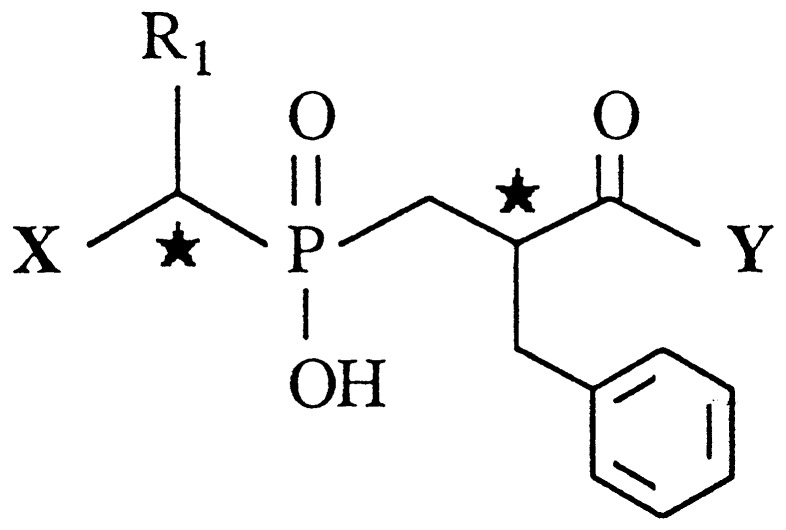
| Compound | X | R1 | Y |
Ki
(nM)*
|
|
|---|---|---|---|---|---|
| NEP† | APN‡ | ||||
| 3(A + B) | NH2 | CH2Ph | (S)Phe | 190 ± 50 | 2.9 ± 0.8 |
| (C + D) | 7,000 ± 900 | 200 ± 40 | |||
| 4(A + B) | NH2 | Ph | (S)Phe | 104 ± 7 | 3.8 ± 0.2 |
| (C + D) | 32,000 ± 1,000 | 320 ± 10 | |||
| 5(A + B) | NH2 | CH3 | (S)Phe | 146 ± 2 | 2.2 ± 0.2 |
| (C + D) | 35,000 ± 1,000 | 670 ± 90 | |||
| 10§ | H | CH2Ph | (S)Phe | 100 ± 7 | >100,000 |
| 11¶ | NH2 | CH2Ph | H | 1,000,000 | 370 ± 80 |
Values are the mean ± SEM from three independent experiments performed in triplicate of five inhibitor concentrations.
NEP activity was measured by using DGNPA as substrate.
APN activity was measured by using Ala-p. NA as substrate.
Compound 10 is a mixture of two stereoisomers.
Compound 11 is a mixture of four stereoisomers.
Compounds 3 to 5 are efficient APN inhibitors with Ki values in the nanomolar range (Table 1), but their inhibitory potencies for NEP were lower with Ki values between 100 and 200 nM. Moreover, the phosphinic inhibitor 11 lacking the C-terminal amino acid was a poor inhibitor of both enzymes whereas the deletion of the free amino group in 10 induced a dramatic decreased activity for APN and a slightly increased NEP affinity. Taking these data into account, various modifications were introduced in the R2 and R3 positions of compounds 3 to 5 to increase NEP recognition (Table 2). Introduction of a biphenyl moiety in position R2 enhanced the recognition of NEP but slightly decreased that of APN (compare 3 with 6). A reduction in the size of R3 (replacement of CH2Ph with CH3) in 7 led to nanomolar potencies on NEP and was responsible for a lower affinity for APN (compare 6 with 7). The same modifications, biphenylmethyl in R2 and methyl in R3, introduced in 4 and 5 led to 8 and 9, which had low nanomolar affinities for NEP and were 10× less potent for APN. It was concluded from these preliminary data that the best compromise for a dual inhibition of NEP and APN consisted of the association of a biphenyl moiety in R2 with a methyl group in R3, whatever the nature of R1 (compounds 7, 8 and 9).
Table 2.
Influence of R1, R2, and R3 substituents of various phosphinic compounds on the in vitro inhibitory potencies for NEP and APN
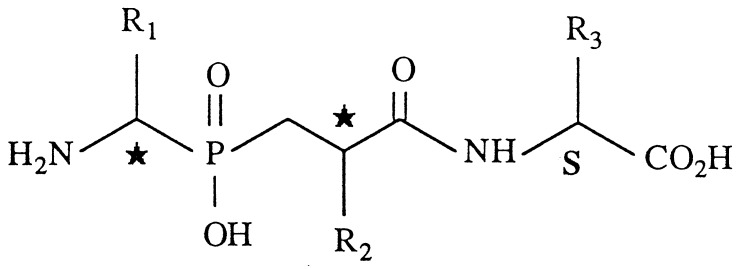
| Compounds | R1 | R2 | R3 |
Ki
(nM)*
|
|
|---|---|---|---|---|---|
| NEP† | APN‡ | ||||
| 3(A + B) | Ch2Ph | CH2Ph | CH2Ph | 189 ± 70 | 2.9 ± 0.8 |
| 6(A + B) | CH2Ph | CH2Ph(p-Ph) | CH2Ph | 150 ± 30 | 17 ± 3 |
| 7(A + B) | CH2Ph | CH2Ph(p-Ph) | CH3 | 2.3 ± 0.1 | 15 ± 2 |
| 8(A + B) | Ph | CH2Ph(p-Ph) | CH3 | 2.9 ± 0.1 | 14 ± 1 |
| 9(A + B) | CH3 | CH2Ph(p-Ph) | CH3 | 1.4 ± 0.2 | 5.2 ± 0.4 |
Values are the mean ± SEM from three independent experiments performed in triplicate of five inhibitor concentrations.
NEP activity was measured by using DGNPA as substrate.
APN activity was measured by using Ala-p.NA as substrate.
The resolution of the α-aminophosphinic acids 1, performed as described (23), allowed a complete separation of the four stereoisomers. The absolute configuration of the carbon bearing the R2 group was assigned by using the 1H NMR chemical shift strategy that has been described (24). The measurement of the inhibitory potencies of the separate isomers, on each enzymes, showed that the most efficient compound corresponded to the (R,S,S) configuration (peak B) in all cases (Table 3).
Table 3.
In vitro inhibitory potencies for NEP and APN and antinociceptive properties in the hot plate test in mice for the optically pure aminophosphinic-inhibitors
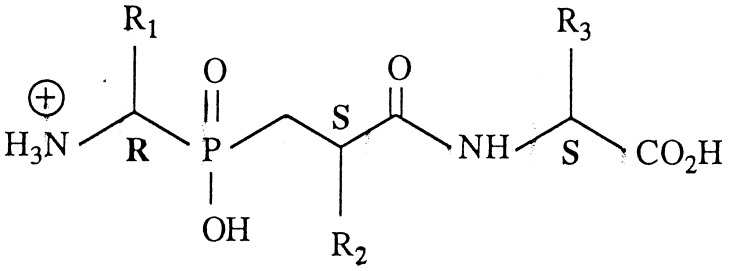
| Compounds | R1 | R2 | R3 |
Ki
(nM)*
|
Antinociceptive properties§ ED50, μg (95% confidence limits) | |
|---|---|---|---|---|---|---|
| NEP† | APN‡ | |||||
| 7(B) | CH2Ph | CH2Ph(p-Ph) | CH3 | 2.2 ± 0.3 | 5.3 ± 0.7 | 11 (6.84–17.7) 19.1 nmol |
| 8(B) | Ph | CH2Ph(p-Ph) | CH3 | 2.0 ± 0.5 | 4.8 ± 0.7 | 10.5 (5.83–18.9) 18.7 nmol |
| 9(B) | CH3 | CH2Ph(p-Ph) | CH3 | 1.2 ± 0.2 | 2.9 ± 0.3 | 6 (3.66–9.84) 12 nmol |
Values are the mean ± SEM from three independent experiments performed in triplicate of five inhibitor concentrations.
NEP activity was measured by using DGNPA as substrate.
APN activity was measured by using Ala-p.NA as substrate.
Inhibitors were administered ICV 15 min before the plate test.
To determine the selectivity of compounds 7B–9B toward the targeted enzymes, their inhibitory potencies also were tested on angiotensin-converting enzymes and aminopeptidase A, two metallopeptidases that have similarities with NEP and APN, respectively. Selectivity factors of ≈100 were obtained between NEP and angiotensin-converting enzymes and ≈1,000 between APN and aminopeptidase A (data not shown).
Analgesic Activity of the Dual Inhibitors.
The antinociceptive properties of the inhibitors 7 to 9 were investigated only on the optically pure B (R,S,S) isomers on the hot plate and writhing tests in mice after ICV administration (Fig. 4 a and b). The dose-response curves obtained in the hot plate test are reported in Fig. 4a. All of the compounds gave higher efficient antinociceptive responses than kelatorphan on the jump latency time. The statistical analysis of the curves showed that they can be considered as parallel, allowing the relative efficiencies of the inhibitors to be compared by using their ED50 values. The order of increasing efficacy was 7B (11 μg) < 8B (10.5 μg) < 9B (6 μg). Compound 9B was the most efficient, giving 100% analgesia at 20 μg. For all compounds, the analgesic responses were antagonized by a pretreatment with naloxone (0.1 mg/kg, s.c.) supporting the involvement of opioid receptors in these responses (data not shown).
Figure 4.

(a) Comparative antinociceptive properties of the most efficient dual inhibitors 7B to 9B and kelatorphan on the hot plate test in mice (55°C ± 0.5) (jump response) measured 15 min after ICV administration of the compounds. Each points represents the percentage of analgesia ± SEM (n = 10 mice). ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001 (Dunnett’s t test). (a) Comparative antinociceptive effects of compound 9B and kelatorphan in the writhing test in mice (n = 10 mice). The drugs were injected ICV at the doses indicated 15 min before testing. The results are expressed as percentage of analgesia ± SEM. ∗∗, P < 0.01; ∗∗∗, P < 0.001 (Dunnett’s t test).
Moreover, when high doses (100–200 mg/kg) of 9B were administered i.v. in mice, they produced significant antinociceptive response (30–40% analgesia) 15 min after injection (data not shown). The antinociceptive properties of 9B and kelatorphan were compared in the writhing test in mice (Fig. 4b) after ICV administration. The ED50 value of the phosphinic inhibitor 9B [3.6 μg (2.57–5.00); 7.2 nmol] was ≈10× lower than that of kelatorphan [22 μg (15.01–32.12); 69.7 nmol].
In Vivo Inhibition of NEP.
Fifteen minutes after i.v. injection of 100 mg/kg compound 9B, mouse brain NEP was shown to be highly inhibited (80%), demonstrating the ability of the dual inhibitor to cross the blood-brain barrier.
DISCUSSION
The aim of this study was to design a dual inhibitor with nanomolar affinities for NEP and APN, which are both involved in the inactivation of the endogenous opioid peptides, enkephalins. The importance of the enkephalins in the control of nociceptive stimuli has been evidenced initially by using inhibitors of both peptidases, resulting in significant analgesic responses (5, 9) and confirmed by deletion of the prepro-enkephalin gene, which was shown to generate a significant decrease in nociceptive thresholds (25). Conversely, homozygote mice in which the gene encoding NEP has been deleted are slightly less sensitive to nociceptive hot stimuli (26). This latter result confirms that inhibition of both NEP and APN are required to obtain physiological analgesic responses for possible clinical use (1).
NEP and APN belong to the superfamily of zinc metallopeptidases, but the former is an endopeptidase whereas the latter belongs to the group of exopeptidases. This was one of the difficulties encountered in designing dual inhibitors because the S1 subsite, probably located at the surface of the active site cleft of NEP (1), has been reported to be hydrophobic and thus was assumed to be unfavorably fitted by side chains containing hydrophilic groups.
In contrast, aminopeptidases require the presence of a free α-amino group for the residue acting as P1 component of substrates to ensure their exopeptidase specificity. This was confirmed by mutagenesis experiments showing that the amino group interacts with the carboxylate of a Glu residue present at the edge of the S1 subsite (13). These experiments also have shown that optimal recognition of APN by inhibitors requires the presence of the NH2 group in the same position as that found in the substrate. Taking these constraints into account, the strategy used to fulfill the criteria requested for optimal recognition of NEP and APN was to design, as inhibitors, transition state analogues to fix the right group in the right position.
The S1 subsite of APN interacts preferentially with hydrophobic or basic aliphatic chains (27). No precise exploration of the S1′ and S2′ subsites of this enzyme has been reported, but, from the structures of bestatin (28), amastatin (29), and various hydroxamate inhibitors (7, 30), they are assumed to be hydrophobic also. The importance of the S2′ subsite occupancy for an optimal inhibition of APN has been shown by using various dual NEP/APN hydroxamate inhibitors (7) and was verified in this series of aminophosphinic compounds by removing the C-terminal amino acid in 11, which induces a large decrease in inhibitory activity for APN (factor 30) as well as for NEP (factor 500) (Table 1).
Therefore, in a first step, compounds derived from the dipeptide Phe-Phe, which have been shown to interact efficiently with the S1′ and S2′ subsites of both enzymes, were selected as templates (7). Various α-amino alkyl phosphinic acids, able to recognize the S1 subsite of APN and chosen from the best described β-amino thiol inhibitors of this exopeptidase (27), were introduced to provide transition state analogues.
As expected, this strategy was effective for APN recognition because compounds 3 to 8 inhibited this enzyme with Ki values in the nanomolar range. As compared with the parent β-amino thiols interacting only with the S1 subsite (27) and the catalytic Zn2+, a 5- to 12-fold increase in affinity was observed with the new inhibitors probably caused, at least partly, by additional interactions with the S1′ and S2′ subsites of APN. However, the inhibitory potencies for NEP of 3 to 5 were in the 0.1–1 μM range. To verify whether this rather weak activity was caused by the presence of the free amino group, the analogue 10, without this function, was synthesized. The improvement in NEP inhibition was very slight when compared with 3 (Table 1), showing that the free amino group was not detrimental for NEP recognition and that the side chains selected to interact with the S1, S1′, and S2′ subsites of NEP were not optimized. It has been shown that a biphenyl group in the P1′ position significantly increases NEP inhibition (31). However, it also has been shown that an accumulation of aromatic or aliphatic bulky side chains in a pseudotripeptide is unfavorable for NEP binding (32). This is shown, once more, with compound 6, whose Ki value remained in the 0.1 μM range. Therefore, to optimize the recognition of NEP subsites, a small residue was introduced as the C-terminal component of the phosphinic inhibitors 7 to 9. As expected, these compounds behave as highly potent dual NEP/APN inhibitors (Table 2).
The resolution of the α-aminophosphinic acid (23) with crystallization of the isomer (R) led to the formation of only isomers B and D for each inhibitors. They were separated easily by preparative HPLC and the stereochemical assignment (R, S, S) of the most active isomer (B) was assessed. The in vitro and in vivo properties of three pure isomers 7B-9B, which have nanomolar activities on both NEP and APN and have low activities on related enzymes, such as angiotensin-converting enzyme and aminopeptidase A, were studied in more details (Table 3).
The antinociceptive activities of these inhibitors, on the hot plate test, are well correlated with their in vitro potencies; a change in IC50 values by a factor of 2 led to the same change in ED50 values in vivo (Fig. 4). Moreover, as compared with the reported hydroxamate dual inhibitor kelatorphan (ED50: 158 nmol) (5), the large improvement in pharmacological potencies of 7B to 9B (Fig. 4 a and b) clearly could be related to their increased APN inhibition (IC50 of kelatorphan: 710 nM for APN), showing once more the importance of APN as an enkephalin metabolizing enzyme.
It was also interesting to compare the in vivo activity of compounds 7B–9B to that of an equimolar association of the two selective inhibitors of NEP (HSCH2CH(CH2Φ)CONHCH (CH2Φ)COOH; IC50: 2 nM) and APN [H3+N—CH(CH2CH2SCH3)CH2SH, IC50: 9 nM], which are linked by a disulfide bridge in the mixed inhibitor prodrug RB 101. An ED50 value of 72 nmol was obtained for this mixture on the jump latency. The difference between this value and the ED50 value of 9B (12 nmol) cannot be caused by only the better recognition of APN by the latter inhibitor but reflects essentially the great advantage of the in vivo administration of a single compound with a dual action as compared with the dual administration of a single compounds.
The aminophosphinic compounds described in this paper are dual inhibitors of NEP and APN with nanomolar affinities. They displayed the best antinociceptive responses of all of the currently reported compounds acting by this mechanism of action. Moreover, these responses are prevented by administration of the opioid antagonist naloxone indicating that, as expected, the antinociceptive effects resulting from the protection of endogenous enkephalins are associated with opioid receptor stimulation. Indeed, despite the relative broad specificities of NEP and APN, a certain in vivo specificity seems to be achieved, governed by both the distribution of the peptidases and that of endogenous enkephalins. On the other hand, probably for conformational reasons, β-endorphin, as well as dynorphin 1–13 and 1–17, appear to be resistant to NEP, and, to a lesser extent, APN (33). The possible involvement of NEP in the physiological inactivation of other neuropeptides has not been confirmed by the use of specific antagonists of their receptors, which were found unable to reverse the effects induced by an NEP inhibitor (reviewed in ref. 1). The antinociceptive responses induced by the ICV administration of the dual inhibitors were not greatly different from those of morphine; on the hot plate and writhing tests the ED50 values of 9B are only 10× and 30× greater than that of morphine (ED50: 1 nmol and 0.24 nmol, respectively). However, even at very high concentrations, at which they completely inhibit enkephalin degradation, mixed inhibitors were unable to produce the maximum analgesic effect induced by morphine in several antinociceptive tests. These observations suggest that the local increase in enkephalin concentration is still too low to saturate opioid binding sites, in agreement with in vivo binding experiments (34). This would eliminate, or at least minimize, receptor overstimulation essentially of μ-type, which is though to be responsible for the major side effects of morphine and surrogates (35). Moreover, because of their higher intrinsic efficacy, enkephalins need to occupy less μ opioid receptors than morphine to give the same pharmacological responses (36) and induced weaker intracellular modifications (37, 38).
Moreover, the main advantage of modifying the concentration of endogenous peptides by use of peptidase inhibitors is that pharmacological effects are induced only at receptors tonically or phasically stimulated by the natural effectors. Furthermore, in contrast to exogenous ligands, chronic administration of mixed enkephalin-degrading enzyme inhibitors does not induce changes in the synthesis of the target peptide precursors or in the secretion of the active peptides (39).
Taken together, these results, associated with the significant analgesic responses and the absence of side effects observed after i.v. administration of high doses (100 to 200 mg/kg in mice) of compound 9B, indicate that this type of compound could cross the blood–brain barrier and, like RB 101, is devoid of the drawbacks of morphine and surrogates. Given these results, the synthesis of prodrugs is now in progress to improve the bioavailability of these dual inhibitors. These could allow a complete characterization of the pharmacological properties of these molecules after systemic administration in various animal models of pain and could lead to new analgesics fulfilling the gap between antalgics and opioids.
Acknowledgments
We thank E. Ruffet, S. Da Nascimento, S. Cornet, and H. Meudal for excellent technical assistance. We gratefully acknowledge A. Beaumont for stylistic revision and C. Dupuis for the expert drafting of the manuscript.
ABBREVIATIONS
- NEP
neutral endopeptidase
- APN
aminopeptidase N
- ICV
intracerebroventricularly
- DGNPA
Dansyl-Gly-(pNO2)Phe-βAla
References
- 1. Roques B P, Noble F, Daugé V, Fournié-Zaluski M-C, Beaumont A. Pharmacol Rev. 1993;45:87–146. [PubMed] [Google Scholar]
- 2.Waksman G, Hamel E, Fournié-Zaluski M-C, Roques B P. Proc Natl Acad Sci USA. 1986;83:1523–1527. doi: 10.1073/pnas.83.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waksman G, Bouboutou R, Devin J, Bourgoin S, Cesselin F, Hamon M, Fournié-Zaluski M-C, Roques B P. Eur J Pharmacol. 1985;117:233–243. doi: 10.1016/0014-2999(85)90608-9. [DOI] [PubMed] [Google Scholar]
- 4.Bourgoin S, Le Bars D, Artaud F, Clot A M, Bouboutou R, Fournié-Zaluski M-C, Roques B P, Hamon M, Cesselin F. J Pharmacol Exp Ther. 1986;238:360–366. [PubMed] [Google Scholar]
- 5.Fournié-Zaluski M-C, Chaillet P, Bouboutou R, Coulaud A, Chérot P, Waksman G, Costentin J, Roques B P. Eur J Pharmacol. 1984;102:525–528. doi: 10.1016/0014-2999(84)90575-2. [DOI] [PubMed] [Google Scholar]
- 6.Kayser V, Fournié-Zaluski M-C, Guilbaud G, Roques B P. Brain Res. 1989;497:94–101. doi: 10.1016/0006-8993(89)90974-8. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt C, Peyroux J, Noble F, Fournié-Zaluski M-C, Roques B P. Eur J Pharmacol. 1991;192:253–262. doi: 10.1016/0014-2999(91)90050-z. [DOI] [PubMed] [Google Scholar]
- 8.Fournié-Zaluski M-C, Coric P, Turcaud S, Lucas E, Noble F, Maldonado R, Roques B P. J Med Chem. 1992;35:2474–2481. doi: 10.1021/jm00091a016. [DOI] [PubMed] [Google Scholar]
- 9.Noble F, Soleilhac J M, Soroca-Lucas E, Turcaud S, Fournié-Zaluski M-C, Roques B P. J Pharmacol Exp Ther. 1992;261:181–190. [PubMed] [Google Scholar]
- 10.Daugé V, Mauborgne A, Cesselin F, Fournié-Zaluski M-C, Roques B P. J Neurochem. 1996;67:1301–1308. doi: 10.1046/j.1471-4159.1996.67031301.x. [DOI] [PubMed] [Google Scholar]
- 11.Noble F, Coric P, Fournié-Zaluski M-C, Roques B P. Eur J Pharmacol. 1992;223:91–96. doi: 10.1016/0014-2999(92)90822-l. [DOI] [PubMed] [Google Scholar]
- 12.Fournié-Zaluski M-C, Gonzalez W, Turcaud S, Pham I, Roques B P, Michel J B. J Neurochem. 1996;67:1301–1308. doi: 10.1046/j.1471-4159.1996.67031301.x. [DOI] [PubMed] [Google Scholar]
- 13.Luciani N, Marie-Claire C, Ruffet E, Beaumont A, Roques B P, Fournié-Zaluski M-C. Biochemistry. 1998;37:686–692. doi: 10.1021/bi971705p. [DOI] [PubMed] [Google Scholar]
- 14.Hagmann W K, Lark M W, Becker J W. In: Annual Reports in Medicinal Chemistry. Bristol J A, editor. Vol. 31. New York: Academic; 1996. pp. 403–420. [Google Scholar]
- 15.Goudreau N, Guis C, Soleilhac J M, Roques B P. Anal Biochem. 1994;219:87–95. doi: 10.1006/abio.1994.1235. [DOI] [PubMed] [Google Scholar]
- 16.Aubry M, Berthelot A, Roques B P, Crine P. Biochem Cell. 1987;65:309–338. doi: 10.1139/o87-050. [DOI] [PubMed] [Google Scholar]
- 17.Alba F, Iribar C, Ramirez M, Arenas C. Arch Neurobiol. 1989;52:169–173. [PubMed] [Google Scholar]
- 18.Cheng Y C, Prussof W H. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 19.Haley T, McCormick W G. Br J Pharmacol. 1957;12:12–16. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddy N B, Leimbach D. J Pharmacol Exp Ther. 1953;107:385–389. [PubMed] [Google Scholar]
- 21.Koster R M, Anderson M, De Beer E J. Fed Proc. 1959;18:412. [Google Scholar]
- 22.Litchfield J T, Wilcoxon F. J Pharmacol Exp Ther. 1949;96:99–113. [PubMed] [Google Scholar]
- 23.Baylis E K, Campbell C D, Dingwall J G. J Chem Soc, Perkin Trans. 1984;1:2845–2853. [Google Scholar]
- 24.Fournié-Zaluski M-C, Lucas-Soroca E, Devin J, Roques B P. J Med Chem. 1986;29:751–757. doi: 10.1021/jm00155a027. [DOI] [PubMed] [Google Scholar]
- 25.Konig M, Zimmer A M, Steiner H, Holmes P V, Crawley J N, Brownstein M J, Zimmer A. Nature (London) 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- 26.Saria A, Hauser K F, Traurig H H, Turbek C S, Hersh L, Gérard C. Neurosci Lett. 1997;234:27–30. doi: 10.1016/s0304-3940(97)00660-5. [DOI] [PubMed] [Google Scholar]
- 27.Fournié-Zaluski M-C, Coric P, Turcaud S, Bruetschy L, Lucas E, Noble F, Roques B P. J Med Chem. 1992;35:1259–1266. doi: 10.1021/jm00085a013. [DOI] [PubMed] [Google Scholar]
- 28.Suda H, Takita T, Aoyagi T, Umezawa H. J Antibiot. 1976;29:100. doi: 10.7164/antibiotics.29.100. [DOI] [PubMed] [Google Scholar]
- 29.Tobe H, Morishima H, Nagasawa H, Takita T, Aoyagi T, Umezawa H. Agric Biol Chem. 1979;43:591–596. [Google Scholar]
- 30.Xie J, Soleilhac J M, Schmidt C, Peyroux J, Roques B P, Fournié-Zaluski M-C. J Med Chem. 1989;32:1497–1503. doi: 10.1021/jm00127a017. [DOI] [PubMed] [Google Scholar]
- 31.De Lombaert S, Blanchard L, Tam J, Sakane Y, Bevry C, Ghai R D. Bioorg Med Chem Lett. 1995;5:145–150. [Google Scholar]
- 32.Gomez-Monterrey I, Turcaud S, Lucas E, Bruetschy L, Roques B P, Fournié-Zaluski M-C. J Med Chem. 1993;36:87–94. doi: 10.1021/jm00053a011. [DOI] [PubMed] [Google Scholar]
- 33.Turner A J, Hooper N M, Kenny A J. In: Mammalian Ectoenzymes. Kenny A J, Turner A J, editors. Amsterdam: Elsevier; 1987. pp. 211–248. [Google Scholar]
- 34.Ruiz-Gayo M, Baamonde A, Turcaud S, Fournié-Zaluski M-C, Roques B P. Brain Res. 1992;571:306–312. doi: 10.1016/0006-8993(92)90669-z. [DOI] [PubMed] [Google Scholar]
- 35.Matthes H W D, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, et al. Nature (London) 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 36.Noble F, Roques B P. Neurosci Lett. 1995;185:75–78. doi: 10.1016/0304-3940(94)11228-b. [DOI] [PubMed] [Google Scholar]
- 37.Abbadie C, Honoré P, Fournié-Zaluski M-C, Roques B P, Besson J M. Eur J Pharmacol. 1994;258:215–227. doi: 10.1016/0014-2999(94)90483-9. [DOI] [PubMed] [Google Scholar]
- 38.Tölle T R, Schadrack J, Castro-Lopes J M, Evan G, Roques B P, Zieglgansberger W. Pain. 1994;56:103–112. doi: 10.1016/0304-3959(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 39.Delay-Goyet P, Kayser V, Zajac J M, Guilbaud G, Besson J M, Roques B P. Neuropharmacology. 1989;28:1341–1348. doi: 10.1016/0028-3908(89)90008-7. [DOI] [PubMed] [Google Scholar]