

Abstract
The bacterial enhancer-binding protein nitrogen regulatory protein C (NtrC) activates transcription by σ54-containing RNA polymerase in a reaction that depends on ATP hydrolysis. Phosphorylation of an aspartate residue in the N-terminal receiver domain of NtrC induces oligomerization of the protein and activates the ATPase activity, which is a function of its central output domain. To study the role of the receiver domain of NtrC, which is known to act positively, we isolated mutant forms of the protein carrying single cysteine residues and derivatized them with a sulfhydryl-specific nitroxide reagent for electron paramagnetic resonance studies. Single cysteines were placed at four positions at which we had obtained constitutive amino acid substitutions, those that yield activity without phosphorylation. In only one case, derivatized C86 in α-helix 4 of the receiver domain, did the motion of the side chain become dramatically slower upon phosphorylation. Importantly, derivatized NtrCD86C (NtrCD86C*) activated transcription normally. Additional experiments indicated that the spectral change observed upon phosphorylation of NtrCD86C* was due to interdomain interactions rather than a conformational change within the N-terminal domain itself. These interactions did not appear to occur within a monomer. Although it is not clear whether the spectral change seen upon phosphorylation of NtrCD86C* is due to an interaction that occurs within a dimer of NtrC or requires the formation of higher-order oligomers, the change indicated that α-helix 4 of the receiver domain probably plays an important role in communication with the remainder of the protein.
In bacteria, so-called “two-component” signal transduction systems are perhaps the most common means of responding to environmental change (1–3). The “second component” in such systems is a receiver domain that becomes phosphorylated on an aspartate residue and modulates the activity of its cognate output domain, often a transcriptional activation domain. The “first component” is a histidine autokinase that functions as the physiological phosphodonor. The phosphoaspartate linkage of receiver domains often is labile, and no structure has been obtained for either an intact phosphorylated receiver protein or an isolated phosphorylated receiver domain. Hence, the nature of the conformational changes that accompany phosphorylation of a receiver domain and the means by which they are transmitted to the cognate output domain remain poorly defined.
The bacterial enhancer-binding protein nitrogen regulatory protein C (NtrC) is a well studied receiver protein (4). When phosphorylated at aspartate-54, NtrC activates transcription by the σ54-holoenzyme form of RNA polymerase. Phosphorylation of NtrC, which is dimeric in solution (110 kDa), induces the formation of large oligomers—octamers or possibly hexamers—that have the capacity to hydrolyze ATP and thereby provide the energy necessary for transcriptional activation. The N-terminal domain of NtrC is known to act positively—that is, removing it does not substitute for phosphorylation. Hence, to bring about transcriptional activation the phosphorylated domain presumably contacts either another receiver domain or the output domain of NtrC, which bears the determinants for ATP hydrolysis and interaction with σ54-holoenzyme. To obtain physical evidence for such an interaction, we constructed forms of the NtrC protein from Salmonella typhimurium containing single cysteine residues at selected positions within the N-terminal domain and the beginning of the output domain, derivatized these with the sulfhydryl-specific (1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl) methanethiosulfonate spin label (MTSSL), and probed for phosphorylation-induced changes in mobility of the nitroxide side chains that were introduced by EPR spectroscopy (5, 6). The positions chosen for placement of single cysteines were three of the positions in the N-terminal domain (13.6 kDa) that had yielded so-called “constitutive” substitutions previously, positions 86, 89, and 115 (Fig. 1), and a similar position at the beginning of the central domain, position 160 (7). Constitutive substitutions are those that result in some degree of activity without a requirement for phosphorylation, and, in fact, two such substitutions in the N-terminal domain, D86N and A89T, have been shown by NMR spectroscopy to cause conformational changes in this domain similar to those caused by substitution of glutamate for aspartate at the site of phosphorylation, D54E (8), the fourth constitutive substitution in the N-terminal domain. Positions 86 and 89 in the N-terminal domain lie in α-helix 4, whereas position 115 lies in α-helix 5. Position 160 in the central domain lies at the end of what is predicted to be a long α-helix that is postulated to function specifically in communication between the N-terminal and output domains (9); the S160F constitutive substitution at this position appears to bypass the requirement for phosphorylation by facilitating formation of active oligomers upon binding of unphosphorylated NtrC to a DNA enhancer (7).
Figure 1.

Ribbon diagram of the N-terminal receiver domain of NtrC (residues 1–124) (8). The side chain of the active-site residue D54 and positions of cysteine substitutions are shown. The N-terminal domain is connected to the remainder of the protein, whose structure has not been determined, by a flexible linker (1). An intact NtrC monomer contains 469 residues (4).
MATERIALS AND METHODS
Mutagenesis to Obtain NtrC Proteins Carrying Single Cysteine Residues and Construction of Vectors Allowing Overproduction of Maltose Binding Protein (MBP) Fusion Proteins Derived from Them.
Plasmid pJES990, which encodes an NtrC protein lacking the two natural cysteines (designated, simply, NtrC hereafter) was derived from pJES608 (8), which encodes NtrCwild-type, in the following steps. Oligonucleotide site-directed mutagenesis was used to effect the substitutions C30V and C364A in plasmids pJES988 and 989, respectively. These substitutions were chosen by comparison with other NtrC proteins (C30V) or with other activators of σ54-holoenzyme (C364A) (9). The appropriate fragments then were combined by standard cloning procedures to yield pJES990. Single-stranded DNA from pJES 990 was used as template for oligonucleotide site-directed mutagenesis to yield plasmids pJES1016 (encodes NtrCD86C), pJES1057 (NtrCA89C), pJES1058 (NtrCV115C), and pJES1017 (NtrCS160C). The presence of the desired mutations was confirmed by dideoxy-nucleotide DNA sequencing.
The KpnI/CspI fragment from plasmid pJES990 was cloned into the corresponding sites of pJES559 (10) to yield pJES994, which encodes MBP-NtrC. The KpnI/CspI fragments from plasmids pJES1016, pJES1057, pJES1058, and pJES1017 were cloned into the corresponding sites of pJES994 to yield, respectively, plasmids pJES1020 (MBP-NtrCD86C), pJES1087(MBP-NtrCA89C), pJES1088 (MBP-NtrCV115C), and pJES1021 (MBP-NtrCS160C).
Plasmid pJES1039, which encodes an MBP fusion to the N-terminal fragment of NtrCD86C (lacking C30), and plasmids pJES1068 and pJES1069, which encode MBP-NtrCS160F and MBP-NtrCD86C, S160F, respectively, were constructed by standard cloning procedures. To construct plasmid pJES1098, which encodes MBP-NtrCD86C, Δ444–469, the 3.2-kb CspI/NdeI fragment from pJES1020 was replaced by the corresponding 2.7-kb fragment from pJES713 (11).
Purification of MBP-NtrC Fusion Proteins and Assays of Their Activities.
MBP-NtrC fusion proteins were purified essentially as described (10) from cells of Escherichia coli strain DH5α carrying the proper overproduction vector, dialyzed against B buffer [50 mM Tris-acetate, pH 8.3/50 mM KCl/5% (vol/vol) glycerol/0.1 mM EDTA/1 mM DTT], and frozen quickly in dry ice and stored at −80°C. They were >95% pure as assessed by visual inspection of SDS-polyacrylamide gels stained with Coomassie brilliant blue R250. Protein concentrations (given as dimer concentrations unless noted otherwise) were determined from absorption at 280 nm in the presence of 6 M guanidine hydrochloride by using an extinction coefficient of 111,310 M−1 cm−1 (12). Proteins were assayed for their capacity to catalyze open-complex formation in a single-cycle transcription assay (13, 14) by using pJES534 (4) as template. Assays for ATPase activity were performed as described (7, 15). Stimulation of ATPase activity by DNA binding was assessed by using synthetic DNA oligomers of 69 bp (7); one oligomer contained a strong enhancer—two strong binding sites for NtrC—and the other lacked specific binding sites. DTT was omitted from all buffers used to study activities of spin-labeled proteins or to compare them with those of the corresponding underivatized proteins.
Gel-Filtration Chromatography to Assess Oligomerization State.
Sephacryl S-300 HR was packed in an XK16/70 column (Pharmacia) to a final bed volume of 105 ml. For sieving unphosphorylated or phosphorylated NtrC, the column was equilibrated in B buffer containing 8 mM MgCl2 or 8 mM MgCl2/10 mM carbamoyl phosphate, respectively. It was run at 4°C at 0.25 ml/min, and protein was detected by monitoring absorption at 280 nm. NtrC proteins were phosphorylated for 10 min at room temperature immediately before loading. A high-molecular-weight gel-filtration kit (Pharmacia) was used to calibrate the column just before sieving MBP-NtrC proteins.
Spin Labeling and EPR Spectroscopy.
For spin labeling, proteins were dialyzed against modified B buffer (pH 7.8, no DTT) to remove DTT and subsequently treated with a 5-fold molar excess of MTSSL at pH 7.8 for 4 hr. Excess spin label was removed by dialysis against modified B buffer. EPR spectra were collected by using a Bruker ESP 300 E spectrometer equipped with a loop-gap resonator (Medical Advances, Milwaukee, WI) and a low-noise amplifier (Miteq, Hauppauge, NY). Spectra were collected by using 1-mW microwave power, and the modulation amplitude was optimized to prevent spectral distortion (≤2 G). Doubly integrated EPR spectra were compared with that for a 100-μM 2,2,6,6-tetramethylpiperidine-N-oxyl standard. To obtain the extent of labeling the spin count was divided by the protein concentration. All the derivatized proteins contained 1.0 label per cysteine (±10%).
Analysis of EPR line shapes yields information on the mobility of the nitroxide side chain (16). Motion of the nitroxide relative to the protein is due to rotations around single bonds within the nitroxide side chain itself or to segmental motion of the backbone.
Accessibility Measurements.
For power saturation measurements, samples (≈5 μl) were placed in TPX capillaries (Medical Advances, Milwaukee, WI), and a series of EPR spectra was collected as the incident microwave power was varied for samples under nitrogen or in the presence of the paramagnetic reagents O2 from air or NiEDDA (5 mM) under nitrogen (17, 18). The saturation curves for the mI = 0 lines of the EPR spectra were analyzed to obtain P1/2, the microwave power at which the first-derivative amplitude is reduced to one-half of the value expected in the absence of saturation. The quantity ΔP1/2 = P1/2(O2 or NiEDDA) − P1/2(N2) is proportional to the collision frequency between the nitroxide and the paramagnetic reagent, but it also depends on the spectral line width and the spectrometer characteristics (17, 19). The change in collision frequency can be obtained by calculating the ratio:
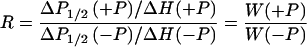 |
where ΔH is the peak-to-peak line width of the mI = 0 line and W(+P) and W(−P) are the quantities proportional to collision frequencies between the paramagnetic reagent and the nitroxide in phosphorylated and unphosphorylated forms of the protein, respectively. W is proportional to the solvent-exposed area of the nitroxide.
RESULTS
Survey of Putative Communication Regions in NtrC by Using EPR Spectroscopy.
To introduce single cysteines at particular positions of NtrCwild-type, we first constructed a cysteine-free NtrC derivative, hereafter called, simply, NtrC (see Materials and Methods). We then introduced single cysteines into NtrC at positions we thought might participate in interdomain communication—see Introduction—and derivatized these cysteine residues with MTSSL. The resulting derivatives, which were purified as MBP-NtrC fusion proteins, are designated NtrCD86C*, NtrCA89C*, NtrCV115C*, and NtrCS160C*. We also derivatized a control protein carrying only the natural cysteine at position 30 to obtain NtrCC30*. Cysteines at all positions chosen were derivatized nearly 100% as assessed by EPR (data not shown). The line shapes of EPR spectra for NtrCD86C* and NtrCA89C* (Fig. 2) revealed that motion of the nitroxides was relatively fast (rotational correlation time τR between 2.5 and 3 ns), indicating that the nitroxides were at surface-exposed sites (20). By contrast, the EPR spectrum for NtrCV115C* showed that the nitroxide was highly immobilized. For NtrCS160C* and NtrCC30*, the EPR spectra revealed that motion of the nitroxides was somewhat slowed (τ ≈ 5 ns), suggesting that they make some tertiary contacts. Phosphorylation of the five spin-labeled proteins resulted in large changes in the EPR spectrum only for NtrCD86C*: motion of the nitroxide side chain became retarded with τR near 10 ns. Therefore, we focused on characterizing NtrCD86C* and the basis for the spectral change. A similar spectral change, but of lesser magnitude, was observed for NtrCA89C*, which also carries a substitution in helix 4 of the N-terminal domain.
Figure 2.

Effect of phosphorylation on EPR spectra of NtrC derivatives. NtrC proteins (25-μM dimer) carried a nitroxide spin label on single cysteine residues at the positions indicated (see text). Dashed and solid lines indicate spectra for unphosphorylated and phosphorylated forms of these proteins, respectively, at 22–23°C.
To complement EPR line-shape analysis for NtrCD86C* we measured the accessibility of the spin label to the small paramagnetic molecules O2 and Ni-EDDA in solution (17, 18). In the unphosphorylated protein the nitroxide was highly accessible to the water-soluble NiEDDA complex. In fact, the accessibility parameter W was comparable to those for fully exposed nitroxide side chains in other proteins and peptides under similar conditions (18, 21). When NtrCD86C* was phosphorylated, W values for NiEDDA and oxygen were reduced by factors of 1.8 and 1.7, respectively. Together with the change in EPR line shape that occurs upon phosphorylation of NtrCD86C*, the decrease in accessibility parameters for small paramagnetic molecules is commensurate with the view that the nitroxide side chain has formed new tertiary contacts that both immobilize it and reduce its accessibility to solvent.
Properties of NtrCD86C and NtrCD86C* and Survey of the Properties of Other Proteins.
To determine the biochemical effects of the cysteine substitution at position 86 before and after spin labeling, we compared the properties of NtrCD86C and NtrCD86C* with those of NtrC (cysteine-free) and NtrCwild-type. Both transcriptional activation and ATPase activities were similar for all of the proteins, and in all cases they depended on phosphorylation (data not shown and Table 1). All of the unphosphorylated proteins behaved as dimers on a gel-filtration column (at 5- and 25-μM dimer) (Fig. 3). When phosphorylated, all yielded a mixture of dimers and larger oligomers at 5 μM and, essentially, all larger oligomers at 25 μM. Thus, neither the amino acid substitutions present in NtrCD86C* nor the spin label resulted in any notable loss of activity or change in oligomerization state.
Table 1.
ATPase activity of spin-labeled NtrC derivatives*
| NtrC derivative | Concentration at 1 μM
|
Concentration at 5 μM
|
||
|---|---|---|---|---|
| Without phosphorylation | With phosphorylation† | Without phosphorylation | With phosphorylation† | |
| Wild type | — | — | 4 | 2,525 |
| NtrC (minus cysteines) | — | — | 21 | 2,195 |
| NtrCD86C | 1 | 699 | 27 | 2,115 |
| NtrCD86C* | 1 | 580 | 4 | 2,023 |
| NtrCD86C, S160F | 908 | 1,813 | — | — |
| NtrCD86C*, S160F | 181 | 1,896 | — | — |
ATPase activity (pmol/10 μl per 10 min) was measured at 37°C.
NtrC proteins were phosphorylated with 10 mM carbamoyl phosphate and 8 mM MgCl2 for 10 min at room temperature before the addition of ATP.
Figure 3.

Gel-filtration chromatography of NtrC derivatives. Spin-labeled proteins at the concentrations indicated were sieved as described in Materials and Methods; the designation + P indicates that they were phosphorylated. Traces indicate absorbance at 280 nm, and the vertical bars indicate positions of elution of molecular mass standards used to calibrate the column (thyroglobulin, 669 kDa; ferritin, 440 kDa; catalase, 232 kDa; BSA, 67 kDa). Larger volumes were used for samples at 5 μM than at 25 μM (200 vs. 50 μl, respectively), and sensitivities of detection were set differently in the two cases.
Because ATP is present for all of our biochemical assays, we demonstrated that ATP did not influence the EPR spectrum of NtrCD86C* before or after phosphorylation (data not shown). We also performed EPR analysis of NtrCD86C* at 37°C and 4°C because our biochemical assays were done at the former temperature and sieving experiments were done at the latter. Spectra at 37°C indicated that motion of the nitroxide was greater than at room temperature but that the extent of immobilization upon phosphorylation was similar at the two temperatures. At 4°C motion of the nitroxide was somewhat retarded but there was a decrease in mobility upon phosphorylation, as at the other two temperatures.
When tested at 20 nM, the phosphorylated forms of NtrCA89C, NtrCV115C, NtrCS160C, and the control protein carrying only the natural cysteine at position 30 activated transcription at least half as well as phosphorylated NtrC (data not shown). After derivatization with MTSSL, phosphorylated NtrCA89C* retained good ability to activate transcription, whereas phosphorylated NtrCV115C* essentially had lost this ability. Phosphorylated NtrCS160C* and NtrCC30*, which were not tested for transcriptional activation, had lost ATPase activity (tested at 0.5- and 1-μM dimer; data not shown).
The EPR Spectral Change of NtrCD86C* upon Phosphorylation Is Not Due to a Conformational Change Within the N-Terminal Domain.
EPR spectra of the isolated, spin-labeled N-terminal domain of NtrCD86C* (MBP-N-terminal domain fusion of 55 kDa) indicated that the nitroxide side chain was very mobile (τR ≈ 1.5 ns) and that its mobility did not change upon phosphorylation (Fig. 4). The motion was slightly faster than in the intact MBP-NtrC fusion protein because of the smaller size of the N-terminal fragment, which is 13.6 kDa by itself. The expected τR for the NtrC N-terminal fragment (13.6 kDa) is ≈6 ns (22), and the τR determined directly by NMR spectroscopy at high concentrations was 9–10 ns (23); hence, had the label become immobilized relative to the fragment upon phosphorylation, the EPR spectrum should have shown a clearly discernible change in line shape that reflected the latter tumbling rates. The results demonstrated that the EPR spectral change observed upon phosphorylation of intact NtrCD86C* was not due to intradomain conformational changes within the N terminus itself.
Figure 4.

Effect of the oligomerization state of NtrC on EPR spectral changes at position 86. All proteins (25-μM dimer) carried a spin label at position 86 (D86C*; not designated). Dashed and solid lines indicate spectra for unphosphorylated and phosphorylated forms, respectively, of the proteins indicated (t = 22–23°C). (a) NtrCD86C*. (b) Isolated N-terminal domain of NtrCD86C*. (c) NtrCD86C*, S160F. (d) NtrCD86C*, S160F + enhancer DNA (12.5-μM fragment). (e) NtrCD86C*, Δ444–469 (50-μM monomer).
Formation of Oligomers Is Not Sufficient for the EPR Spectral Change.
To determine whether the putative tertiary or quaternary interactions responsible for the change in EPR signal of NtrCD86C* upon phosphorylation could be accounted for by interactions between dimers that occurred upon formation of large NtrC oligomers, we introduced a second amino acid substitution, S160F (see Introduction), into NtrCD86C to yield NtrCD86C, S160F. Previous studies had indicated that unphosphorylated NtrCS160F could form large, active oligomers upon binding to an enhancer (7). Hence, in the presence of enhancer DNA, the S160F substitution in the central domain appears to bypass the requirement for phosphorylation. Preliminary characterization of NtrC D86C, S160F indicated that it had at least as much ATPase activity without phosphorylation as did phosphorylated NtrCD86C and that phosphorylation stimulated this activity (Table 1).
Although unphosphorylated, spin-labeled NtrCD86C*, S160F had lower ATPase activity than the underivatized protein, this activity was still 20–30% that of phosphorylated NtrCD86C* and, hence, of phosphorylated NtrCD86C or phosphorylated NtrC (Table 1). Moreover, the transcriptional activation capacity of unphosphorylated NtrCD86C*, S160F was at least as great as that of phosphorylated NtrCD86C* (Fig. 5). When phosphorylated, NtrCD86C*, S160F had ATPase activity comparable to that of the underivatized protein and its transcriptional activation capacity was increased.
Figure 5.

Effect of phosphorylation on transcriptional activation by NtrC derivatives. Catalysis of open-complex formation by phosphorylated (+P) and unphosphorylated (−P) forms of spin-labeled NtrCD86C, S160F (D86C*S160F) and NtrCD86C (D86C*) was assessed on plasmid pJES534 (1 nM) in a single-cycle transcription assay as described in Materials and Methods.
As was observed for NtrCS160F (7), enhancer DNA greatly stimulated the ATPase activity of unphosphorylated NtrCD86C*, S160F and the corresponding underivatized protein (5-fold in each case, at 200 nM protein and 100 nM DNA fragment; see Materials and Methods), indicating that the enhancer stimulated the formation of active oligomers by these proteins at low protein concentrations. As expected, DNA lacking binding sites for NtrC had no effect (1.3-fold stimulation). At the high protein concentrations used to collect EPR spectra (5 or 25 μM), NtrCD86C*, S160F sieved as all large oligomers on a gel-filtration column, and this was also the case after phosphorylation (Fig. 3).
As was true for NtrCD86C*, EPR spectra indicated that the nitroxide side chain was mobile in unphosphorylated NtrCD86C*, S160F and immobilized in the phosphorylated protein (Fig. 4). Taken together with the results of activity assays and behavior on gel-filtration columns, the result for the unphosphorylated protein indicates that formation of active oligomers by a bypass mechanism is not sufficient to yield the full EPR spectral change observed upon phosphorylation. To strengthen this conclusion, we showed that even binding to enhancer DNA was not sufficient to yield the full spectral change. When NtrCD86C*, S160F was mixed with an enhancer-bearing DNA fragment at molar ratios of protein to NtrC-binding sites of 1:0.25, 1:0.5, 1:1, or 1:2, EPR spectra indicated that the nitroxide side chain remained mobile (Fig. 4 and data not shown). When NtrCD86C*S160F was phosphorylated, the characteristic change in the EPR line shape again was observed, which is indicative of the formation of tertiary contacts by the nitroxide side chain. Spectra in the two cases were similar to those of unphosphorylated NtrCD86C* and phosphorylated NtrCD86C*, respectively.
The EPR Spectral Change Does Not Occur Within a Monomer.
To determine whether the EPR spectral change occurred within a monomer, we prepared a truncated form of NtrCD86C lacking the helix-turn-helix DNA-binding motif, which makes a large contribution to dimerization (10), and derivatized it with MTSSL. When phosphorylated, NtrCD86C*, Δ444–469 showed little EPR spectral change (Fig. 4). To determine whether this change could be accounted for by residual dimer at the high concentrations used for EPR spectroscopy, we compared spectra at 50-μM monomer (25-μM potential dimer) with those at 10-μM monomer (5-μM potential dimer). In contrast to the case for NtrCD86C*, which showed the same change in spectrum at 5 μM, the degree of spectral change for the truncated protein was less at the lower concentration (data not shown), providing preliminary evidence that the interdomain interactions that occur upon phosphorylation do not occur within a monomer.
DISCUSSION
Phosphorylated NtrCD86C* essentially has a normal ability to oligomerize, hydrolyze ATP, and activate transcription. Because phosphorylated NtrC has a rapid autophosphatase activity and, therefore, phosphorylation must be maintained in situ (24), EPR spectroscopy is a technique of choice for monitoring phosphorylation-induced conformational changes. EPR spectra indicate that the nitroxide side chain at position 86 becomes less mobile as a consequence of tertiary interactions when NtrCD86C* is phosphorylated and that the same is true to a lesser extent at position 89. Taken together, the additional results presented indicate that (i) the EPR spectral change observed upon phosphorylation of NtrCD86C* and, probably, NtrCD89C* is due to interdomain interactions rather than to conformational changes within the N-terminal receiver domain; (ii) interdomain interactions apparently do not occur within a monomer; and (iii) formation of large oligomers by a bypass mechanism that does not depend on phosphorylation (see Results) is not sufficient to yield the relevant interdomain interactions, and, hence, they are not a trivial consequence of increased molecular mass. We do not know whether the phosphorylation-induced change in the EPR spectrum of NtrCD86C* is due to interdomain interactions between the monomers of an NtrC dimer or requires the formation of larger oligomers. Precedent for phosphorylation-induced conformational changes affecting the former is provided by yeast and muscle glycogen phosphorylases (25, 26).
Given that the phosphorylated N-terminal domain of NtrCwild-type shows no tendency to dimerize even at concentrations as high as 0.3 mM (D. Kern and D. Wemmer, personal communication) and that there is independent evidence that the oligomerization determinants of the protein lie outside this domain (7), we hypothesize that the EPR spectral change observed upon phosphorylation of NtrCD86C* reflects an interaction of the N-terminal domain with the output domain. If this is the case, helix 4 of the N-terminal domain, which lies on the activating surface of the domain—the so-called 3445 face (8)—is likely to play a specific role in communication with the remainder of the protein. Preliminary results obtained by derivatizing C86 of NtrCD86C with an iron-EDTA chelate, a reagent that is capable of hydrolyzing the protein backbone under appropriate conditions (27), indicate that there is a phosphorylation-dependent cleavage of the backbone of NtrC that occurs outside its N-terminal domain (J.L., J. Owens, S.K., and C. Meares, unpublished data).
Acknowledgments
We are grateful to Jeffrey Pelton for the preparation of Fig. 1. This work was supported by National Institutes of Health Grants GM38361 and GM51290 to S.K. and Y.-K.S., respectively, a Searle Scholarship to Y.-K.S., and a National Institutes of Health training grant to J.L.
ABBREVIATIONS
- MTSSL
methanethiosulfonate spin label
- MBP
maltose binding protein
References
- 1.Parkinson J S, Kofoid E C. Annu Rev Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- 2.Mizuno T, Kaneko T, Tabata S. DNA Res. 1996;3:407–414. doi: 10.1093/dnares/3.6.407. [DOI] [PubMed] [Google Scholar]
- 3.Mizuno T. DNA Res. 1997;4:161–168. doi: 10.1093/dnares/4.2.161. [DOI] [PubMed] [Google Scholar]
- 4.Porter S C, North A K, Kustu S. In: Two-Component Signal Transduction. Hoch J A, Silhavy T J, editors. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 147–158. [Google Scholar]
- 5.Milhauser G L. Trends Biochem Sci. 1992;17:448–452. doi: 10.1016/0968-0004(92)90486-s. [DOI] [PubMed] [Google Scholar]
- 6.Hubbell W L, Altenbach C. Curr Opin Struct Biol. 1994;4:566–573. [Google Scholar]
- 7.Flashner Y, Weiss D S, Keener J, Kustu S. J Mol Biol. 1995;249:700–713. doi: 10.1006/jmbi.1995.0330. [DOI] [PubMed] [Google Scholar]
- 8.Nohaile M, Kern D, Wemmer D, Stedman K, Kustu S. J Mol Biol. 1997;273:299–316. doi: 10.1006/jmbi.1997.1296. [DOI] [PubMed] [Google Scholar]
- 9.Osuna J, Soberón X, Morett E. Protein Sci. 1997;6:543–555. doi: 10.1002/pro.5560060304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klose K E, North A K, Stedman K M, Kustu S. J Mol Biol. 1994;241:233–245. doi: 10.1006/jmbi.1994.1492. [DOI] [PubMed] [Google Scholar]
- 11.North A K, Kustu S. J Mol Biol. 1997;267:17–36. doi: 10.1006/jmbi.1996.0838. [DOI] [PubMed] [Google Scholar]
- 12.Edelhoch H. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 13.Popham D L, Szeto D, Keener J, Kustu S. Science. 1989;243:629–635. doi: 10.1126/science.2563595. [DOI] [PubMed] [Google Scholar]
- 14.Wedel A, Weiss D, Popham D, Dröge P, Kustu S. Science. 1990;248:486–490. doi: 10.1126/science.1970441. [DOI] [PubMed] [Google Scholar]
- 15.Weiss D S, Batut J, Klose K E, Keener J, Kustu S. Cell. 1991;67:155–167. doi: 10.1016/0092-8674(91)90579-n. [DOI] [PubMed] [Google Scholar]
- 16.Scheider D J, Freed J H. In: Biological Magnetic Resonance. Berliner L J, Ruben J, editors. Vol. 8. New York: Plenum; 1989. pp. 1–76. [Google Scholar]
- 17.Altenbach C, Flitsch S L, Khorana H G, Hubbell W L. Biochemistry. 1989;28:7806–7812. doi: 10.1021/bi00445a042. [DOI] [PubMed] [Google Scholar]
- 18.Yu Y G, Thorgeirsson T E, Shin Y-K. Biochemistry. 1994;33:14221–14226. doi: 10.1021/bi00251a034. [DOI] [PubMed] [Google Scholar]
- 19.Farahbakhsh Z T, Altenbach C, Hubbell W L. Photochem Photobiol. 1992;56:1019–1033. doi: 10.1111/j.1751-1097.1992.tb09725.x. [DOI] [PubMed] [Google Scholar]
- 20.Mchaourab H S, Lietzow M A, Hideg K, Hubbell W L. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 21.Kim C-H, Macosko J C, Shin Y-K. Biochemistry. 1998;37:137–144. doi: 10.1021/bi971982w. [DOI] [PubMed] [Google Scholar]
- 22.Cantor C R, Schimmel P R. Biophysical Chemistry II. San Francisco: Freeman; 1980. [Google Scholar]
- 23.Nohaile M J. Ph.D. Thesis. Berkeley: Univ. of California; 1996. [Google Scholar]
- 24.Keener J, Kustu S. Proc Natl Acad Sci USA. 1988;85:4976–4980. doi: 10.1073/pnas.85.14.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin K, Hwang P K, Fletterick R J. J Biol Chem. 1995;270:26833–26839. doi: 10.1074/jbc.270.45.26833. [DOI] [PubMed] [Google Scholar]
- 26.Johnson L N, O’Reilly M. Curr Opin Struct Biol. 1996;6:762–769. doi: 10.1016/s0959-440x(96)80005-4. [DOI] [PubMed] [Google Scholar]
- 27.Greiner D P, Miyake R, Moran J K, Jones A D, Negishi T, Ishihama A, Meares C F. Bioconjugate Chem. 1997;8:44–48. doi: 10.1021/bc9600731. [DOI] [PubMed] [Google Scholar]