

Abstract
Resolution of acute lung inflammation and injury is an active process; it is not merely the absence of proinflammatory signals. Restoration of homeostasis is coordinated by specific mediators and cellular events. In response to injury and inflammatory stimuli, infiltrating leukocytes and tissue-resident cells interact to generate lipoxins (LXs), which are bioactive eicosanoids derived from arachidonic acid. In contrast to proinflammatory leukotrienes and prostaglandins, LXs display potent antiinflammatory actions. LXA4 interacts with a G protein–coupled receptor, termed ALX, that transduces counter-regulatory signals in part via intracellular polyisoprenyl phosphate remodeling. Presqualene diphosphate (PSDP) is a polyisoprenyl phosphate in human neutrophils that is rapidly converted to presqualene monophosphate (PSMP) upon cell activation. PSDP, but not PSMP, directly inhibits phospholipase D, phosphoinositol-3 kinase, and superoxide anion generation. LXs block PSDP turnover in neutrophil membranes to prevent proinflammatory responses. Hence, LX and polyisoprenyl phosphate signaling provide a counter-regulatory circuit to promote resolution of acute lung inflammation. LXA4 and PSDP mimetics have been prepared with potent protective actions in murine models of asthma and acute lung injury.
Keywords: acute inflammation, lipid mediators, resolution
LEUKOCYTE ACTIVATION IN ACUTE LUNG INJURY GENERATES BIOACTIVE LIPID MEDIATORS
CLINICAL RELEVANCE
This article reviews recent advances in uncovering endogenous signaling circuits that promote the resolution of lung injury and inflammation.
Acute lung injury (ALI) initiates polymorphonuclear leukocyte (PMN) recruitment and activation (1). During these responses to tissue injury, PMN metabolic and membrane perturbations often lead to aberrant release of reactive oxygen species (ROS), serine proteases, and other potentially toxic products from PMN into the extracellular space (2). Thus, if persistent and unregulated, a leukocytic infiltrate may result in tissue injury—a process that underlies many human lung diseases, including ALI/acute respiratory distress syndrome (ARDS). Lipid mediators play important roles in regulating leukocyte function (3), and ALI/ARDS is characterized in part by excess production of eicosanoids (4–6). Leukocyte activation initiates the rapid generation of eicosanoids via release of arachidonic acid (C20:4) from cell membranes by the actions of cytosolic phospholipase A2 for subsequent metabolic conversion to cyclooxygenase (COX)-derived products, such as prostaglandins (PGs), or lipoxygenase (LO)-derived products, such as leukotrienes (LTs) (3). LTB4 is a potent leukocyte chemoattractant and secretagogue that may serve as an effector molecule in host defense (7–9). Given their high potency as proinflammatory mediators, LT overproduction during ALI can contribute to formation and maintenance of the diffuse inflammatory infiltrates of ARDS (10). Mice deficient in cytosolic phospholipase A2 are protected from the early manifestations of ALI with marked decreases in LTs (11).
Similar to LTs, lipoxins (LXs) and 15-epi-LXs are LO-derived eicosanoids, yet their biological actions differ dramatically from LTs (3). The major pathway for transcellular LX biosynthesis in respiratory tissues involves 5- and 15-LO (12). A distinct pathway for 15-epi-LX biosynthesis during cell–cell interactions involves COX-2 in cytokine-primed endothelia or epithelia and 5-LO in leukocytes. Aspirin acetylated COX-2 or cytochrome p450 enzymes catalyze the formation of 15R-HETE for subsequent conversion to 15-epi-LXs by leukocyte 5-LO (3). Formation of distinct classes of pro- and anti-inflammatory eicosanoids is tightly regulated with early COX-2–derived products critical to LX and 15-epi-LX production at later time points concurrent with resolution of acute inflammation and lung injury (13, 14). In the nM range, LXs inhibit PMN superoxide anion generation, chemotaxis, transmigration across endothelial and epithelial cells, diapedesis from postcapillary venules, and entry into inflamed tissues in animal models (3, 15). In contrast to their inhibition of granulocytes, LXs recruit monocytes and dendritic cells to sites of inflammation, infection, or injury. LX activation of monocyte-derived macrophages serves as a potent trigger for phagocytosis of apoptotic PMN and does not lead to the release of ROS (3, 16, 17). Inhibition of PMN tissue entry and promotion of apoptotic cell clearance provides critical effector mechanisms for resolution of inflammation. Moreover, LXs inhibit cytokine-driven inflammatory responses in a SOCS-2–dependent manner and block edema, vascular endothelial growth factor–mediated angiogenesis, and metalloproteinase production (3, 18, 19), providing additional mechanisms for these compounds to promote resolution of inflammation. LXs enhance mucosal host defense by controlling pathogen-induced inflammatory responses (16, 19) and inducing bactericidal permeability–inducing protein expression in gastrointestinal epithelia (20). Bactericidal permeability–inducing protein is an antimicrobial peptide expressed in human PMNs and epithelia that act via direct cytotoxicity to gram-negative bacterial membranes, binding of bacterial lipopolysaccharide, and bacterial opsonization to promote phagocytosis. In some respiratory diseases characterized by inflammation, including aspirin-sensitive and severe asthma and cystic fibrosis, LX generation is deficient and may contribute to the chronic inflammation in these conditions (21–24).
POLYIOSPRENYL PHOSPHATES AS INTRACELLULAR MEDIATORS FOR RECEPTOR-OPERATED BLOCKAGE OF PMN RESPONSES
LTB4 and LXA4 interact with highly specific and distinct G protein–coupled membrane receptors (25, 26) to evoke opposing PMN responses, including LXA4 inhibition of LTB4-initiated chemotaxis, adhesion, and transmigration (3). In addition to LXA4, 15-epimer-LXs are more potent than LXs, and LXA4 and 15-epi-LXA4 interact with the same LXA4 receptor (ALX) on PMN (25). LTB4 receptor activation triggers a rapid (i.e., within seconds) and significant decrease in the levels of presqualene diphosphate (PSDP) (∼ 30%) in PMN cell membranes (27) that returns to baseline within 300 s, concurrent with LTB4's kinetics for PMN activation and deactivation (8). The agonist-initiated decrease in PSDP is associated with turnover to its monophosphate form presqualene monophosphate (PSMP) (28). PSDP remodeling in PMNs is also initiated by other receptor-mediated stimuli, such as the chemotactic peptide formyl-methionyl-leucyl-phenylalanine and granulocyte monocyte colony-stimulating factor (Figure 1) (28). PSDP, but not PSMP, displays significant inhibition of PMN 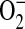 generation (28), highlighting a novel signaling role for these polyisoprenyl phosphates as regulators of PMN responses. Alone, a 15-epi-LXA4 analog does not affect the rate of polyisoprenyl phosphate remodeling in PMN, but coactivation of ALX and LTB4 receptors prevents LTB4-initiated decrements in PSDP (27). These results indicate that LXA4 and a 15-epi-LXA4 analog, which inhibit LTB4 responses (27, 29), dramatically switch LTB4-initiated polyisoprenyl phosphate signaling via ALX activation (Figure 2). Together, these findings point to a pivotal diphosphate phosphatase that upon cell activation rapidly converts PSDP to PSMP to unlock proinflammatory signals from counter-regulation by PSDP and serve as a new intracellular target for LXA4 regulation of LTB4 signaling. Recently, we identified the first PSDP phosphatase, now termed polyisoprenyl diphosphate phosphatase 1 (30).
generation (28), highlighting a novel signaling role for these polyisoprenyl phosphates as regulators of PMN responses. Alone, a 15-epi-LXA4 analog does not affect the rate of polyisoprenyl phosphate remodeling in PMN, but coactivation of ALX and LTB4 receptors prevents LTB4-initiated decrements in PSDP (27). These results indicate that LXA4 and a 15-epi-LXA4 analog, which inhibit LTB4 responses (27, 29), dramatically switch LTB4-initiated polyisoprenyl phosphate signaling via ALX activation (Figure 2). Together, these findings point to a pivotal diphosphate phosphatase that upon cell activation rapidly converts PSDP to PSMP to unlock proinflammatory signals from counter-regulation by PSDP and serve as a new intracellular target for LXA4 regulation of LTB4 signaling. Recently, we identified the first PSDP phosphatase, now termed polyisoprenyl diphosphate phosphatase 1 (30).
Figure 1.

Polyisoprenyl phosphate remodeling. PMN activation by G-protein–coupled (LTB4, formyl-methionyl-leucyl-phenylalanine) and growth factor (granulocyte monocyte colony-stimulating factor) receptors can trigger PSDP remodeling to PSMP.
Figure 2.

Polyisoprenyl phosphate remodeling in PMN: a rapid signaling pathway engaged during LX–ALX interactions. LXA4 and 15-epi-LXA4 are produced during inflammation as “stop signals” for agonist-triggered PMN responses. Interactions with ALX block agonist-induced PSDP turnover to PSMP. PSDP inhibits PLD and PI3K to reduce prophlogistic cellular responses.
Human PMNs have a natural deficiency in cholesterol biosynthesis, yet polyisoprenyl phosphate production is preserved (31). In addition to sterols, polyisoprenyl phosphates are pivotal intermediates for several vital outcomes, including ubiquinone, protein prenylation, and dolichol (32). Synthesis of the polyisoprenyl phosphate PSDP is considered essential because mice that lack squalene synthase are embryonic lethal at midgestation (33). Of interest for the regulation of inflammation, Hyper-IgD syndrome and periodic fever leads to generalized inflammation and is associated with mevalonate kinase deficiency and low isoprenoid levels (34), indicating counter-regulatory roles for select isoprenoids.
POTENTIAL INTRACELLULAR TARGETS FOR PSDP-MEDIATED COUNTER-REGULATION
Phospholipase D (PLD) hydrolyzes membrane phosphatidylcholine to generate phosphatidic acid and regulate cell function (35). In PMN, LTB4-stimulated PLD activity is associated with morphologic change, degranulation, and 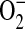 production (36). LTB4-triggered PLD is maximal within 60 s with an inverse relationship to the time course for PSDP turnover to PSMP (27). ALX activation by a 15-epi-LXA4 analog blocks LTB4-stimulated PLD activity (27). In activated PMN cavitates and isolated in vitro experiments with plant, microbial, or human PLD, PSDP is a potent and direct inhibitor (27, 37). Structure—activity relationships have been established with PSDP conformers and truncated human PLD1b, indicating at least two interaction domains for PSDP with PLD1b (37). Compared with PSDP, PSMP is over 2 log orders less potent as a PLD inhibitor. PSDP can also interact in vitro with Src-homology 2 domains on the adaptor protein Grb2 (38). The inverse relationship between PSDP levels and PLD activity in human PMNs and its direct inhibition of PLD support a role for PSDP as an endogenous regulator of PMN PLD.
production (36). LTB4-triggered PLD is maximal within 60 s with an inverse relationship to the time course for PSDP turnover to PSMP (27). ALX activation by a 15-epi-LXA4 analog blocks LTB4-stimulated PLD activity (27). In activated PMN cavitates and isolated in vitro experiments with plant, microbial, or human PLD, PSDP is a potent and direct inhibitor (27, 37). Structure—activity relationships have been established with PSDP conformers and truncated human PLD1b, indicating at least two interaction domains for PSDP with PLD1b (37). Compared with PSDP, PSMP is over 2 log orders less potent as a PLD inhibitor. PSDP can also interact in vitro with Src-homology 2 domains on the adaptor protein Grb2 (38). The inverse relationship between PSDP levels and PLD activity in human PMNs and its direct inhibition of PLD support a role for PSDP as an endogenous regulator of PMN PLD.
Phosphoinositide signaling initiated by phosphatidylinositol-3 kinase (PI3K) is another critical early signaling event in PMN activation and functional responses, such as phagocytosis and chemotaxis (39), and PI3K contributes to ALI pathophysiology (40). In addition to PSDP remodeling, LTB4 initiates PI3K activation in PMN to stimulate PLD, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase assembly, and ROS production (41). Agonist-triggered activation of class IA and IB PI3Ks is transient, lasting only seconds (29). Recently, lipid extracts from p110γ-PI3K immunoprecipitates obtained from PMN 30 s after exposure to LTB4 uncovered a physical interaction between p110γ-PI3K and PSDP, concomitant with PI3K deactivation (29). In vitro experiments demonstrated potent inhibition of p110γ-PI3K by PSDP, but not PSMP, in a concentration-dependent manner (29). The LTB4-triggered PMN remodeling of PSDP corresponds to the concentration range observed for regulation of p110γ PI3K. Together, these findings indicate that receptor-mediated agonists for PMN remodel PSDP in time frame and amounts consistent with functional impact on PI3K activity and cellular responses.
LIPID MEDIATORS CAN REGULATE ALI
Select eicosanoids can promote resolution of acute inflammation in vivo in several models of chest disease, including carrageenan-induced pleurisy (42), allergic pleuritis (18), and allergic airway inflammation and hyper-reactivity (43, 44). Recently, we developed a murine experimental model of mild ALI from acid aspiration that spontaneously resolves without supplemental oxygen or mechanical ventilation, allowing for the identification of endogenous resolution mechanisms in the lung (13). Early after acid injury, levels of COX-2 in murine lung markedly increase, gradually returning to baseline within 48 h. Specific inhibition of COX-2 by pharmacologic or gene disruption led to significant decrements in early PMN trafficking to the lung, but paradoxically dramatic increases in inflammation were present at later time points (13). COX-2–derived PGD2 and PGE2 can serve as pivotal signals for switching from LTB4 to LXA4 biosynthesis in acute exudative inflammation (14), and after ALI, COX-2 inhibition blocked the increased LXA4 production, resulting in an exacerbation of ALI with longer recovery times (13). Transgenic mice with myeloid targeted expression of human ALX (via a component of the CD11b promoter) were protected from acid-induced ALI (13). Thus, endogenous ligands, such as LXA4, that are generated in response to airway injury and acute inflammation can interact with ALX to speed ALI resolution (Figure 3).
Figure 3.

Resolution of acute lung inflammation by LXs. Acid aspiration is a common cause of ALI. During host responses to injury, LXs are generated locally to block PMN transmigration and activation, decrease edema and leakage of plasma proteins into the airways, promote restitution of injured airway epithelium, and block cytokine and chemokine release.
Aspiration of gastric acid directly injures respiratory tract epithelium, leading to disruption of the epithelial barrier, cell shedding, and acute inflammation (45). Transient HCl exposure in vitro leads to rapid and marked upregulation of COX-2 (within 2 h) and PGE2 generation by differentiated normal human bronchial epithelial cells (13, 46). COX-2–derived PGE2 generation also induces ALX expression by differentiated airway epithelial cells (46). In an ALX-dependent manner, LXA4 directly regulates several functional responses by the injured human bronchial epithelial cells, including inhibition of acid-induced IL-6 release and basal to apical PMN transmigration across well-differentiated bronchial epithelial cells. Similar to eye injury, LXA4 stimulates basal airway epithelial cell proliferation, an early event in restoring airway epithelial integrity (46, 47). LXA4 also inhibits growth factor–induced proliferation of lung fibroblasts (48). Together, these findings indicate that acid-initiated ALI triggers the generation of specific autacoid lipid mediators and receptors to establish counter-regulatory signaling pathways that promote epithelial restitution and resolution of inflammation and injury.
PSDP REMODELING IN VIVO DURING TISSUE INJURY AND INFLAMMATION
In response to acid-initiated ALI, expression of class IA and IB PI3Ks are increased in mouse lung (29). Despite increased PMN numbers, PSDP levels are significantly lower in the acid-injured lung. Similar to PMN responses in vitro, experimental lung injury led to an inverse relationship in vivo between lung PSDP and inflammation, with decrements in PSDP concomitant with increased PMN accumulation (29). Polyisoprenyl phosphate mimetics have been prepared to resist rapid inactivation and to investigate the impact of PSDP in vivo in models of acute inflammation. Recently, PSDP and select conformers were determined to dampen murine PMN trafficking elicited during zymosan A–induced peritonitis (37), and a diphosphonate PSDP structural mimetic (designed to resist phosphatase-based inactivation) administered before HCl-initiated ALI diminished PMN infiltration and PI3K activity (29). Thus, PSDP can regulate PMN activation, tissue accumulation, and PLD and PI3K activities in vivo during experimental models of acute inflammation. These new findings support a signaling relationship between polyisoprenyl phosphates and intracellular enzymes in the control of leukocyte functions during inflammation.
CONCLUSION
Resolution of acute inflammation is an active and tightly regulated process that is orchestrated by specific counter-regulatory signaling pathways. Here, we have reviewed recent findings that have uncovered LXs as potent agonists for resolution of lung inflammation and injury. In response to noxious stimuli, activated leukocytes and lung tissue–resident cells interact to generate LXs that block PMN infiltration and activation and promote restitution of injured-airway epithelium. LXs can regulate cell responses, in part, via polyisoprenyl phosphate remodeling, a newly identified intracellular signal transduction pathway. ALX activation blocks PSDP conversion to PSMP, and PSDP (not PSMP) is a potent and direct inhibitor of PLD and PI3K. LXA4 and PSDP mimetics have been prepared and display potent counter-regulation in vivo for acute inflammation and ALI. Together, these findings suggest that the natural properties of counter-regulatory lipid mediators, such as LXs and polyisoprenyl phosphates, can serve as windows into pathophysiology and potential templates for new therapeutic strategies for inflammatory lung diseases.
This work was supported in part by HL68669, DE016191, and AI068084, and by a postdoctoral fellowship from La Fondation de la Recherche Médicale.
Originally Published in Press as DOI: 10.1165/rcmb.2006-0269TR on September 21, 2006
Conflict of Interest Statement: C.B. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. B.D.L. has participated as a speaker in scientific meetings or courses organized and financed by various pharmaceutical companies (Critical Therapeutics, Kyorin Pharmaceutical, Banyu Pharmaceutical), has served as a consultant (Critical Therapeutics), and has received patent licensing fees (< $5,000) from Schering AG.
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–1349. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Points of control in inflammation. Nature 2002;420:846–852. [DOI] [PubMed] [Google Scholar]
- 3.Serhan CN. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 2005;73:141–162. [DOI] [PubMed] [Google Scholar]
- 4.Bernard GR, Korley V, Chee P, Swindell B, Ford-Hutchinson AW, Tagari P. Persistent generation of peptido leukotrienes in patients with the adult respiratory distress syndrome. Am Rev Respir Dis 1991;144:263–267. [DOI] [PubMed] [Google Scholar]
- 5.Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Synergism between leukotriene B4 and thromboxane A2 in mediating acid-aspiration injury. Surgery 1992;111:55–61. [PubMed] [Google Scholar]
- 6.Ohara M, Sawa T, Kurahashi K, Wiener-Kronish JP, Doshi V, Kudoh I, Gropper MA. Induction of cyclooxygenase-2 in alveolar macrophages after acid aspiration: selective cyclooxygenase-2 blockade reduces interleukin-6 production. Anesthesiology 1998;88:1014–1022. [DOI] [PubMed] [Google Scholar]
- 7.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J Immunol 1996;157:5221–5224. [PubMed] [Google Scholar]
- 8.Borgeat P, Naccache PH. Biosynthesis and biological activity of leukotriene B4. Clin Biochem 1990;23:459–468. [DOI] [PubMed] [Google Scholar]
- 9.Haribabu B, Verghese MW, Steeber DA, Sellars DD, Bock CB, Snyderman R. Targeted disruption of the leukotriene B(4) receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J Exp Med 2000;192:433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN. Preventing injury from within, using selective cPLA2 inhibitors. Nat Immunol 2000;1:13–15. [DOI] [PubMed] [Google Scholar]
- 11.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol 2000;1:42–46. [DOI] [PubMed] [Google Scholar]
- 12.Levy BD. Lipoxins and lipoxin analogs in asthma. Prostaglandins Leukot Essent Fatty Acids 2005;73:231–237. [DOI] [PubMed] [Google Scholar]
- 13.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE, Levy BD. Cyclooxygenase-2 plays a pivotal role in the resolution of acute lung injury. J Immunol 2005;174:5033–5039. [DOI] [PubMed] [Google Scholar]
- 14.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2001;2:612–619. [DOI] [PubMed] [Google Scholar]
- 15.Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, Szczeklik W, Drazen JM, Serhan CN. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat Med 2002;8:1018–1023. [DOI] [PubMed] [Google Scholar]
- 16.Bafica A, Scanga CA, Serhan C, Machado F, White S, Sher A, Aliberti J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest 2005;115:1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J Immunol 2000;164:1663–1667. [DOI] [PubMed] [Google Scholar]
- 18.Bandeira-Melo C, Serra MF, Diaz BL, Cordeiro RS, Silva PM, Lenzi HL, Bakhle YS, Serhan CN, Martins MA. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: relationship with concurrent eosinophilia. J Immunol 2000;164:1029–1036. [DOI] [PubMed] [Google Scholar]
- 19.Machado FS, Johndrow JE, Esper L, Dias A, Bafica A, Serhan CN, Aliberti J. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat Med 2006;12:330–334. [DOI] [PubMed] [Google Scholar]
- 20.Canny G, Levy O, Furuta GT, Narravula-Alipati S, Sisson RB, Serhan CN, Colgan SP. Lipid mediator-induced expression of bactericidal/permeability-increasing protein (BPI) in human mucosal epithelia. Proc Natl Acad Sci USA 2002;99:3902–3907. (comment). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonnans C, Vachier I, Chavis C, Godard P, Bousquet J, Chanez P. Lipoxins are potential endogenous antiinflammatory mediators in asthma. Am J Respir Crit Care Med 2002;165:1531–1535. [DOI] [PubMed] [Google Scholar]
- 22.Karp CL, Flick LM, Park KW, Softic S, Greer TM, Keledjian R, Yang R, Uddin J, Guggino WB, Atabani SF, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat Immunol 2004;5:388–392. [DOI] [PubMed] [Google Scholar]
- 23.Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med 2005;172:824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanak M, Levy BD, Clish CB, Chiang N, Gronert K, Mastalerz L, Serhan CN, Szczeklik A. Aspirin-tolerant asthmatics generate more lipoxins than aspirin-intolerant asthmatics. Eur Respir J 2000;16:44–49. [DOI] [PubMed] [Google Scholar]
- 25.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med 1997;185:1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 1997;387:620–624. [DOI] [PubMed] [Google Scholar]
- 27.Levy BD, Fokin VV, Clark JM, Wakelam MJ, Petasis NA, Serhan CN. Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: a ‘stop’ signaling switch for aspirin-triggered lipoxin A4. FASEB J 1999;13:903–911. [DOI] [PubMed] [Google Scholar]
- 28.Levy BD, Petasis NA, Serhan CN. Polyisoprenyl phosphates in intracellular signalling. Nature 1997;389:985–990. [DOI] [PubMed] [Google Scholar]
- 29.Bonnans C, Fukunaga K, Keledjian R, Petasis NA, Levy BD. Regulation of phosphatidylinositol 3-kinase by polyisoprenyl phosphates in neutrophil-mediated tissue injury. J Exp Med 2006;203:857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukunaga K, Arita M, Takahashi M, Morris AJ, Pfeffer M, Levy BD. Identification and functional characterization of a presqualene diphosphate phosphatase. J Biol Chem 2006;281:9490–9497. [DOI] [PubMed] [Google Scholar]
- 31.Shechter I, Fogelman AM, Popjak G. A deficiency of mixed function oxidase activities in the cholesterol biosynthetic pathway of human granulocytes. J Lipid Res 1980;21:277–283. [PubMed] [Google Scholar]
- 32.Holstein SA, Hohl RJ. Isoprenoids: remarkable diversity of form and function. Lipids 2004;39:293–309. [DOI] [PubMed] [Google Scholar]
- 33.Tozawa R, Ishibashi S, Osuga J, Yagyu H, Oka T, Chen Z, Ohashi K, Perrey S, Shionoiri F, Yahagi N, et al. Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J Biol Chem 1999;274:30843–30848. [DOI] [PubMed] [Google Scholar]
- 34.Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, Beckmann JS, van der Meer JW, Delpech M. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999;22:178–181. [DOI] [PubMed] [Google Scholar]
- 35.Billah MM, Eckel S, Mullmann TJ, Egan RW, Siegel MI. Phosphatidylcholine hydrolysis by phospholipase D determines phosphatidate and diglyceride levels in chemotactic peptide-stimulated human neutrophils: involvement of phosphatidate phosphohydrolase in signal transduction. J Biol Chem 1989;264:17069–17077. [PubMed] [Google Scholar]
- 36.Zhou HL, Chabot-Fletcher M, Foley JJ, Sarau HM, Tzimas MN, Winkler JD, Torphy TJ. Association between leukotriene B4-induced phospholipase D activation and degranulation of human neutrophils. Biochem Pharmacol 1993;46:139–148. [DOI] [PubMed] [Google Scholar]
- 37.Levy BD, Hickey L, Morris AJ, Larvie M, Keledjian R, Petasis NA, Bannenberg G, Serhan CN. Novel polyisoprenyl phosphates block phospholipase D and human neutrophil activation in vitro and murine peritoneal inflammation in vivo. Br J Pharmacol 2005;146:344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levy BD, Serhan CN. Polyisoprenyl phosphates: natural antiinflammatory lipid signals. Cell Mol Life Sci 2003;59:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 2000;287:1049–1053. [DOI] [PubMed] [Google Scholar]
- 40.Yum HK, Arcaroli J, Kupfner J, Shenkar R, Penninger JM, Sasaki T, Yang KY, Park JS, Abraham E. Involvement of phosphoinositide 3-kinases in neutrophil activation and the development of acute lung injury. J Immunol 2001;167:6601–6608. [DOI] [PubMed] [Google Scholar]
- 41.Ito N, Yokomizo T, Sasaki T, Kurosu H, Penninger J, Kanaho Y, Katada T, Hanaoka K, Shimizu T. Requirement of phosphatidylinositol 3-kinase activation and calcium influx for leukotriene B4-induced enzyme release. J Biol Chem 2002;277:44898–44904. [DOI] [PubMed] [Google Scholar]
- 42.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med 1999;5:698–701. [DOI] [PubMed] [Google Scholar]
- 43.Gavett SH, Madison SL, Chulada PC, Scarborough PE, Qu W, Boyle JE, Tiano HF, Lee CA, Langenbach R, Roggli VL, et al. Allergic lung responses are increased in prostaglandin H synthase-deficient mice. J Clin Invest 1999;104:721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peebles RS Jr, Hashimoto K, Morrow JD, Dworski R, Collins RD, Hashimoto Y, Christman JW, Kang KH, Jarzecka K, Furlong J, et al. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med 2002;165:1154–1160. [DOI] [PubMed] [Google Scholar]
- 45.Wynne JW, Ramphal R, Hood CI. Tracheal mucosal damage after aspiration: a scanning electron microscope study. Am Rev Respir Dis 1981;124:728–732. [DOI] [PubMed] [Google Scholar]
- 46.Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. Am J Pathol 2006;168:1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gronert K, Maheshwari N, Khan N, Hassan IR, Dunn M, Laniado Schwartzman M. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J Biol Chem 2005;280:15267–15278. [DOI] [PubMed] [Google Scholar]
- 48.Wu SH, Wu XH, Lu C, Dong L, Chen ZQ. Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol 2006;34:65–72. [DOI] [PubMed] [Google Scholar]