

Summary
Synaptic potentials originating at distal dendritic locations are severely attenuated when they reach the soma and, thus, are poor at driving somatic spikes. Nonetheless, distal inputs convey essential information, suggesting that such inputs may be important for compartmentalized dendritic signaling. Here we report a new plasticity rule in which stimulation of distal perforant path inputs to hippocampal CA1 pyramidal neurons induces long-term potentiation at the CA1 proximal Schaffer collateral synapses when the two inputs are paired at a precise interval. This subthreshold form of heterosynaptic plasticity occurs in the absence of somatic spiking but requires activation of both NMDA receptors and IP3 receptor-dependent release of Ca2+ from internal stores. Our results suggest that direct sensory information arriving at distal CA1 synapses through the perforant path may serve a novel function by providing compartmentalized, instructive signals that assess the saliency of mnemonic information propagated through the hippocampal circuit to proximal synapses.
Introduction
Neurons extend complex dendritic arbors at which they transform the inputs from thousands of synapses to produce a meaningful output. However, synaptic potentials initiated along the dendritic tree can be greatly attenuated and slowed when they arrive at the site of action potential initiation near the soma due to the passive cable properties of the dendrites (Gulledge et al., 2005; Williams and Stuart, 2003). To help compensate for this distortion, which increases with increasing distance of an input from the soma, dendrites employ a number of active mechanisms to boost the amplitude and speed the time course of synaptic potentials in a distance-dependent manner, thereby normalizing the integration of synaptic potentials at the soma (Hausser et al., 2000; Magee, 2000). However, neurons may also exploit the signaling constraints of their dendritic architecture to perform unique computations that are adapted to the function of the circuit in which they are embedded (London and Hausser, 2005). Here we report a novel heterosynaptic learning rule in which the delay caused by the propagation of distal synaptic potentials along the dendrites of a hippocampal CA1 pyramidal neuron is used to assess the appropriate timing of information propagated through the hippocampal circuit that arrives at proximal synapses.
The view that CA1 neurons may exploit their dendritic architecture to perform computations important for the hippocampal circuit dates to studies of Cajal (Cajal, 1911), who first demonstrated that information from the entorhinal cortex arrives at CA1 pyramidal neurons through two main pathways that are segregated to distinct regions of the apical dendrites (Amaral and Witter, 1989; Steward, 1976). The perforant path inputs, which provide direct sensory information from the entorhinal cortex, terminate on the distal CA1 dendrites in stratum lacunosum-moleculare (SLM). In contrast, the Schaffer collateral inputs from hippocampal CA3 pyramidal neurons, which provide indirect information from the entorhinal cortex that has been processed through the hippocampal trisynaptic circuit, terminate on more proximal CA1 dendrites in stratum radiatum (SR). The trisynaptic path introduces a delay-line architecture so that information arising from entorhinal cortex arrives at the distal CA1 dendrites 10-20 ms prior to the arrival of information at the proximal CA1 dendrites (Yeckel and Berger, 1990). This circuitry has led to the postulate that CA1 neurons compare direct sensory information with memory traces from associative hippocampal networks (Lisman, 1999), although the nature of the comparison is unclear.
Several recent studies have provided evidence that the direct cortical input to CA1 neurons conveys essential spatial information to the hippocampus (Brun et al., 2002; Fyhn et al., 2004; Steffenach et al., 2005) that is sufficient for some forms of spatial memory (Brun et al., 2002) and necessary for memory consolidation (Remondes and Schuman, 2004). Moreover, enhanced long-term synaptic plasticity in this pathway is correlated with enhanced spatial learning (Nolan et al., 2004). The behavioral importance of the distal synapses, however, contrasts with their weak influence on the somatic voltage. The excitatory drive of the distal inputs is reduced by the passive cable properties of the dendrites, robust feedforward inhibition (Colbert and Levy, 1993; Empson and Heinemann, 1995; Levy et al., 1995a) and the high levels of expression in the distal dendrites of ion channels that decrease local membrane resistance (Lörincz et al., 2002; Magee, 1999; Notomi and Shigemoto, 2004). Moreover, in vivo single unit recordings have revealed that the neurons in the superficial layers of the entorhinal cortex that give rise to the perforant path fire low frequency, isolated action potentials that may not be sufficient for driving somatic spiking in CA1 (Frank et al., 2001).
The relatively weak influence of the perforant path synapses on somatic output has led to the idea that these distal inputs primarily serve a modulatory function (Levy et al., 1995b; Sherman and Guillery, 1998). For example, the feedforward inhibition driven by strong repetitive stimulation of distal inputs inhibits the induction of long-term plasticity at proximal synapses (Colbert and Levy, 1993; Remondes and Schuman, 2002; Steward et al., 1990). In contrast, depolarization generated by strong bursts of rhythmic activity at distal synapses can enhance the induction of synaptic plasticity at proximal inputs by regulating the timing and rate of spikes driven by proximal inputs (Buzsáki, 2002; Judge and Hasselmo, 2004; Levy et al., 1995b). Coincident activity at perforant path and Schaffer collateral synapses has been found to boost the size of distal EPSP signals arriving at the soma, although the boosting requires strong repetitive firing of the distal inputs (Ang et al., 2005; Jarsky et al., 2005).
The strong bursts of PP stimuli required to elicit the modulatory effects seen in these previous studies are in contrast to the sparse firing patterns and highly specific spatial information conveyed by inputs from the entorhinal cortex (McNaughton et al., 2006). This raises the question as to whether distal modulatory mechanisms exist that are more consistent with the physiological characteristics of these inputs. A clue as to the nature of one possible modulatory function comes from classic (Rall and Rinzel, 1973; Rinzel and Rall, 1974) and more recent theoretical studies (Golding et al., 2005) suggesting that individual distal synaptic events, although inefficient at triggering somatic spikes, can produce significant depolarization in the proximal dendrite. We therefore hypothesized that isolated distal excitatory inputs may provide precisely timed subthreshold signals for compartmentalized computations at more proximal synapses. Here we show that the pairing of single distal synaptic inputs with proximal synaptic events at an interval matched to the 20 ms timing delay inherent in the hippocampal circuit leads to a long-lasting potentiation of proximal CA1 synapses. This form of synaptic enhancement, which we term input-timing-dependent plasticity (ITDP), requires activation of NMDA receptors and IP3 receptor-mediated release of Ca2+ from intracellular stores. Our results thus suggest that distal synaptic inputs can indirectly influence somatic output by acting as temporally specific instructive signals for the induction of plasticity at proximal synapses. This may provide a mechanism by which CA1 neurons and the hippocampal circuit select and stabilize mnemonic information that bears the appropriate temporal relationship to direct sensory input.
Results
Pairing of proximal and distal CA1 inputs leads to NMDA receptor-dependent long-term potentiation of the proximal inputs
We obtained whole-cell current-clamp recordings of distal and proximal EPSPs from the soma of CA1 pyramidal neurons in mouse hippocampal slices. Focal extracellular stimulation in stratum lacunosum moleculare (SLM) and in stratum radiatum (SR) were used to recruit, respectively, distal perforant path inputs (PP) and proximal Schaffer collateral (SC) inputs (see Methods, schematic in Figure 1A). The two pathways were stimulated independently (> 300 ms apart) to monitor synaptic strength.
Figure 1. Pairing of perforant path and Schaffer collateral inputs to a CA1 neuron induces input-timing-dependent plasticity (ITDP) of the Schaffer collateral EPSPs.
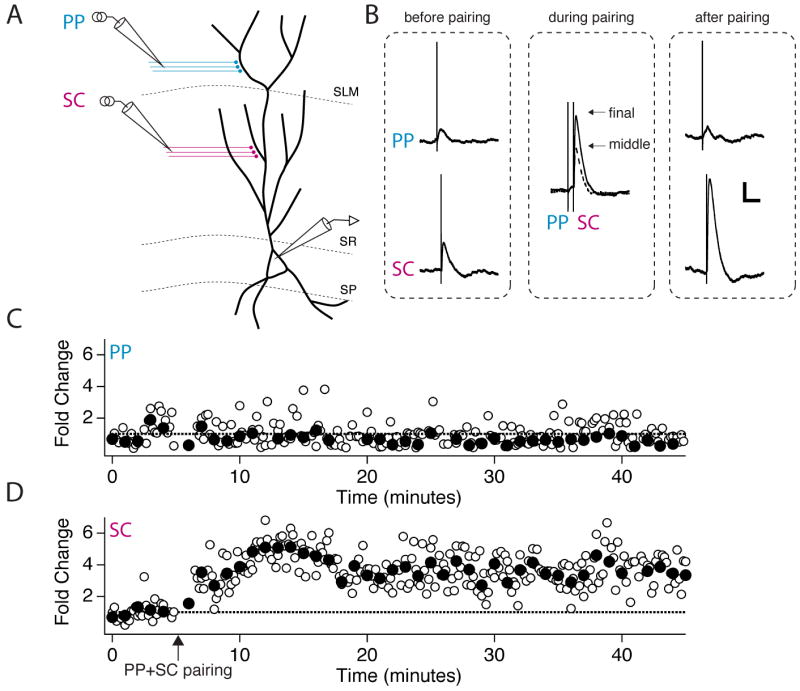
(A) Schematic of experimental setup. Whole-cell recordings were obtained from the soma of CA1 pyramidal neurons. One stimulating electrode was placed in the middle third of stratum radiatum (SR), the site of Schaffer collateral (SC) synapses, and one stimulating electrode was placed in the inner half of stratum lacunosum-moleculare (SLM), the site of perforant path (PP) synapses. (B) EPSPs in response to PP (distal) and SC (proximal) stimulation. Each trace shows the average of several EPSPs. Left (before pairing), PP or SC EPSPs obtained at the end of the baseline period. Middle (during pairing), two superimposed paired PP and SC EPSPs elicited during a −20 ms pairing protocol (SC stimulation occurs 20 ms after PP stimulation). The smaller EPSP pair (dotted trace) was recorded during the middle third of the 90 s, 1 Hz ITDP pairing protocol. The larger EPSP pair was elicited at the end of the pairing protocol (solid trace). Right (after pairing), PP or SC EPSPs elicited after the pairing protocol. Scale bars: 2 mV, 100 ms. (C-D) Time course of the PP (C) and SC (D) EPSPs during the entire experiment. Open symbols show fold change in peak amplitude of individual EPSPs; filled symbols show boxcar averages of fold change in EPSP amplitude during 1 minute (6 stimuli) windows. Arrow indicates start of 90 s pairing protocol.
To test the interaction of distal and proximal synaptic inputs we first applied pairs of stimuli in which the distal stimulus preceded the proximal stimulus by 20 ms (defined as a −20 ms interval), matching the expected propagation delay (Yeckel and Berger, 1990). Pairing of small subthreshold distal and proximal synaptic inputs at this temporal offset for 90 s at 1 Hz induced a large and sustained potentiation of the proximal Schaffer collateral EPSPs (3.1 ± 0.3 fold potentiation, n=12, P < 0.0001, paired t-test; Figures 1, 2B), with no change in the average magnitude of perforant path EPSPs (0.99 ± 0.14 fold potentiation, P = 0.94, paired t-test; Figures 1, 2C, S1). We refer to this form of heterosynaptic plasticity as associative input-timing-dependent plasticity (ITDP). Importantly, the small EPSPs evoked during the pairing protocol failed to evoke a somatic action potential, indicating that the signal for plasticity occurs below the threshold for somatic output.
Figure 2. ITDP is precisely tuned to timing interval and requires NMDA receptor activation.

(A) Summary data showing effects on the SC EPSP amplitude of PP + SC pairing protocols using variable timing intervals. The average fold change in EPSP measured 15-20 minutes after pairing protocol is plotted versus pairing interval, defined as the time of PP stimulation minus the time of SC stimulation during pairing; negative values reflect stimulation of the PP prior to stimulation of the SC pathway. Open circles show data from individual experiments. Filled circles show averages (and bars show S.E.M.) of points binned in 10 ms intervals (only bins with data are plotted). All pairing protocols applied for 90 s at 1 Hz. (B) Time course of average SC EPSP size during pairing with −20 ms interval (n=12 slices). EPSP amplitude was normalized to the average EPSP prior to pairing. Data from different experiments were averaged at each time point and then binned in a boxcar average (1 min window). (C) Time course of average PP EPSP size during pairing with −20 ms interval (c, n=12). (D) Time course of SC EPSP size during pairing with +20 ms interval (n=6). (E) Time course of SC EPSP size during pairing with −20 ms interval in presence of 50 μM D-APV (n=6).
The lack of change in distal EPSPs following pairing suggests that ITDP expression is restricted to proximal synapses and thus does not represent a cell-wide change in dendritic excitability. To examine whether the enhancement in the SC EPSP was specific to the paired pathway, we performed a two-pathway experiment in which a PP EPSP was paired with one of two independent SC inputs. We found that the SC EPSP in the paired pathway was significantly potentiated whereas the SC EPSP in the unpaired pathway was not enhanced (p>0.1). However, there was a trend towards potentiation in the unpaired pathway (which could indicate either that the two pathways were not always fully independent or that a component of ITDP lacks synapse specificity; see Figure S2).
We next assessed the dependence of ITDP on the timing interval between the paired PP and SC stimuli (Figure 2). There was a sharp drop off in potentiation for pairing intervals surrounding the ideal 20 ms interval. Indeed, we observed a small depression in Schaffer collateral EPSPs at the other timing intervals and a highly significant dependence of potentiation upon pairing interval (ANOVA(interval), F = 12.04, P < 0.0001; Figure 2A). Importantly, the potentiation at the −20 ms interval was highly significant when compared with the small depression at the inverse +20 ms pairing interval in which the proximal afferents were stimulated 20 ms before distal afferents (P < 0.0005).
The timing of ITDP and its independence from somatic action potential firing differ strikingly from the more classical Hebbian forms of plasticity previously characterized for the Schaffer collateral synapses, such as spike timing dependent plasticity (STDP) (Dan and Poo, 2004). In STDP the presynaptic cell must fire an action potential to elicit an EPSP prior to a strong associative depolarization of the postsynaptic cell (e.g. an action potential); that is, activity in the presynaptic cell must be causal for activity in the postsynaptic cell (Hebb, 1949). In contrast, an active Schaffer collateral synapse is potentiated by the ITDP protocol only if it is preceded (by a 20 ms interval) by activation of the postsynaptic CA1 cell by a distal EPSP. Thus, ITDP implements a predictive learning rule functionally distinct from STDP.
The dependence of Hebbian plasticity on timing interval is thought to reflect the properties of NMDA receptors, which require that the membrane must be depolarized while the receptors are activated by glutamate to expel Mg2+ and permit Ca2+ influx. Does ITDP also require activation of NMDA receptors, even though its timing dependence is the opposite of STDP? Indeed, ITDP was completely suppressed when the pairing protocol (-20 ms time window) was applied with NMDARs blocked by 50 μM D-APV (Figure 2E). We explored the mechanism for the reversed timing dependence of ITDP in a computational model discussed below (see Figure 7).
Figure 7. Simulation of NMDA receptor currents at a proximal synapse predicts the timing dependence of ITDP.

A simple, three-compartment model (“Model 1” in Experimental Procedures) was used to simulate the effect of injected voltage waveforms on the EPSC at a representative proximal synapse. Current was injected into the dendritic shaft to simulate input from a backpropagating action potential (“bAP”; A1-C1), distal synaptic input (“PP”; A2-C2) or independent proximal synaptic input (“SC”; A3-C3). (A1-3) Spine head voltage traces. (A1) bAP waveforms were injected either 5 ms before (magenta) or 5 ms after (cyan) start of proximal EPSP to simulate key intervals typically used to induce STDP. Dashed black trace shows unpaired spine EPSP. (A2) PP waveforms were injected either 20 ms before (magenta) or 20 ms after (cyan) the proximal EPSP to simulate intervals used to induce ITDP. (A3) SC waveforms were injected at intervals of 20 ms (solid traces) and 5 ms (dotted traces) before (magenta) and after (cyan) independent proximal EPSP. Dashed black trace shows unpaired EPSP. (B1-3) NMDA-receptor mediated synaptic currents in spine head during negative (magenta) and positive pairing intervals (cyan) associated with voltage traces in A1-3. Dashed black trace shows unpaired NMDAR EPSC. (C1-3) Spine [Ca2+] as a function of time in response to the different pairing protocols, calculated by convolving the NMDA receptor current with the impulse response of the intracellular calcium transient (Sabatini et al., 2002; see Experimental Procedures). The spine [Ca2+] signal during pairing was normalized by the peak [Ca2+] with an unpaired EPSP (shaded regions). Insets, Ratio of the peak [Ca2+] in response to a given stimulus pair divided by the peak [Ca2+] in response to the proximal EPSC alone plotted against the pairing interval.
ITDP requires activation of distal AMPA and NMDA receptors but is independent of inhibitory synaptic transmission
Distal stimulation could recruit proximal LTP through the activation of non-glutamatergic modulatory inputs whose axons pass through the SLM region of CA1 (Otmakhova and Lisman, 1999, 2000). To distinguish between the importance of modulatory inputs versus the perforant path glutamatergic inputs, we locally applied CNQX (a blocker of AMPA receptors) and D-APV to distal synapses in SLM to block their ionotropic glutamate receptors (Figure 3). We first verified that the local application of these inhibitors was effective in selectively blocking fast glutamatergic transmission at distal synapses while sparing glutamatergic synaptic transmission at proximal Schaffer collateral synapses (Figure 3B,C). Importantly, local application of the antagonists to distal synapses also blocked the ability of the pairing protocol to induce ITDP (Figure 3C,D). Since application of the blockers in SLM should not alter the direct firing of any modulatory axons in response to the distal stimulus, we conclude that ITDP requires the specific activation of distal glutamatergic inputs.
Figure 3. ITDP requires activation of glutamate receptors at perforant path inputs to CA1 neurons.

(A) Schematic of setup for PP + SC ITDP experiment testing effects of local blockade of distal fast glutamatergic synaptic transmission. A large patch pipette filled with APV (50 μM) and CNQX (10 μM) was placed in SLM near the expected location of the distal dendritic arbor of the patch-clamped CA1 cell. Blockers were puffed onto the slice in this region for the final 2.5 minutes of the baseline period and during the PP+SC pairing protocol (-20 ms interval), after which blocker application was stopped. (B) Representative experiment showing that perforant path EPSPs (bottom traces) were reversibly blocked during local application of antagonists (middle trace) whereas Schaffer collateral EPSPs (top traces) were unaffected, providing a check of the efficacy and specificity of the restricted blocker application. Scale bars: 2 mV, 100 ms. (C) Time course of fold change in amplitude of Schaffer collateral EPSPs (top graph) and perforant path EPSPs (bottom graph) during ITDP experiment with −20 ms pairing interval delivered during local application of glutamate receptor blockers to distal synapses. Bars show duration of application of CNQX + D-APV. Arrow shows time of delivery of 90 s pairing protocol. Top, Distal perfusion of CNQX and D-APV does not affect the SC EPSP but blocks the enhancement of the SC EPSP elicited by the ITDP pairing protocol. Bottom, The PP EPSP is transiently blocked by dstal application of inhibitors, and then slowly recovers upon drug wash out. (D) Summary data showing that distal CNQX plus D-APV blocks ITDP, whereas global bath application of GABAA and GABAB receptor blockers does not inhibit ITDP. Bars show magnitude of ITDP under control conditions, when pairing was performed during local CNQX plus D-APV perfusion, or during bath application of CGP55845 (2 μM) and SR95531 (1 μM) to block GABAA and GABAB receptors, respectively.
In addition to activating the perforant path inputs from entorhinal cortex, distal stimulation also recruits strong feedforward GABAergic inhibition. Thus, we wondered whether ITDP could result from a change in inhibitory transmission (Wöhrl et al., 2007). However, when the optimal pairing protocol was delivered in the presence of SR95531 (1 μM) and CGP55845 (2 μM), which block GABAA and GABAB receptors, respectively, we still observed a normal-sized ITDP (Figure 3D), supporting the specific role of distal glutamatergic transmission.
Pairing of distal and proximal inputs induces ITDP without recruiting a dendritic spike
How does the distal glutamatergic EPSP interact with the proximal EPSP to induce ITDP? The importance of NMDA receptor activation suggests that the depolarization due to the distal EPSP may sum with the local EPSP at proximal synapses to relieve Mg2+ block of the NMDA receptors, thus enhancing Ca2+ influx into the proximal spine to induce synaptic plasticity. However, in contrast to ITDP, most forms of NMDAR-dependent long-term potentiation at Schaffer collateral synapses require somatic spiking or strong somatic depolarization. Might the ITDP protocol elicit local spikes in the proximal dendrites that do not propagate to the soma, as previously observed during the induction of LTP at distal inputs using tetanic stimulation (Golding et al., 2002)? To determine whether ITDP requires dendritic spiking, we performed whole-cell patch clamp recordings from the trunk of the apical dendrite, at distances up to 200 μm from the soma, approaching the outer third of SR (Figure 4A,B).
Figure 4. ITDP induction does not elicit dendritic spikes in stratum radiatum.

(a) Schematic of dendritic recording experiments. (B) Representative epifluorescence image of cell filled with Alexa 594 (25μM) during whole-cell recording from a CA1 neuron dendrite. Scale bar: 20 μm. (C) Example current clamp response to depolarizing and hyperpolarizing dendritic current steps. A suprathreshold depolarizing current step elicits somatic spikes, identified by their characteristic sharp inflection (inset) (Golding and Spruston, 1998), that backpropagate into the dendrite. (D) Voltage recorded in the dendrite in response to stimulation of distal and proximal inputs at intervals of -40 (cyan), −20 (black) or +20 ms (magenta). Stimulus intensities were matched to those used during ITDP pairing protocol. Pairing was limited to a few stimuli to prevent induction of ITDP. (E) Average fold summation for the proximal EPSP (peak amplitude of paired EPSP divided by peak amplitude of proximal EPSP alone) across a range of pairing intervals. (F-G) Putative proximal dendritic spikes elicited by strong bursts of proximal synaptic stimulation. Dendritic voltage responses to 10 stimuli applied at 100 Hz to SC inputs with increasing stimulus current. (F) Example of dendritic spikes observed in SR close to the soma (inner third of SR, ∼75 μm from soma). (G) Spikes in SR dendrites near the SR-SLM border (∼250 μm from soma). At all dendritic locations fast and large spikes evoked with SC burst stimulation were apparent. Scale bars: 20 mV, 50 ms.
Stimulation of individual proximal inputs or paired proximal and distal inputs produced local depolarizing responses in the dendrites of 8–20 mV, several fold larger than those typically recorded in the soma. However, these responses were always subthreshold; dendritic spikes were never observed during paired stimulation of distal and proximal synapses for a wide range of pairing intervals (Figure 4D). Although subthreshold, the paired distal and proximal EPSPs did summate to enhance the peak depolarization over that produced by the proximal EPSP alone (Figure 4D,E). Interestingly, maximal summation was observed using an interstimulus interval of −20 ms, the same interval that produced ITDP, although the curve relating EPSP size to pairing interval was broader than that of ITDP.
The lack of dendritic spiking during the pairing of distal and proximal inputs does not reflect a general lack of excitability of the mouse CA1 neuron dendrites. Thus, we were able to detect both backpropagating dendritic action potentials in response to direct depolarizing current injections (Figure 4C) as well as putative dendritic spikes in response to repetitive SC stimulation (Figure 4F,G). Moreover, in Ca2+ imaging experiments we observed large Ca2+ transients in both proximal and distal dendrites in response to strong bursts of proximal or distal synaptic stimulation. Importantly, such dendritic Ca2+ “spikes” were not elicited in either the proximal or distal dendrites during the relatively weak ITDP pairing protocol (Figure S3). Thus, ITDP does not require large, forward- or back-propagating dendritic spikes, in contrast to other forms of long-term potentiation in the hippocampus (Bi and Poo, 1998; Golding et al., 2002; Magee and Johnston, 1997).
Pairing of distal and proximal inputs enhances the Ca2+ transient at proximal dendritic spines
The lack of pairing-induced dendritic action potentials raises the question as to whether a subthreshold interaction between distal and proximal EPSPs during the pairing protocol might be capable of enhancing the proximal spine Ca2+ transient for the induction of ITDP. We examined this possibility using two-photon microscopy to image Ca2+ at single spines on proximal dendrites in response to distal and proximal synaptic stimulation (Figure 5). We first identified proximal spines that were directly activated in response to Schaffer collateral stimulation based on the presence of a Ca2+ transient localized to the spine head. The Ca2+ transient in the active spine was significantly greater than the Ca2+ transient in the neighboring dendritic shaft or in neighboring spines (Figure S4) and showed probabilistic successes and failures in response to successive single Schaffer collateral inputs (Figure S5). Such properties indicate that the Ca2+ transients resulted from activation of that spine's own synaptic input and were not caused by a local dendritic spike.
Figure 5. Two-photon Ca2+ imaging during synaptic pairing.

(A) Structural fluorescence image (Alexa 594) of a proximal CA1 dendrite with superimposed position of linescan used to obtain data plotted in (B). White arrowhead indicates the active spine. (B) Example optical linescan recordings from activated spine. Upper panel shows spine Fluo-5F (Ca2+) fluorescence intensity as a function of time (same as graph) during a single pairing of distal and proximal inputs at a −20 ms interval (top trace) or a +20 ms interval (bottom trace). Arrowheads indicate time of SC and PP stimulation. Bottom graph plots the change in Ca2+ fluorescence signal as measured by ΔG/R × 100% for the −20 ms (black) and +20 ms (gray) intervals. Three to five linescans were averaged and smoothed (Savitzky-Golay, 9.17 ms window). (C) Peak spine Ca2+ transients (ΔG/R × 100%) plotted against the interstimulus interval for PP-SC pairing (n=7-9). Individual, paired results from single spines for the -20 and +20 ms interval are shown as open symbols (gray lines) and mean responses are plotted as filled symbols (black) with standard error bars. Mean data is plotted for -30, -20, +20 and +30 ms pairing intervals. (D) Enhancement (defined as [{max(ΔG2/R2)-max(ΔG1/R1)}/max(ΔG1/R1)]*100 percent) of the Ca2+ transient using a −20 ms pairing interval as compared to +20 and -30 ms intervals. Both the -20/+20 comparison (P<0.0005, paired t-test) and the -20/-30 comparison (P=0.04, unpaired t-test) were significantly different. (E) Application of 50 μm D-APV reduced spine Ca2+ transients in response to SC stimulation alone by ∼65% (n=3).
A single proximal stimulus elicited a sizable Ca2+ transient with a mean peak fluorescence increase (ΔG/R × 100%, see Experimental Procedures) of 77.2 ± 23.0% (n=3). In contrast, proximal spines showed no Ca2+ response to a brief burst of distal synaptic stimulation (data not shown). We next delivered isolated pairs of distal and proximal stimuli at different interstimulus intervals (Figure 5B-D). Delivery of a single pair of PP and SC stimuli at a pairing interval of +20 ms elicited a spine Ca2+ transient whose amplitude (100 ± 15.7%; n=5) was not significantly different from the transient caused by a single SC stimulus (P=0.45). In contrast, when a pair of PP and SC stimuli were applied at a -20 pairing interval, the Ca2+ transient was markedly increased to a value of 155.98 ± 13.75%. On average, the peak Ca2+ transient elicited by stimuli paired at the −20 ms interval was 65.9 ± 13.9% greater than the transient elicited with the +20 ms interval (P<0.0005, paired t-test). The Ca2+ transient at the -20 ms pairing interval was also significantly greater than the transient elicited with a -30 ms pairing interval, whose peak ΔG/R value was 97.65 ± 23.81% (P<0.05 relative to the -20 ms interval; Figure 5C,D). Finally, the vast majority of the spine Ca2+ transient was blocked by 50 μM D-APV (Figure 5E), indicating the importance of NMDARs and consistent with the pharmacology of ITDP.
These results clearly demonstrate that an NMDA receptor-dependent Ca2+ signal evoked by a SC synaptic input in a proximal spine can be significantly enhanced by an appropriately timed distal EPSP. Since the enhancement of the proximal Ca2+ transient occurs in response to a single pair of stimuli separated by only 20 ms, the enhancement probably is too rapid to be mediated by a biochemical signaling cascade. Rather, as suggested by the computational model discussed below, the enhancement in spine Ca2+ is likely to involve the temporal summation of the SC and PP EPSPs, leading to the depolarization-dependent relief of Mg2+ block of the NMDA receptors at the proximal spines. Moreover, the finding that the enhancement in the spine Ca2+ transient depends on pairing interval in a manner similar to that of ITDP strongly suggests that this NMDAR-dependent Ca2+ signal is important for the induction of ITDP (Figure 2A).
Pairing of two SC inputs fails to induce ITDP or to potentiate Ca2+ influx at proximal spines
Does ITDP represent a specific mechanism by which distal inputs influence CA1 neuron function? We examined this question by determining whether subthreshold pairing of two Schaffer collateral inputs could induce a similar form of plasticity (schematic in Figure 6A). In fact, repeated pairing of two SC inputs at a 20 ms pairing interval failed to induce long-term changes in synaptic efficacy (Figure 6B-D). The lack of plasticity could reflect the fact that the 20 ms pairing interval is suboptimal for temporal integration of two proximal EPSPs. We therefore repeated the pairing protocol using a 5 ms interval, which, according to our calculations using a realistic computer model (Poirazi et al., 2003), falls within the 0-10 ms timing window required for maximal summation of proximal inputs (see also Figure S6). Nonetheless, even when we paired subtreshold SC inputs at an optimal 5 ms interval, we failed to induce any significant potentiation (Figure 6D; 1.21 ± 0.08 and 1.22 ± 0.10 fold change, respectively; ANOVA (interval) F=1.98, P=0.16). These results thus suggest that ITDP requires a specific interaction between distal and proximal excitatory inputs.
Figure 6. ITDP specifically requires activation of perforant path inputs to CA1 neurons.

(A) Schematic of experimental in which two SC pathways were stimulated. Stimulus intensities were adjusted so that paired responses were subthreshold. (B) Representative EPSPs from two independent SC pathways before, during and after pairing for 90 s at 1 Hz using a −20 ms interval (defined as time of SC1 − time of SC2). Scale bars 4 mV, 100 ms. (C) Time course of EPSPs elicited by the two SC stimuli during the pairing experiment shown in B. (D) Fold change in EPSP amplitude induced by pairing protocols with intervals of 5 ms or 20 ms. Mean values shown as large circles; individual experiments shown as small circles. A small, reciprocal potentiation is observed in the 5 ms pairing condition that is not statistically significant. Note: pairing SC inputs at a 0 ms interval leads to a suprathreshold response that induces LTP (data not shown). (E-G) Results from Ca2+ imaging experiments during SC-SC pairing. (E) Examples of individual EPSPs (left) elicited by stimulating two SC pathways and their summed responses at 5 ms and 20 ms interstimulus intervals (right). Scale bars: 0.1 s, 5 mV. (F) Average Ca2+ transients recorded at a proximal spine for the experiment shown in E. Responses at the -20, -5 and +20 ms pairing intervals were indistinguishable. Scale bars: 20% ΔG/R, 0.2 s. (G) Population data for a set of proximal pairing experiments at three different pairing intervals (n=6). No significant differences between time points were detected.
The lack of ITDP with paired subthreshold SC inputs is surprising as dendritic recordings indicate that paired proximal EPSPs should produce a similar or greater dendritic depolarization than optimally paired distal and proximal EPSP (data not shown). To probe the reason for the failure of SC-SC pairing to induce ITDP, we examined whether pairing of subthreshold SC inputs could enhance the proximal spine Ca2+ transient, similar to the effect of pairing PP and SC inputs seen above (Figure 5). However, pairing of two SC inputs (only one of which elicited a Ca2+ transient in the spine that was imaged) at a variety of intervals failed to enhance the Ca2+ transient at the imaged spine (Figure 6E-G). This finding supports the view that the enhancement in spine Ca2+ transient during PP-SC pairing plays an important role in the induction of ITDP. Moreover, because the PP-SC pairing-induced enhancement in the spine Ca2+ transient is likely to depend on the summation of the PP and SC EPSP waveforms (see above), the inability of the SC EPSP to enhance the Ca2+ transient at an independent SC synapse implies that optimal integration may require the slower voltage waveform associated with a PP EPSP relative to a SC EPSP (Figure S7). We next use a simple computational model to explore the mechanism underlying the pairing-dependent enhancement in spine Ca2+.
A computational model explains the efficacy and timing dependence by which distal EPSPs facilitate Ca2+ influx through NMDARs at a proximal synapse
How does the pairing of a distal EPSP with a proximal EPSP enhance the proximal Ca2+ transient? Why is the optimal timing dependence for induction of ITDP and the enhancement of the proximal spine Ca2+ signal the reverse of the Hebbian timing dependence of STDP despite the shared involvement of NMDA receptors? Why does pairing of two SC EPSPs fail to enhance the proximal spine Ca2+ signal or induce ITDP? To gain insight into these questions, we implemented a simplified computational model of a proximal dendritic spine including a detailed kinetic model of the NMDA receptor, the voltage-dependent relief of its block by Mg2+ (Kampa et al., 2004), and the resultant rise in spine Ca2+ concentration due to the NMDAR-mediated Ca2+ influx ((Sabatini et al., 2002), see Experimental Procedures).
We first used the model to examine the STDP induction protocol, in which a fast dendritic excitatory conductance similar to a proximal input is paired with a voltage waveform similar to a backpropagating action potential (bAP). Maximal temporal integration of the two signals occured when the bAP rapidly followed (within 5 ms) the EPSP, rather than when it preceded the EPSP (Figure 7A1). The temporal integration enhanced the voltage-dependent relief of Mg2+ block of the NMDAR, which enhanced the NMDAR-dependent ionic current (Figure 7B1) and resultant spine Ca2+ transient (Figure 7C1). By contrast when we examined a protocol used to induce ITDP, in which we paired a fast proximal EPSP with a slower voltage waveform similar to a PP EPSP, maximal temporal integration (Figure 7A2), NMDAR current (Figure 7B2) and spine Ca2+ (Figure 7C2) occurred when the PP EPSP preceded the proximal input, similar to the timing dependence of ITDP. We observed a similar timing dependence using a morphologically accurate model of a CA1 pyramidal neuron (Figure S6). Finally, pairing a SC input with a second SC input using a fast, transient EPSP waveform produced maximal temporal summation (Figure 7A3), enhancement in the NMDAR current (Figure 7B3) and spine Ca2+ (Figure 7C3) at positive pairing intervals but had little effect at negative pairing intervals. Importantly, the magnitude of the enhancement in the Ca2+ signal with SC-SC pairing, even at the optimal interval, was significantly less than that observed with PP-SC pairing, consistent with the lack of Ca2+ enhancement or ITDP with SC-SC pairing.
These results help explain several important aspects of ITDP versus STDP. First, the rapid time course of a bAP limits its effectiveness in summating with a SC EPSP to when the bAP follows the EPSP (with a temporal offset determined by the kinetics of the NMDA receptor (Kampa et al., 2004)), explaining the Hebbian timing dependence of STDP. Second, the slow rising phase of the PP EPSP when it reaches a proximal spine, due in part to dendritic filtering, restricts the time window for its optimal temporal summation with a SC EPSP to when the PP EPSP precedes the SC input by ∼20 ms, accounting for the non-Hebbian timing dependence of ITDP. Third, the superiority of the PP EPSP relative to the SC EPSP in enhancing NMDAR current and spine Ca2+ at a proximal synapse is due to the prolonged depolarization produced by summation of the PP and SC EPSPs, which leads to a more prolonged relief of Mg2+ block of the NMDAR.
Ca2+ release from internal stores is required for ITDP and the pairing-induced enhancement in spine Ca2+ transient
The model presented above provides evidence that under certain conditions slow temporally integrated waveforms may preferentially enhance local calcium entry at the synapse. However, the 33% enhancement seen in the model cannot fully explain the larger 65% increase in Ca2+ signal during the -20 ms pairing interval at proximal synapses. We therefore explored the possibility that enhanced Ca2+ influx through the NMDARs during pairing may be amplified through the release of Ca2+ from intracellular stores, a process that has been implicated in other forms of synaptic plasticity that are induced by relatively sparse, low-frequency synaptic activation (Doi et al., 2005; Emptage et al., 2001; Miyata et al., 2000; Wang et al., 2000).
To examine the importance of intracellular Ca2+ stores for the pairing-dependent enhancement in spine Ca2+, we repeated the pairing protocol in the presence of cyclopiazonic acid (CPA), an inhibitor of the smooth endoplasmic reticulum Ca2+ (SERCA) pump that depletes the stores of Ca2+. Indeed, addition of CPA caused a significant inhibition in the ability of synaptic pairing to enhance the Ca2+ transient (Fig. 8A-C). Thus, in the presence of CPA, the Ca2+ transient elicited with the −20 ms pairing interval was now only 33.2 ± 6.6% greater than the transient elicited using the +20 ms interval, half as large as the 66% increase observed above with intact Ca2+ stores (Figure 8B-C, P<0.05, unpaired t-test) but in good agreement with the increase in spine Ca2+ due to influx through the NMDARs predicted from our modeling results. This reduction is detectable despite a trend of CPA to increase the amplitude and time constant of decay of the Ca2+ transients at all intervals (Figure 8D), an effect expected from the blockade of Ca2+ reuptake into internal stores by the SERCA pump.
Figure 8. Inhibitors of Ca2+ signaling through intracellular stores block ITDP but not STDP and reduce the pairing-dependent enhancement in spine Ca2+.

(A) Schematic of experimental setup. (B) Representative fluorescent line-scan images (top) and plots of spine Ca2+ transients (bottom) during pairing of PP and SC inputs at a −20 ms interval (top image; black) or +20 ms interval (bottom image; gray) in the presence of CPA (1 μM). Arrowheads indicate time of SC and PP stimulation. (C) Mean enhancement of Ca2+ transients with pairing at the −20 ms interval relative to the +20 ms interval. CPA significantly reduced the enhancement of the Ca2+ transient (P<0.05, unpaired t-test). (D) The time constant of decay of the spine Ca2+ transient is significantly increased in the presence of CPA (P<0.05, unpaired t-test). (E) Representative perforant path and Schaffer collateral EPSPS before, during, and after the −20 ms ITDP pairing protocol delivered in presence of 0.01% DMSO (control, E1) or 1 μM thapsigargin (E2). Scale bars: 5mV, 100 ms. (F) Time course of fold change in SC EPSP peak amplitude during individual pairing experiments in DMSO (F1) or thapsigargin (THAP; F2). (G-H) Summary of effects of antagonists of Ca2+ signaling on ITDP (G) or STDP (H). (G) Average fold change in EPSP amplitude when −20 ms ITDP pairing protocol was delivered: 1. Under control conditions (CNTRL, n=23); 2. During blockade of Group I mGluRs with 20 μM MPEP and 100 μM LY367385 (n=7); 3. During bath application of THAP (1 μM, gray circles, n=7) or cyclopiazonic acid (CPA; 1 μM, open circles, n=4) (the CPA and THAP results were obtained from separate experiments but combined into a single bar); 4. In presence of intracellular heparin (HEPARIN; 400U/mL, n=9) an IP3 receptor antagonist; and 5. In presence of intracellular ruthenium red (RUTH RED, 30 μM, n=3) a ryanodine receptor antagonist. (H) Average fold change in SC EPSP amplitude during spike-timing-dependent plasticity (STDP) in presence of DMSO (closed circles, n=6) or CPA (1 μM, open circles, n=6). STDP was induced by pairing SC EPSPs (1 Hz, 90 stimuli) with a brief, suprathreshold current pulse (2 nA, 2 ms) at a +5 ms offset.
Does the release of Ca2+ from intracellular stores also contribute to ITDP? We first examined the effects of CPA or thapsigargin, another inhibitor of the SERCA pump and store release. Application of either inhibitor was highly effective in blocking the induction of ITDP (Figure 8E-G). In contrast, CPA did not block homosynaptic STDP evoked by pairing Schaffer collateral stimulation with the firing of postsynaptic action potentials in the CA1 neuron (Figure 8H), consistent with previous results (Nishiyama et al., 2000). Production of biochemical signals necessary for release of calcium from intracellular stores often depends on signaling through metabotropic glutamate receptors (mGluRs), as reported previously for other forms of plasticity (Anwyl, 1999; Berridge, 1998; Ito, 2001; Okubo et al., 2004). Indeed, we found that blockade of Group I mGluRs (mGluR1 and mGluR5) was also effective in blocking the induction of ITDP (Figure 8G).
CPA and thapsigargin inhibit both IP3 receptor-mediated Ca2+ release as well as Ca2+-induced Ca2+ release from stores through ryanodine receptors. To distinguish between these mechanisms, we included in the whole cell recording solution either heparin or ruthenium red, membrane-impermeant selective inhibitors of IP3 receptors and ryanodine receptors, respectively. Intracellular loading with heparin (400U/mL) significantly reduced the magnitude of ITDP (1.40 ± 0.11 fold potentiation; P<0.01), whereas inclusion of ruthenium red had little effect (Figure 8G). These results thus support the view that the induction of ITDP requires the group I mGluR-dependent activation of a phospholipase C-mediated signaling cascade, leading to the IP3-dependent release of Ca2+ from intracellular stores.
The above electrophysiological, computational and imaging results suggest a relatively simple model for how the interaction between distal and proximal inputs underlies the induction of ITDP. First, the slowly rising, broad depolarization produced by a distal synaptic input maximally summates with a proximal SC EPSP when distal stimulation precedes proximal stimulation by 20 ms (Figures 4, 7, S6, and S7), leading to a marked increase in Ca2+ influx through NMDA receptors at the proximal Schaffer collateral synapse (Figures 5 and 7). Second, the enhanced Ca2+ entry then induces the IP3 receptor-dependent release of Ca2+ from intracellular stores, amplifying the spine Ca2+ signal and leading to the induction of ITDP (Figure 8). However, the slow depolarization produced by the distal EPSP, although necessary for induction of ITDP, may not be sufficient. Thus, we found that pairing a slow PP-EPSP-like depolarization produced by direct somatic current injection with a proximal SC EPSP failed to potentiate the proximal EPSP (Figure S8). This implies that PP stimulation may also be required to recruit the mGluR-depedent biochemical signal that is required for the induction of ITDP.
Discussion
Our data reveal a novel mechanism by which distal perforant path inputs influence CA1 neuron activity: the distal synapses act as instructive signals that induce a heterosynaptic form of subthreshold, long-term plasticity, ITDP, at more proximal, Schaffer collateral synapses. This form of associative synaptic plasticity represents a synaptic learning rule that is functionally distinct from Hebbian forms of synaptic plasticity. Unlike Hebbian plasticity, ITDP occurs in the absence of a large postsynaptic depolarization or somatic spiking, consistent with the low level of basal hippocampal activity (Henze et al., 2000). Moreover, the temporal order of presynaptic and postsynaptic activity required to induce ITDP, in which the distal EPSP must precede activation of the Schaffer collateral inputs, is the opposite of the temporal order required to induce Hebbian plasticity, in which activity in the presynaptic cell must precede activity in the postsynaptic cell. Importantly, the pairing interval for ITDP appears finely tuned to the expected 20 ms latency of propagated activity in the hippocampal circuit (Yeckel and Berger, 1990). Finally, the pattern of activity used to induce ITDP (low frequency, isolated spikes) is consistent with the firing properties of superficial entorhinal cortex neurons (Frank et al., 2001; Fyhn et al., 2004; Hargreaves et al., 2005) and CA3 neurons (Frerking et al., 2005).
Specific role of distal inputs in the induction of ITDP
The finding that perforant path inputs are efficient at inducing ITDP whereas subthreshold proximal inputs are ineffective could reflect a number of differences between distal and proximal synapses. For example, the larger NMDA component (Nicholson et al., 2006; Otmakhova et al., 2002) and distal location of the perforant path inputs (Gulledge et al., 2005) result in postsynaptic potentials with a relatively slow time course compared to SC EPSPs. Our computational modeling indicates that long-lasting subthreshold depolarizations are particularly effective in enhancing NMDA receptor mediated Ca2+ influx. In addition the slow time course of the distal EPSP may be well matched to the kinetic properties of voltage-gated channels that underlie local nonlinearities responsible for boosting the Ca2+ signal in proximal oblique branches (Losonczy and Magee, 2006) and individual spines (Bloodgood and Sabatini, 2007). However, our failure to observe large nonlinearities in the apical trunk suggests that local spikes, if they do occur, are likely modest in amplitude and fail to forward propagate.
Mechanisms for boosting proximal spine Ca2+ signals during induction of ITDP
One surprising feature of ITDP is the relatively large size of the potentiation that is induced at the proximal synapses, despite the relatively modest depolarization that the distal EPSPs produce in the proximal dendrite. Our Ca2+ imaging studies show that the appropriate pairing of distal EPSPs also elicits a surprisingly large enhancement in the proximal spine Ca2+ transient. Importantly, we find that this large enhancement is due in part to a boosting effect caused by Ca2+ release from intracellular stores in the proximal spines. These results are consistent with a number of previous studies that have detected store-dependent Ca2+ signals at proximal CA1 synapses (Emptage et al., 1999; Emptage et al., 2001; Nakamura et al., 1999; Nakamura et al., 2002; Nakamura et al., 2000; Raymond and Redman, 2002, 2006; Svoboda and Mainen, 1999), which can contribute very rapid Ca2+ transients with a time course similar to what we report here (Emptage et al., 1999; Emptage et al., 2001; Raymond and Redman, 2002, 2006). The importance of Ca2+ release from internal stores is further emphasized by our finding that ITDP is blocked by heparin, an antagonist of IP3 receptors, as well as by antagonists of Group I mGluRs (mGluR1 and mGluR5), which signal through phospholipase C and recruitment of IP3.
Based on these results, we favor a model in which distal stimulation produces two effects that are required for the induction of ITDP. First the propagation of the distal PP EPSP to proximal dendrites leads to temporal summation with the SC EPSP, resulting in relief of Mg2+ block of proximal NMDARs and enhanced Ca2+ entry into proximal spines. Second, the distal inputs recruit a specific biochemical signal, possibly through activation of distal group I mGluRs. The interaction of this signal with the enhanced spine Ca2+ transient that would then induce ITDP.
As discussed above, the enhancement in proximal spine Ca2+ upon delivery of a single pair of PP and SC stimuli is probably too rapid to result from the activation of mGluRs in response to the synaptic pairing at a 20 ms offset. This suggests that activation of mGluRs over the slower time scale of the 60-90 s duration of the ITDP pairing protocol may be what is critical for the induction of synaptic plasticity. However, we cannot rule out the possibility that basal levels of mGluR activation (Losonczy et al., 2003), perhaps due to the spontaneous agonist-independent activity of the receptor (Losonczy et al., 2003), might contribute to the pairing-induced enhancement in proximal spine Ca2+. Such basal mGluR activity could produce a small elevation in resting levels of IP3 (Abdul-Ghani et al., 1996) that then might be sufficient to stimulate Ca2+ release through the IP3 receptors during synaptic pairing, due to the known synergistic interaction between IP3 and cytoplasmic Ca2+ (Berridge, 2002). As our results do not permit a definitive localization of the biochemical signaling during the induction of ITDP, a challenging goal in the future will be to identify the nature of this biochemical messenger.
Function of ITDP in the context of the architecture of a CA1 pyramidal neuron and the hippocampal circuit
Although the primary function of dendrites is to integrate diverse excitatory and inhibitory inputs to trigger an action potential output from a neuron (Hausser et al., 2000; London et al., 2002; Magee, 2000; Williams and Stuart, 2003), our results suggest that neurons may also exploit the biophysical constraints of their dendritic morphology to perform unique subthreshold computations. Thus, low frequency excitation by distal synaptic inputs (Frank et al., 2001) may play a relatively minor role in the ongoing conversation of propagated spiking activity in the hippocampal circuit. Rather, we propose that in addition to driving somatic spikes in a suprathreshold regime during strong synaptic activation, isolated distal synaptic inputs can provide instructive signals for the induction of plasticity at proximal synaptic inputs in a subthreshold regime. The coexistence of complementary suprathreshold and subthreshold forms of plasticity would dramatically increase the computational power of CA1 pyramidal neurons. For example, the indirect influence of distal synaptic inputs via ITDP may serve to implement a supervised learning rule that assesses the salience of the transformed hippocampal input arriving at proximal CA1 synapses, enhancing the transmission of those signals that bear the proper temporal relation to the original sensory context conveyed by the direct input from entorhinal cortex.
One striking finding from these experiments is how the cellular mechanisms for the induction of ITDP appear finely tuned to match the functional properties of the hippocampal circuit in which this form of synaptic plasticity is embedded. Thus, the electrotonic architecture and biophysical properties of CA1 pyramidal neuron dendrites introduce a timing delay in the propagation of a distal EPSP to more proximal synapses that matches the temporal dynamics of the delay-line architecture in the hippocampal trisynaptic circuit. The fact that circuits throughout the brain have defined anatomical arrangements that impose distinct temporal constraints on signal propagation and are composed of neurons with unique dendritic architectures and biophysical properties raises the intriguing possibility that the tuning of cellular plasticity to circuit dynamics may be a more general organizing principle in the nervous system.
Experimental Procedures
Electrophysiology and analysis
Horizontal brain slices were prepared from P30-P50 mice. The standard ACSF had the following composition (mM): NaCl (125), NaH2PO4 (1.25), KCl (2.5), NaHCO3 (25), Glucose (25), CaCl2 (2), MgCl2 (1), pyruvate (0-2), continuously bubbled with 95%/5% O2/CO2. Patch pipettes (2.5-5 MΩ for somatic recordings and 7-10 MΩ for dendritic recordings) were filled with “intracellular solution” containing (mM): KMeSO4 (130), KCl (10), HEPES (10), NaCl (4), EGTA (0.1), MgATP (4), Na2GTP (0.3), Phosphocreatine (10). Focal stimulating electrodes (patch pipettes coated with AgCl paint and filled with ACSF or 1M NaCl) were used to apply single, unipolar shocks of 0.1-0.2 ms in duration using a constant current stimulator. All experiments were performed at 32°C-34°C (see Supplementary procedures).
The “fold change” in EPSP size in response to induction of plasticity was determined from the average peak EPSP 15-30 minutes post-induction divided by the average EPSP prior to the induction protocol. For time course plots, data was converted to fold change for each experiment and averaged. A boxcar average was then taken of 6-12 consecutive individual responses (1-2 minute). All error bars are standard errors of the population mean or boxcar mean. In most experiments drugs (from Sigma or Tocris-Cookson) were added to the bath solution by dilution from stock solutions (500-1000-fold concentrated). In a subset of experiments drugs were locally superfused through a patch-pipette touching the surface of the slice in SLM. Statistical tests were performed using Excel (Microsoft, Redmond, WA) and SigmaStat (Systat Software, Inc., San Jose, CA).
Two-photon Ca2+ Imaging
Two-photon Ca2+ imaging (BioRad Radiance 2100 MP; Zeiss, Jena, Germany) in CA1 proximal spines was performed using a preloading procedure in which the whole cell pipette was withdrawn 5-7 minutes after obtaining a whole cell configuration to minimize washout of synaptic plasticity (Lamsa et al., 2005). The EGTA in the pipette solution was replaced with the low-affinity Ca2+ dye, Fluo-5F (500 μM); a structural dye, Alexa 594 cadaverine (25 μM) (Invitrogen, Carlsbad, CA), was also included. The Ca2+-related fluorescence change was expressed as the ratio: (green fluorescence intensity at a given time minus the resting green fluorescence intensity) divided by the red fluorescence intensity, expressed as a percentage (ΔG/R × 100%). Further details are provided in Supplementary Materials.
Computational Modeling
Model 1
A simple multicompartment model was implemented in NEURON (NEURON version 5.9, available at http://www.neuron.yale.edu/neuron) to simulate a mixed AMPA/NMDA receptor synapse on a dendritic spine attached to the shaft through a narrow neck (see Supplemental Materials for details). Spine [Ca2+] due to influx through the NMDAR was determined by convolving the NMDAR EPSC with the spine Ca2+ impulse response function (Sabatini et al., 2002).
Model 2
The model previously described by Poirazi and colleagues (Poirazi et al., 2003) was used to simulate interactions between proximal and distal synapses in a morphologically reconstructed CA1 dendrite.
Supplementary Material
Acknowledgments
We thank Richard Axel, Robert Hawkins, Eric Kandel, Bina Santoro, Kevin Franks, Rebecca Piskorowski, and Anthony DeCostanzo for helpful comments on previous versions of the manuscript. This work was supported by a Graduate Research Fellowship from the NSF (JTD), a NIH postdoctoral training grant (T32 MH015174-30; JTD) and the Howard Hughes Medical Institute (SAS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Ghani MA, Valiante TA, Carlen PL, Pennefather PS. Metabotropic glutamate receptors coupled to IP3 production mediate inhibition of IAHP in rat dentate granule neurons. J Neurophysiol. 1996;76:2691–2700. doi: 10.1152/jn.1996.76.4.2691. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Ang CW, Carlson GC, Coulter DA. Hippocampal CA1 circuitry dynamically gates direct cortical inputs preferentially at theta frequencies. J Neurosci. 2005;25:9567–9580. doi: 10.1523/JNEUROSCI.2992-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Araya R, Jiang J, Eisenthal KB, Yuste R. The spine neck filters membrane potentials. Proc Natl Acad Sci U S A. 2006;103:17961–17966. doi: 10.1073/pnas.0608755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Brun VH, Otnass MK, Molden S, Steffenach HA, Witter MP, Moser MB, Moser EI. Place cells and place recognition maintained by direct entorhinal-hippocampal circuitry. Science. 2002;296:2243–2246. doi: 10.1126/science.1071089. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Cajal SRy. Histologie du Système Nerveux de l'Homme et des Vertebrés. 1911. [Google Scholar]
- Colbert CM, Levy WB. Long-term potentiation of perforant path synapses in hippocampal CA1 in vitro. Brain Res. 1993;606:87–91. doi: 10.1016/0006-8993(93)91573-b. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–23. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Doi T, Kuroda S, Michikawa T, Kawato M. Inositol 1,4,5-trisphosphate-dependent Ca2+ threshold dynamics detect spike timing in cerebellar Purkinje cells. J Neurosci. 2005;25:950–961. doi: 10.1523/JNEUROSCI.2727-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Empson RM, Heinemann U. Perforant path connections to area CA1 are predominantly inhibitory in the rat hippocampal-entorhinal cortex combined slice preparation. Hippocampus. 1995;5:104–107. doi: 10.1002/hipo.450050203. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Frank LM, Brown EN, Wilson MA. A comparison of the firing properties of putative excitatory and inhibitory neurons from CA1 and the entorhinal cortex. J Neurophysiol. 2001;86:2029–2040. doi: 10.1152/jn.2001.86.4.2029. [DOI] [PubMed] [Google Scholar]
- Frerking M, Schulte J, Wiebe SP, Staubli U. Spike timing in CA3 pyramidal cells during behavior: implications for synaptic transmission. J Neurophysiol. 2005;94:1528–1540. doi: 10.1152/jn.00108.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyhn M, Molden S, Witter MP, Moser EI, Moser MB. Spatial representation in the entorhinal cortex. Science. 2004;305:1258–1264. doi: 10.1126/science.1099901. [DOI] [PubMed] [Google Scholar]
- Golding NL, Mickus T, Katz Y, Kath WL, Spruston N. Factors mediating powerful voltage attenuation along CA1 dendrites. J Physiol. 2005;568:69–82. doi: 10.1113/jphysiol.2005.086793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Spruston N. Dendritic sodium spikes are variable triggers of axonal action potentials in hippocampal CA1 pyramidal neurons. Neuron. 1998;21:1189–1200. doi: 10.1016/s0896-6273(00)80635-2. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Kampa BM, Stuart GJ. Synaptic integration in dendritic trees. J Neurobiol. 2005;64:75–90. doi: 10.1002/neu.20144. [DOI] [PubMed] [Google Scholar]
- Hargreaves EL, Rao G, Lee I, Knierim JJ. Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science. 2005;308:1792–1794. doi: 10.1126/science.1110449. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- Hebb DO. Organization of Behavior: A Neuropsychological Theory. John Wiley and Sons; 1949. [Google Scholar]
- Henze DA, Borhegyi Z, Csicsvari J, Mamiya A, Harris KD, Buzsaki G. Intracellular features predicted by extracellular recordings in the hippocampus in vivo. J Neurophysiol. 2000;84:390–400. doi: 10.1152/jn.2000.84.1.390. [DOI] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Jahr CE, Stevens CF. Voltage dependence of NMDA-activated macroscopic conductances predicted by single-channel kinetics. J Neurosci. 1990;10:3178–3182. doi: 10.1523/JNEUROSCI.10-09-03178.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsky T, Roxin A, Kath WL, Spruston N. Conditional dendritic spike propagation following distal synaptic activation of hippocampal CA1 pyramidal neurons. Nat Neurosci. 2005;8:1667–1676. doi: 10.1038/nn1599. [DOI] [PubMed] [Google Scholar]
- Judge SJ, Hasselmo ME. Theta rhythmic stimulation of stratum lacunosum-moleculare in rat hippocampus contributes to associative LTP at a phase offset in stratum radiatum. J Neurophysiol. 2004;92:1615–1624. doi: 10.1152/jn.00848.2003. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart GJ. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol. 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa K, Heeroma JH, Kullmann DM. Hebbian LTP in feed-forward inhibitory interneurons and the temporal fidelity of input discrimination. Nat Neurosci. 2005;8:916–924. doi: 10.1038/nn1486. [DOI] [PubMed] [Google Scholar]
- Levy WB, Colbert CM, Desmond NL. Another network model bites the dust: entorhinal inputs are no more than weakly excitatory in the hippocampal CA1 region. Hippocampus. 1995a;5:137–140. doi: 10.1002/hipo.450050209. [DOI] [PubMed] [Google Scholar]
- Levy WB, Desmond NL, Zhang DX. Perforant path activation modulates the induction of long-term potentiation of the schaffer collateral-hippocampal CA1 response: theoretical and experimental analyses. Learn Mem. 1995b;4:510–518. doi: 10.1101/lm.4.6.510. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Relating hippocampal circuitry to function: recall of memory sequences by reciprocal dentate-CA3 interactions. Neuron. 1999;22:233–242. doi: 10.1016/s0896-6273(00)81085-5. [DOI] [PubMed] [Google Scholar]
- London M, Hausser M. Dendritic computation. Annu Rev Neurosci. 2005;28:503–532. doi: 10.1146/annurev.neuro.28.061604.135703. [DOI] [PubMed] [Google Scholar]
- London M, Schreibman A, Hausser M, Larkum ME, Segev I. The information efficacy of a synapse. Nat Neurosci. 2002;5:332–340. doi: 10.1038/nn826. [DOI] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:291–307. doi: 10.1016/j.neuron.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Somogyi P, Nusser Z. Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J Neurophysiol. 2003;89:1910–1919. doi: 10.1152/jn.00842.2002. [DOI] [PubMed] [Google Scholar]
- Magee J. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:848. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- McNaughton BL, Battaglia, Jensen O, Moser EI. Path integration and the neural basis of the ‘cognitive map’. Nat Rev Neurosci. 2006;7:663–678. doi: 10.1038/nrn1932. [DOI] [PubMed] [Google Scholar]
- Miyata M, Finch EA, Khiroug L, Hashimoto K, Hayasaka S, Oda SI, Inouye M, Takagishi Y, Augustine GJ, Kano M. Local calcium release in dendritic spines required for long-term synaptic depression. Neuron. 2000;28:233–244. doi: 10.1016/s0896-6273(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lasser-Ross N, Nakamura K, Ross WN. Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol. 2002;543:465–480. doi: 10.1113/jphysiol.2002.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Nakamura K, Lasser-Ross N, Barbara JG, Sandler VM, Ross WN. Inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release evoked by metabotropic agonists and backpropagating action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 2000;20:8365–8376. doi: 10.1523/JNEUROSCI.20-22-08365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DA, Trana R, Katz Y, Kath WL, Spruston N, Geinisman Y. Distance-dependent differences in synapse number and AMPA receptor expression in hippocampal CA1 pyramidal neurons. Neuron. 2006;50:431–442. doi: 10.1016/j.neuron.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Okubo Y, Kakizawa S, Hirose K, Iino M. Cross talk between metabotropic and ionotropic glutamate receptor-mediated signaling in parallel fiber-induced inositol 1,4,5-trisphosphate production in cerebellar Purkinje cells. J Neurosci. 2004;24:9513–9520. doi: 10.1523/JNEUROSCI.1829-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. Dopamine selectively inhibits the direct cortical pathway to the CA1 hippocampal region. J Neurosci. 1999;19:1437–1445. doi: 10.1523/JNEUROSCI.19-04-01437.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. Dopamine, serotonin, and noradrenaline strongly inhibit the direct perforant path-CA1 synaptic input, but have little effect on the Schaffer collateral input. Ann N Y Acad Sci. 2000;911:462–464. doi: 10.1111/j.1749-6632.2000.tb06746.x. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Lisman JE. Pathway-specific properties of AMPA and NMDA-mediated transmission in CA1 hippocampal pyramidal cells. J Neurosci. 2002;22:1199–1207. doi: 10.1523/JNEUROSCI.22-04-01199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirazi P, Brannon T, Mel BW. Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron. 2003;37:977–987. doi: 10.1016/s0896-6273(03)00148-x. [DOI] [PubMed] [Google Scholar]
- Rall W, Rinzel J. Branch input resistance and steady attenuation for input to one branch of a dendritic neuron model. Biophys J. 1973;13:648–687. doi: 10.1016/S0006-3495(73)86014-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol. 2002;88:249–255. doi: 10.1152/jn.2002.88.1.249. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol. 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remondes M, Schuman EM. Direct cortical input modulates plasticity and spiking in CA1 pyramidal neurons. Nature. 2002;416:736–740. doi: 10.1038/416736a. [DOI] [PubMed] [Google Scholar]
- Remondes M, Schuman EM. Role for a cortical input to hippocampal area CA1 in the consolidation of a long-term memory. Nature. 2004;431:699–699. doi: 10.1038/nature02965. [DOI] [PubMed] [Google Scholar]
- Rinzel J, Rall W. Transient response in a dendritic neuron model for current injected at one branch. Biophys J. 1974;14:759–790. doi: 10.1016/S0006-3495(74)85948-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca(2+) ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. On the actions that one nerve cell can have on another: distinguishing “drivers” from “modulators”. Proc Natl Acad Sci U S A. 1998;95:7121–7126. doi: 10.1073/pnas.95.12.7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffenach HA, Witter M, Moser MB, Moser EI. Spatial memory in the rat requires the dorsolateral band of the entorhinal cortex. Neuron. 2005;45:301–313. doi: 10.1016/j.neuron.2004.12.044. [DOI] [PubMed] [Google Scholar]
- Steward O. Topographic organization of the projections from the entorhinal area to the hippocampal formation of the rat. J Comp Neurol. 1976;167:285–314. doi: 10.1002/cne.901670303. [DOI] [PubMed] [Google Scholar]
- Steward O, Tomasulo R, Levy WB. Blockade of inhibition in a pathway with dual excitatory and inhibitory action unmasks a capability for LTP that is otherwise not expressed. Brain Res. 1990;516:292–292. doi: 10.1016/0006-8993(90)90930-a. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Mainen ZF. Synaptic $[$Ca2+$]$: intracellular stores spill their guts. Neuron. 1999;22:427–430. doi: 10.1016/s0896-6273(00)80698-4. [DOI] [PubMed] [Google Scholar]
- Wang SS, Denk W, Hausser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Role of dendritic synapse location in the control of action potential output. Trends Neurosci. 2003;26:147–154. doi: 10.1016/S0166-2236(03)00035-3. [DOI] [PubMed] [Google Scholar]
- Wöhrl R, Haebler Dv, Heinemann U. Low-frequency stimulation of the direct cortical input to area CA1 induces homosynaptic LTD and heterosynaptic LTP in the rat hippocampal-entorhinal cortex slice preparation. Eur J Neurosci. 2007;25:251–258. doi: 10.1111/j.1460-9568.2006.05274.x. [DOI] [PubMed] [Google Scholar]
- Yeckel MF, Berger TW. Feedforward excitation of the hippocampus by afferents from the entorhinal cortex: redefinition of the role of the trisynaptic pathway. Proc Natl Acad Sci U S A. 1990;87:5832–5836. doi: 10.1073/pnas.87.15.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.