

Abstract
To determine the location of the proteinase in the covalent serpin-proteinase complex we prepared seven single-cysteine-containing variants of the Pittsburgh variant of the serpin α1-proteinase inhibitor, and we labeled each cysteine with the dansyl fluorophore. The dansyl probes were used to determine proximity of the proteinase trypsin in covalent and noncovalent complexes with the serpin, both by direct perturbation and by fluorescence energy transfer from tryptophans in trypsin to dansyl. Large direct effects on dansyl fluorophores were seen for only two positions in covalent complex and one position in noncovalent complex. Distances ranging from <14 Å to 64 Å were used to severely constrain possible structures for the complex. The structure consistent with both distance constraints and direct perturbations of the dansyl fluorophores placed the proteinase at the distal end of the serpin from the initial docking site. This position for the proteinase requires complete translocation of the proteinase from one end of the serpin to the other and full insertion of the reactive center loop into β-sheet A to form the kinetically trapped complex. The consequent tight juxtapositioning of serpin and proteinase could explain how distortion of the proteinase active site can occur and hence how many combinations of serpin and proteinase can be inhibited by a common conformational change mechanism.
Keywords: α1-proteinase inhibitor, Pittsburgh variant, serpin covalent complex, fluorescence resonance energy transfer
Elucidation of the structure of the serpin-proteinase complex remains the holy grail in trying to understand how serpins inhibit serine proteinases by a kinetic trap mechanism. Serpins inhibit proteinases by a branched pathway, suicide substrate inhibition mechanism (Fig. 1) in which a peptide bond in the exposed reactive center loop is initially recognized as an appropriate proteolytic cleavage site by proteinase, which thereby forms an initial noncovalent Michaelis complex. Formation of this complex is followed by attack by the proteinase active site on the peptide bond (1). Although there may be special cases of particular serpin-proteinase pairs where reaction stops at the Michaelis complex (2), formation of the serpin-proteinase complex under most circumstances involves progression of this initial noncovalent complex to the covalent acyl enzyme intermediate (E-I in Fig. 1), release of the newly formed amino terminus (P1′ residue) (3), and insertion of the now unconstrained reactive center loop into β-sheet A. Because the proteinase is covalently linked to the P1 residue of the serpin through an ester linkage, any insertion of the reactive center loop must involve concomitant proteinase translocation. At a point during insertion of the reactive center loop into β-sheet A, a physical interaction is thought to occur between the serpin and the proteinase that is sufficiently large enough to alter the properties of the proteinase (4) and thereby render it catalytically incompetent. The resulting structure represents the kinetically trapped covalent serpin-proteinase complex (E-I†).
Figure 1.

Branched pathway mechanism for serpins as suicide substrate inhibitors. E, proteinase; I, serpin; E⋅I, the noncovalent Michaelis complex; E-I, the covalent acyl enzyme intermediate prior to loop insertion, E-I† the kinetically trapped covalent acyl enzyme intermediate; and I*, cleaved serpin. The rate constants k3 and k4 are for the competing substrate and inhibition pathway and determine the stoichiometry of inhibition (SI) as SI = k4/(k3 + k4).
Several studies support such a general scheme by demonstrating movement of the proteinase as a necessary part of formation of this complex, though without definitively showing where the proteinase is located in the final complex (5–7), and also by providing evidence for structural changes within the proteinase in the final complex (8–11). One study, using a combination of chemical cross-linking and fluorescence energy transfer, concluded that only partial insertion of the reactive center loop into β-sheet A of the serpin occurs and that the proteinase consequently moves only part way down the body of the serpin to a final resting place flanking helix F (Fig. 2, right-hand structure, green trypsin) (6). A recent study from this laboratory was, however, very much at variance with this conclusion (7). Fluorescent 7-nitrobenz-2-oxa-1,3-diazole (NBD) reporter groups were used to indicate proximity of the proteinase in the complex and strongly implied that the proteinase was not in the vicinity of helix F, but instead at the bottom of the serpin. Although some qualitative fluorescence resonance energy transfer measurements were also attempted, they used nonspecifically localized labels on the proteinase and could therefore not be used to accurately triangulate the position of the proteinase. Because of this remaining fundamental difference in conclusions between these two studies (6, 7) we sought to more definitively determine the location of the proteinase in covalent complex with a serpin. The present report presents the results of this study, using trypsin as the proteinase and the Pittsburgh variant of α1PI (12) as the serpin.
Figure 2.

Orthogonal views of α1-proteinase inhibitor (α1PI; yellow), with rotation about vertical axis, showing location of cysteines used for labeling. The reactive center loop is in cyan. In the right-hand view trypsin is also shown in two different locations. At top, in the position it is expected to occupy in the noncovalent Michaelis complex (cyan), and on the side, in the position proposed for the covalent complex in an earlier study (6) (green). In the Michaelis complex trypsin has been docked with the reactive center loop, with the proteinase active site centered over the P1–P1′ bond. In the earlier proposed structure for the covalent complex (6) trypsin is docked against the flank of the F helix (bottom right). The location of residues 159, 121, and 314 with respect to trypsin in this model should be noted, since 159 should be in contact with or very close to the proteinase (8.8 Å to the mean trypsin tryptophan position), whereas residues 121 and 314 are well removed (31.5 and 43 Å, respectively, to the mean trypsin tryptophan position).
MATERIALS AND METHODS
Site-Directed Mutagenesis.
Site-directed mutagenesis was carried out on a double-stranded pET16b plasmid (Novagen) containing α1PI cDNA, using the QuikChange method (Stratagene), as described (5). All α1PI variants carried the M358R mutation and all except the unaltered Pittsburgh variant carried the C232S mutation. All mutations were confirmed by dideoxynucleotide sequencing in the host plasmid.
Expression and Refolding of Recombinant α1-PI Variants.
Expression, protein purification, and refolding of solubilized inclusion bodies of recombinant α1PI variants were carried out as previously described (7). An extinction coefficient of 27,000 M−1⋅cm−1 (13) was used for all α1PI variants.
Preparation of Anhydrotrypsin and β-Trypsin.
Anhydrotrypsin was prepared from commercial crystallized trypsin (Sigma) by alkaline β-elimination of the PMSF adduct (14). Following reaction, the solution was treated with Phe-Phe-Arg chloromethyl ketone (20 μM) to inhibit any remaining or regenerated active trypsin and acidified to pH 3.0. β-Anhydrotrypsin was purified from the reaction mixture by chromatography on a soybean trypsin inhibitor affinity matrix. β-Trypsin was prepared from TPCK-treated commercial trypsin by affinity chromatography using the same soybean trypsin inhibitor affinity matrix.
Labeling of α1-PI with 5-[2-(2-Iodoacetamido)ethylamino]-1-naphthalenesulfonic Acid (I-AEDANS) and Preparation of Complexes and Cleaved Species.
Reactions and all subsequent experiments were carried out in 20 mM sodium phosphate buffer, pH 7.4, containing 100 mM NaCl, 0.1 mM EDTA, and 0.1% PEG 8000. α1PI (5–20 μM) was reacted with a 2- to 3-fold excess of DTT for 15 min at room temperature. A 10- to 20-fold excess of I-AEDANS in the same buffer was reacted on ice in the dark for 3–8 hr. A 100-fold excess of DTT was added to quench the reaction. The sample was dialyzed overnight against 2,000 vol of buffer and filtered through a 0.22-μm-pore filter, and the extent of labeling was determined spectrophotometrically, using an extinction coefficient of 5700 M−1⋅s−1 for dansyl at 340 nm, and the protein absorbance at 280 nm, corrected for the contribution of dansyl at this wavelength, which was determined empirically to be 22.7% of the absorbance at 340 nm. The labeling ratios of samples used in this study varied between 0.75 and 1.16 label per molecule of α1PI.
Cleaved α1PI species were made by reaction of 5 μM α1PI variants with 0.15 μM papain at 37°C for 10 min followed by addition of 10 mM iodoacetamide to quench the reaction. Noncovalent complex was made by mixing α1PI and anhydrotrypsin in a 1:1 ratio at 5 μM each. Covalent complex was formed by reaction of 1:1 α1PI variant (5 μM) with β-trypsin for 45 sec (determined empirically to be sufficient to complete the reaction, which indicates that labeling had not reduced the rate of reaction greatly for any of the variants) followed by inhibition of any free trypsin by addition of a small excess of Phe-Phe-Arg chloromethyl ketone.
Fluorescence Measurements.
All fluorescence measurements were made on an SLM8000 scanning fluorometer (Urbana, IL). Spectra were recorded at 25°C in a thermostated cuvette. Dansyl emission spectra in the absence of energy transfer from tryptophan used excitation at 340 nm, with emission recorded from 400 to 600 nm in 2-nm steps. Slits were 4 nm for both excitation and emission. For measurement of energy transfer between tryptophan and dansyl, excitation was at 292 nm, to avoid excitation of tyrosine, with emission recorded from 300 to 600 nm to include emission from both tryptophan and dansyl.
Energy transfer from tryptophan to dansyl was determined in two ways. One used increase in dansyl emission and the second used reduction in tryptophan emission. Each method has different limitations and advantages in this system. Calculation of the efficiency of transfer by increase in dansyl emission used the relationship
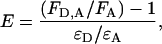 |
1 |
where FD,A and FA are the fluorescence of the acceptor (dansyl) in the presence and absence, respectively, of donor (tryptophan), and ɛD and ɛA are the extinction coefficients of donor and acceptor, respectively, at the wavelength of excitation (292 nm) (14,310 and 1,910 M−1⋅cm−1, respectively). FD,A can be determined accurately from measurements on the appropriate complex. Measurement of FA was carried out on either cleaved α1PI, as structurally representative of the serpin moiety in the covalent complex, or native α1PI, as representative of the serpin moiety in the noncovalent complex. In each case, the fluorescence FA was adjusted for changes in quantum yield caused by the presence of the proteinase by multiplying by the ratio of the quantum yields in the absence and presence of proteinase (see Table 1). This method is accurate as long as there is not significant energy transfer from the two tryptophans internal to α1PI. For positions 121 and 314, for which the largest dansyl enhancements are seen, energy transfer from tryptophans in α1PI is unlikely to be significant, given the large separations involved (42 and 53 Å to 121 and 46 and 50 Å to 314) and the small value for R0, the separation for 50% efficiency of transfer, (22.6 Å). Similarly, it is not expected to be significant for positions 85 and 159 (39 and 42 Å, and 33 and 43 Å, respectively). Measurements by this method for positions 232 and 360 are most likely to be in error, because the separations are much less (9 and 21 Å for 232 and 14.8 and 25.6 Å for 360). This proximity would lead to an underestimate for the efficiency of resonance energy transfer.
Table 1.
Fluorescence properties of dansyl-labeled α1PIPittsburgh variants, alone and in covalent and noncovalent complexes
| Position | λmax, nm
|
Quantum yield
|
||||||
|---|---|---|---|---|---|---|---|---|
| Native | Cleaved | NCC | CC | Native | Cleaved | NCC | CC | |
| 85 | 492 | 492 | 492 | 492 | 0.128 | 0.125 | 0.130 | 0.131 |
| 111 | 494 | 494 | 494 | 494 | 0.242 | 0.224 | 0.238 | 0.236 |
| 121 | 494 | 494 | 494 | 484 | 0.294 | 0.284 | 0.293 | 0.396 |
| 159 | 492 | 496 | 492 | 496 | 0.251 | 0.221 | 0.251 | 0.220 |
| 232 | 492 | 492 | 492 | 492 | 0.302 | 0.278 | 0.296 | 0.288 |
| 314 | 492 | 496 | 492 | 472 | 0.305 | 0.280 | 0.305 | 0.537 |
| 360 | 494 | 492 | 488 | 492 | 0.186 | 0.183 | 0.236 | 0.169 |
Large changes are in boldface type. NCC, noncovalent complex with anhydrotrypsin; CC, covalent complex with β-trypsin.
The second method for determining efficiency of transfer was from reduction in intensity of tryptophan emission in comparison to the intensity of the non-dansyl-labeled complex. This is not subject to errors due to energy transfer from tryptophans within α1PI, but is subject to errors from the need to make corrections for degrees of labeling of less than 1.0 dansyl per α1PI. This is not the case for the first method, since dansyl emission is observed only for labeled complexes. Efficiency of transfer by this method is calculated as
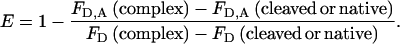 |
2 |
The correction in both numerator and denominator for the contribution from tryptophans within α1PI [FD,A(cleaved or native) and FD(cleaved or native)] is so that the percentage reduction relates only to the tryptophans of β-trypsin. Because of the greater uncertainties of this method, as applied to this system with tryptophans in both serpin and proteinase, efficiencies used for distance calculation were estimated by the first method in all cases except for the noncovalent complex with the I360C variant.
The value for R0 used in distance calculations was determined by using the relationship given in Eq. 3.
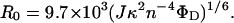 |
3 |
Here, J is the overlap integral between the emission spectrum of tryptophans in trypsin and the absorption spectrum of dansyl in the serpin. This was calculated, for the complex, to be 6.24 × 10−15 M−1⋅cm3. ΦD is the quantum yield for the donor tryptophans in trypsin, determined here to be 0.146 from comparison with the emission intensity of quinine sulfate. The refractive index term, n, is routinely taken to be 1.4. This value was used here. The orientation factor, κ2, is potentially the largest source of error, since it can have values between 0 and 4 depending on the motional constraints and relative orientations of the donor and acceptor fluorophores. If both donor and acceptor have isotropic motion, a value of 2/3 can be used. In other situations, measurements of fluorescence anisotropy can be used (15) to place upper and lower limits on the orientation factor and consequently on the calculated R0 value. We therefore measured fluorescence anisotropies for dansyl at positions 121, 314, 111, and 159, in complex with trypsin and for dansyl at position 360 in complex with anhydrotrypsin. The values that were calculated ranged from 0.12 for positions 121 and 314 and 0.13 for position 360, to 0.09 for position 111 and 0.06 for position 159, indicating significant motional freedom, though not isotropic rotation. Since the crystal structure of trypsin shows the four tryptophans to be partially or completely buried, a value for the anisotropy of 0.4, representing complete immobilization, was used. Using these limiting anisotropies, we calculated upper and lower values for κ2, and hence for R0. The mean value of R0, calculated by using these upper and lower bounds, was 23.3 ± 2.8 Å, which differs only minimally from the value of 22.6 Å calculated assuming a κ2 value of 2/3. The value of 22.6 Å for R0 was therefore used in distance calculations. Quantum yields for all dansyl fluorophores were calculated by comparison with the emission intensity of quinine sulfate.
In all cases a sample was taken from the cuvette immediately after recording the fluorescence spectra and analyzed by SDS/PAGE to confirm the integrity and composition of the sample. Gels were analyzed both by dansyl fluorescence intensity and by Coomassie blue staining. The stoichiometry of inhibition (SI) for every labeled variant was estimated by scanning densitometry of the fluorescence of the gels, to determine the relative amounts of covalent complex versus cleaved α1PI in the mixture. Most labeled variants gave SI values in the range 1.05–1.36. Q111C gave an SI of 1.76. In each case, the changes in fluorescence intensity were corrected for the percentage of complex present.
Construction of a Molecular Model of Covalent Complex.
The model shown in Fig. 4 represents a juxtapositioning of structures of cleaved α1PI and β-trypsin to best account for the experimental distance determinations obtained here. The model was constructed by using the Swiss PDB viewer molecular visualization program. The following criteria were used in constructing the model. (i) The distances between the tryptophans of trypsin and the engineered cysteines of α1PI were brought into closest self-consistent agreement with experimental values. (ii) Obvious backbone steric clashes were avoided, though no attempt at energy minimization was made. (iii) The active site of trypsin was positioned close enough to the P1 residue of α1PI, with the active site facing this, to allow for the existence of a covalent acyl ester linkage between the serine γ-O of trypsin and the backbone carboxyl of the P1 residue. The model in Fig. 4 makes the assumption that the structure of the α1PI moiety in the complex is close to that of cleaved α1PI, and that the structure of the trypsin moiety is close to that of free trypsin. The former assumption is justified by the self-consistency of all experiments with the model based on this, whereas the assumption with regard to trypsin is true only to a first approximation, since we acknowledge that there must be some distortion of the structure caused by impingement against the serpin. However, this approximation will not qualitatively alter the nature of our proposed model.
Figure 4.

Proposed structure for the trypsin-α1PIPittsburgh covalent complex based on fluorescence resonance energy transfer measurements, showing the same views as in Fig. 2. The serpin structure used (yellow) is of cleaved α1PI (17) and the structure of trypsin (red) is for β-trypsin (18). The reactive center loop is in cyan. The active-site serine is in white. The sites of covalent labeling (85, 111, 121, 159, 232, 314, and 360) are in green. The four tryptophans of trypsin (blue) and two tryptophans of α1PI (cyan) are shown (note the approximately symmetrical disposition of the four trypsin tryptophans around the active site of trypsin and the large separation between the two tryptophans of α1PI and residues 121 and 314).
RESULTS AND DISCUSSION
Proximity Perturbation of Fluorophores.
The most direct measure of the effect of proximity of proteinase to one or other of the labeled positions was from change in fluorescence properties of the dansyl-labeled α1PI (wavelength maximum of emission and quantum yield) upon complex formation with trypsin (Table 1). For each covalent complex an appropriate control of reactive center loop-cleaved serpin was used to estimate changes due solely to conformational change.
For dansyl at position 121 there was a 35% increase in quantum yield and a 10-nm blue shift upon forming covalent complex, but no change upon forming noncovalent complex or cleaved serpin. Even more striking was the perturbation in the covalent complex at position 314. This gave a 24-nm blue shift and a 76% increase in quantum yield. Formation of cleaved serpin had very much smaller effects (8% reduction in quantum yield and 2-nm blue shift). Because the serpin moiety in the complex is almost certainly cleaved, the changes seen for the covalent complex must be direct effects of the proteinase and cannot be ascribed to loop insertion, because they differ completely from the changes for cleaved serpin. Dansyl at position 360 showed a 27% increase in quantum yield and a 6-nm blue shift upon forming noncovalent complex, and an 11% reduction in quantum yield and 2-nm blue shift upon forming covalent complex. In contrast, dansyl emission spectra for label at position 85 showed no sensitivity to formation of covalent or noncovalent complex or of cleaved serpin. Spectra for labels at positions 111 and 232 showed no alteration in wavelength maximum for the two types of complex and for cleaved serpin, but small changes of intensity (2–7%) upon complex formation. For label at position 159, there was no effect of forming noncovalent complex, but a 12% reduction in intensity and a 4-nm red shift upon forming covalent complex. This reduction was, however, due solely to loop insertion rather than any proximity effect of the proteinase, because the spectrum for covalent complex was identical to that for reactive center loop-cleaved serpin.
These proximity perturbations are consistent with the proteinase in the covalent complex being localized to the bottom of the serpin, closest to position 314, but still close to position 121, and of the anhydroproteinase in the noncovalent complex being at the distal end, docked with the reactive center loop and therefore very close to P2′. The interpretation for the noncovalent complex is as expected for a simple docking interaction, and it therefore serves as a useful positive control for the method by demonstrating that the dansyl group is sensitive to effects of proximity of the proteinase. These measurements are also in complete agreement with our earlier study, using the same approach with a different fluorophore, NBD, on a smaller subset of the cysteine variants used here (7).
Mapping the Proteinase Location.
To independently and more definitively localize the proteinase in both covalent and noncovalent complexes, we measured the efficiency of resonance energy transfer between donor fluorophore on the proteinase and acceptor fluorophore on the serpin for the same complexes examined above by direct perturbation effects. These measurements were used to calculate the separation between these fluorophores and consequently between the proteins to which they are attached, as described in Materials and Methods. To overcome the problem of obtaining site-specifically attached donor fluorophore on the proteinase, we used the endogenous fluorescence of the four tryptophans of trypsin for all measurements. The acceptor in all cases was the dansyl fluorophore covalently bound to a single cysteine on the serpin.
Measurements were made for seven dansyl-labeled α1PIPittsburgh variants, with cysteines located at positions 85 (helix C1), 111 (near end of strand 2 of β-sheet A), 121 (far end of strand 2 of β-sheet A), 159 (outer face of helix F), 232 (helix F2), 314 (loop between strands 6 and 5 of β-sheet A), and 360 (P2′ position of the reactive center loop) (Fig. 2). Efficiencies were calculated, as described, to eliminate any effects of conformational change.
Because the R0 value for the tryptophan–dansyl pair is relatively small (22.6 Å), only those complexes in which the dansyl group was very close gave energy transfer >10%. This is a consequence of the 1/R6 dependence of the efficiency of transfer, and it ensures that observation of high efficiency of transfer necessitates close proximity. For the covalent complexes, that with dansyl at position 121 gave 46% efficiency of transfer (Table 2 and Fig. 3), and that with dansyl at position 314 gave ≈100% efficiency of transfer (Table 2 and Fig. 3) (both measured from increase in dansyl emission). All other covalent complexes gave very small efficiency of transfer (Table 2), including that for position 159 (Fig. 3). Even with the limitations on the accuracy of the method imposed by a small uncertainty in R0, by the treatment of four discretely placed tryptophans within trypsin as though they were all located at a single effective position (Reff), and by errors in the magnitude of the fluorescence changes (see footnote ∗ to Table 2), these findings unequivocally localize the proteinase to the immediate vicinity of residues 314 and 121, but closer to 314, and at the same time exclude the proteinase from any space within at least 32 Å (expected efficiency <10%) of positions 85, 111, 159, 232, and 360. The only structure compatible with these severe constraints places the proteinase fully at the bottom of the serpin (Fig. 4), with the reactive center loop fully inserted into β-sheet A, to permit sufficient travel of the proteinase to the new location. This placement is in full agreement with the conclusions above from direct perturbation of the fluorophores.
Table 2.
Fluorescence resonance energy transfer parameters for covalent complexes of dansyl-labeled α1PI with trypsin, corrected for effects of conformational change
| Position | Experimental
|
Present structure
|
Ref. 6
|
|||
|---|---|---|---|---|---|---|
| E, %* | R2/3 exp† | Reff calc‡ | E calc, %§ | Reff calc¶ | E calc, %¶ | |
| 85 | 7 | 34.7 | 34.0 | 8 | 52.8 | 0.6 |
| 111 | 2 | 43.2 | 48.4 | 1 | 19.4 | 72 |
| 121 | 46 | 23.2 | 23.8 | 41 | 31.5 | 12 |
| 159 | 5 | 36.8 | 35.1 | 6 | 8.8 | 99.6 |
| 232 | 2 | 41.8 | 53.3 | 5 | 44.3 | 1.7 |
| 314 | 101 | <13.7‖ | 13.6 | 95 | 43 | 2 |
| 360 | 0.2 | 63.6 | 70.1 | 0.1 | 43.1 | 2 |
Measured efficiency of transfer from increase in acceptor emission. The estimated error in recording spectra is ±9% (SD of spectra recorded on different days), which leads to progressively larger maximum errors in the efficiency of energy transfer calculated by using Eq. 1, depending on the magnitude of the transfer. Thus, for small transfer efficiencies (<10%) the maximum error will be <3%; for 100% efficiency, the maximum error would be <20%. Even these uncertainties do not lead to large changes in the measured distances. Thus, if the efficiency for position 314 were 80%, the calculated distance would increase only to 17.9 Å. Similarly, ±10% change of the 46% measured for position 121 would alter the separation to 23.2 ± 1.5 Å. These small uncertainties in distance do not significantly alter the final structure.
Interfluorophore separation calculated from measured efficiency of transfer and using κ2 of 2/3. However, as indicated in Materials and Methods, the R0 value calculated with this value for κ2 (22.6 Å) is almost identical to the value of R0 that is the mean between upper and lower permissible values of R0 (23.2 Å).
Measured interfluorophore separation from structure in Fig. 4, using single effective position for tryptophan calculated using as Reff = [4/∑i (1/Ri6)]1/6, where Ri is the separation of each of the four trypsin tryptophans from the site of labeling.
Calculated efficiency of transfer using model-based Reff and κ2 of 2/3.
Based on the best-guess placement of trypsin relative to the serpin deducted from the proposed model of the complex in figure 3 of Wilczynska et al. (6) for the serpin PAI 1 and the proteinase uPA.
Calculated for efficiency of transfer of >95%.
Figure 3.

Fluorescence emission spectra for dansyl-labeled α1PI variants with primary excitation of tryptophan at 292 nm. Evidence for fluorescence resonance energy transfer between tryptophan and dansyl in covalent (121C and 314C variants) and noncovalent (360C variant) complexes, from increase in emission intensity for complex compared with uncomplexed α1PI. The 159C variant shows minimal enhancement for either covalent or noncovalent complex formation. For each variant the solid line is the spectrum of native α1PI alone, the dot–dashed line is cleaved α1PI, the dotted line is noncovalent complex with anhydrotrypsin, and the dashed line is covalent complex with trypsin. All spectra are scaled to the same effective dansyl concentration.
Although all four tryptophans of trypsin were treated as being located at one average position in our analysis, the effect of this simplification can be evaluated by using the structure in Fig. 4 to back-calculate both the weighted mean position for such an average donor fluorophore and the corresponding efficiency of transfer. For all of the covalent complexes, there is excellent agreement between the expected efficiency of transfer and that observed (Table 2), showing that our conclusions are not compromised by this simplification. Importantly, while resolving the location of the proteinase in the complex, these findings at the same time show that the earlier model of Wilczynska and colleagues (6), which placed the proteinase only part way down the flank of the serpin and abutting helix F, cannot be correct. Indeed, the discordance between the expected efficiencies of energy transfer based on that model (6) and what is observed here experimentally is striking for almost all of the positions (Table 2). Whereas it is possible that the different conclusions between the present study and that of Wilczynska et al. result from the use of different serpin-proteinase pairs in the two studies (PAI1–uPA and α1PI–elastase), it should be noted that these authors concluded that both complexes had the same structure, suggesting a common structural basis for all serpin inhibition, and therefore an independence from the identity of the particular pair used. We have also found previously that both thrombin and trypsin appear to be located in the same region in complex with α1PIPittsburgh (7), again indicating similar behavior of different serpin-proteinase pairs. It should also be realized, however, that the single distance constraint used in defining their model (60 Å between label at positions P1′ and P3 of PAI1) is also compatible with the model proposed here, so that of the two types of data used in their study, only the chemical cross-linking results require a model different from that proposed here. The question is, thus, how their chemical cross-linking results could have led to a very different structural conclusion. A possibility is that only a small fraction of all of the serpin-proteinase complexes were chemically cross-linked, and that conclusions based on analysis of these species are therefore not representative of the structure of the normal complex.
With the exception of label at position P2′ (residue 360), for which 32% efficiency of transfer was measured (from decrease in trypsin tryptophan emission), very little energy transfer was detected for label at any of the other six positions (0–3%) in the noncovalent complexes between anhydrotrypsin and α1PI. This result is expected for a noncovalent complex in which the anhydroproteinase is docked with the reactive center loop, with the P1 residue in the S1 specificity pocket (see Fig. 2). This experiment thus serves as a useful positive control for the resonance energy transfer approach used here. The P2′ residue and its attached dansyl label must be very close-by and consequently give a high efficiency of fluorescence resonance energy transfer. Conversely, the distances between label at the other six positions and the weighted mean position of the tryptophans in anhydrotrypsin are all relatively large (29–73 Å), which should result in correspondingly low efficiency of energy transfer (0.1–2.4%, with the exception of 232, for which 19% efficiency is predicted), as observed. The observed efficiency of transfer to P2′ (32%) is lower than expected for the structure in Fig. 2, but this may result from steric crowding that directs the dansyl group away from the intimate anhydrotrypsin-reactive center loop interface and thereby increases the separation by the full length of the spacer connecting the cysteine sulfur to the dansyl moiety.
Consequences for the Mechanism of Inhibition.
Our conclusion, from two independent sets of measurements, that the structure of the serpin-proteinase complex involves a fully inserted reactive center loop and a proteinase that abuts the underside of the serpin, tethered by a reactive center that is just sufficiently long to hold it there without slack, provides experimental confirmation of the model of Wright and Scarsdale (16). This model is the only one that can readily explain why proteinases of widely different size, shape, and composition can form a common type of complex with serpins that results in a common perturbation of the active site. Such a perturbation could be induced by compressing the proteinase against the bottom of the serpin and hence provide a common mechanism of kinetic trapping of the reaction intermediate. As such, our present findings on a specific serpin-proteinase pair are likely to be generally true for all pairs in which proteinase inhibition uses the full irreversible serpin mechanism. Separate evidence for distortion of the proteinase as the basis of the kinetic trap has been obtained from NMR (9), calorimetric (11), and proteolytic cleavage (8) studies, but without establishing the basis for such distortion. The present findings establish a structural basis for inducing this distortion, though it still remains to be resolved what the nature of the distortion actually is.
Summary.
Quantitative measurements of the separations between dansyl fluorophores at specific sites in α1PI and tryptophans in trypsin in the covalent complex formed between these two proteins have been presented that represent a major advance over our earlier study (7) by permitting localizing of the proteinase in such serpin-proteinase complexes with much greater precision. The resulting model of the complex, with the proteinase at the distal end of the serpin from the initial docking site, is consistent both with direct fluorescence perturbations also reported here and with other fluorescence perturbations reported previously (7). This model provides an experimentally determined structural basis to account for the distortion of the proteinase that appears to be critical for the kinetic trap mechanism by which serpins inhibit proteinases.
Acknowledgments
We thank Dr. Steven Olson for generous access to his SLM8000 fluorometer, help with the anisotropy measurements, and useful discussions on the experiments and the manuscript. This work was supported by Grant HL49234 from the National Institutes of Health (P.G.W.G.) and by a University Fellowship from the University of Illinois at Chicago (E.S.).
ABBREVIATIONS
- α1PI
α1-proteinase inhibitor
- SI
stoichiometry of inhibition
- NBD
7-nitrobenz-2-oxa-1,3-diazole
References
- 1.Gettins P G W, Patston P A, Olson S T. Serpins: Structure, Function and Biology. Austin, TX: Landes; 1996. [Google Scholar]
- 2.Shieh B-H, Potempa J, Travis J. J Biol Chem. 1989;264:13420–13423. [PubMed] [Google Scholar]
- 3.Lawrence D A, Ginsburg D, Day D E, Berkenpas M B, Verhamme I M, Kvassman J-O, Shore J D. J Biol Chem. 1995;270:25309–25312. doi: 10.1074/jbc.270.43.25309. [DOI] [PubMed] [Google Scholar]
- 4.Lawrence D A, Olson S T, Palaniappan S, Ginsburg D. J Biol Chem. 1994;269:27657–27662. [PubMed] [Google Scholar]
- 5.Stratikos E, Gettins P G W. Proc Natl Acad Sci USA. 1997;94:453–458. doi: 10.1073/pnas.94.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilczynska M, Fa M, Karolin J, Ohlsson P I, Johansson L B Å, Ny T. Nat Struct Biol. 1997;4:354–357. doi: 10.1038/nsb0597-354. [DOI] [PubMed] [Google Scholar]
- 7.Stratikos E, Gettins P G W. J Biol Chem. 1998;273:15582–15589. doi: 10.1074/jbc.273.25.15582. [DOI] [PubMed] [Google Scholar]
- 8.Kaslik G, Patthy A, Bálint M, Gráf L. FEBS Lett. 1995;370:179–183. doi: 10.1016/0014-5793(95)00816-r. [DOI] [PubMed] [Google Scholar]
- 9.Plotnick M I, Mayne L, Schechter N M, Rubin H. Biochemistry. 1996;35:7586–7590. doi: 10.1021/bi960233w. [DOI] [PubMed] [Google Scholar]
- 10.Stavridi E S, O’Malley K, Lukacs C M, Moore W T, Lambris J D, Christianson D W, Rubin H, Cooperman B S. Biochemistry. 1996;35:10608–10615. doi: 10.1021/bi9605806. [DOI] [PubMed] [Google Scholar]
- 11.Kaslik G, Kardos J, Szabó L, Závodszky P, Westler W M, Markley J L, Gráf L. Biochemistry. 1997;36:5455–5474. doi: 10.1021/bi962931m. [DOI] [PubMed] [Google Scholar]
- 12.Owen M C, Brennan S O, Lewis J H, Carrell R W. N Engl J Med. 1983;309:694–698. doi: 10.1056/NEJM198309223091203. [DOI] [PubMed] [Google Scholar]
- 13.Pannell R, Johnson D, Travis J. Biochemistry. 1974;13:5439–5445. doi: 10.1021/bi00723a031. [DOI] [PubMed] [Google Scholar]
- 14.Ako H, Foster R J, Ryan C A. Biochem Biophys Res Commun. 1972;47:1402–1407. doi: 10.1016/0006-291x(72)90228-8. [DOI] [PubMed] [Google Scholar]
- 15.Dale R E, Eisinger J, Blumberg W E. Biophys J. 1979;26:161–194. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wright H T, Scarsdale J N. Proteins. 1995;22:210–225. doi: 10.1002/prot.340220303. [DOI] [PubMed] [Google Scholar]
- 17.Löbermann H, Tokuoka R, Deisenhofer J, Huber R. J Mol Biol. 1984;177:731–757. [PubMed] [Google Scholar]
- 18.Marquart M, Walter J, Deisenhofer J, Bode W, Huber R. Acta Crystallogr B. 1983;39:480–490. [Google Scholar]