

Abstract
Recent research regarding the structure and function of Bacillus anthracis lethal (LeTx) and edema (ETx) toxins provides growing insights into the pathophysiology and treatment of shock with this lethal bacteria. These are both binary-type toxins composed of protective antigen necessary for their cellular uptake and either lethal or edema factors, the toxigenic moieties. The primary cellular receptors for protective antigen have been identified and constructed and key steps in the extracellular processing and internalization of the toxins clarified. Consistent with the lethal factor's primary action as an intracellular endopeptidase targeting mitogen-activated protein kinase kinases, growing evidence indicates that shock with this toxin does not result from an excessive inflammatory response. In fact, the potent immunosuppressive effects of LeTx may actually contribute to the establishment and persistence of infection. Instead, shock with LeTx may be related to the direct injurious effects of lethal factor on endothelial cell function. Despite the importance of LeTx, very recent studies show that edema factor, a potent adenyl cyclase, has the ability to make a substantial contribution to shock caused by B. anthracis and works additively with LeTx. Furthermore, ETx may contribute to the immunosuppressive effects of LeTx. Therapies under development that target several different steps in the cellular uptake and function of these two toxins have been effective in in vitro and in vivo systems. Understanding how best to apply these agents clinically and how they interact with conventional treatments should be goals for future research.
Keywords: anthrax, toxin, shock, treatment
Inhalational Bacillus anthracis is a serious bioterrorism-related health threat today (1). In the 2001 outbreak in the United States, the uniform fatality in patients with shock despite aggressive hemodynamic support as well as the pronounced hemoconcentration and recurrent pleural effusions noted support observations that the pathophysiology of shock with this infection may differ from more common types of sepsis (1, 2). Whether supportive measures conventionally applied in septic shock are as effective for B. anthracis is unclear. It is apparent, however, that lethal toxin (LeTx), composed of lethal factor (LF) and protective antigen (PA), and edema toxin (ETx), made up of edema factor (EF) and PA, are important for this microbe's pathogenesis (3, 4). Past and recent work directed at the structure and function of these two toxins provides increasing insights into both the mechanisms and treatment of B. anthracis shock (5). Some of the work presented in this article has been presented previously in abstract form (6).
THE BINARY NATURE OF B. anthracis LETHAL AND EDEMA TOXINS
Smith and Keppie first showed that plasma from B. anthracis–infected animals produced lethality in normal animals (7). Crude preparations of toxin prepared from such plasma were associated with extravascular edema, hypothermia, and hemoconcentration similar to that in infected animals. Antiserum developed against these preparations prevented lethality. In experiments by others, the serum of rhesus monkeys infected with anthrax was progressively more toxic to rats as bacterial count in donor animals rose, and was also lethal when given to uninfected monkeys (8).
In the first experiments describing the three B. anthracis toxin components, EF, initially designated factor I, produced local skin edema and, in larger doses, lethality, but only when given with a second factor (factor II), now known as PA (9). LF, first designated factor III, was highly lethal when combined with PA but did not produce skin edema. The combination of all three factors was most lethal and produced many of the characteristics of unfractionated toxin and bacterial infection. Vaccination with the combination of components was protective during bacterial infection (10). Later studies in mice with mutant B. anthracis strains supported the relative roles of these toxins during infection (3). Mutant strains lacking either PA or LF were not lethal. Strains lacking EF still produced lethality but this was not as great as with the parental strain producing all three components.
On the basis of these and other studies, lethal and edema toxins are now recognized to be binary or A-B–type toxins (11). PA is the “B” component that initiates cell binding and uptake of each toxin (Figure 1). LF and EF are the “A,” or toxigenic, components, which attach to PA on the cell surface and are then delivered into the cytosol where they exert their effects. These three proteins are encoded on the pXO1 plasmid, which is required for bacterial virulence.
Figure 1.

Summary of the fundamental steps in lethal and edema toxin uptake by host cells, the intracellular effects of the toxins, and the points in the process that have provided targets for antitoxin therapies. ATR = anthrax toxin receptor (tumor endothelial marker-8); CMG2 = capillary morphogenesis gene 2; DNI = dominant-negative inhibitor; EF = edema factor; Hexa-d-Arg = hexa-d-arginine; LF = lethal factor; MAPKK = mitogen-activated protein kinase kinase; PA = protective antigen; PVI = polyvalent inhibitor. Adapted by permission from Reference 5.
PA AND ITS HOST CELLULAR RECEPTORS
PA is an 83-kD (PA83) four-domain protein (12). Circulating PA binds via domain IV to one of at least two different cellular receptors, including tumor endothelial marker 8 (TEM8) and capillary morphogenesis gene 2 (CMG2) (13, 14) (Figure 1). The PA precursor molecule then undergoes furin protease–mediated cleavage in domain I into 20 kD (PA20) and 63 kD (PA63) proteins (15). After PA20 release, remaining PA63 units form a heptamer, also termed a prepore, that localizes to lipid raft regions in host cell membranes (16). Between one and three molecules of circulating LF or EF bind competitively to high-affinity sites on individual heptamers (17). Oligomerization is necessary for LF/EF binding. This combination then undergoes endocytosis and is progressively acidified. At a pH of 5.5, the prepore forms a barrel-like pore in the endosomal membrane through which LF and EF are translocated into the cytosol (18).
Both TEM8 and CMG2, the two host cellular receptors to which PA binds, have been associated with a variety of normal tissues, including heart, lung, small intestine, spleen, liver, kidney, skeletal muscle, and skin (13, 14, 19). Common to both proteins and with a high degree of homology are von Willebrand factor A or integrin inserted (VWA/I) extracellular domains (20). These domains contain metal ion–dependent adhesion sites (MIDAS) normally involved in binding to adhesion molecules or extracellular matrix proteins. Domain IV of PA competes with natural ligands for these MIDAS binding sites.
LF AND TOXIN
Structure and Function
LF is a 90-kD Zn2+ protease with four folding domains (21). Domain I binds to PA prior to cell uptake (Figure 1). The only presently recognized substrates for LF in the cytosol are the mitogen-activated protein kinase kinases (MAPKKs or MEKs) 1 to 4, 6, and 7, which are bound by domains II and III of LF and cleaved at the N-terminal ends by domain IV (5, 22). This cleavage prevents activation of each of the MAPKKs and has the potential to disrupt downstream signaling, which is essential for a variety of normal cell functions, including activation of immune and stress responses.
Cardiovascular Effects of LeTx
Despite LeTx's association with the pathogenesis of B. anthracis, few studies have investigated its cardiovascular effects. In early studies in guinea pigs, LeTx was reported to produce extravasation of fluid and a shocklike state, but hemodynamics were not measured (23). Bolus administration of toxin in rats caused pulmonary edema, pleural fluid collections, hemoconcentration, hypoxemia, and rapid lethality (24–26). In one such study, heart rate and blood pressure decreased immediately before death, reportedly secondary to respiratory distress (26). Intravenous administration of LeTx in rhesus monkeys reduced blood pressure and heart rate immediately before death, reportedly secondary to central respiratory depression (27). After these early investigations, no additional studies describing this toxin's hemodynamic effects were reported over the ensuing 30 years. However, aspects of the disease noted during the 2001 outbreak, while consistent with some of these prior observations, such as the hemoconcentration and extravasation of fluid, were not consistent with others. For example, shock in patients occurred well before death in nonsurvivors and was not initially a result of respiratory failure.
After the 2001 outbreak, our laboratory developed a rat model to further investigate LeTx-induced shock (28). Rather than administering toxin as a bolus, however, it was infused over 24 hours to better simulate the pattern of release during bacterial infection. Using LeTx doses designed to produce 50% lethality rates, death occurred in some rats starting at 10 to 12 hours and rats continued to die over the next 24 to 48 hours. Compared with control rats, LeTx produced reductions in blood pressure and heart rate several hours before initial lethality, which worsened in nonsurvivors but recovered in survivors (Figures 2A and 2B). Although toxin was associated with evidence of hemoconcentration, pleural fluid collections, and tissue hypoperfusion, respiratory failure and arterial hypoxemia were not primary contributors to death (28) (Figure 2C). Histologic studies did not demonstrate evidence of pulmonary or myocardial injury. In subsequent studies, inhibition of PA with monoclonal antibody administered up to 6 hours after the initiation of LeTx significantly increased blood pressure and heart rate, reduced evidence of hemoconcentration, and improved survival (29) (Figure 3). Thus, in contrast to earlier studies, findings in this rat model suggest that LeTx can produce shock independent of pulmonary dysfunction.
Figure 2.

Serial (A) mean arterial blood pressures, (B) heart rates, and (C) alveolar arterial oxygen gradients in surviving and nonsurviving rats after initiation of a 24-h lethal toxin (LeTx) infusion or in animals receiving diluent only (controls). Reductions in blood pressure and heart rate were first evident in nonsurvivors 4 to 5 h before initial lethality at 10 h. Arterial hypoxemia was not evident. *p values are for the time and group interaction.
Figure 3.

The effects of treatment with protective antigen–directed monoclonal antibody (PA-mAb) compared with placebo started at the time of or up to 12 h after the start of a 24-h LeTx infusion on (A) mean arterial blood pressure (MBP) and (B) the hazard ratio of survival in rats. Blood pressures were measured and averaged over the 12 h after treatment, and survival was monitored for 168 h. PA-mAb increased blood pressure and survival either significantly or in trends approaching it even if delayed for up to 12 h after the start of LeTx (29).
Potential Mediators and Mechanisms of LeTx-induced Shock
The mediators and mechanisms underlying shock with LeTx remain unclear. Earlier studies suggested that consistent with the excessive inflammatory response some bacterial toxins elicit, LeTx stimulated macrophage production of IL-1 and tumor necrosis factor (TNF) that ultimately led to cell lysis and cytokine release (30). Mice depleted of macrophages with silica were resistant to the lethal effects of LeTx, whereas supplementation with cultured macrophages restored these animals' sensitivity. Finally, inhibition of IL-1, and to a lesser extent TNF, improved survival with LeTx. Subsequent studies, however, have indicated that neither excessive cytokine nor nitric oxide (NO) production contributes to the pathogenesis of LeTx. In fact, consistent with its inhibitory effects on MAPKK function, LeTx may suppress the inflammatory response. In in vitro studies in murine macrophages, LeTx did not stimulate TNF-α or IL-1β production by macrophages, and inhibited cytokines, IL-1β mRNA, and NO production after LPS challenge (31, 32). In murine and human macrophages, MAPKK cleavage by LeTx inhibited LPS-stimulated IFN-regulatory factor 3 (IRF3) (33). In T cells, LeTx blocked IL-2 production through MAPKK inhibition (34). LeTx-treated murine dendritic cells that were stimulated with LPS did not up-regulate costimulatory molecules, secreted greatly diminished inflammatory cytokines, and did not stimulate T cells in vivo (35). In in vivo studies in BALB/c and C57BL/6 mice, lethal LeTx doses produced small increases in IL-1β in the former but none in the latter strain, whereas TNF levels were not altered in either strain (36). In our rat model, compared with 24-hour infusions of LPS, similarly lethal doses of LeTx were associated with significantly reduced inflammatory cytokine and NO production (Figure 4) (28). In further studies, sublethal LeTx doses inhibited LPS and Escherichia coli–stimulated cytokine (Figure 5A) and NO production (37).
Figure 4.

A comparison of the effects of similarly lethal 24-h infusions of lipopolysaccharide (LPS) or LeTx on mean serum (A) tumor necrosis factor (TNF), (B) interleukin (IL)-1β, (C) IL-6, and (D) nitric oxide (NO) levels measured from 3 to 9 h. Control animals (data not shown) were challenged with diluent only. Compared with LPS, LeTx was associated with greatly reduced measures of each of these mediators and TNF levels were actually reversed (28).
Figure 5.

The effects of sublethal doses of LeTx compared with placebo administered 3 h before intravenous LPS or intratracheal E. coli challenge on (A) 13 different plasma cytokine levels, (B) MBP, and (C) the odds ratio of survival. Cytokines were measured at 2, 8, and 24 h; blood pressure was averaged over the 24 h after challenge; and survival was assessed after 168 h. With both LPS and E. coli, sublethal LeTx had variable effects on cytokines at 2 and 24 h but consistently reduced all 13 levels at 8 h. The consistent decreases caused by LeTx in all cytokines at 8 h was significantly different from the more variable changes noted at 2 and 24 h with both challenges (p = 0.001 and 0.02 as shown in A). These antiinflammatory effects of sublethal LeTx, while increasing MBP and survival with LPS challenge, produced smaller MBP increases and reduced survival with E. coli in patterns that were significantly different comparing the two challenges (37).
Growing evidence does suggest that direct effects of LeTx on endothelial cell function could contribute to shock. Cultured human endothelial cells exposed to LeTx underwent apoptosis possibly via extracellular signal-regulated protein kinase (ERK) pathway inhibition (38). Also, LeTx decreased the barrier function of human lung microvascular endothelial cells as assessed with transendothelial electrical resistance and labeled albumin measures (39). This was accompanied by altered distribution of actin fibers and vascular endothelial cadherin necessary for normal barrier function. Finally, intradermal LeTx administration resulted in vascular leak within 15 to 25 minutes in a mouse model using the Miles Evans blue assay (40). Consistent with this evidence, LeTx challenge in animal models is associated with extravascular fluid collections and hemoconcentration similar to that observed during live bacterial infection (28, 36, 40, 41).
LeTx may also contribute to shock via its effects on glucocorticoid receptor (GR) function. In in vitro studies, LeTx inhibited transactivation of dexamethasone-induced GR in both a transfection system and in cells endogenously expressing GR (42). In in vivo studies, LeTx inhibited dexamethasone-induced liver tyrosine aminotransferase activity in BALB/c mice. Despite these suppressive effects, however, dexamethasone treatment worsened outcome in LeTx-challenged BALB/c mice.
Immunosuppressive Effects of LeTx
Although LeTx may not produce shock via stimulation of inflammatory host mediators, its inhibition of these same mediators as well as other components in host defense may contribute to the establishment and persistence of B. anthracis infection. In higher doses, LeTx causes macrophage lysis, although this appears species and strain specific and is not required to produce lethality or shock (28, 36). The sensitivity of murine macrophages to lysis by LeTx is determined by the gene Nalp1b on chromosome 11 (43). Nalp1b activates the caspase-1 response to LeTx, which in turn leads to macrophage death. In sublytic doses, LeTx can also cause apoptosis of activated macrophages via MAPKK or caspase pathways (44, 45).
In addition to its inhibitory effects on cytokines associated with the innate immune response outlined above, LeTx impairs neutrophil function. Neutrophils exposed to LeTx generated significantly less superoxide anion 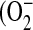 ) in response to LPS compared with LeTx-free cells (46). LeTx was also shown to interfere with actin assembly and to impair neutrophil chemotaxis (47). This is consistent with the finding that LeTx challenge causes intravascular aggregation of neutrophils in mice (36).
) in response to LPS compared with LeTx-free cells (46). LeTx was also shown to interfere with actin assembly and to impair neutrophil chemotaxis (47). This is consistent with the finding that LeTx challenge causes intravascular aggregation of neutrophils in mice (36).
Studies in our rat model support the potent inhibitory effect LeTx may have on innate immune responses and protective host defense mechanisms during infection (37). Reductions in inflammatory cytokines and NO release with sublethal LeTx doses in animals challenged with LPS or intratracheal E. coli were associated with increased blood pressures, although these were greater with the former than the latter (Figures 5A and 5B). However, whereas sublethal LeTx doses improved survival with LPS, a noninfectious challenge, it decreased survival with E. coli in patterns that were significantly different (Figure 5C). Worsened outcome with E. coli infection was also associated with evidence of reduced alveolar leukocyte recruitment that would normally be critical for extravascular microbial clearance (37).
LeTx alters adaptive immunity as well. In T-cell lymphocytes, LeTx disrupted antigen receptor signaling through the CD3 and CD28 receptors (48, 49). Furthermore, in human CD4+ T cells, LeTx inhibited IL-2 production and IL-2–dependent T-cell proliferation after T-cell receptor (TCR) stimulation (49). In other studies, MAPKK cleavage by LeTx inhibited B-cell proliferation and IgM production (50).
EF AND TOXIN
Structure and Function
EF, like LF, is transported into the cell via the PA heptamer (Figure 1) (51). The binding site on EF for PA is a seven–amino-acid (residues 136–142) chain identical to residues 147–153 on LF. EF mutations in Tyr 137, Tyr 138, Ile 140, and Lys 142 block interaction with PA (52). EF is an adenyl cyclase that leads to increased cAMP in host cells (53). It is distinct from mammalian adenyl cyclase but shares a common two–metal-ion catalytic mechanism (54). EF's catalytic activity requires the binding of calmodulin, a eukaryotic calcium binding protein (54). This leads to a change of calmodulin activity: two C-terminal Ca2+ binding sites show increased affinity and loss of cooperativity, whereas the N-terminal domain has a reduction in affinity and an increase in cooperativity (55). The EF–calmodulin complex catalyzes the synthesis of cAMP in host cells (56). The production of cAMP is dependent on an influx of calcium, and cAMP accumulation is prevented by calcium channel antagonists or the absence of calcium. EF has also now been shown to stimulate PA receptor function (57).
Cardiovascular Effects of ETx
After early findings that ETx was less lethal than LeTx, there were no further studies assessing its in vivo effects until two very recent ones (10, 58, 59). In one, administration of ETx in BALB/cJ mice produced pathologic lesions, including lymph node and focal gastrointestinal tract hemorrhage, adrenal damage, and intestinal fluid accumulation, similar to those reported in human series (58). Pleural effusions were absent, however. Compared with prior studies with LeTx in the same model, comparable doses of ETx produced greater lethality. Hemodynamic measures close to the time of death with a 100% lethal dose of ETx showed hypotension and, despite this toxin's potent adenyl cyclase activity, bradycardia.
Using the rat model developed in our lab, the effects of recombinant preparations of ETx and LeTx both alone and together as 24-hour infusions were compared (59). The range of doses of ETx alone that produced increasing lethality (0–100%) was tenfold greater than that of LeTx. However, similarly lethal doses of ETx produced greater and earlier hypotension than LeTx and increased rather than decreased heart rates (Figures 6A and 6B). Lethal doses of ETx were not associated with arterial hypoxemia. Furthermore, ETx and LeTx, when combined in doses that were either similar in weight or lethality, had effects that were additive (Figure 7). These and other findings therefore suggest that ETx may be as important as LeTx in the development of shock during B. anthracis infection and should be viewed as a possible therapeutic target.
Figure 6.

The effects of similarly lethal 24-h infusions of LeTx and ETx in rats on (A) MBP and (B) heart rate, early (2 to 10 h) and late (12 to 24 h), compared with animals receiving diluent only (data not shown). ETx produced earlier and greater reductions in MBP and increased rather than decreased heart rate compared with LeTx (59).
Figure 7.

The effects in rats on (A) survival, (B) MBP, and (C) heart rate (HR) of 24-h infusions of ETx and LeTx alone or together using doses that were similar either in weight or lethality. Control animals received diluent only. Changes in MBP and HR were averaged from 12 to 24 h during the infusions and survival was measured at 168 h. The two toxins together, whether in doses similar in weight or lethality, increased mortality rates and reduced MBP in patterns not different from the ones predicted based on their effects alone. Likewise, the effects of the toxins together on HR were always intermediate between their effects alone. Thus, ETx and LeTx appeared to have additive effects in this model (59).
Potential Mediators and Mechanisms of ETx-induced Shock
In light of its effects on tissue edema after local administration, extravasation of fluid is one possible mechanism for the shock occurring with ETx. However, in a mouse model, although highly lethal ETx doses produced intestinal intraluminal fluid accumulation and hemoconcentration, histologic analysis of several organs did not show significant extravasation of fluid (58). In our rat model, despite studying a range of ETx doses, hemoconcentration was not evident (59). Thus, to what extent shock with ETx is related to extravasation of fluid is unclear. What is evident, however, is that ETx has potent adenyl cyclase activity (53–56). This characteristic provides a strong basis for the cardiovascular dysfunction it has been shown to produce in recent in vivo models (60). Increases in cellular cAMP and activation of cAMP-dependent protein kinase could result both in arterial relaxation and shock as well as the tachycardia noted in our model.
ETx, similar to LeTx and consistent with its effects on intracellular cAMP levels, has also not been associated with the excessive inflammatory cytokine or NO production typically associated with other types of sepsis (58, 59). In fact, ETx may have immunosuppressive effects similar to LeTx. In in vitro or in vivo studies, ETx appeared to inhibit TNF-α, IFN-γ, IL-12p70, monocyte chemoattractant protein (MCP)–1, macrophage inflammatory protein (MIP)–1α, and MIP-1β (49, 58, 61–63). However, ETx has been associated with increases in IL-6, granulocyte colony–stimulating factor, exotaxin, keratinocyte-derived chemokine (KC), IL-10, IL-1 and monocyte-selective chemokine (JE)/MCP-1 (49, 58, 62, 63). This toxin has also been shown to inhibit activation and proliferation of CD4+ T cells (49). Neutrophils exposed to ETx were unable to phagocytize Sterne strain B. anthracis (64). In our rat model, ETx was not associated with any consistent increase in cytokine or NO release (59). However, circulating neutrophil numbers were increased and lymphocytes decreased over the 24 hours during which ETx was infused.
THERAPEUTIC IMPLICATIONS
Emerging evidence about the effects of LeTx and ETx raises several issues and questions regarding the conventional treatment of B. anthracis–related shock. Although antibiotics are clearly important in management of B. anthracis, their efficacy might not only be due to bacterial clearance. It is possible that agents such as clindamycin or rifampin, used in the 2001 outbreak, might also inhibit protein or RNA synthesis necessary for toxin production (65). Later in infection, however, when toxin levels are likely the highest, the immunosuppressive effects of both LeTx and ETx could inhibit microbial clearance and impair the efficacy of antibiotics. It is noteworthy that, in the absence of antibiotics, PA–monoclonal antibody (PA-mAb) treatment in a spore-challenged rabbit model was associated with the clearance of bacteria from blood cultures compared with placebo-treated animals (66).
It is also currently unknown how the vascular effects of LeTx and ETx might alter responsiveness to conventional hemodynamic support. As noted, the recurrent pleural fluid accumulation and marked hemoconcentration observed in patients with B. anthracis are consistent with emerging data regarding abnormalities of endothelial cell function associated with LeTx as well as the original effects associated with ETx on local edema formation (1, 4, 7, 38–41). In this context, whether standard volume support is as efficacious during B. anthracis infection as it is during other types of sepsis is unknown. In our rat model, in contrast to E. coli and LPS challenge, fluid support worsened outcome with LeTx challenge (6). Furthermore, review of all reported inhalational anthrax cases over the past 100 years has suggested that aggressive and frequent drainage of the pleural effusions with B. anthracis may be associated with improved outcome (1). Whether LeTx and ETx are also capable of altering the response to conventional vasopressor treatments is unknown. Certainly, disruption of intracellular calcium metabolism by the adenyl cyclase activity of ETx could alter the effects of such therapy (53, 56, 60). However, whether any one vasopressor is more effective than others for shock with LeTx and ETx requires study.
Two other therapies used with increasing frequency clinically during sepsis deserve mention (67). Inhibition of GR function by LeTx raises the possibility that corticosteroid therapy might be beneficial during shock with B. anthracis (42). However, in the limited studies conducted to date, dexamethasone administration was not effective in LeTx-challenged animals models (42). Finally, although not clearly attributable to LeTx or ETx, the mediastinal hemorrhagic necrosis and recurrent hemorrhagic pleural effusions noted in patients with B. anthracis may make the use of recombinant human activated protein C, with its associated bleeding risks, problematic (68).
Despite such questions, growing knowledge of the structure and function of both LeTx and ETx has led to the design of an increasing number of agents capable of neutralizing these toxins or their effects (Figure 1). Even before the availability of effective antibiotics, antiserum developed against B. anthracis was used in the acute treatment of patients with inhalational anthrax and, in some reports, may have prevented death (1). Such agents may be even more important because antibiotics, although quickly clearing bacteremia in patients during the 2001 outbreak, still did not prevent lethality in some (69). Agents inhibiting PA may have an advantage because they block both LF and EF. Antibodies against PA have been obtained from animal and human sources and humanized mAbs have been synthesized (70–77). PA antibodies prevent toxin entry into cells and macrophage lysis (73). These antibodies have been shown to be effective after the onset of shock with LeTx challenge and established infection with spore challenge (29, 70). PA antibodies appear to be safe in human volunteers (72). On the basis of such data, the U.S. military has now purchased 20,000 doses of a PA-directed mAb (Human Genome Sciences, Rockville, MD) (78). In addition, the U.S. Department of Health and Human Services is acquiring 10,000 doses of anthrax immune globulin (79). Animal data suggest, however, that antibodies like these will be most effective if administered early after shock is recognized (29).
Several other treatments directed against PA have also appeared to be effective in in vitro or in vivo models. Mutant forms of the PA molecule (dominant-negative inhibitors) with abnormal 2β2–2 β3 loops that form dysfunctional heptamers with wild-type PA prevented the uptake and transport of LF or EF and were effective with LeTx challenge in cell culture and Fisher 344 rats (80, 81). Hexa-d-arginine, a furin inhibitor that blocks PA cleavage and heptamer formation, was protective with LeTx challenge in murine alveolar macrophages and Fisher 344 rats (82). Finally, soluble PA receptor protected against LeTx in in vitro studies (13).
Agents have also been developed to directly inhibit LF. An mAb (LF8) directed against the PA binding site of LF was protective in macrophage and mouse models with LeTx (83). An antibody to domain III of LF prevented macrophage lysis in vitro and was protective in LeTx-challenged Fisher 344 rats (84). Small-molecule inhibitors of LF have been identified by chemical screening, including one that was effective with spore challenge when added to ciprofloxacin (85–87). Polyphenols from green tea, such as catechin, were protective with LeTx challenge in vitro and in rats (88). Finally, peptide molecules that act as substrate inhibitors of LF metalloproteolytic activity were protective in vitro (89, 90).
Treatments against EF have also been identified and may be useful if combined with anti-LF treatments. Adefovir, a drug used for chronic hepatitis B infection, inhibited EF with great affinity in murine macrophages and prevented EF-induced increases in cAMP (91). A quinazoline selectively inhibited EF without affecting mammalian adenyl cyclases (92).
CONCLUSIONS AND SUGGESTIONS FOR FUTURE RESEARCH
There has been considerable progress toward understanding the structure and function of LeTx and ETx and their possible contributions to shock with B. anthracis. At this time, however, important questions remain regarding the precise nature and mechanisms underlying the cardiovascular dysfunction these toxins produce. For example, the extent to which peripheral vascular dysfunction as opposed to myocardial injury contributes to shock with these toxins requires investigation. Furthermore, the question of how responsive this dysfunction is to conventional support has received little attention. Clearly defining the effects of fluid support both with toxin and live bacterial infection is essential at this time as is clarifying the effectiveness of conventional vasopressor support with agents such as norepinephrine and vasopressin. It is increasingly clear, however, that these toxins may have substantial suppressive effects on normal host defense function. Investigating whether these suppressive effects participate in the establishment of infection or contribute to the high bacterial loads noted in patients dying of B. anthracis will be important. It will also be necessary to determine whether other components of B. anthracis besides LeTx and ETx contribute to the shock occurring with infection. In light of the homology of TEM8 and CMG2 to von Willebrand factor, whether the interaction of these receptors with PA contributes to endothelial injury and to even the diseminated intravascular coagulation that has been observed during B. anthracis infection in humans and animals requires study (69, 93). Encouragingly, recent work directed at LeTx and ETx has resulted in a range of new agents capable of reducing their injurious effects. Understanding how best to apply these agents and how they interact with conventional treatments should be additional goals for future research.
Acknowledgments
The authors thank Ms. Jennifer Candotti for preparation of the manuscript.
Supported by the Intramural Program of the National Institutes of Health, Clinical Center, Critical Care Medicine Department.
Originally Published in Press as DOI: 10.1164/rccm.200608-1239CP on November 9, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Holty JE, Bravata DM, Liu H, Olshen RA, McDonald KM, Owens DK. Systematic review: a century of inhalational anthrax cases from 1900 to 2005. Ann Intern Med 2006;144:270–280. [DOI] [PubMed] [Google Scholar]
- 2.Grinberg RM, Abramova FA, Yampolskaya OV, Walker DH, Smith JH. Quantitative pathology of inhalational anthrax I: quantitative microscopic findings. Mod Pathol 2001;5:482–495. [DOI] [PubMed] [Google Scholar]
- 3.Pezard C, Berche P, Mock M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect Immun 1991;59:3472–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leppla SH. Anthrax toxin. In: Aktories K, Just I, editors. Bacterial proteins. New York: Springer; 2000. pp. 445–472.
- 5.Rainey GJA, Young JAT. Antitoxins: novel strategies to target agents of bioterrorism. Nat Rev Microbiol 2004;2:721–726. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Cui X, Haley M, Li X, Leppla S, Moayeri M, Fitz Y, Eichacker PQ. Fluids have opposing effects on survival comparing lipopolysaccharide (LPS) and Bacillus anthracis lethal toxin challenge in rats [abstract]. Proc Am Thorac Soc 2005;2:A39. [Google Scholar]
- 7.Smith H, Keppie J. Observations on experimental anthrax: demonstration of a specific lethal factor produced in vivo by Bacillus anthracis. Nature 1954;173:869–870. [DOI] [PubMed] [Google Scholar]
- 8.Klein F, Hodges DR, Mahlandt BG, Jones WI, Haines BW, Lincoln RE. Anthrax toxin: causative agent in the death of rhesus monkeys. Science 1962;138:1331–1333. [DOI] [PubMed] [Google Scholar]
- 9.Stanley JL, Smith H. Purification of factor I and recognition of a third factor of the anthrax toxin. J Gen Microbiol 1961;26:49–63. [DOI] [PubMed] [Google Scholar]
- 10.Hambleton P, Carman JA, Melling J. Anthrax: the disease in relation to vaccines. Vaccine 1984;2:125–132. [DOI] [PubMed] [Google Scholar]
- 11.Collier RJ, Young JA. Anthrax toxin. Annu Rev Cell Dev Biol 2003;19:45–70. [DOI] [PubMed] [Google Scholar]
- 12.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature 1997;385:833–838. [DOI] [PubMed] [Google Scholar]
- 13.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JAT. Identification of the cellular receptor for anthrax toxin. Nature 2001;414:225–229. [DOI] [PubMed] [Google Scholar]
- 14.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA 2003;100:5170–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect Immun 1995;63:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol 2003;160:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci USA 2002;99:7045–7048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wesche J, Elliot JL, Falnes PO, Olsnes S, Collier RJ. Characterization of membrane translocation by anthrax protective antigen. Biochemistry 1998;37:15737–15746. [DOI] [PubMed] [Google Scholar]
- 19.Bonuccelli G, Sotgia F, Frank PG, Williams TM, de Almeida CJ, Tanowitz HB, Scherer PE, Hotchkiss KA, Terman BI, Rollman B, et al. ATR/TEM8 is highly expressed in epithelial cells lining Bacillus anthracis' three sites of entry: implications for the pathogenesis of anthrax infection. Am J Physiol Cell Physiol 2005;288:C1402–C1410. [DOI] [PubMed] [Google Scholar]
- 20.Lacy DB, Wigelsworth DJ, Scobie HM, Young JA, Collier RJ. Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc Natl Acad Sci USA 2004;101:6367–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pannifer AD, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy BD, Collier RJ, Park S, Leppla SH, et al. Crystal structure of the anthrax lethal factor. Nature 2001;414:229–233. [DOI] [PubMed] [Google Scholar]
- 22.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998;280:734–737. [DOI] [PubMed] [Google Scholar]
- 23.Smith H, Keppie J, Stanley JL. The chemical basis of the virulence of Bacillus anthracis. V. The specific toxin produced by B. anthracis in vivo. Br J Exp Pathol 1955;36:460–472. [PMC free article] [PubMed] [Google Scholar]
- 24.Eckert NJ, Bonventre PF. In vivo effects of B. anthracis culture filtrates. J Infect Dis 1963;112:226–232. [Google Scholar]
- 25.Beall FA, Dalldorf FG. The pathogenesis of the lethal effect of anthrax toxin in the rat. J Infect Dis 1966;116:377–389. [DOI] [PubMed] [Google Scholar]
- 26.Fish DC, Klein F, Lincoln RE, Walker JS, Dobbs JP. Pathophysiological changes in the rat associated with anthrax toxin. J Infect Dis 1968;118:114–124. [DOI] [PubMed] [Google Scholar]
- 27.Vick JA, Lincoln RE, Klein F, Mahlandt BG, Walker JS, Fish DC. Neurological and physiological responses of the primate to anthrax toxin. J Infect Dis 1968;118:85–96. [DOI] [PubMed] [Google Scholar]
- 28.Cui X, Moayeri M, Li Y, Li X, Haley M, Fitz Y, Correa-Araujo R, Banks SA, Leppla SH, Eichacker PQ. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am J Physiol Regul Integr Comp Physiol 2004;286:R699–R709. [DOI] [PubMed] [Google Scholar]
- 29.Cui X, Li Y, Moayeri M, Choi GH, Subramanian GM, Li X, Haley M, Fitz Y, Feng J, Banks SM, et al. Late treatment with a protective antigen derived monoclonal antibody improves hemodynamic function and survival in a lethal toxin-infused rat model of anthrax sepsis. J Infect Dis 2005;191:422–434. [DOI] [PubMed] [Google Scholar]
- 30.Hanna PC, Acosta D, Collier RJ. On the role of macrophages in anthrax. Proc Natl Acad Sci USA 1993;90:10198–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNγ-induced release of NO and TNFα. FEBS Lett 1999;462:199–204. [DOI] [PubMed] [Google Scholar]
- 32.Erwin JL, DaSilva LM, Bavari S, Little SF, Friedlander AM, Chanh TC. Macrophage-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect Immun 2001;69:1175–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang O, Navarro L, Anderson K, David M. Cutting edge: anthrax lethal toxin inhibits activation of IFN-regulatory factor 3 by lipopolysaccharide. J Immunol 2004;172:747–751. [DOI] [PubMed] [Google Scholar]
- 34.Fang H, Cordoba-Rodriguez R, Lankford CSR, Frucht DM. Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4+ T cells. J Immunol 2005;174:4966–4971. [DOI] [PubMed] [Google Scholar]
- 35.Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A. Quinn C, Pulendran B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003;424:329–334. [DOI] [PubMed] [Google Scholar]
- 36.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-α-independent hypoxia-mediated toxicity in mice. J Clin Invest 2003;112:670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui X, Li Y, Li X, Haley M, Moayeri M, Fitz Y, Leppla SH, Eichacker PQ. Sublethal doses of Bacillus anthracis lethal toxin inhibit inflammation with lipopolysaccharide and Escherichia coli challenge but have opposite effects on survival. J Infect Dis 2006;193:829–840. [DOI] [PubMed] [Google Scholar]
- 38.Kirby JE. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect Immun 2004;72:430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warfel JM, Steele AD, D'Agnillo F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am J Pathol 2005;166:1871–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gozes Y, Moayeri M, Wiggins JF, Leppla SH. Anthrax lethal toxin induces ketotifen-sensitive intradermal vascular leakage in certain inbred mice. Infect Immun 2006;74:1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Culley NC, Pinson DM, Chakrabarty A, Mayo MS, LeVine SM. Pathological manifestations in mice exposed to anthrax lethal toxin. Infect Immun 2005;73:7006–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moayeri M, Webster JI, Wiggins JF, Leppla SH, Sternberg EM. Endocrine perturbation increases susceptibility to anthrax lethal toxin. Infect Immun 2005;73:4238–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 2006;38:240–244. [DOI] [PubMed] [Google Scholar]
- 44.Popov SG, Villasmil R, Bernardi J, Grene E, Cardwell J, Wu A, Alibek D, Bailey C, Alibek K. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem Biophys Res Commun 2002;293:349–355. [DOI] [PubMed] [Google Scholar]
- 45.Park JM, Greten FR, Wong A, Westrick RJ, Arthur JSC, Otsu K, Hoffman A, Montiminy M, Karin M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis-CREB and NF-kB as key regulators. Immunity 2005;23:319–329. [DOI] [PubMed] [Google Scholar]
- 46.Wright GC, Mandell GI. Anthrax toxin blocks priming of neutrophils by lipopolysaccharide and by muramyl dipeptide. J Exp Med 1986;164:1700–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.During RL, Li W, Hao B, Koenig JM, Stephens DS, Quinn CP, Southwick FS. Anthrax lethal toxin paralyzes neutrophil actin-based motility. J Infect Dis 2005;192:837–845. [DOI] [PubMed] [Google Scholar]
- 48.Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D'Elios MM, Tang E, Montecucco C, Baldari C. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J Exp Med 2005;201:325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Comer JE, Chopra AK, Peterson JW, Konig R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect Immun 2005;73:8275–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang H, Xu L, Chen TY, Cyr JM, Frucht DM. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J Immunol 2006;176:6155–6161. [DOI] [PubMed] [Google Scholar]
- 51.Guidi-Rontani C, Weber-Levy M, Mock M, Cabiaux V. Translocation on B. anthracis lethal and oedema factors across endosome membranes. Cell Microbiol 2000;2:259–264. [DOI] [PubMed] [Google Scholar]
- 52.Kumar P, Ahuja N, Bhatnagar R. Purification of anthrax edema factor from Escherichia coli and identification of residues required for binding to anthrax protective antigen. Infect Immun 2001;69:6532–6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leppla SH. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations in eukaryotic cells. Proc Natl Acad Sci USA 1982;79:3162–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen Y, Zhukovskaya NL, Guo Q, Florian J, Tang W. Calcium-independant calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J 2005;24:929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ulmer TS, Soelaiman S, Li S, Klee CB, Tang W, Bax A. Calcium dependence of the interaction between calmodulin and anthrax edema factor. J Biol Chem 2003;278:29261–29266. [DOI] [PubMed] [Google Scholar]
- 56.Ahuja N, Kumar P, Bhatnagar R. The adenylate cyclase toxins. Crit Rev Microbiol 2004;30:187–196. [DOI] [PubMed] [Google Scholar]
- 57.Maldonado-Arocho FJ, Fulcher JA, Benhur L, Bradley KA. Anthrax edema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol Microbiol 2006;61:324–337. [DOI] [PubMed] [Google Scholar]
- 58.Firoved AM, Miller GF, Moayeri M, Kakkar R, Shen Y, Wiggins JF, McNally EM, Tang W, Leppla SH. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol 2005;167:1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui X, Li Y, Li X, Laird MW, Subramanian M, Moayeri M, Leppla SH, Fitz Y, Su J, Sherer K, et al. B. anthracis edema and lethal toxin have different hemodynamic effects but function together to worsen shock and outcome in a rat model. J Infect Dis (In press) [DOI] [PubMed]
- 60.Murray KJ. Cyclic AMP and mechanisms of vasodilation. Pharmacol Ther 1990;47:329–345. [DOI] [PubMed] [Google Scholar]
- 61.Tournier J, Quesnel-Hellmann A, Mathieu J, Montecucco C, Tang W, Mock M, Vidal DR, Goossens PL. Anthrax edema toxin cooperated with lethal toxin to impair cytokine secretion during infection of dendritic cells. J Immunol 2005;174:4934–4941. [DOI] [PubMed] [Google Scholar]
- 62.Krakauer T, Little SF, Stiles BG. Bacillus anthracis edema toxin inhibits Staphylococcus aureas enterotoxin B effects in vitro: a potential protein therapeutic? Infect Immun 2005;73:7069–7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoover DL, Friedlander AM, Rogers LC, Yoon IK, Warren RL, Cross AS. Anthrax edema toxin differentially regulates lipopolysaccharide-induced monocyte production of tumor necrosis factor alpha and interleukin-6 by increasing intracellular cyclic AMP. Infect Immun 1994;62:4432–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Brien J, Friedlander A, Dreier T, Ezzell J, Leppla S. Effects of anthrax toxin components on human neutrophils. Infect Immun 1985;47:306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka M, Hasegawa T, Okamoto A, Torii K, Ohta M. Effect of antibiotics on group A Streptococcus exoprotein production analyzed by two-dimensional gel electrophoresis. Antimicrob Agents Chemother 2005;49:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beeb E, Zhong J, Clagett M, Subramanian M, Choi G. Protection against inhalation anthrax-induced lethality by a human monoclonal antibody to protective antigen in rabbits and cynomologous monkeys [abstract B-33]. In: Program and abstracts of the 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy (Chicago). Washington, DC: American Society for Microbiology; 2003. p. 47.
- 67.Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, Gea-Banacloche J, Keh D, Marshall JC, Parker MM, et al.; Surviving Sepsis Campaign Management Guidelines Committee. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004;32:858–873. [DOI] [PubMed] [Google Scholar]
- 68.Haley M, Cui X, Minneci PC, Deans KJ, Natanson C, Eichacker PQ. Recombinant human activated protein C in sepsis: assessing its clinical use. Am J Med Sci 2004;328:215–219. [DOI] [PubMed] [Google Scholar]
- 69.Jernigan DB, Raghunathan PL, Bell BP, Brechner R, Bresnitz EA, Butler JC, Cetron M, Cohen M, Doyle T, Fischer M, et al.; National Anthrax Epidemiologic Investigation Team. Investigation of bioterrorism-related anthrax, United States, 2001: epidemiologic findings. Emerg Infect Dis 2002;8:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mohamed N, Clagett M, Li J, Jones S, Pincus S. D'AliaG, Nardone L, Babin M, Spitalny G, Casey L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect Immun 2005;73:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peterson JW, Comer JE, Noffsinger DM, Wenglikowski A, Walberg KG, Chatuev BM, Chopra AK, Stanberry LR, Kang AS, Scholz WW, et al. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect Immun 2006;74:1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Subramanian GM, Cronin PW, Poley G, Weinstein A, Stoughton SM, Zhong J, Ou Y, Zmuda JF, Osborn BL, Freimuth WW. A phase 1 study of PAmAb, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clin Infect Dis 2005;41:12–20. [DOI] [PubMed] [Google Scholar]
- 73.Maynard JA, Maassen CBM, Lepple SH, Brasky K, Patterson JL, Iverson BL, Georgiou G. Protection against anthrax toxin by recombinant antibody fragments correlates with antigen affinity. Nat Biotechnol 2002;20:597–601. [DOI] [PubMed] [Google Scholar]
- 74.Chen Z, Moayeri M, Zhou Y, Leppla S, Emerson S, Sebrell A, Yu F, Svitel J, Schuck P, St. Claire M, et al. Efficient neutralization of anthrax toxin by chimpanzee monoclonal antibodies against protective antigen. J Infect Dis 2006;193:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mabry R, Rani M, Geiger R, Hubbard GB, Carrion R, Brasky K, Patterson JL, Georgiou G, Iverson BL. Passive protection against anthrax by using a high-affinity antitoxin antibody fragment lacking an fc region. Infect Immun 2005;73:8362–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sawada-Hirai R, Jiang I, Wang F, Sun SM, Nedellec R, Ruther P, Alvarez A, Millis D, Morrow PR, Kang AS. Human anti-anthrax protective antigen neutralizing monoclonal antibodies derived from donors vaccinated with anthrax vaccine adsorbed. J Immune Based Ther Vaccines 2004;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wild MA, Xin H, Maruyama T, Nolan MJ, Calveley PM, Malone JD, Wallace MR, Bowdish KS. Human antibodies from immunized donors are protective against anthrax toxin in vivo. Nat Biotechnol 2003;21:1305–1306. [DOI] [PubMed] [Google Scholar]
- 78.Rosenwald MS. US to buy anthrax treatment from HGS. Washington Post June 20, 2006. Available from: www.washingtonpost.com/wp-dyn/content/article/2006/06/19/AR2006061901135_pf.html (accessed August 10, 2006).
- 79.Cangene Corp. Winnipeg, MB, Canada. Available from: www.hhs.gov/news/press/2006pres/20060728.html-9k (accessed Aug 10, 2006).
- 80.Singh Y, Khanna H, Chopra AP, Mehra V. A dominant-negative mutant of Bacillus anthracis protective antigen inhibits anthrax toxin in vivo. J Biol Chem 2001;276:22090–22094. [DOI] [PubMed] [Google Scholar]
- 81.Sellman BR, Mourez M, Collier RJ. Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science 2001;292:695–697. [DOI] [PubMed] [Google Scholar]
- 82.Sarac MS, Peinado JR, Leppla SH, Lindberg I. Protection against anthrax toxemia by hexa-D-arginine in vitro and in vivo. Infect Immun 2004;72:602–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao P, Liang X, Kalbfleisch J, Koo H, Cao B. Neutralizing monoclonal antibody against anthrax lethal factor inhibits intoxication in a mouse model. Hum Antibodies 2003;12:129–135. [PubMed] [Google Scholar]
- 84.Lim N, Kim J, Oh MS, Lee S, Kim S, Kim K, Kang H, Hong HJ, Inn K. An anthrax lethal factor-neutralizing monoclonal antibody protects rats before and after challenge with anthrax toxin. Infect Immun 2005;73:6547–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Min D, Tang W, Mrksich M. Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat Biotechnol 2004;22:717–723. [DOI] [PubMed] [Google Scholar]
- 86.Panchal RG, Hermone AR, Nguyen TL, Wong TY, Schwarzenbacher R, Schmidt J, Lane D, McGrath C, Turk BE, Burnett J, et al. Identification of small molecule inhibitors of anthrax lethal factor. Nat Struct Mol Biol 2004;11:67–72. [DOI] [PubMed] [Google Scholar]
- 87.Forino M, Johnson S, Wong TY, Rozanov DV, Savinov AY, Li W, Fattorusso R, Becattini B, Orry AJ, Jung D, et al. Efficient synthetic inhibitors of anthrax lethal factor. Proc Natl Acad Sci USA 2005;102:9499–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.DellAica I, Dona M, Tonello F, Piris A, Mock M, Montecucco C, Garbisa S. Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep 2004;5:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tonello F, Seveso M, Marin O, Mock M, Montecucco C. Screening inhibitors of anthrax lethal factor. Nature 2002;418:386. [DOI] [PubMed] [Google Scholar]
- 90.Turk BE, Wong TY, Schwarzenbacher R, Jarrell ET, Leppla SH, Collier RJ, Liddington RC, Cantley LC. The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat Struct Mol Biol 2004;11:60–66. [DOI] [PubMed] [Google Scholar]
- 91.Shen Y, Zhukovskaya NL, Zimmer MI, Soelaiman S, Bergson P, Wang C, Gibbs CS, Tang W. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc Natl Acad Sci USA 2004;101:3242–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Soelaiman S, Wei BQ, Bergson P, Lee Y, Shen Y, Mrksich M, Shoichet BK, Tang W. Structure-based inhibitor discovery against adenyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J Biol Chem 2003;278:25990–25997. [DOI] [PubMed] [Google Scholar]
- 93.Stearns-Kurosawa DJ, Lupu F, Taylor FB Jr, Kinasewitz G, Kurosawa S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am J Pathol 2006;169:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]