

Abstract
We determined the effects of peroxynitrite (ONOO−) on cardiac myosin, actin and thin filaments in order to more clearly understand the impact of this reactive compound in ischemia/reperfusion injury and heart failure. Actin filaments, native thin filaments and α-cardiac myosin from rat hearts were exposed to ONOO− in the presence of 2 mM bicarbonate. Filament velocities over myosin, calcium sensitivity, and relative force generated by myosin were assessed in an in vitro motility assay in the absence of reducing agents. ONOO− concentrations ≥10 μM significantly reduced the velocities of thin filaments or bare actin filaments over α-cardiac myosin when any of these proteins were exposed individually. These functional deficits were linearly related to the degree of tyrosine nitration, with myosin being the most sensitive. However, at 10 μM [ONOO−] the calcium sensitivity of thin filaments remained unchanged. Co-treatment of myosin and thin filaments, analogous to the in vivo situation, resulted in a significantly greater functional deficit. The load supported by myosin after ONOO− exposure was estimated using mixtures experiments to be increased 3-fold. These data suggest that nitration of myofibrillar proteins can contribute to cardiac contractile dysfunction in pathologic states where ONOO− is liberated.
Keywords: myosin, actin, oxidative stress, cardiac muscle
INTRODUCTION
Reactive oxygen species (ROS) cause damage to DNA, proteins, lipids, and other macromolecules in a variety of cell types. An abundance of ROS engenders oxidative stress, which is thought to play a role in a number of pathologies, including neurodegenerative diseases [19,44], inflammation [12,22], cancer [1], and cardiovascular disease [47]. The direct effects of ROS on cardiac muscle are of particular interest because ROS concentrations increase in ischemia/reperfusion injury [43,56,62] and heart failure [30,34]. Indeed, oxidative stress can lead to cardiac myocyte apoptosis [3,26] and necrosis [20].
Peroxynitrite (ONOO−) is a strong oxidant formed in vivo via the near diffusion-limited reaction between nitric oxide and superoxide anion. It is considerably more reactive and damaging to cells than its precursors [6] and can aggravate myocardial ischemia/reperfusion injury in rats [29]. A number of potentially harmful protein modifications result from ONOO− pathways, including tyrosine nitration [4] to form 3-nitrotyrosine (3-NT), cysteine oxidation, methionine oxidation, and carbonylation of lysine, arginine and proline [18]. Under physiological conditions, ONOO− reacts with carbon dioxide to form a reactive nitrosoperoxycarbonate intermediate that favors nitration reactions over carbonylation or methionine oxidation [54]. Protein 3-NT levels are generally accepted as a biomarker of ONOO− activity [8], and increased levels are associated with both coronary artery disease [49] and heart disease [14,27,34–36,58,60,62].
The most important targets of ONOO− in myocardium are unknown, though ONOO− activation of poly (ADP ribose) polymerase [61] and matrix metaloproteases [40], as well as inhibition of creatine kinase [33] have all been proposed as mediating myocardial dysfunction. Age-related increases in tyrosine nitration have been observed in cardiac proteins related to metabolism, and of particular interest here, the cytoskeleton [21]. The functional consequences of nitration in those proteins, however, are unknown. The direct effects of ONOO− on the myofibrillar apparatus have been examined in skinned cardiac [13,36] and diaphragm muscles [52], where it reduces the maximum isometric tension at high (μM) but not low (50 nM) concentrations, though other ROS have been shown to increase submaximal force in skeletal muscle [2]. However, studies of the effects of ONOO− and other naturally-arising oxidants on purified myofibrillar and cytoskeletal protein function are few [10,11,24,53]. To date there is only one report of the effects of ONOO− on myosin. Specifically, exposure of myosin subfragment 1 (S1) to ONOO− may lead to partial unfolding of the protein and decreases its Mg2+-ATPase activity [53]. However, ATPase activity is not always a faithful reflection of the ability of myosin to generate force and motion. We therefore determined the consequences of ONOO− exposure on purified cardiac myosin and thin filament function in an in vitro assay of motility.
MATERIALS AND METHODS
Purification of Proteins
Myosin was prepared from rat cardiac muscle as described in [50] immediately before use. Each sample (~15mg wet weight) was homogenized using a glass-bead tissue grinder in 0.5mL of 0.3 M KCl, 10 mM HEPES, 10 mM Na4P2O7, 4 mM MgCl2, 1 mM ATP, 10 mM DTT, and TPCK/TLCK as protease inhibitors (pH 6.45). The homogenate was centrifuged at 140,000xg for 60 minutes in a Sorvall M120XG Ultracentrifuge to remove muscle residue and actin. The supernatant was diluted with 0.01M DTT and allowed to stand on ice for 60 minutes to precipitate myosin. The myosin was collected by centrifugation at 20,000xg for 20 minutes, and the pellet was dissolved in “myosin buffer” (0.3M KCl, 25 mM imidazole/EGTA, 4 mM MgCl2, and 10 mM DTT, pH 7.4). This preparation contains myosin heavy chain and light chains, and myosin binding protein C (MyBPC). Myosin to be used in experiments testing the effects of ONOO− on myosin was prepared with no DTT in the myosin buffer.
Native thin filaments were isolated immediately before use from cardiac muscle according to Lehman et al. [25] Each sample (~15mg wet weight) was homogenized using a glass-bead tissue homogenizer in 0.5 mL of 25 mM imidazole 1mM EGTA, 100 mM KCl, 5 mM MgCl2, 5 mM ATP, 10 mM DTT, TPCK/TLCK, and 1% Triton-X (pH 6.45). The homogenate was centrifuged for 20min at 40,000xg and the resulting supernatant was clarified by centrifugation for 45min at 200,000xg. The resulting pellet was resuspended in 0.2mL of 25 mM imidazole, 1mM EGTA, 100 mM KCl, 5 mM MgCl2, 5 mM ATP, and 10 mM DTT (pH 7.9). The resulting mixture was centrifuged for 5 min at 40,000xg for clarification followed by a second 45 min centrifugation at 200,000xg. The pellet was allowed to swell overnight at 4°C in 30 μl of 25 mM Imidazole, 1 mM EGTA, 100 mM KCl, 5 mM MgCl2, 10 mM DTT, and TRITC phallodin (Sigma, St. Louis, MO) (pH 6.45). On day 2, the pellet was resuspended in 200 μl of 25 mM Imidazole, 1 mM EGTA, 100 mM KCl, 4 mM MgCl2, 5 mM ATP, and 10 mM DTT (pH 7.9) and the thin filaments were collected by centrifugation for 10 min at 40,000xg. Thin filaments were prepared with no DTT in the TRITC phallodin solution and the final buffer.
Actin was purified as described by Pardee and Spudich [41] and labeled with TRITC-phalloidin. To remove DTT from the actin to be used in experiments testing the effects of ONOO− on actin, purified actin was centrifuged 15 min at 20,000xg and resuspended in DTT-free buffer. This washing procedure was then repeated. Protein concentrations were determined with Advanced Protein Assay Reagent (Cytoskeleton, Denver, CO) according to the manufacturer’s instructions.
Treatment of Proteins with Peroxynitrite
NaONOO (Caymen Chemical, Ann Arbor, MI) was stored in liquid nitrogen, thawed, and diluted in 100 μM KOH immediately before use. Proteins were treated with a range of ONOO− concentrations by adhering a droplet of diluted ONOO− to the side of the treatment tube containing protein and 2 mM bicarbonate to mimic physiologic pCO2 (which favors nitration over other potential reactions of ONOO−) and applying the tube to a vortexer for 2 seconds to ensure rapid mixing. Control proteins were treated with 100 μM KOH using the same method.
In Vitro Motility Assay
Motility was measured at 30°C with treated and control myosin, thin filaments, and actin using standard in vitro motility assay methods [15,23,57] modified to be performed without reducing agents. All buffers were degassed under vacuum for 20 min in small aliquot (100–200 μl) to remove oxygen. These aliquot were handled gently so as not to break surface tension. Solutions were re-degassed after 3 flow cells or 20 minutes, whichever came first.
Treated or control myosin at a concentration of ~50 μg/ml was applied to a flow cell constructed from a nitrocellulose-coated coverslip and a glass slide. After 1 min incubation, the flow cell was blocked with 0.5% Tween 20 (Sigma) in “actin buffer” (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 4mM MgCl2, pH 7.4) for 1 minute. TRITC-phalloidin labeled actin filaments or thin filaments were introduced, followed by two washes with actin buffer, and one volume of “motility buffer” (25 mM KCl, 25 mM imidazole, 1 mM EGTA, 4mM MgCl2, 1 mM ATP, 0.5% methylcellulose, pH 7.4). The motility buffer also contained an oxygen scavenger system (0.125 mg/ml glucose oxidase, 0.0225 mg/ml catalase, and 2.87 mg/ml glucose). In thin filament assays, CaCl2 was added to the motility buffer to achieve a saturating free [Ca2+] of 10 μM.
Thin filament calcium sensitivity was measured by changing the concentration of calcium in the motility buffer. Appropriate calcium levels from pCa 8.0- pCa 5.0 were calculated using MaxChelator software [42]. Calculations were based on a buffer pH of 7.4 and an ionic strength of 60 using 1mM EGTA as a Ca2+/Mg2+ chelator. Due to the highly viscous nature of motility buffer containing 0.5% methylcellulose, aliquots of motility buffer were dispensed using a positive-displacement pipette (Rainin) and weighed. The volume of 100 mM CaCl2 to be added was determined based upon that weight and the density of motility buffer (1.007 g/cm3 at 20°C). Density was determined using an analytical balance and a vendor-supplied density measuring kit (Mettler-Toledo). Filaments were imaged on an Olympus IX70 inverted microscope with a heated 100x objective and a PTI IC-200 intensified CCD camera. Movies were made in NIH Image using a Scion frame grabber and analyzed as described below.
Relative Force Measured by Mixtures Assay
In an in vitro motility assay, multiple myosin molecules pull simultaneously but asynchronously on actin filaments, generating continuous motion at a velocity characteristic of the isoform and the experimental conditions. When the assay is performed with two different populations of myosin, the different myosin molecules pulling on the same actin filament are essentially in a ‘tug-of-war.’ If the two myosins propel actin at different velocities, then the velocity of an actin filament will depend on the relative proportions of the two myosins, and the relative pulling strengths or kinetics of the two myosins; in a mixture, filaments will tend to run disproportionately closer to the velocity of the stronger myosin. Myosin mixture assays can therefore be used to estimate the relative difference in force or detachment kinetics of two different populations of myosin.
We performed mixtures assays to determine the effect of ONOO− treatment on the force generating capacity of cardiac myosin by mixing in different proportions untreated myosin with myosin treated with 10 μM ONOO−. The total myosin concentration added to the flow cell remained constant, while the fraction of each myosin type was varied from 0 to 1. Filament velocities were measured, and two models applied to derive relevant functional parameters. First, the “cross-bridge interactions model” relates the velocity of actin filaments sliding over mixtures of two types of myosin to the force-generating capacity or kinetics of the two myosins. In this model, the actin filament velocity observed when two myosins are attempting to move the same filament is a result of mechanical interactions between the two myosin types [16,57].
Actin filament velocity (Vactin) over the myosin mixture can be derived from a quadratic function of Vactin as follows:
| (1) |
where:
k is the fraction of the faster myosin, af/Pof and as/Pos are hyperbolic constants defining the curvature of the force-velocity curves of the faster and slower myosin, respectively [16,57]. Vmaxf and Vmaxs are the maximum actin filament velocity produced by the faster and slower myosin, respectively, and Pos/Pof is the ratio of the maximum average force produced by the slower relative to the faster myosin. Equation 1 was fitted to the data, making the assumption that the curvatures of the force-velocity relationships for the two types of myosin (untreated and treated, in our case) are equal to 0.26. From the fit, the relative average cross-bridge force generated or supported by each type of myosin was estimated.
An alternative interpretation derives from the original two-state model proposed by Huxley [17]. Cuda at al. [9] suggested that sliding velocity is primarily determined by the detachment rate from the strongly bound state for mixtures involving only actively cycling cross-bridges. The cross-bridge was modeled as having a single detached state and a single, strongly bound, attached state. Assuming linearly elastic cross-bridges with an elastic force constant κ, the positive force produced by cross-bridges during the power stroke can by approximated as F+ = κh2/2 and the force opposing contraction following completion of the power stroke can be approximated as F− = −κV2/g2, where h is the displacement of the power stroke, g is the rate constant for cross-bridge detachment, and V is sliding velocity. For unloaded shortening velocity, as in the in vitro motility assay, F+ + F− = 0. In a mixture of a slower and faster myosin, F+s + F+f + F−s + F−f = 0, where subscripts s and f denote slower and faster myosin species. From this, with the fraction of slower myosin denoted by σ and the ratio of the slower myosin to faster myosin elastic force constant denoted by η,
| (2) [9] |
Fitting the equation for V to mixtures assay velocity data allows the detachment rate constants gf and gs, and the elastic force constant (η) to be estimated.
Tracking Algorithm
A novel approach to the energy minimization technique of Crocker and Grier [7] was implemented to track actin filaments. The algorithm computes every possible track for all particles within a user-defined maximum distance and finds the lowest sum squared displacement for all particles. The complete track of every particle in a field of view is thus determined by an “energy” minimization approach.
The algorithm was implemented as a plugin to ImageJ [46]. Briefly, a built-in method is called for each image (slice) in a sequence (stack) to acquire the centroids of individual actin filaments. For every centroid in a particular slice, those centroids that lie within a user-defined maximum distance are identified, both in the current and the subsequent slice. Segmenting the image in this manner helps reduce the computational load. Next, the sum-squared displacement of every possible centroid combination, one time point to the next, is calculated using a recursive function. It is assumed that the combination of centroid matches with the lowest sum-squared displacement (analogous to energy) is the correct match of the centroids from one time point to those at the next [7].
Since the tracking algorithm is called for every filament (centroid) in the field of view, and each may have neighboring filaments that are included in the regional energy minimization, some might be identically or differently matched in subsequent, overlapping regions. If the previously matched filaments have different matches in a subsequent region, the sum-squared displacements for those combinations are compared. The pairing with the lower value is kept. At the end, a 3-D array contains the x and y positions of matched filaments arranged by index number for each point in time. All the filament positions (centroids) in that array that share a common index number represent the complete path of a filament. These are subsequently tallied to determine the velocity averaged over each filament track, along with the fraction of filaments that move. This “moving fraction” is measured by assuming that the overall displacement of the filament must be above some user-defined threshold value to be considered moving. Filaments that cross cause the termination of one of the two filament paths, since crossed filaments are interpreted as having a single centroid. However, the terminated path resumes as a new path once the filaments clear one another. An “overall velocity” is calculated as the product of the mean velocity and the moving fraction.
On a Pentium4 1.7GHz computer, the plugin requires only 5 seconds to track ~100 actin filaments per frame for 25 frames. The speed is highly dependent upon the number of filaments per unit area, since widely-spaced filaments cause the algorithm to behave like a nearest-neighbor matching algorithm. The values generated by this algorithm do not differ significantly from those generated by manually tracking every filament in a field of view using cross-correlation [45].
Tyrosin Nitration and Cysteine Accessibility
To assess the nitration levels of proteins exposed to ONOO−, SDS-PAGE and total protein staining was performed on control and ONOO−-treated (10 μM and 100 μM) thick and thin filament proteins as well as purified actin using pre-cast NuPage-MOPS 12% bis-tris gels (Invitrogen) for electrophoresis. Total protein concentrations were measured to ensure consistent protein loading. Gels were washed and fixed before staining, and were imaged using a Bio-Rad FX fluoresecent scanner. 16-bit images of each gel stain were collected and analyzed using the ImageJ gel analysis toolkit.
Relative protein nitration was determined by western blot using antibodies against 3-NT (Upstate). Nitration was assessed in control and ONOO−-treated myosin, thin filaments, and actin. Blots were developed using the Western Breeze chromogenic system (Invitrogen).
The availability of reactive cysteines in proteins was assayed by incubating ONOO−-treated and untreated myosin, thin filaments, and actin with an excess of 5-iodoacetomido fluorescein (5-IAF; MGT Inc), a thiol-reactive fluorescent dye. An increase in the number of solvent-accessible sulfhydryls would imply protein unfolding, whereas a decrease would indicate cysteine oxidation. 5-IAF was added to protein samples at a 4:1 molar excess. The reaction was allowed to take place in the dark for 30 min at room temperature. A 10:1 molar excess of DTT was added to the mixture to quench excess 5-IAF. 5-IAF reacted proteins were then run on SDS-PAGE in the dark. Gels were imaged using a Bio-Rad FX Molecular Imager using a 488 nm wavelength scanning laser and 530 nm emission filter. Gels were subsequently stained using Simply Blue coomassie total protein gel stain and imaged. 5-IAF:total protein ratios were determined using ImageJ software for band quantification.
RESULTS
Filament velocities
Actin filament velocity over control rat cardiac myosin was 3.7 ± 0.1 μm/s, consistent with previously reported values [51,59]. The function of cardiac myosin was significantly reduced when myosin was exposed to [ONOO−] of 10 μM and above (Figure 1A). Both the number of filaments moving and the velocity of moving filaments were affected. Mean velocity fell 18 % from control at 10 μM ONOO−, and 45 % from control at 100 μM ONOO−. Treatment with 10 μM ONOO− caused the fraction of filaments moving to drop 23 % relative to control, and even more (79%) with 100 μM ONOO−. Because ONOO− affected both the velocity measured for all moving filaments, and the fraction of filaments moving, we calculated the “overall velocity” as the product of these two values. Overall velocity was significantly reduced at [ONOO−] ≥ 10μM. An exponential fit to the overall velocity data allowed us to determine the [ONOO−] that resulted in 50% reduction in overall velocity (ED50) - 18.6 ± 3.8 μM when cardiac myosin alone was treated. The deficits were not reversible by exposure to DTT, eliminating disulfide bond formation and nitrosation as mechanisms leading to reduced function.
Figure 1.

Changes in actin filament velocity (gray bars), moving fraction (white bars), and overall function (black bars) as a function of [ONOO−]. Error bars indicate standard error of the mean. Curves are exponential fits to overall velocity data. A: Myosin is treated with ONOO−. B: Actin is treated with ONOO−. C: Native, regulated thin filaments are treated with ONOO−. D: Co-treatment of myosin and thin filaments. *p<0.05 compared to control.
Actin filaments exhibited a similar pattern of ONOO− sensitivity (Figure 1B). Mean velocity of treated actin filaments fell 14 % and 43% relative to controls when treated with 10 μM ONOO− and 100 μM ONOO−, respectively. The ED50 was calculated to be 18.5 ± 3.6 μM.
Native, regulated thin filaments were isolated from rat myocardium and treated with ONOO−. The velocity of control (untreated) thin filaments over cardiac myosin at saturating (10−5M) Ca2+ was 3.6 μm/s ± 0.2 (Figure 1C). The mean velocity fell by 6 % and 34 % at 10 μM and 100 μM ONOO−, respectively. An exponential fit to the overall velocity data yielded an ED50 of 55.6 ± 13.7 μM, significantly higher than that required for inhibition of actin or myosin.
Exposing both thin filaments and myosin to ONOO− did not lower the threshold [ONOO−] necessary to elicit a deficit, but did make the deficit more severe (Figure 1D). An exponential fit to the overall velocity data yielded an ED50 of 4.7 ± 1.3 μM when both proteins were treated. We thus find ONOO− to have a detrimental effect on all myofibrillar proteins in terms of sliding velocity and the fraction of moving filaments. Functional decline is evident at ONOO− levels in the low μM range when myosin and intact thin filaments are both exposed to the oxidant.
Calcium sensitivity
We also tested the effects of ONOO− on the Ca2+ sensitivity of thin filaments. A conservative [ONOO−] of 10 μM was chosen for these experiments because this is the lowest concentration that yielded consistent, though not always significant, deficits in overall protein function. Ca2+ sensitivity curves were constructed using overall velocity plotted against [Ca2+] in the motility buffer between 10−8 and 10−5 M (Figure 3). Measured pCa50 values were 6.78 ± 0.18 and 6.75 ± 0.02 for control and treated thin filaments, respectively, with corresponding Hill coefficients of 3.2 ± 0.9 and 2.7 ± 0.2. Thus there was no significant change in calcium sensitivity between treated and untreated thin filaments, though velocity at saturating [Ca2+] was reduced by 10 % according to fits of the Hill equation.
Figure 3.

Calcium sensitivity for control (●) and 10 μM ONOO−-treated (○) native thin filaments. Lines indicate fits of the Hill equation to each data set. Neither the Hill coefficients nor the pCa50 are significantly altered. Error bars indicate standard error of the mean.
Relative force
The mixtures assay resulted in a concave-up relationship between the overall velocity and the fraction of untreated myosin (Figure 2), meaning that in a mixture of treated and untreated myosin, the actin filaments run closer to the velocity of the treated myosin. There are two possible interpretations of this result. Interpreted with the cross-bridge interaction model, fits of equation 1 suggest that ONOO− exposure increases the average force generated or supported by myosin by a factor of 3.3 ± 0.2 relative to untreated myosin. Alternatively, fitting the two-state cross-bridge detachment model (equation 2) to the data allowed the detachment rate constants (gs, gf) of the treated and untreated myosin to be estimated. The fit yielded values of 78.4 ± 3.3 s−1 (gs) for the treated and 200.2 ± 4.1 s−1 (gf) for the untreated myosin detachment rates and a value of 1.04 ± 0.22 for the ratio of myosin elastic force constants. These data suggest ONOO− exposure results in an increase in force generated or supported by myosin and/or a decrease in the detachment rate of myosin from actin.
Figure 2.

Mixtures assay of untreated myosin mixed with 10 μM ONOO− treated myosin. The line represents the fit of the cross-bridge interaction model to the data. Error bars indicate standard error of the mean.
Post-translational modifications
Protein nitration was assayed by western blot and 3-nitrotyrosine-specific antibody, and compared to the relative change in filament velocity (Figure 4A). Nitration of myosin, myosin binding protein C (MyBPC), actin in thin filaments, and bare actin all increased with increasing levels of ONOO− treatment. Further, the relative degrees of nitration were linearly related to the measured functional deficits at those concentrations (Figure 4B). Tropomyosin showed no nitration until a [ONOO−] of 100 μM, while troponin bands were not visible.
Figure 4.
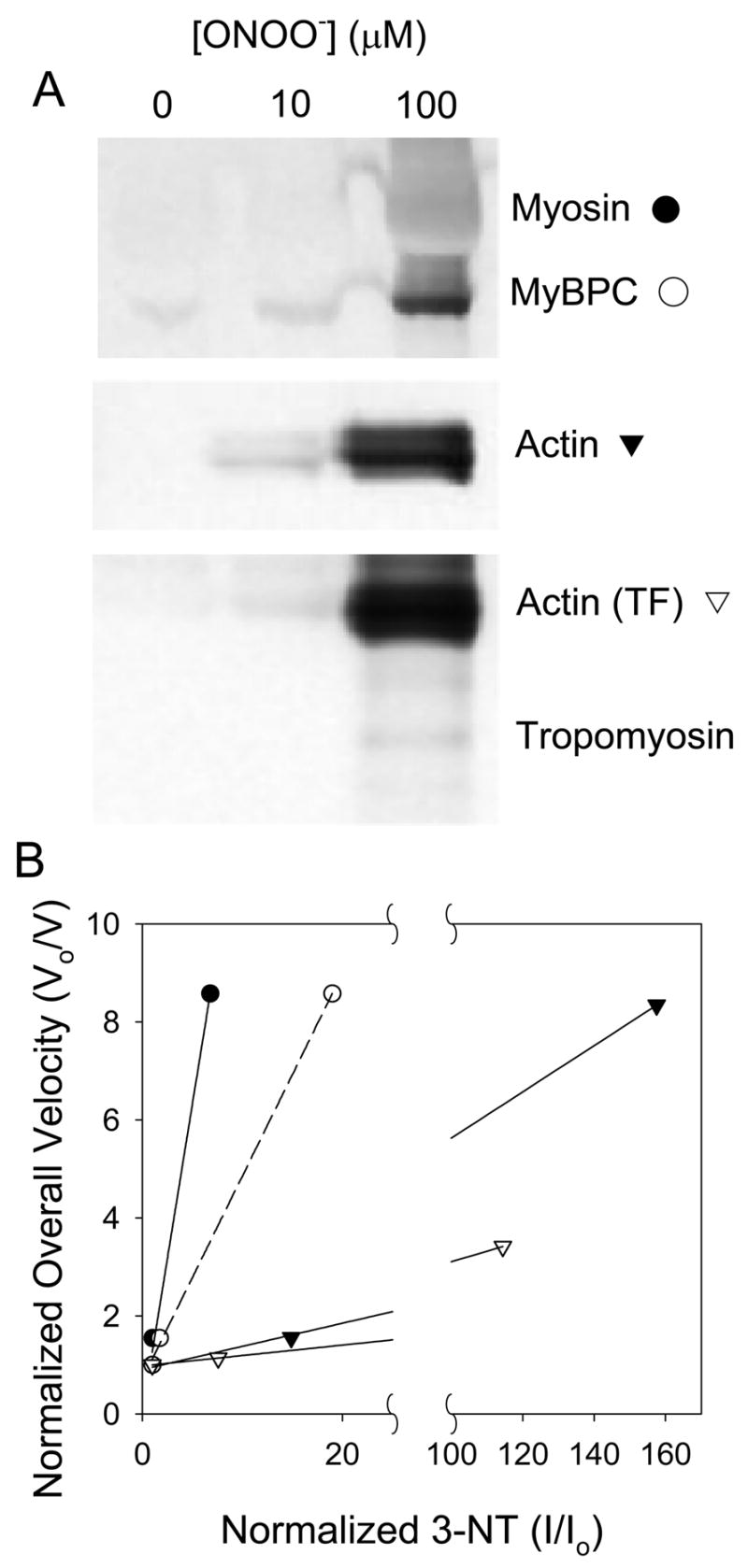
Nitration of myofibrillar proteins. A: Western blot against 3-NT for myosin, myosin binding protein C (MyBPC), purified actin, and thin filaments. Nitrated troponins are not evident. B: Nitration sensitivity curves. Fold-changes in overall velocity are plotted relative to changes in 3-NT for myosin, MyBPC, purified actin, and actin from thin filaments treated with 10 or 100 μM ONOO−. Lines indicate linear fits to the data. Vo/V is the inversely normalized velocity (i.e. fold-change in velocity) relative to control; an increase in Vo/V indicates that velocity has fallen. I/Io is the corresponding normalized band intensity from the western blot relative to control. The linear fit for MyBPC is dashed to indicate that it is present along with myosin when myosin is treated, and so it is not possible to completely separate the effects on these two proteins.
SDS-PAGE gels of 5-IAF-stained proteins were used to assess the number of accessible sulfhydryls. An increase in the number of solvent-accessible sulfhydryls would imply protein unfolding, whereas a decrease would indicate cysteine oxidation. Myosin demonstrated an increase in label-accessible cysteine-sulfhydryls with increasing levels of ONOO− treatment (Figure 5), suggesting unfolding of the protein. The magnitude of the observed increase relative to control was 2.2-fold and 4.7-fold for myosin treated with 10 μM and 100 μM ONOO−, respectively. A similar though less pronounced trend was seen with ONOO−-treated actin in both purified form and in thin filaments, as well as Troponin I. Other thin filament regulatory proteins showed no change. Limited digestion of 5-IAF stained myosin with trypsin yielded the expected fragments near 75, 50, 25 and 20 kDa. In contrast to a previous study [53], the 20 kDa fragment (which contains the so-called “reactive sulfhydryls”) showed no apparent change in cysteine availability.
Figure 5.
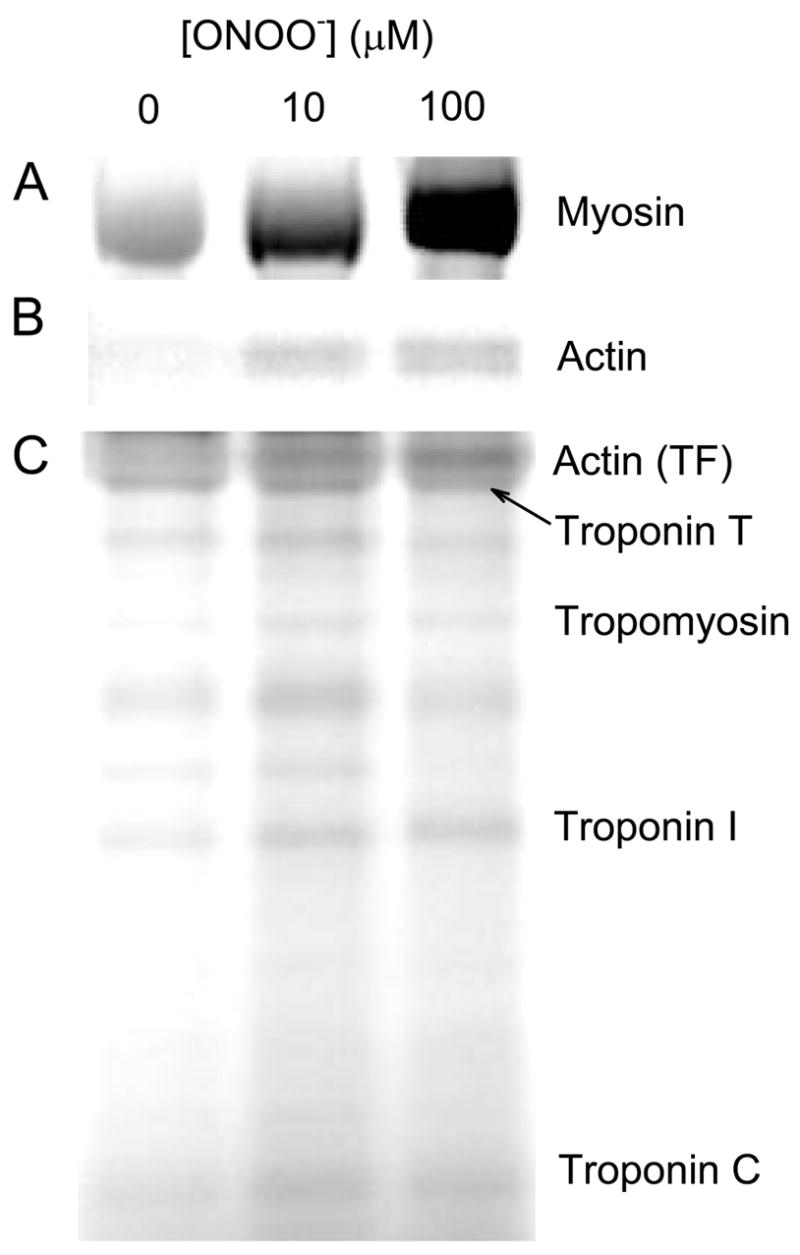
Solvent-accessible cysteines. Myosin (A), purified actin (B), and thin filaments (C) were treated with 10 or 100 μM ONOO− and subsequently reacted with 5-IAF to label reactive thiol groups. Solvent-exposed thiols in each protein increase with ONOO− treatment.
DISCUSSION
While previous studies of the effects of ONOO− on muscle function have been conducted in such systems as isolated papillary muscle [13], permeablized ventricular myocytes [5], and skeletal muscle fibers [1], the current study provides a more direct examination of its effects on specific proteins. We have shown that ONOO− concentrations believed to be possible in local environments in the heart lead to dramatic decreases in myofibrillar protein function. Upon co-treatment of myosin and thin filaments with ONOO− (analogous to the in vivo situation), at 10 μM the overall velocity of thin filaments over myosin decreased to less than 20 % of control. At these concentrations ONOO− appears to cause nitration and partial unfolding of myofibrillar proteins and may therefore play a significant role in conditions such as ischemia/reperfusion injury and heart failure - conditions in which elevated levels of ROS and 3-nitrotyrosine have been found.
The in vivo concentration of ONOO− in an infracted heart is not definitively known, though ONOO− levels in the heart have been shown to increase greater than 10-fold in the first minute of reperfusion following ischemia [56]. Concentrations in the low μM range are generally thought to be feasible under conditions of high oxidative stress [13,28]. However, because tyrosine nitration and some other ONOO−-induced post-translational modifications are essentially irreversible, they may accumulate even under low concentrations of ONOO−. This is especially likely given the slow turnover of myofibrillar proteins in myocytes [31]. Low levels of ONOO− may therefore result in functional deficits when generated continuously.
Interpretation of mixtures experiments
Results from the mixtures assay suggest a decrease in myosin detachment rate for ONOO−- treated myosin according to the two-state cross-bridge detachment model. A decreased detachment rate is unsurprising given the decreased mean velocity observed for myosin treated with ONOO−. Indeed, the magnitudes of decrease in mean velocity and detachment rate are reasonably similar, with a 41% drop in measured mean velocity and a 61% drop in calculated detachment rate.
The mixtures data may also be interpreted as an increased generation of force or support of load for ONOO−-treated myosin compared to control. This contrasts with previous studies [5,13] that showed a decrease in isometric force, though neither of these studies were done with purified cardiac myosin. However, the two interpretations of these data are not mutually exclusive. Force is proportional to duty cycle, d, which is the fraction of the total cycle time that myosin is attached to actin. It is given by gon/(gon+ goff), where gon is the attachment rate, and goff is the detachment rate. A decreased detachment rate will therefore contribute to an increase in force if gon is increased or unchanged. Direct measurements of force in purified actomyosin systems will clarify the interpretation. Whatever the biophysical interpretation, a slower myosin capable of supporting greater force may impose a significant negative load on the contractile apparatus and contribute to contractile dysfunction.
Nitration of myosin and myosin binding protein C
While it is not possible to assign the functional deficits to modifications of specific amino acid residues, the inclusion of 2 mM bicarbonate in our ONOO− treatments will bias reactions strongly toward tyrosine nitration. Indeed, we observed no net loss of reactive cysteines in any myofibrillar proteins, but instead observed an increase. This suggests partial unfolding of the proteins and exposure of buried cysteines subsequent to nitration. This is similar to the results of Tiago et al. [53], though we found no evidence for oxidation of cys707.
The steep slope of the sensitivity curve given in Figure 4B suggests that myosin is highly sensitive to nitration relative to the other myofibrillar proteins. However, there are two alternative interpretations of these data. First, the minimal nitration generated by 10 μM ONOO−, along with the significant decrease in the overall velocity of myosin treated with this same [ONOO−], suggests that other oxidative modifications may contribute significantly to the decline in myosin function (e.g. methionine oxidation to methionine sulfoxide or sulfone). It is also possible that the functional deficit in myosin at 10 μM ONOO− does not result from oxidative modification of myosin itself, but instead of myosin-binding protein C. Because myosin and MyBPC copurify in our preparation, they are treated together and are both present in the in vitro motility assay.
The role of MyBPC in the regulation of contraction is still unclear, but the C1-C2 region of MyBPC is thought to bind with myosin-S2 [39]. Phosphorylation of MyBPC inhibits the MyBPC-S2 interaction, leading to changes in ATPase activity, Ca2+ sensitivity [38], and maximum force generation [32]. It was recently shown that C1-C2 decreases actin and thin filament sliding velocity in the motility assay even in the absence of myosin S2, suggesting a novel mechanism involving C1-C2 binding to actin and/or myosin S1 [48]. The MyBPC nitration induced by ONOO− in the current study has the potential to affect the interaction of MyBPC with any of its potential binding-partners, and may contribute to the observed functional deficits even in the absence of myosin nitration. Thus it isn’t possible to definitively say whether modifications to only one or to both of these proteins are leading to the observed functional changes; this will be the subject of further study. One approach to addressing this question is to purify MyBPC apart from myosin, expose each separately to ONOO−, and reconstitute them into the motility assay in a four-way combinatorial experiment.
Our data show a minimal increase in myosin nitration upon exposure to 10 μM ONOO−, but a 6.8-fold nitration increase in myosin exposed to 100 μM ONOO−. Indeed, others have observed a lack of myosin heavy chain nitration after exposure to ONOO− concentrations similar to those used here, only to observe nitration after exposure to very high ONOO− concentrations of 250 μM and above [5,21]. One possible explanation for why 10 μM ONOO− results in minimal myosin nitration is that nitration of myosin is poorly assayed by immunoblotting. Epitopes are not generally as small as a single amino acid. It is therefore reasonable to assume that “anti-3-NT antibodies” actually recognize 3-NT and neighboring amino acids as their epitopes. Since available anti-3-NT antibodies are raised against nitrated albumin, it is possible that none of the recognized epitopes will be found in a randomly chosen protein. This would lead to an unpredictable lack of sensitivity and/or specificity. Supporting this possibility is our observation that the band covering the range of myosin heavy chain on our anti-3-NT western blots is very diffuse - an observation that can be made of published blots of muscle [5].
It is also possible that myosin is nitration-resistant, perhaps by design. This is an exciting possibility for which we are aware of no precedent. However, more reliable measures of nitration in myosin must be developed before this possibility can be explored.
Effects of ONOO− on actin and thin filaments
Actin, whether bare or in native thin filaments, was nitrated at both 10 and 100 μM ONOO−. Further, functional deficits were linearly related to the degree of actin nitration in filaments (Figure 4B), suggesting a causative relationship. However, bare actin filaments suffer a greater functional deficit at similar levels of actin nitration when compared to thin filaments. There are three potential explanations for the decreased functional sensitivity of thin filaments to nitration. First, tropomyosin and the troponins act as a sink for nitration reactions, essentially diluting the effect on actin. Arguing against this possibility, we found that similar levels of actin nitration in bare filament and thin filaments still give rise to different functional deficits. Further, tropomyosin and the troponins showed no nitration at 10 μM and only minimal nitration at 100 μM. A second explanation is that during treatment with ONOO−, thin filaments are in the “blocked state” due to the absence of calcium. In this case, tropomyosin may mask sites on actin that would otherwise be available for nitration or other ONOO−-induced modifications, in particular the myosin binding site. Conducting similar experiments in the presence of calcium is one approach to testing this possibility.
Finally, we must take into account that thin filaments are cooperatively activated by myosin; that is, even in the presence of calcium, tropomyosin will partially overlay the myosin binding sites on actin. The binding of a small number of myosins is necessary to achieving full activation of the thin filament. As a result, at some surface densities of myosin one can measure forces generated by myosin in excess of bare, unregulated actin [55]. Though these differences do not necessarily translate to differences in filament velocities, the myosin concentration used in the current study corresponds well with those concentrations showing cooperative effects. Repeating the current experiments at different myosin concentrations will reveal whether or not cooperative activation is allowing nitrated actin in native thin filaments to retain greater function as compared to similarly nitrated bare actin.
There is as yet no consensus regarding the effect of ONOO− (or ROS in general) on intact cardiac thin filament Ca2+ sensitivity, though the current study suggests a minimal effect. Our results show no significant change in thin filament Ca2+ sensitivity at a ONOO− concentration (10 μM) where both average force generation and overall velocity were effected. This is in contrast to a recent study in skeletal muscle that suggests a ROS-mediated decline in Ca2+ sensitivity [37].
While we can predict that ONOO− modification of myofibrillar proteins will indeed contribute to contractile dysfunction, it is not yet possible to say to what degree the observed functional changes will contribute to altered contractile function in vivo. In fact, it is not possible to do so for any cardiac proteins known to be oxidatively modified. Aside from the magnitude of functional deficit for a given degree of chemical modification, one must know the rates of chemical modification for each protein, the rate of ONOO− generation, and the localization of ONOO− within the cell. Once these parameters are more precisely known, a systems biological approach may shed light on the relative contribution of oxidative modifications to cellular dysfunction.
Acknowledgments
We’d like to thank Kenneth Hensley and Kelly Williamson of the Oklahoma Medical Research Foundation for their input and assistance in measuring protein nitration. We also acknowledge the generous support of the National Institutes of Health (EB002185, AR45604, and HL071958) and the Department of Biomedical Engineering at the University of Virginia.
This work was supported by NIH grants AR45604, EB002185, and HL071958.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade FH, Reid MB, Westerblad H. Contractile response of skeletal muscle to low peroxide concentrations: myofibrillar calcium sensitivity as a likely target for redox-modulation. FASEB J. 2001;15:309–311. doi: 10.1096/fj.00-0507fje. [DOI] [PubMed] [Google Scholar]
- 3.Arstall MA, Sawyer DB, Fukazawa R, Kelly RA. Cytokine-mediated apoptosis in cardiac myocytes - The role of inducible nitric oxide synthase induction and peroxynitrite generation. Circulation Research. 1999;85:829–840. doi: 10.1161/01.res.85.9.829. [DOI] [PubMed] [Google Scholar]
- 4.Beckmann JS, Ye YZ, Anderson PG, Chen J, Accavitti MA, Tarpey MM, White CR. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- 5.Borbely A, Toth A, Edes I, Virag L, Papp JG, Varro A, Paulus WJ, van dV, Stienen GJ, Papp Z. Peroxynitrite-induced alpha-actinin nitration and contractile alterations in isolated human myocardial cells. Cardiovasc Res. 2005;67:225–233. doi: 10.1016/j.cardiores.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Brunelli L, Crow JP, Beckman JS. The comparative toxicity of nitric oxide and peroxynitrite to Escherichia coli. Arch Biochem Biophys. 1995;316:327–334. doi: 10.1006/abbi.1995.1044. [DOI] [PubMed] [Google Scholar]
- 7.Crocker JC, Grier DG. Methods of digital video microscopy for colloidal studies. Journal of Colloid and Interface Science. 1996;179:298–310. [Google Scholar]
- 8.Crow JP, Ischiropoulos H. Detection and quantitation of nitrotyrosine residues in proteins: in vivo marker of peroxynitrite. Methods Enzymol. 1996;269:185–194. doi: 10.1016/s0076-6879(96)69020-x. [DOI] [PubMed] [Google Scholar]
- 9.Cuda G, Fananapazir L, Epstein ND, Sellers JR. The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J Muscle Res Cell Motil. 1997;18:275–283. doi: 10.1023/a:1018613907574. [DOI] [PubMed] [Google Scholar]
- 10.Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di Simplicio P, Colombo R, Milzani A. Methionine oxidation as a major cause of the functional impairment of oxidized actin. Free Radical Biology and Medicine. 2002;32:927–937. doi: 10.1016/s0891-5849(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 11.Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Lusini L, Milzani A, Di Simplicio P, Colombo R. Actin carbonylation: from a simple marker of protein oxidation to relevant signs of severe functional impairment. Free Radic Biol Med. 2001;31:1075–1083. doi: 10.1016/s0891-5849(01)00690-6. [DOI] [PubMed] [Google Scholar]
- 12.Dedon PC, Tannenbaum SR. Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 13.Digerness SB, Harris KD, Kirklin JW, Urthaler F, Viera L, Beckman JS, Darley-Usmar V. Peroxynitrite irreversibly decreases diastolic and systolic function in cardiac muscle. Free Radic Biol Med. 1999;27:1386–1392. doi: 10.1016/s0891-5849(99)00184-7. [DOI] [PubMed] [Google Scholar]
- 14.Ferdinandy P, Danial H, Ambrus I, Rothery RA, Schulz R. Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure. Circ Res. 2000;87:241–247. doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- 15.Guo B, Guilford WH. The tail of myosin reduces actin filament velocity in the in vitro motility assay. Cell Motil Cytoskeleton. 2004;59:264–272. doi: 10.1002/cm.20040. [DOI] [PubMed] [Google Scholar]
- 16.Harris DE, Work SS, Wright RK, Alpert NR, Warshaw DM. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15:11–19. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- 17.Huxley AF. Muscle structure and theories of contraction. Prog Biophys Biophys Chem. 1957;7:255–318. [PubMed] [Google Scholar]
- 18.Ischiropoulos Hal, Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- 19.Ischiropoulos H, Beckman JS. Oxidative stress and nitration in neurodegeneration: cause, effect, or association? J Clin Invest. 2003;111:163–169. doi: 10.1172/JCI17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 21.Kanski J, Behring A, Pelling J, Schoneich C. Proteomic identification of 3-nitrotyrosine-containing rat cardiac proteins: effects of biological aging. Am J Physiol Heart Circ Physiol. 2005;288:H371–H381. doi: 10.1152/ajpheart.01030.2003. [DOI] [PubMed] [Google Scholar]
- 22.Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:471–479. doi: 10.2174/1568010054526359. [DOI] [PubMed] [Google Scholar]
- 23.Kron SJ, Spudich JA. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc Natl Acad Sci U S A. 1986;83:6272–6276. doi: 10.1073/pnas.83.17.6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landino LM, Hasan R, McGaw A, Cooley S, Smith AW, Masselam K, Kim G. Peroxynitrite oxidation of tubulin sulfhydryls inhibits microtubule polymerization. Arch Biochem Biophys. 2002;398:213–220. doi: 10.1006/abbi.2001.2729. [DOI] [PubMed] [Google Scholar]
- 25.Lehman W, Vibert P, Uman P, Craig R. Steric-blocking by tropomyosin visualized in relaxed vertebrate muscle thin filaments. J Mol Biol. 1995;251:191–196. doi: 10.1006/jmbi.1995.0425. [DOI] [PubMed] [Google Scholar]
- 26.Levrand S, Vannay-Bouchiche C, Pesse B, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite is a major trigger of cardiomyocyte apoptosis in vitro and in vivo. Free Radic Biol Med. 2006;41:886–895. doi: 10.1016/j.freeradbiomed.2006.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu P, Hock CE, Nagele R, Wong PY. Formation of nitric oxide, superoxide, and peroxynitrite in myocardial ischemia-reperfusion injury in rats. Am J Physiol. 1997;272:H2327–H2336. doi: 10.1152/ajpheart.1997.272.5.H2327. [DOI] [PubMed] [Google Scholar]
- 28.Liu P, Xu B, Quilley J, Wong PY. Peroxynitrite attenuates hepatic ischemia-reperfusion injury. Am J Physiol Cell Physiol. 2000;279:C1970–C1977. doi: 10.1152/ajpcell.2000.279.6.C1970. [DOI] [PubMed] [Google Scholar]
- 29.Ma XL, Lopez BL, Liu GL, Christopher TA, Ischiropoulos H. Peroxynitrite aggravates myocardial reperfusion injury in the isolated perfused rat heart. Cardiovasc Res. 1997;36:195–204. doi: 10.1016/s0008-6363(97)00179-x. [DOI] [PubMed] [Google Scholar]
- 30.Malinski T. Understanding nitric oxide physiology in the heart: a nanomedical approach. Am J Cardiol. 2005;96:13i–24i. doi: 10.1016/j.amjcard.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 31.Martin AF, Rabinowitz M, Blough R, Prior G, Zak R. Measurements of half-life of rat cardiac myosin heavy chain with leucyl-tRNA used as precursor pool. J Biol Chem. 1977;252:3422–3429. [PubMed] [Google Scholar]
- 32.McClellan G, Kulikovskaya I, Winegrad S. Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C. Biophys J. 2001;81:1083–1092. doi: 10.1016/S0006-3495(01)75765-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mihm MJ, Bauer JA. Peroxynitrite-induced inhibition and nitration of cardiac myofibrillar creatine kinase. Biochimie. 2002;84:1013–1019. doi: 10.1016/s0300-9084(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 34.Mihm MJ, Coyle CM, Schanbacher BL, Weinstein DM, Bauer JA. Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc Res. 2001;49:798–807. doi: 10.1016/s0008-6363(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 35.Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, Bauer JA. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation. 2001;104:174–180. doi: 10.1161/01.cir.104.2.174. [DOI] [PubMed] [Google Scholar]
- 36.Mihm MJ, Yu F, Weinstein DM, Reiser PJ, Bauer JA. Intracellular distribution of peroxynitrite during doxorubicin cardiomyopathy: evidence for selective impairment of myofibrillar creatine kinase. Br J Pharmacol. 2002;135:581–588. doi: 10.1038/sj.bjp.0704495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moopanar TR, Allen DG. Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 degrees C. J Physiol. 2005;564:189–199. doi: 10.1113/jphysiol.2005.083519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oakley CE, Chamoun J, Brown LJ, Hambly BD. Myosin binding protein-C: Enigmatic regulator of cardiac contraction. Int J Biochem Cell Biol. 2007 doi: 10.1016/j.biocel.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Oakley CE, Chamoun J, Brown LJ, Hambly BD. Myosin binding protein-C: Enigmatic regulator of cardiac contraction. Int J Biochem Cell Biol. 2007 doi: 10.1016/j.biocel.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto T, Akaike T, Nagano T, Miyajima S, Suga M, Ando M, Ichimori K, Maeda H. Activation of human neutrophil procollagenase by nitrogen dioxide and peroxynitrite: a novel mechanism for procollagenase activation involving nitric oxide. Arch Biochem Biophys. 1997;342:261–274. doi: 10.1006/abbi.1997.0127. [DOI] [PubMed] [Google Scholar]
- 41.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85(Pt B):164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 42.Patton C, Thompson S, Epel D. Some precaustions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 43.Powell SR, Gurzenda EM, Wahezi SE. Actin is oxidized during myocardial ischemia. Free Radic Biol Med. 2001;30:1171–1176. doi: 10.1016/s0891-5849(01)00514-7. [DOI] [PubMed] [Google Scholar]
- 44.Rao AV, Balachandran B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr Neurosci. 2002;5:291–309. doi: 10.1080/1028415021000033767. [DOI] [PubMed] [Google Scholar]
- 45.Rao V, La Bonte LR, Xu Y, Yang Z, French BA, Guilford WH. Alterations to myofibrillar protein function in non-ischemic regions of the heart early after myocardial infarction. American Journal of Physiology In Revision. 2007 doi: 10.1152/ajpheart.01314.2006. Ref Type: Journal (Full) [DOI] [PubMed] [Google Scholar]
- 46.Rasband WS. NIH Image, ImageJ. Bethesda: Maryland National Institutes of Health; 1997. [Google Scholar]
- 47.Rojas A, Figueroa H, Re L, Morales MA. Oxidative stress at the vascular wall. Mechanistic and pharmacological aspects. Arch Med Res. 2006;37:436–448. doi: 10.1016/j.arcmed.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 48.Shaffer JF, Razumova MV, Tu AY, Regnier M, Harris SP. Myosin S2 is not required for effects of myosin binding protein-C on motility. FEBS Lett. 2007;581:1501–1504. doi: 10.1016/j.febslet.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Shishehbor MH, Aviles RJ, Brennan ML, Fu X, Goormastic M, Pearce GL, Gokce N, Keaney JF, Jr, Penn MS, Sprecher DL, Vita JA, Hazen SL. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA. 2003;289:1675–1680. doi: 10.1001/jama.289.13.1675. [DOI] [PubMed] [Google Scholar]
- 50.Shiverick KT, Thomas LL, Alpert NR. Purification of cardiac myosin. Application to hypertrophied myocardium. Biochim Biophys Acta. 1975;393:124–133. doi: 10.1016/0005-2795(75)90222-6. [DOI] [PubMed] [Google Scholar]
- 51.Sugiura S, Kobayakawa N, Momomura S, Chaen S, Omata M, Sugi H. Different cardiac myosin isoforms exhibit equal force-generating ability in vitro. Biochim Biophys Acta. 1996;1273:73–76. doi: 10.1016/0005-2728(95)00149-2. [DOI] [PubMed] [Google Scholar]
- 52.Supinski G, Stofan D, Callahan LA, Nethery D, Nosek TM, DiMarco A. Peroxynitrite induces contractile dysfunction and lipid peroxidation in the diaphragm. J Appl Physiol. 1999;87:783–791. doi: 10.1152/jappl.1999.87.2.783. [DOI] [PubMed] [Google Scholar]
- 53.Tiago T, Simao S, Aureliano M, Martin-Romero FJ, Gutierrez-Merino C. Inhibition of skeletal muscle S1-myosin ATPase by peroxynitrite. Biochemistry. 2006;45:3794–3804. doi: 10.1021/bi0518500. [DOI] [PubMed] [Google Scholar]
- 54.Tien M, Berlett BS, Levine RL, Chock PB, Stadtman ER. Peroxynitrite-mediated modification of proteins at physiological carbon dioxide concentration: pH dependence of carbonyl formation, tyrosine nitration, and methionine oxidation. Proc Natl Acad Sci U S A. 1999;96:7809–7814. doi: 10.1073/pnas.96.14.7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.VanBuren P, Palmiter KA, Warshaw DM. Tropomyosin directly modulates actomyosin mechanical performance at the level of a single actin filament. Proc Natl Acad Sci U S A. 1999;96:12488–12493. doi: 10.1073/pnas.96.22.12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang P, Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. Evidence for peroxynitrite-mediated reperfusion injury. J Biol Chem. 1996;271:29223–29230. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- 57.Warshaw DM, Desrosiers JM, Work SS, Trybus KM. Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J Cell Biol. 1990;111:453–463. doi: 10.1083/jcb.111.2.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinstein DM, Mihm MJ, Bauer JA. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther. 2000;294:396–401. [PubMed] [Google Scholar]
- 59.Yamashita H, Sugiura S, Fujita H, Yasuda S, Nagai R, Saeki Y, Sunagawa K, Sugi H. Myosin light chain isoforms modify force-generating ability of cardiac myosin by changing the kinetics of actin-myosin interaction. Cardiovasc Res. 2003;60:580–588. doi: 10.1016/j.cardiores.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 60.Yasmin W, Strynadka KD, Schulz R. Generation of peroxynitrite contributes to ischemia-reperfusion injury in isolated rat hearts. Cardiovasc Res. 1997;33:422–432. doi: 10.1016/s0008-6363(96)00254-4. [DOI] [PubMed] [Google Scholar]
- 61.Zingarelli B, Cuzzocrea S, Zsengeller Z, Salzman AL, Szabo C. Protection against myocardial ischemia and reperfusion injury by 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase. Cardiovasc Res. 1997;36:205–215. doi: 10.1016/s0008-6363(97)00137-5. [DOI] [PubMed] [Google Scholar]
- 62.Zweier JL, Fertmann J, Wei G. Nitric oxide and peroxynitrite in postischemic myocardium. Antioxid Redox Signal. 2001;3:11–22. doi: 10.1089/152308601750100443. [DOI] [PubMed] [Google Scholar]