

Abstract
We present the first evidence for a fast activation of the nuclear protein poly(ADP-ribose) polymerase (PARP) by signals evoked in the cell membrane, constituting a novel mode of signaling to the cell nucleus. PARP, an abundant, highly conserved, chromatin-bound protein found only in eukaryotes, exclusively catalyzes polyADP-ribosylation of DNA-binding proteins, thereby modulating their activity. Activation of PARP, reportedly induced by formation of DNA breaks, is involved in DNA transcription, replication, and repair. Our findings demonstrate an alternative mechanism: a fast activation of PARP, evoked by inositol 1,4,5,-trisphosphate–Ca2+ mobilization, that does not involve DNA breaks. These findings identify PARP as a novel downstream target of phospholipase C, and unveil a novel fast signal–induced modification of DNA-binding proteins by polyADP-ribosylation.
Keywords: poly(ADP-ribose) polymerase; calcium signaling; inositol 1,4,5-trisphosphate; electrical stimulation; brain neurons
Introduction
Membrane depolarization influences neuronal development (Oppenheim 1991; Spitzer 1991) and prevents apoptotic cell death of neurons in cultures deprived of growth factors (Brenneman et al. 1990; Franklin and Johnson 1992; D'Mello et al. 1993; Galli et al. 1995) by a poorly understood mechanism. These phenomena prompted us to examine the possible effect of membrane depolarization on the activity of the nuclear protein poly(ADP-ribose) polymerase (PARP). PARP is an abundant and highly conserved chromatin bound protein (113 kD), found only in eukaryotes, which catalyzes exclusively polyADP-ribosylation of DNA-binding proteins (Udea 1990; Lautier et al. 1993; Lindahl et al. 1995; D'Amours et al. 1999). Reportedly, PARP, activated by binding to free DNA-endings, acts as an ADP-ribose transferase, adding ADP-ribose to carboxyls of aspartic and glutamic residues. This reaction proceeds by a short-lived (t 1/2 = 1 min) polymerization of ADP-riboses, i.e., polyADP-ribosylation (Kupper et al. 1990; Satoh and Lindahl 1992; Satoh et al. 1994). Activated PARP is auto-polyADP-ribosylated (Desmarais et al. 1991; Satoh et al. 1994; Kim et al. 1997). PolyADP-ribosylation is terminated by the release of extensively polyADP-ribosylated (negatively charged) PARP molecules from DNA (Satoh et al. 1994). ADP-ribose polymers are then instantaneously subjected to partial degradation by polyADP-ribose–glycohydrolase, and completely degraded by a relatively slow process (20–30 min; Satoh et al. 1994; Lin et al. 1997).
Known substrates of PARP include topoisomerase I (Ferro and Olivera 1984; Kasid et al. 1989), RNA-polymerase II (Hanawalt et al. 1994, Li Oei et al. 1998), DNA polymerases (Simbulan et al. 1993), transcription factors (Rawling and Alvarez-Gonzalez 1997; Li Oei et al. 1998), histones (Boulikas 1990; D'Amours et al. 1999), high mobility group proteins (Tsai et al. 1992; D'Amours et al. 1999), p53 (Li Oei et al. 1998), and DNA-dependent kinase (Ruscetti et al. 1998). PolyADP-ribosylation modulates their activity, influencing DNA replication (Cesarone et al. 1990), transcription (Meisterernst et al. 1997, D'Amours et al. 1999), and repair (Satoh and Lindahl 1992; Lazebnik et al. 1994; Nicholson et al. 1995; Schreiber et al. 1995; Martinou 1996; Trucco et al. 1998).
The findings presented here demonstrate a fast signal–induced activation of PARP in brain cortical neurons, mediated by inositol 1,4,5,-trisphosphate (IP3)–induced Ca2+ mobilization, which does not involve DNA damage. Thus, PARP acts as a downstream target of phospholipase C.
Materials and Methods
Primary Culture of Rat Brain Cortical Neurons
Primary culture of rat brain cortical neurons was prepared from 18–19-d-old embryos of Sprague Dawley rats. Brain cortex was dissociated mechanically and plated in MEM (Biological Industries), containing 8% horse serum, 8% FCS, 0.6% glucose, 2 mM glutamine, and 15 μg/ml gentamicin. Plating density was 106 cells per 35-mm-diameter Nunc plates, precoated with 50 μg/ml poly-l-lysine. Glial cell proliferation was blocked by the addition of 20 μg/ml 5-fluoro-2-deoxyuridine and 50 μg/ml uridine on the third day after plating. Experiments were performed on the fifth and sixth days. Neurons survived in these cultures for 15–18 d.
Crude Nuclei
Crude nuclei were isolated from lysed brain cortical neurons (Cohen-Armon et al. 1996). Cultured cortical neurons were homogenized on ice in isotonic 0.32 M sucrose containing PMSF (0.1 mM), using a glass/glass homogenizer, and were centrifuged at 900 g for 10 min at 4°C. Cells in the resulting pellet were lysed in hypotonic solution (50 mM Tris-Cl, pH 7.4) and centrifuged as described above. This procedure was repeated in 0.32 M sucrose (900 g for 10 min at 4°C) and in 50 mM Tris-Cl, pH 7.4 (12,000 g for 10 min, 4°C). The resulting pellet contained isolated crude nuclei (see electromicrograph in Fig. 8 a).
Figure 8.
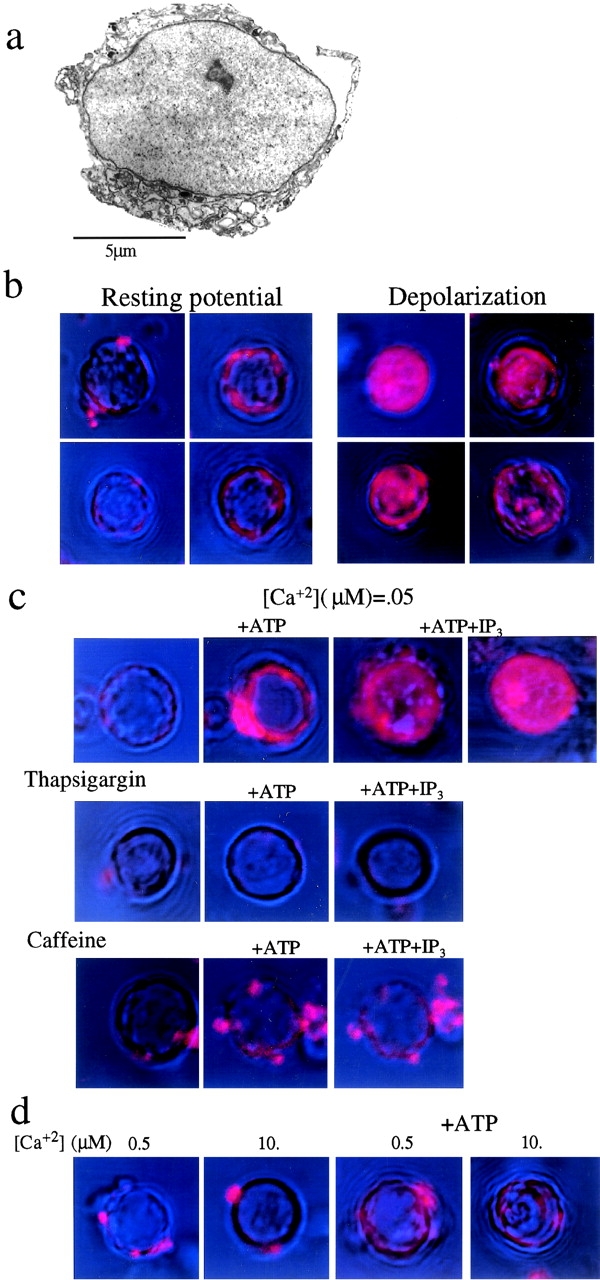
Ca2+ mobilization in crude nuclei isolated from brain cortical neurons. (a) Electromicrograph of a crude nucleus isolated from lysed brain cortical neuron (Materials and Methods). (b–d) Confocal microscopy showing Ca2+ redistribution in crude nuclei of cortical neurons as indicated by changes in the fluorescence of rhod-2 AM (Materials and Methods). (b) Ca2+ detected in the nucleoplasm of depolarized (high-[K+] depolarization, 5 min) and unstimulated neurons. (c) Ca2+ redistribution, visualized instantaneously during application of ATP (2.5 mM) and IP3 (1 μM) to crude nuclei of unstimulated neurons in the presence or absence of 5 mM caffeine, or to nuclei of neurons pretreated by 3 μM thapsigargin (10 min, 37°C). (d) Ca2+ redistribution in crude nuclei, evoked by increased extranuclear [Ca2+] in the presence or absence of ATP (2.5 mM).
Recording of Membrane Potential during Depolarizing Stimulation
Cultured cortical neurons were depolarized by raising the extracellular [K+] from 4.7 mM to 60 mM (high-[K+]) in the absence of extracellular Ca2+. The added KCl always replaced NaCl, thus preserving the physiological osmolarity and ionic strength of the original solutions (Cohen-Armon and Sokolovsky 1991). Changes in the resting potential of the cultured neurons were measured by the accumulation of the permeant-labeled cation, tetraphenyl-phosphonium ([3H]TPP+; Cohen-Armon and Sokolovsky 1991). Alternatively, cortical neurons were depolarized by pulsed electrical stimulation, using a pulse generator (Gruss Medical Instruments) and Pt electrodes installed in 2 ml/plate of either MEM or bath solution (defined below). There was no direct contact between neurons and stimulating electrodes (bath-stimulation). Membrane potential was recorded in individual neurons during stimulation by the patch-clamp technique, using the “whole cell” configuration in the current-clamp mode (Hamill et al. 1981), with Axopatch amplifier 200A and pCLAMP6.0 software (Axon Instruments, Inc.). Signals were filtered at 2 kHz (−3dB point) and digitized at a rate of 50 kHz. The solution in the patch pipette contained (mM): 146 KCl, 5 NaCl, 10 Hepes, 1 MgATP, 1 CaCl2, 2 BAPTA (pH 7.2) and 310 mOsm. Bath solution contained (mM): 130 NaCl, 5 KCl, 30 Glucose, 25 Hepes, 1 MgCl2, 2 CaCl2 (pH 7.4) and 300 mOsm.
Immunoprecipitation
PolyADP-ribosylated proteins were immunoprecipitated from nuclear protein extracts by monoclonal antibody directed against ADP-ribose polymers containing >10 ADP-riboses (10H; Lamarre et al. 1988; Shah et al. 1995) (see Materials). PARP was immunoprecipitated from the nuclear protein extracts by an affinity-purified goat polyclonal antibody raised against amino acids 1–20 at the NH2 terminus of human PARP (N-20; see Materials). For immunoprecipitation, nuclear proteins (∼400 μg protein/sample) were extracted during incubation of crude nuclei (30 min, 4°C) with 50 μl buffered solution containing 500 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-Cl (pH 7.4). Samples were then centrifuged (10,000 g, 5 min) and the supernatants were diluted in buffered solution containing 1.5 mM MgCl2 and 10 mM Tris-Cl. Nuclear proteins were exposed in this solution (overnight, 4°C) to the first antibody (dilution 1:20). Proteins bound to the antibody were precipitated during overnight incubation with protein G–conjugated agarose beads at 4°C, and then extracted from the beads after several washes with PBS by boiling for 2 min in sample buffer.
In Situ Immunofluorescent Labeling of PolyADP-ribosylated Proteins in Cultured Cortical Neurons
Tissue cultures were prepared on coverslips. Monoclonal 10H antibody (dilution 1:10) was introduced into rapidly fixed neurons (fixed for 10 min in ice-cold methanol/acetone 1:1, vol/vol). After overnight incubation with the first antibody at 4°C, neurons were washed with PBS containing 0.1% Tween 20 and exposed to the secondary antibody (dilution 1:500) for 3 h at room temperature. ADP-ribose polymers bound to the nuclear proteins were visualized by the FITC-conjugated affinity pure goat anti–mouse IgG3κ secondary antibody, using a fluorescence confocal inverted microscope (ZEISS LSM 410).
[32P]PolyADP-ribosylation of Proteins in Isolated Crude Nuclei
Unless indicated otherwise, crude nuclei isolated from cortical neurons were incubated for 1–5 min with [32P]NAD (1,000 Ci/mmol; 1μ Ci/sample) and 2.3 mM MgATP at 37°C in a solution containing (mM): 0.045 EDTA, 60 Tris-Cl, 1 MgCl2, and 0.8 DTT (pH 7.4). Deionized water contained 25–30 nM Ca2+ (determined by atomic absorbtion). [32P]Poly-ADP-ribosylation was terminated by high salt extraction of the nuclear proteins (500 mM NaCl, 10 mM Tris-Cl [pH 7.4], 4°C, 30 min). [32P]polyADP-ribosylated PARP was immunoprecipitated from the nuclear proteins extracts, subjected to SDS-PAGE, electroblotted, and autoradiographed. [32P]PolyADP-ribosylation was quantified by densitometry.
Thymidine Incorporation into DNA during Stimulation
Cultured neurons were incubated with [3H]thymidine (1 μCi/ml) for 1 h before stimulation. 4 h after stimulation, neurons were lysed and harvested onto filters (GF/C, Whatman). The tritium β emission of incorporated [3H]thymidine was counted in scintillation mixture (Friedberg et al. 1995).
Incorporation of modified thymidine, 5-bromodeoxyuridine (BrdUrd) was measured by immunolabeling with anti-BrdUrd monoclonal antibody IU-4 (Caltag Laboratories). BrdUrd (50 μM) was added to cultured neurons 1 h before stimulation. 6 h after stimulation, the neurons were fixed and treated with RNase A. A limited DNA denaturation was performed to allow access of anti-BrdUrd antibody into the DNA (Selden and Dolbeare 1994). Immunolabeled neurons were then incubated with 5 μg/ml propidium iodide, which intercalates into native DNA (Selden and Dolbeare 1994). The amount of incorporated BrdUrd labeled by FITC-conjugated secondary antibody (green fluorescence), indicating DNA synthesis, and the amount of intercalated propidium iodide (red fluorescence), indicating the amount of double stranded DNA, were measured by flow cytometry (FACSort machine operated by CellQest software; Becton Dickinson).
Single Strand DNA Breaks Examined by Alkaline Gel Electrophoresis
This method provides a sensitive and rapid method for direct quantitation of breaks in DNA single strands (Sutherland et al. 1999). DNA was isolated from the nuclei of cortical neurons using the Hirt procedure (Hirt 1967). The migration of equivalent amounts of DNA was analyzed by electrophoresis on 1% alkaline agarose gel (Sutherland et al. 1999). DNA was stained with ethidium bromide (1 μg/ml) and photographed under UV illumination.
Selective Extraction of Fragmented DNA from Nuclei
Fragmented DNA was selectively extracted from prefixed nuclei in high molarity phosphate–citrate buffer (Darzynkiewicz and Juan 1999). High molecular weight DNA and DNA attached to the nuclear matrix resisted extraction, but fragmented DNA was extracted from the nuclei and identified on agarose gel by staining with ethidium bromide (1 μg/ml).
Displacement of Bound [3H]IP3 by IP3
Samples (20 μl) of crude nuclei (1.5 mg protein/ ml) were incubated (10 min, 4°C) with [3H]IP3 (200 pmol/20 μl sample) in the solution used for [32P]polyADP-ribosylation. Crude nuclei were then rapidly washed under pressure on Whatman GF/B glass-fiber filters, with ice-cold solution containing 25 mM Tris-Cl, 5 mM NaHCO3 and 1 mM EDTA, pH 8.0 (Challiss et al. 1990). The amount of [3H]IP3 bound to the crude nuclei was assayed by counting their β emission in scintillation fluid. Nonspecific binding of [3H]IP3 was determined in the presence of 10 μM IP3.
Topoisomerase I Activity
Topoisomerase I activity was measured in nuclear protein extracts as described previously (Liu and Miller 1984). Extracted nuclear proteins (0.1 μg/sample) were added to a reaction mixture containing, at a final volume of 25 μl (mM): 20 Tris-Cl (pH 8.1), 1 DTT, 20 KCl, 10 MgCl2, 0.5 EDTA, 20 μg/ml BSA, and (as substrate) 250 ng of pUC-19, a supercoiled DNA plasmid (Promega). After incubation at 37°C for 30 min, the reaction was terminated by the addition of 5 μl of buffer containing: 50 mM EDTA (pH 8.0), 1% SDS, 15% glycerol, and 0.05% bromophenol blue. The reaction products were analyzed by electrophoresis on 1% agarose gel. Under these experimental conditions topoisomerase II is not activated (Liu and Miller 1984).
Electron Microscopy
Nuclei isolated from cultured brain cortical neurons were fixed with glutaraldehyde/paraformaldehyde (3:1%) in Krebs-Henseleit buffer (pH 7.4) containing 30% BSA. They were then washed at 4°C with 0.1 M PBS (pH 7.4) and postfixed with 1% OsO4 and 1.5% potassium ferricyanide in PBS at 4°C for 2 h. The samples were examined under a Jeol Jem-100CX electron microscope.
Simultaneous Recording of Rhod-2 Fluorescence
Isolated crude nuclei were loaded with the Ca2+ indicator rhod-2/AM (4.5 μM, 30 min incubation, 25°C, at dark), washed, and attached to poly-l-lysine–coated coverslips. Ca2+-induced fluorescent signal of rhod-2 (excitation, 540 nm; emission, >570 nm) was collected through appropriate filters above 520 nm and monitored by confocal inverted microscope (ZEISS LSM 410), equipped with a 25 mW krypton–argon laser (488- and 568-nm lines) and 10 mW He–Ne laser (633-nm line). A 40× NA/1.2 C-apochromat water-immersion lens (Axiovert 135 M, ZEISS) was used for imaging.
DNAse I Activity in Nuclei Isolated from Cultured Neurons
DNAse I activity in nuclei isolated from cultured neurons was assayed according to the procedure described by Boulikas 1990. Nuclei were incubated with DNAse I (RNAse-free; D 7291, Sigma-Aldrich) in buffered solution containing 20 mM Mn2+, 10% glycerol, 10 mM Tris-Cl, and 1 mM DTT (pH 7.4). The reaction was terminated by the addition of 25 mM EDTA (pH 8.0). Fragmented DNA was examined by gel agarose electrophoresis.
Materials
[Adenylate-32P]nicotinamide-adenindinucleotide, di(tri-ethyl-ammonium) salt ([32P]NAD) (1,000 Ci/mmol) was purchased from DuPont or from Amersham Pharmacia Biotech. D-myo-[3H]Inositol 1,4,5,-trisphosphate, potassium salt ([3H]IP3) (20–60 Ci/mmol) was from Amersham Pharmacia Biotech. [Methyl-3H]thymidine 5′-triphosphate, tetrasodium salt (70–90 Ci/mmol) and [phenyl-3H]tetraphenyl phosphonium bromide ([3H]TPP+) (35 Ci/mmol) were from DuPont. IP3 (hexapotassium salt) was from BIOMOL. Ethylenediamine-tetraacetic acid (EDTA) and ethyleneglycol-bis(β-amino-ethyl) N,N,N′,N′-tetraacetic acid (EGTA) were from Merck. D(−)-2-amino-5-phosphovaleric acid (APV) was from Cambridge Research Biochemicals. Ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl) ester (BAPTA AM) and rhod-2 AM were from Molecular Probes. (+)-MK-801 hydrogen maleate was from Biotrend. The polyclonal anti–human PARP antibody Vic-5 and monoclonal antibody 10H, directed against ADP-ribose polymers, were kind gifts from Dr. Sugimura, Tokyo Cancer Center, Japan. Anti–human PARP antibody (N-20) and secondary antibodies were from Santa Cruz Biotechnology. Other materials were from Sigma-Aldrich.
Results
Membrane Depolarization Induces PolyADP-ribosylation of Nuclear Proteins in Rat Cortical Neurons
We examined the effect of membrane depolarization on PARP activity in rat brain cortical neurons. Enhanced activity of PARP in depolarized neurons was indicated by: in situ immunolabeling of polyADP-ribosylated proteins; auto-polyADP-ribosylation of PARP; and inhibition of topoisomerase I activity (Ferro and Olivera 1984; Kasid et al. 1989).
In Situ Immunolabeling of PolyADP-ribosylated Nuclear Proteins by Antibody Directed against ADP-ribose Polymers.
PolyADP-ribosylated proteins were immunolabeled in situ by monoclonal antibody directed against ADP-ribose polymers (10H; Shah et al. 1995) in neurons, permeabilized by a rapid fixing procedure (see Materials and Methods). Immunolabeling of ADP-ribose polymers indicated an increased polyADP-ribosylation of proteins in the nuclei of depolarized neurons, relative to that in nuclei of unstimulated neurons (Fig. 1 a). Moreover, in situ polyADP-ribosylated PARP was immunoprecipitated by 10H antibody from nuclear extracts of depolarized or electrically stimulated neurons, indicating its enhanced polyADP-ribosylation during depolarization (Fig. 1 b). A significantly higher polyADP-ribosylation of PARP was observed in nuclei of neurons pretreated by H2O2, an agent producing DNA breaks (Dizdaroglu 1992; de Murcia et al. 1994; Fig. 1 b). PARP in nuclei of unstimulated neurons was not immunoprecipitated by 10H antibody (Fig. 1 b).
Figure 1.

Membrane depolarization induces polyADP-ribosylation of nuclear proteins in brain cortical neurons. (a) PolyADP-ribosylated proteins in the nuclei of prefixed cultured rat cortical neurons were immunolabeled in situ with monoclonal antibody directed against ADP-ribose polymers (10H; see Materials and Methods). Confocal images of neurons, labeled with fluorescein-conjugated secondary antibody (top), were also visualized in transmitted light (bottom). The four frames, from left to right, show neurons depolarized by high-[K+] for 5 min; unstimulated neurons; neurons pretreated with H2O2 (1 mM, 10 min); and depolarized neurons labeled only with the secondary antibody (n = 4). (b) Western blots of polyADP-ribosylated PARP immunoprecipitated by 10H antibody from nuclei of unstimulated (lane 1) and depolarized (lanes 2–4) cortical neurons. Neurons were depolarized by high-[K+] (lane 2), or stimulated by a 2-min train of repetitive (100 Hz) 30-volt, 0.1 ms pulses (lane 3), or by a 10-min train of repetitive (10 Hz) 30-volt, 0.1 ms pulses (lane 4). (Lane 5) Neurons pretreated with H2O2. Immunoprecipitated PARP was immunolabeled by anti-PARP, Vic-5 antibody (n = 6). (c, left) Autoradiograms presenting [32P]polyADP-ribosylated PARP (5 min, 37°C) in isolated nuclei of unstimulated neurons (lane 2) and depolarized neurons (high-[K+]; lane 1, stimulated by a 2-min train of repetitive [100 Hz] 30-volt, 0.1 ms pulses; lane 3). [32P]polyADP-ribosylated PARP was immunoprecipitated from the nuclear protein extracts by N-20 antibody (see Materials and Methods), subjected to SDS-PAGE, autoradiographed, electroblotted (Western blot), and immunolabeled (on right) by anti-PARP, Vic-5 antibody (n = 6).
The Extent of In Situ PolyADP-ribosylation of PARP in Cortical Neurons, Determined by its Subsequent [32P]polyADP-ribosylation in Their Isolated Nuclei (“back [32P]polyADP-ribosylation”).
Despite evidence indicating an enhanced polyADP-ribosylation of PARP in depolarized neurons (Fig. 1, a and b), the [32P]polyADP-ribosylation of PARP in their isolated nuclei was significantly lower than that in nuclei isolated from unstimulated neurons (Fig. 1 c). This could not be explained by NAD depletion in nuclei isolated from depolarized neurons; increasing the extra-nuclear concentration of NAD (which permeates the nuclear membrane) did not enhance the [32P]polyADP-ribosylation of PARP in those nuclei (Fig. 2 a). Furthermore, the dose-dependent effect of added NAD on [32P]polyADP-ribosylation of PARP indicated that the ratio between the concentrations of NAD and [32P]NAD in nuclei of depolarized and unstimulated neurons was similarly altered by adding NAD (Fig. 2 a), indicating a similar concentration of endogenous NAD in both preparations (103–104 higher than the concentration of [32P]NAD, 10−8 M).
Figure 2.
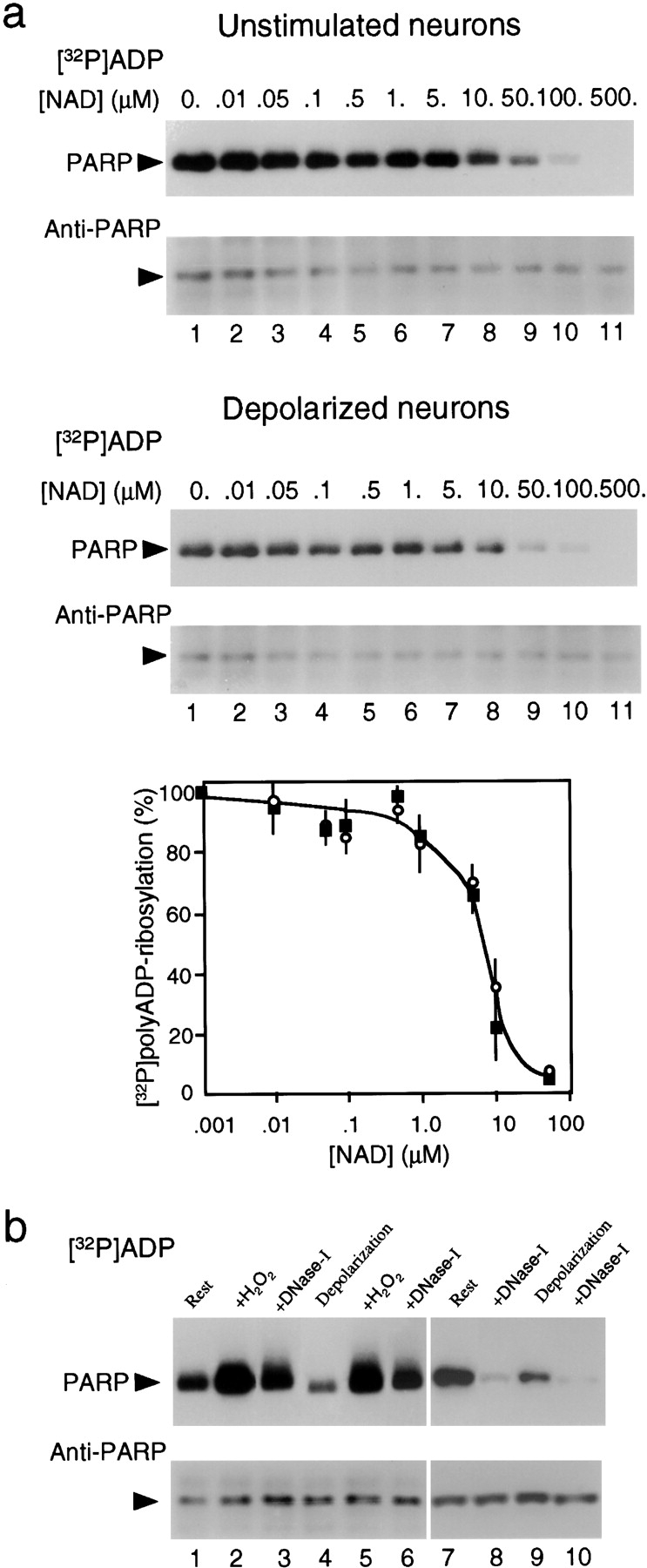
[32P]polyADP-ribosylation of PARP in nuclei isolated from unstimulated and depolarized neurons. (a) [32P]polyADP-ribosylation of PARP (2 min, 37°C) in crude nuclei isolated from unstimulated (top) and depolarized (bottom) neurons, in the presence of increasing concentrations of added NAD. Nuclear protein extracts were analyzed by SDS-PAGE and electroblotted. [32P]polyADP-ribosylated PARP in the nuclear protein extracts of both preparations was autoradiographed and immunolabeled by N-20 antibody. The curve presents average values ± SD of the depletion (%) in PARP [32P]polyADP-ribosylation in nuclei isolated from unstimulated (○) and depolarized (•) neurons due to increasing extranuclear NAD concentration (n = 6). (b) [32P]polyADP-ribosylation of PARP (2 min, 37°C) was conducted in nuclei isolated from unstimulated (lanes 1–3, 7, 8) and depolarized (lanes 4–6, 9, 10) neurons. (Lanes 2 and 5) Neurons treated by H2O2 (1 mM, 10 min, 25°C). (Lanes 3 and 6) Nuclei treated with DNAse I (20 μg/ml, 2 min, 37°C) during [32P]polyADP-ribosylation. (Lanes 8 and 10) Nuclei treated with DNase I (80 μg/ml, 2 min, 37°C) before [32P]polyADP-ribosylation (n = 4).
The possibility that depolarization renders PARP inactive or refractory to [32P]polyADP-ribosylation in the isolated nuclei was also excluded, since PARP was similarly activated by agents inducing formation of DNA breaks (H2O2 and DNAse I) in unstimulated or depolarized neurons (Fig. 2 b). However, although PARP was extensively [32P]polyADP-ribosylated in nuclei subjected to a mild DNA fragmentation by DNAse I (see Fig. 6 d) during [32P]polyADP-ribosylation (Fig. 2 b, lanes 3 and 6), PARP was scarcely [32P]polyADP-ribosylated in nuclei treated with DNAse I before [32P]polyADP-ribosylation (Fig. 2 b, lanes 8 and 10; see Fig. 6 c). As elaborated below, this effect could be attributed to an endogenous polyADP-ribosylation of PARP, evoked by DNA-nicks formation (D'Amours et al. 1999) but prevented in the presence of H2O2 (Fig. 2 b, lanes 2 and 5). PolyADP-ribosylation of PARP is suppressed in the presence of H2O2 (data not shown), apparently due to the destruction of its zinc-fingers by this oxidizing agent (Wu et al. 1996; Park et al. 1999).
Figure 6.
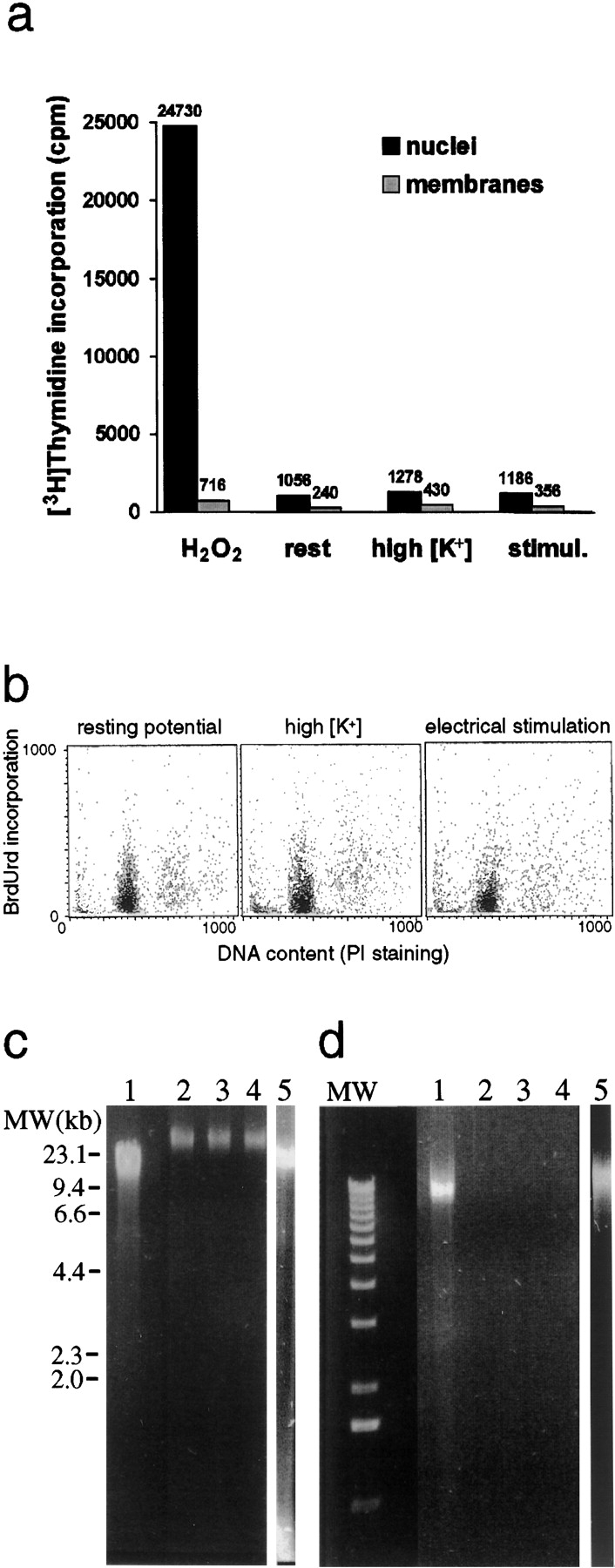
DNA repair examined in depolarized and unstimulated cortical neurons. (a) [3H]Thymidine incorporation in the DNA of cortical neurons pretreated with H2O2, unstimulated neurons (rest), high-[K+]-depolarized neurons (5 min), and electrically stimulated neurons (2-min train of repetitive [100 Hz] 30-volt, 0.1 ms pulses). Numbers above columns indicate the average values (SD < 10%) of tritium β emission (cpm) from [3H]thymidine incorporated into DNA (black) and from nonspecifically bound [3H]thymidine to neuronal cells membranes (gray) (n = 5). (b) Incorporation of BrdUrd into the DNA of cortical neurons, detected by flow cytometry (see Materials and Methods). Neurons were either unstimulated or depolarized by high-[K+], or by a 10-min train of repetitive (10 Hz) 30-volt, 0.1 ms pulses. Incorporated BrdUrd was detected by immunolabeling with anti-BrdUrd monoclonal antibody (IU-4) (Methods) and visualized by FITC-conjugated secondary antibody. The content of double stranded DNA in the preparations was indicated by propidium iodide (PI) intercalation (n = 3). (c) Alkaline gel electrophoresis of DNA extracted from nuclei of neurons pretreated with H2O2 (1 mM, 10 min; lane 1) or from unstimulated neurons (lane 2), neurons depolarized for 5 min by high-[K+] (lane 3) or stimulated by a 10-min train of repetitive (10 Hz) 30-volt, 0.1 ms pulses (lane 4), and from nuclei treated for 2 min at 37°C with 80 μg/ml DNAse I (lane 5). DNA was stained with ethidium bromide (1 μg/ml) in alkaline gel agarose (1%) and photographed under UV illumination (n = 3). (d) A selective extraction of fragmented DNA from nuclei of neurons pretreated with H2O2 (1 mM, 10 min; lane 1) or DNAse I (20 μg/ml, 2 min, 37°C; lane 5). DNA fragments were not extracted from unstimulated neurons (lane 2) or from neurons depolarized by high-[K+] (lane 3) or by electrical stimulation (10-min train of repetitive [10 Hz] 30-volt, 0.1 ms pulses; lane 4). DNA fragments were stained with ethidium bromide (1 μg/ml) in 1% gel agarose and photographed under UV illumination. Markers on left: 1-kb DNA ladder (0.5–10 kb) (n = 3).
The observations described in Fig. 2 b led us to suggest a sensitive method for determining changes in the activity of PARP in intact cells by measuring the extent of its [32P]polyADP-ribosylation in their isolated nuclei. The concept underlying this method was first introduced by Nestler and Greengard 1980 for assaying in situ phosphorylation of proteins by measuring their in vitro [32P] phosphorylation (back-[32P]phosphorylation).
PolyADP-ribosylation of PARP in intact cells can be assayed by its [32P]polyADP-ribosylation in their isolated nuclei (back-[32P]polyADP-ribosylation), since PARP activity is preserved in the isolated nuclei, and only DNA-bound PARP is polyADP-ribosylated (Satoh et al. 1994). Extensively polyADP-ribosylated PARP is released from DNA, and its ADP-ribose polymers are immediately subjected to a partial degradation (Satoh et al. 1994). PARP carrying partially degraded ADP-ribose polymers is not rebound to DNA (Udea 1990; Satoh et al. 1994; Lindahl et al. 1995). Since a complete degradation of ADP-ribose polymers, which would enable PARP de novo binding to DNA, is very slow relative to the time course of its [32P]polyADP-ribosylation (see Materials and Methods; Satoh et al. 1994), extensively polyADP-ribosylated PARP in situ may not undergo further [32P]polyADP-ribosylation. Thus, although PARP was extensively [32P]polyADP-ribosylated during DNA-nicks formation by DNAse I, it was scarcely [32P]polyADP-ribosylated in nuclei pretreated with DNAse I before [32P]polyADP-ribosylation (Fig. 2, compare lanes 3 and 6 with lanes 8 and 10).
Thus, for DNA-bound PARP undergoing [32P]polyADP-ribosylation in the isolated nuclei, the more extensive the PARP endogenous polyADP-ribosylation, the lower its measured [32P]poly-ADP-ribosylation. This is illustrated in Fig. 3, based on the schematic presentation of polyADP-ribosylation by Satoh et al. 1994. The low extent of PARP [32P]polyADP-ribosylation in nuclei isolated from depolarized neurons (Fig. 1 c) is in accordance with its high endogenous polyADP-ribosylation (Fig. 1, a and b, and 3).
Figure 3.

A schematic illustration of the concept underlying back polyADP-ribosylation. The extent of polyADP-ribosylation of PARP in intact cells reflected in the [32P]polyADP-ribosylation of PARP in their isolated nuclei. ○, ADP-ribose; •, [32P]ADP-ribose.
Since NAD does not permeate cell membranes, we determined the activity of PARP in intact neurons by measuring the extent of its back-[32P]polyADP-ribosylation in their isolated nuclei. Changes in PARP activity during electrical stimulation were examined by this method.
Cortical neurons in culture were stimulated by pulsed electrical stimuli (see Materials and Methods). Evoked action potentials and postsynaptic potentials were recorded in individual neurons during stimulation by using the patch-clamp whole cell configuration (see Materials and Methods; Hamill et al. 1981) (Fig. 4 a). Immediately after stimulation, neurons were lysed and their nuclei were isolated (see Materials and Methods). [32P]polyADP-ribosylation was conducted in the isolated nuclei (see Materials and Methods). Generally, a continuous electrical activity in the cortical neurons resulted in a low back-[32P]polyADP-ribosylation of PARP in their isolated nuclei (Fig. 4 b). This was consistent with the directly assayed enhanced polyADP-ribosylation of PARP in depolarized neurons (Fig. 1, a and b). The effect of depolarization on PARP activity was reversed by repolarization (Fig. 4 b); the more effective the stimulation, the longer the repolarization period required for reversal (Fig. 4 b). Stimulated neurons preserved their resting potential (Fig. 4 a), evidence that they were not damaged by the depolarizing stimulations. Also, depolarized neurons survived in their cultures for 10 d after stimulation, similar to the survival period of unstimulated neurons.
Figure 4.

Electrical activity inducing PARP activation in brain cortical neurons. (a) Evoked action potentials and synaptic potentials, recorded by the patch-clamp whole cell technique in the current clamp mode in cultured cortical neurons during electrical stimulation (n = 4). (Top) stimulation inducing polyADP-ribosylation of nuclear proteins (see b): 2-s repetitive (100 Hz) 30-volt, 0.1 ms pulses. Evoked action potentials, underlined by a bar, are presented on an expanded time-base to the right. The break in the trace represents a gap of 12 s. (Bottom) A stimulation which did not induce polyADP-ribosylation: 2-s repetitive (1 Hz) 30-volt, 0.1 ms pulses. (b) (Top) Autoradiograms of [32P]polyADP-ribosylated PARP in crude nuclei isolated from cortical neurons pretreated as follows: lane 1, unstimulated neurons; lanes 3, 5, 7, and 9, neurons repolarized for 20 min after the following depolarizations (respectively): high-[K+] for 5 min (lane 2), a 10-min train of repetitive (10 Hz) 30-volt, 0.1 ms pulses (lane 4), a 2-min train of repetitive (100 Hz) 30-volt, 0.1 ms pulses (lane 6), and repetitive (100 Hz) 30-volt, 0.1 ms pulses, applied for 2 s every minute for 10 min (lane 8). PARP was immunoprecipitated from the nuclear protein extracts by N-20 antibody, subjected to SDS-PAGE, autoradiographed, and electroblotted (Western blot). (Bottom) Immunolabeling of [32P]polyADP-ribosylated PARP by anti-PARP Vic-5 antibody in the immunoprecipitates (n = 8).
Inhibition of Topoisomerase I Activity in Depolarized Neurons Due to polyADP-ribosylation.
The activation of PARP in depolarized neurons was further examined by measuring the activity of topoisomerase I, a known substrate of PARP (Ferro et al. 1983) inhibited by polyADP-ribosylation (Ferro and Olivera 1984; Kasid et al. 1989). Topoisomerase I catalyzes the relaxation of supercoiled DNA, initiating DNA transcription and replication in eukaryotes (Wang 1996). We therefore used the relaxation of a supercoiled DNA-plasmid (related inversely to its mobility in gel agarose electrophoresis; Liu and Miller 1984) to assay topoisomerase I activity. A lower mobility indicated plasmid relaxation and, by inference, the activation of topoisomerase I.
We examined the effect of membrane depolarization on both activity and polyADP-ribosylation of topoisomerase I. Incubation of the supercoiled DNA-plasmid with proteins extracted from nuclei of depolarized neurons resulted in a significantly reduced topoisomerase I activity (Fig. 5 a, lanes 7–11), as compared with its activity in protein extracts of unstimulated or repolarized neurons (Fig. 5 a, lanes 3–6 and 12). Moreover, inhibition of topoisomerase I activity in depolarized neurons was prevented by suppression of PARP activity with 3-aminobenzamide (3-AB; Udea 1990) (Fig. 5 a, lanes 8 and 10). This result was in line with polyADP-ribosylation of topoisomerase I in the depolarized neurons (Fig. 5 b), thereby indicating that topoisomerase I is inhibited in depolarized neurons by polyADP-ribosylation.
Figure 5.
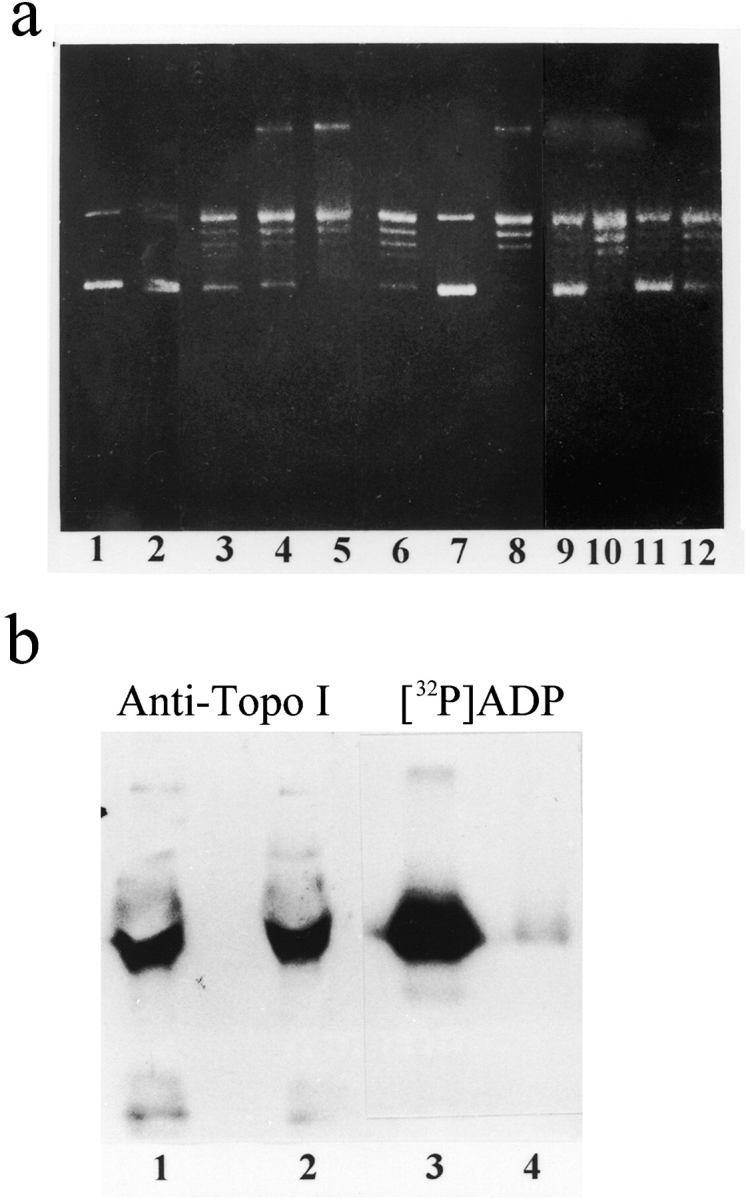
Suppressed activity of topoisomerase I in depolarized neurons. (a) Mobility in gel agarose of supercoiled plasmid (pUC-19) before (lanes 1 and 2) and after (lanes 3–12) incubation with nuclear proteins of unstimulated (lanes 3–6) or depolarized (lanes 7–11) cortical neurons, neurons treated with the PARP inhibitor, 3-AB (0.5 and 1.0 mM: lanes 4 and 5, respectively), neurons depolarized for 5 min by high-[K+] (lanes 7 and 8), neurons depolarized by 2-min train of repetitive (100 Hz) 30-volt, 0.1 ms pulses (lanes 9 and 10), neurons pretreated with 0.5 mM 3-AB (lanes 8 and 10), and neurons depolarized by 10-min train of repetitive (10 Hz) 30-volt 0.1 ms pulses (lanes 11 and 12) and repolarized for 20 min (lane 12). Each lane contained 100 ng protein (n = 5). (b) Topoisomerase I (110 kD) immunolabeled with anti–human topoisomerase I polyclonal antibody in Western blots (lanes 1 and 2) of [32P]polyADP-ribosylated nuclear proteins (5 min, 37°C), separated by SDS-PAGE and autoradiographed (lanes 3 and 4). Proteins were extracted from nuclei of unstimulated (lanes 1 and 3) and high-[K+] depolarized (lanes 2 and 4) cortical neurons. Each lane contained 200 μg protein (n = 3).
No Evidence of DNA Breaks Formation in Depolarized Neurons
Since PARP activation is reportedly induced by binding to free DNA endings in nicked DNA (Menissier-de Murcia et al. 1989; Kupper et al. 1990; Satoh and Lindahl 1992), we examined the possibility that membrane depolarization induces polyADP-ribosylation of PARP due to the formation of DNA breaks.
Depolarizing stimulation induced a transient polyADP-ribosylation of PARP, which disappeared as the resting potential was restored (Fig. 4 b). Therefore, we used methods suitable for detecting DNA repair in intact neurons during this transient effect. Induction of DNA breaks should be reflected in an increased DNA repair in depolarized neurons (Friedberg et al. 1995). We therefore examined DNA synthesis in the stimulated neurons by measuring the incorporation of thymidine (Friedberg et al. 1995) or the thymidine analogue BrdUrd (Selden and Dolbeare 1994) into DNA (see Materials and Methods).
[3H]Thymidine was incorporated only in nicked DNA of neurons pretreated with H2O2 (Fig. 6 a). There was no significant incorporation of [3H]thymidine or BrdUrd into DNA of depolarized or unstimulated neurons (Fig. 6, a and b, respectively).
Lack of DNA breaks in the depolarized neurons was further confirmed by two sensitive methods for DNA breaks detection: (i) alkaline gel electrophoresis of DNA, for detecting breaks in single stranded DNA (Sutherland et al. 1999) and (ii) selective extraction of fragmented DNA from isolated nuclei (Darzynkiewicz and Juan 1999) (see Materials and Methods).
The results depicted in Fig. 6 c show no evidence of breaks in single DNA strands of depolarized or unstimulated neurons. Moreover, there was no evidence of DNA fragmentation in either unstimulated or depolarized neurons (Fig. 6 d). Nicked DNA single strands or fragmented DNA were extracted only from neurons pretreated by H2O2 or from nuclei pretreated with DNAse I (Fig. 6c and Fig. d). These results strongly suggest that the enhanced polyADP-ribosylation of PARP in depolarized neurons is not derived from the formation of DNA breaks.
Lack of breaks in the DNA of depolarized neurons (Fig. 6) is consistent with the lack of NAD consumption in the depolarized neurons (Fig. 2 a) (Satoh and Lindahl 1992).
Evidence Associating Activation of PARP with IP3-mobilized Ca2+
Extranuclear Ca2+ Promotes Activation of PARP.
An increased intracellular Ca2+ concentration ([Ca2+]) is measured in neurons during membrane depolarization (Al-Mohanna et al. 1994). We therefore examined the possibility that Ca2+ is a mediator of depolarization-induced PARP activation. The effect of extranuclear [Ca2+] on [32P]polyADP-ribosylation of nuclear proteins was examined in isolated nuclei of cortical neurons in the presence of ATP (Methods). Nuclei were exposed to increasing [Ca2+], added before or after the addition of [32P] NAD, which initiates [32P]polyADP-ribosylation.
Increasing the extranuclear [Ca2+] during [32P]polyADP-ribosylation enhanced, by a dose-dependent manner, the [32P]polyADP-ribosylation of PARP (Fig. 7, a and b). The effect of Ca2+ on polyADP-ribosylation was very fast. It was therefore identified better at 25°C (rather than at 37°C; Fig. 7 a, lanes 1–6). Accordingly, when Ca2+ was added to the nuclei before [32P]polyADP-ribosylation, the [32P]polyADP-ribosylation of PARP decreased in a dose-dependent manner by increasing extranuclear [Ca2+] (Fig. 7 a, lanes 7–12), indicating a decreased back-[32P]polyADP-ribosylation of the activated PARP (see Fig. 1 and Fig. 3).
Figure 7.
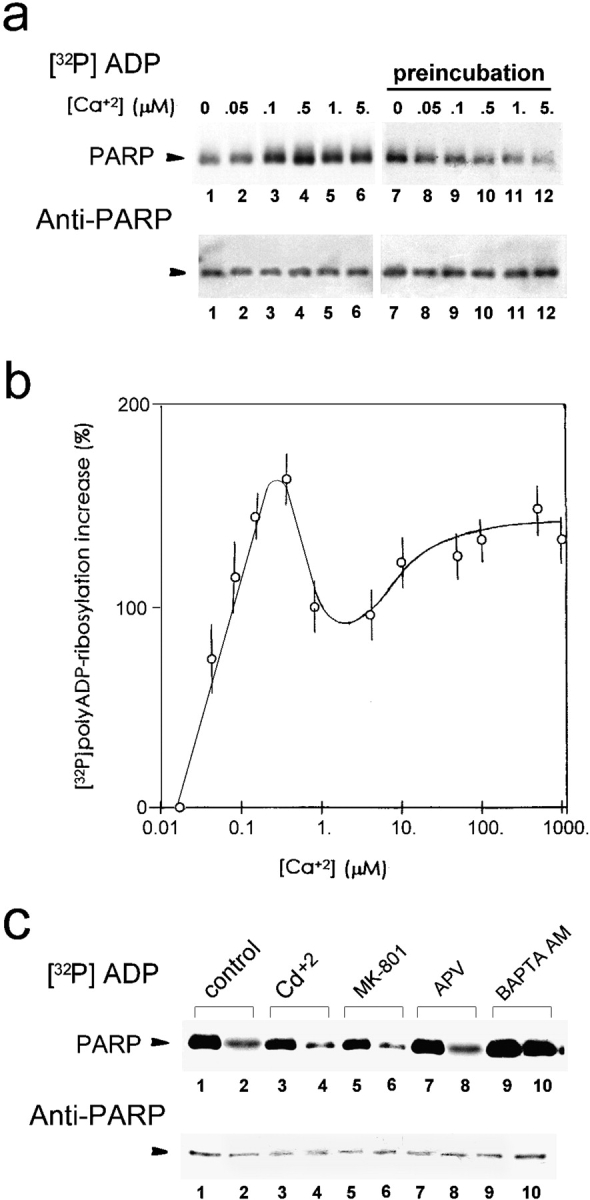
Extra-nuclear Ca2+ promotes polyADP-ribosylation of PARP. (a) PolyADP-ribosylation of PARP in crude nuclei isolated from unstimulated neurons, exposed to increasing [Ca2+], 1 min before (preincubation) or during [32P]polyADP-ribosylation (2 min, 25°C). [32P]polyADP-ribosylated PARP was immunoprecipitated, electroblotted (Western blot), autoradiographed, and immunolabeled by Vic-5 antibody. (b) The curve shows the average increase (±SD) in [32P]polyADP-ribosylation of PARP (measured by densitometry) versus the increase in extranuclear [Ca2+], relative to PARP [32P]polyADP-ribosylation in extranuclear [Ca2+] = 25 nM (see Materials and Methods) (n = 9). (c) Back-[32P]polyADP-ribosylation of PARP, in nuclei of depolarized or unstimulated neurons treated with Ca2+ influx blockers and the permeant chelator BAPTA AM. (Top) Autoradiograms of [32P]polyADP-ribosylated PARP in crude nuclei of unstimulated neurons (lanes 1, 3, 5, 7, and 9), and neurons depolarized by a 10-min train of repetitive (10 Hz) 30-volt, 0.1 ms pulses (lanes 2, 4, 6, 8, and 10). Cultured neurons were preincubated for 10 min at 25°C with 1 mM CdCl2 (lanes 3 and 4) or with antagonists of NMDA-glutamate receptors, MK-80l (20 μM; lanes 5 and 6) and APV (500 μM; lanes 7 and 8), or preincubated (30 min, 25°C) with the permeant Ca2+ chelator, BAPTA AM (50 μM; lanes 9 and 10). PARP was immunoprecipitated from the nuclear protein extracts of these neurons by N-20 antibody, subjected to SDS-PAGE, electroblotted (Western blot), and autoradiographed. (Bottom) Immunolabeling of [32P]-polyADP-ribosylated PARP by Vic-5 antibody in the immunoprecipitates (n = 6).
The stimulatory effect of extra-nuclear [Ca2+] on PARP activity was further examined in depolarized neurons, loaded with the permeant Ca2+-chelator, BAPTA AM (Hardingham et al. 1997; Al-Mohanna et al. 1994). Capture of intracellular Ca2+ by BAPTA AM completely abolished the increase in polyADP-ribosylation of PARP in depolarized neurons (Fig. 7 c). However, neither depletion of extracellular Ca2+ during high-[K+]–induced depolarization (see Materials and Methods; Fig. 1, Fig. 4 b, and 5) nor blocking Ca2+ influx prevented the polyADP-ribosylation of PARP in depolarized neurons. Its polyADP-ribosylation was neither suppressed by blocking of voltage-dependent Ca2+ channels (Olivera et al. 1994) nor by blocking of Ca2+ influx, evoked by stimulation of NMDA-glutamate receptors (Sharkey et al. 1996) (Fig. 7 c). These findings strongly suggest that PARP activation in depolarized neurons is mediated by Ca2+ release from intracellular stores (Zacchetti et al. 1991; Ehrlich et al. 1994; Ehrlich 1995). It should be noted that in vitro conducted polyADP-ribosylation of PARP is similarly enhanced by Mg2+ (10 mM; Ferro and Olivera 1982).
Ca2+ Release into the Nucleoplasm in Isolated Nuclei of Cortical Neurons.
We next examined the possibility that Ca2+, mobilized from intracellular stores, is released into the nucleoplasm. Crude nuclei (Fig. 8 a; see Materials and Methods) were isolated from brain cortical neurons and loaded with the permeant fluorescent Ca2+ indicator rhod-2 AM (Minta et al. 1989) in the absence of extranuclear Ca2+ (see Materials and Methods). Capture of Ca2+ by rhod-2 was visualized by confocal microscopy (see Materials and Methods; Fig. 8, b–d). [Ca2+] was markedly increased in the nucleoplasm of nuclei isolated from depolarized neurons (Fig. 8 b), in line with the transient increase in nuclear [Ca2+] in depolarized neurons (Al-Mohanna et al. 1994; Hardingham et al. 1997).
An increase of extranuclear [Ca2+] did not induce Ca2+ release into the nucleoplasm unless ATP (2.5 mM) was added (Fig. 8 d); extranuclear Ca2+, in its physiological concentration range, was instantaneously accumulated in perinuclear compartments by adding ATP (Fig. 8c and Fig. d). Under these experimental conditions, Ca2+ was instantaneously released into the nucleoplasm by the addition of IP3 (1–2 μM; Fig. 8 c). In the presence of ATP, Ca2+ was also moderately released into the nucleoplasm when extranuclear [Ca2+] was elevated (Fig. 8 d). cADP-ribose (5–20 μM), reportedly inducing Ca2+-dependent Ca2+ release from perinuclear stores (Gerasimenko et al. 1995), had a very small effect on Ca2+ release into the nucleoplasm under these experimental conditions (data not shown). These findings are consistent with a growing body of evidence indicating that extranuclear Ca2+ permeates the nuclear membrane mainly via Ca-ATPase–induced Ca2+ accumulation in IP3-gated perinuclear stores (Gerasimenko et al. 1995; Hennager et al. 1995; Malviya and Rogue 1998) and with evidence indicating phosphatidylinositol signaling in the nucleus (Boronenkov et al. 1998).
The release of Ca2+ into the nucleoplasm was prevented by caffeine, added to the crude nuclei at concentrations suppressing IP3-induced Ca2+ mobilization (Ehrlich et al. 1994) (Fig. 8 c). Release of Ca2+ into the nucleoplasm was also prevented in nuclei isolated from neurons pretreated by thapsigargin that inhibits Ca-ATPase activity (Takemura et al. 1989), thereby preventing Ca2+ accumulation in the perinuclear stores (Malviya and Rogue 1998) (Fig. 8 c).
A Fast Activation of PARP by IP3 in Isolated Nuclei of Cortical Neurons.
We next examined the possibility that PARP is polyADP-ribosylated by IP3-induced Ca2+ mobilization. [32P]polyADP-ribosylation of PARP was examined in the presence of IP3 added to nuclei isolated from unstimulated neurons. EDTA was omitted from the incubation solution (see Materials and Methods), to avoid chelation of free Ca2+. In addition, [32P]polyADP-ribosylation was carried out at 25°C to enable detection of fast changes in the activity of PARP. For the same reason, IP3 was added after the addition of [32P]NAD. [32P]polyADP-ribosylated proteins were extracted 1 min after the addition of IP3. IP3 (at concentrations of 50 nM to 5 μM) enhanced the [32P]polyADP-ribosylation of PARP in a dose-dependent manner. Maximal 10-fold enhancement was measured with a half maximal effect induced by 100 ± 30 nM IP3 (Fig. 9 a). At the same concentration range, IP3 displaced specifically bound [3H]IP3 from its receptors in the crude nuclei (IC50 = 30 ± 5 nM; Fig. 9 b).
Figure 9.

IP3 induces a fast [32P]polyADP-ribosylation of PARP in crude nuclei of brain cortical neurons. (a) Autoradiograms of [32P]polyADP-ribosylated PARP in crude nuclei of unstimulated brain cortical neurons in the absence (lane 1) or presence of IP3 at the indicated concentrations (lanes 2–10). [32P]polyADP-ribosylation (2 min, 25°C) was terminated 1 min after the addition of IP3. Nuclear proteins were extracted, separated by SDS-PAGE, and electroblotted (Western blot). PARP was immunolabeled by N-20 antibody (n = 7). (b) Left ordinate shows displacement of bound [3H]IP3 by IP3 in crude nuclei of cortical neurons (○). Maximal specific binding of [3H]IP3 (10.5 nM) was 18,500–20,500 cpm/mg protein. Nonspecific binding of [3H]IP3 (∼60,000 cpm/mg protein) was determined in the presence of 10 μM IP3. The data show the amount of specifically bound [3H]IP3 (as a percentage of its maximal specific binding), determined as the mean of three experiments performed in triplicates, varying by <15%. Right ordinate shows enhancement in [32P]polyADP-ribosylation of PARP (measured by densitometry; see Materials and Methods) by IP3 (▪). Values are means of seven experiments, expressed for each experiment as a percentage of the maximal enhancement in [32P]polyADP-ribosylation of PARP. (c, top) Autoradiograms of [32P]polyADP-ribosylated PARP in crude nuclei (2 min at 25°C) in the absence (lanes 1, 6, 9–11, and 15) or presence (lanes 2–5, 7, 8, 12–14, and 16) of IP3, BAPTA (lanes 6 and 8), or caffeine (lanes 10, 11, 13, and 14), and in crude nuclei of neurons pretreated with thapsigargin (10 min, 37°C, lanes 15 and 16). Preincubation with BAPTA or caffeine lasted 5 min at 25°C. [32P]polyADP-ribosylated PARP was immunoprecipitated from nuclear proteins extracts by N-20 antibody, subjected to SDS-PAGE, electroblotted (Western blot), autoradiographed, and immunolabeled (bottom) with anti-PARP, Vic-5 antibody (n = 3). (d, top) Autoradiograms of [32P]polyADP-ribosylated PARP (2 min at 25°C) in crude nuclei of cortical neurons in the absence (lanes 1 and 8) and presence (lanes 2–7 and 9–13) of IP3 or FK-506 (lanes 8–13). Since FK-506 was dissolved in ethanol, all the samples contained 0.03% ethanol. [32P]polyADP-ribosylated PARP was immunoprecipitated from nuclear protein extracts by N-20 antibody, subjected to SDS-PAGE, electroblotted (Western blots), autoradiographed, and immunolabeled (bottom) with anti-PARP Vic-5 antibody (n = 3).
Agents suppressing IP3-induced Ca2+-mobilization also suppressed IP3-induced PARP activation; addition of the Ca2+-chelator, BAPTA AM, to the isolated nuclei completely suppressed the IP3-induced [32P]polyADP-ribosylation of PARP (Fig. 9 c, lanes 6–8). Addition of caffeine (3–5 mM; Ehrlich et al. 1994; Ehrlich 1995) had a similar effect (Fig. 9 c, lanes 12–14). Heparin (100 mg/ml, Grade 1-A; Sigma-Aldrich) (Ehrlich et al. 1994) acted similarly to caffeine (data not shown). Neither BAPTA nor caffeine prevented the basal [32P]polyADP-ribosylation of PARP in the absence of IP3 (Fig. 9 c, lanes 6 and 9–11, respectively). IP3-induced [32P]poly-ADP-ribosylation of PARP was also suppressed in nuclei isolated from neurons pretreated by thapsigargin (Fig. 9 c, lanes 15 and 16). IP3-gated Ca2+ stores turn “leaky” by interaction with FK-506 (Cameron et al. 1995; Mikoshiba 1997; Mackrill 1999). We therefore examined the effect of FK-506 on [32P] polyADP-ribosylation of PARP.
Addition of FK-506 (1 μM) altered the dose-dependent effect of IP3 on [32P]polyADP-ribosylation of PARP; the concentration of IP3 required for enhancement of [32P]polyADP-ribosylation in the presence of FK-506 was 10 times lower than that required in untreated nuclei (Fig. 9 d). Hence, the fast polyADP-ribosylation of PARP by IP3 (Fig. 9, a and b), and its modulation by agents affecting IP3-gated Ca2+ release (Fig. 8 c and 9, c and d), strongly suggest that PARP in the isolated nuclei was activated via IP3-induced Ca2+ mobilization.
IP3-induced [32P]polyADP-ribosylation of PARP in isolated nuclei was neither affected by the addition of calmodulin (10–20 μM; Mackrill 1999; data not shown), nor by preventing Ca-calmodulin binding to CAM-kinase II in the presence of saturating amounts (1.5 μM) of the Ca-calmodulin binding peptide on CAM-kinase II (Payne et al. 1988; data not shown). cADP-ribose (5–20 μM) did not alter the basal [32P]poly-ADP-ribosylation of PARP in the crude nuclei (data not shown).
Discussion
The results of this study indicate a fast activation of PARP by electrical activity in brain cortical neurons. This is directly demonstrated by in situ immunolabeling of polyADP-ribosylated proteins in depolarized neurons (Fig. 1, a and b) and, indirectly, by inhibition of topoisomerase I activity due to polyADP-ribosylation in depolarized neurons (Fig. 5). PARP activation was quantified by the extent of its back-[32P]polyADP-ribosylation in isolated nuclei of depolarized neurons (Fig. 1 c and 4 b). These findings constitute the first evidence for a fast activation of PARP by physiological signals in the cell membrane.
High-[K+]–induced membrane depolarization promoted polyADP-ribosylation of nuclear proteins in the absence of extracellular Ca2+ (Fig. 1, Fig. 4 b, and 5). Findings indicating that PARP is activated by intracellular Ca2+ mobilization in the depolarized neurons include: a fast dose-dependent activation of PARP by extranuclear [Ca2+] (Fig. 7, a and b), independent of extracellular Ca2+ influx (Fig. 7 c); and a fast dose-dependent PARP activation by physiological concentrations of IP3 (Fig. 9, a and b), modulated by agents affecting IP3-induced Ca2+ mobilization (Fig. 9c and Fig. d).
IP3-induced Ca2+ release into the nucleoplasm (Fig. 8 c) may underlie the depolarization-induced activation of PARP (Fig. 1 and Fig. 4 b). This is supported by data indicating Ca2+ release into the nucleoplasm of depolarized cortical neurons (Fig. 8 b; Al-Mohanna et al. 1994; Hardingham et al. 1997) and IP3-induced Ca2+ release into the nucleoplasm of cortical neurons (Fig. 8 c), also reported in other cell types (Malviya and Rogue 1998).
An enhanced IP3 production has been measured in depolarized neurons (Gusovsky et al. 1986). It may be attributed to an accelerated phosphoinositide turnover (Gusovsky et al. 1986; Gurwitz and Sokolovsky 1987), as well as to the stimulation of receptor tyrosine kinases (Castren et al. 1992; Huang et al. 1999), or activation of trimeric G-proteins (Banno et al. 1987; Berridge and Irvine 1989; Exton 1990; Sierro et al. 1992; Anis et al. 1999).
The fast polyADP-ribosylation of PARP by IP3 in the isolated nuclei (Fig. 9) is compatible with the time course of Ca2+ release from IP3-gated stores (Ferris and Snyder 1992). Moreover, IP3 stimulated polyADP-ribosylation in the isolated nuclei of cortical neurons (Fig. 9 b) at concentrations compatible with the affinity of IP3-receptors IP3-R1 and IP3-R2 (Mignery et al. 1992; Miyakawa et al. 1999), identified in the brain (Mignery et al. 1992; Ross et al. 1992). IP3-gated Ca2+ stores have been identified in the inner nuclear membrane (Nicotera et al. 1990; Gerasimenko et al. 1995; Malviya and Rogue 1998). Phosphatidylinositol signaling pathways have been identified in the nuclei of several cell types (Boronenkov et al. 1998).
The enhanced activity of PARP in depolarized neurons was independent of extracellular [Ca2+] (see Materials and Methods; Fig. 1, Fig. 4 b, and 5) and resisted Ca2+ influx blockers, including agents suppressing NMDA-induced Ca2+ influx (Fig. 7 c). We therefore consider it unlikely that PARP is activated in depolarized neurons by DNA damage, caused by nitric oxide formation (Zhang et al. 1994; Shah et al. 1996). It is also unlikely that the fast signal–induced activation of PARP was mediated by Ca2+-induced activation of endonucleases, producing DNA breaks (Arends et al. 1990). The activation of Ca,Mg-endonuclease would require extranuclear Ca2+ concentrations 100–1,000-fold higher (Peitsch et al. 1993; Peitsch et al. 1994) than those inducing PARP activation (Fig. 7, a and b). Endonuclease activity at [Ca2+] <1 μM has a much slower time course (>30 min; Jones et al. 1989). Accordingly, DNA breaks or NAD depletion (Satoh and Lindahl 1992) were not detected in the depolarized cortical neurons (Fig. 6 and Fig. 2 a, respectively).
A fast signal–induced PARP activation via IP3-induced Ca2+ mobilization constitutes a novel mode of signaling to the cell nucleus: PARP, being a downstream target of phospholipase C, modulates by polyADP-ribosylation the activity of nuclear proteins in response to signals promoting phosphoinositides turnover and phosphatidyl-inositol 4,5-bisphosphate (PIP2) hydrolysis (Berridge and Irvine 1989; Fruman et al. 1998; Toker 1998). A fast modification of transcription factors by polyADP-ribosylation (Li Oei et al. 1998) during electrical activity in brain cortical neurons may associate depolarization-induced polyADP-ribosylation with “memory storage” (Kandel 1997).
The role of PARP in DNA repair and transcription (Satoh and Lindahl 1992; Oliver et al. 1998; Trucco et al. 1998) may underlie the effect of depolarization in protecting growth factor–deprived neurons from apoptotic cell death (D'Mello et al. 1993; Galli et al. 1995). This mechanism suggests a crucial influence of neuronal activity in preserving the viability of brain cortical neurons, thereby implementing the rule of “use it or lose it.”
Acknowledgments
We would like to express our thanks to Dr. Gilbert de Murcia for enlightening discussions; to Professors James H. Schwartz and Zvi Selinger for their extensive revision of the manuscript; to Dr. Naomi Feinstein for preparing electromicrographs of the isolated nuclei; and to Professors Itzhak Parnas, Menahem Segal, and Arie Moran for their helpful suggestions.
This work was supported by grants that Dr. Cohen-Armon received from the Israel Academy of Science, Adams Super-Center for Brain Research (Tel-Aviv University), and the Israel Ministry of Science.
Footnotes
Abbreviations used in this paper: BrdUrd, 5-bromodeoxyuridine; IP3, inositol 1,4,5-triphosphate; PARP, poly(ADP-ribose) polymerase.
References
- Al-Mohanna F.A., Caddy K.W., Bolsover S.R. The nucleus is insulated from large cytosolic calcium ion changes. Nature. 1994;367:745–750. doi: 10.1038/367745a0. [DOI] [PubMed] [Google Scholar]
- Anis Y., Nurnberg B., Visochek L., Reiss N., Naor A., Cohen-Armon M. Activation of Go-proteins by membrane depolarization traced by in-situ photoaffinity labeling of Gαo-proteins with [α32P]GTP-azidoanilide. J. Biol. Chem. 1999;274:7431–7440. doi: 10.1074/jbc.274.11.7431. [DOI] [PubMed] [Google Scholar]
- Arends M.J., Morris R.G., Wyllie A.H. Apoptosisthe role of the endonuclease. Am. J. Pathol. 1990;136:593–608. [PMC free article] [PubMed] [Google Scholar]
- Banno Y., Nagao S., Katada T., Nagata K., Ui M., Nozawa Y. Stimulation by GTP-binding proteins (Gi, Go) of partially purified phospholipase C activity from human platelet membranes. Biochem. Biophys. Res. Commun. 1987;146:861–869. doi: 10.1016/0006-291x(87)90610-3. [DOI] [PubMed] [Google Scholar]
- Berridge M.J., Irvine R.F. Inositol phosphates and cell signaling. Nature. 1989;341:197–204. doi: 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- Boronenkov I.V., Loijens J.C., Umeda M., Anderson R.A. Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre-mRNA processing factors. Mol. Biol. Cell. 1998;9:3547–3560. doi: 10.1091/mbc.9.12.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulikas T. Poly(ADP-ribosylated) histones in chromatin replication. J. Biol. Chem. 1990;265:14638–14647. [PubMed] [Google Scholar]
- Brenneman D.E., Yu C., Nelson P.G. Multi-determinate regulation of neuronal survivalneuropeptides, excitatory amino acids and bioelectric activity. Int. J. Dev. Neurosci. 1990;8:371–378. doi: 10.1016/0736-5748(90)90070-i. [DOI] [PubMed] [Google Scholar]
- Cameron A.M., Steiner J.P., Roskams A.J., Ali S.M., Ronnett G.V., Snyder S.H. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor FKBP12 complex modulates Ca+2 flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- Castren E., Zafra F., Thoenen H., Lindholm D. Light regulates expression of brain-derived neurotrophic factor mRNA in rat visual cortex. Proc. Natl. Acad. Sci. USA. 1992;89:9444–9448. doi: 10.1073/pnas.89.20.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarone C.F., Scarabelli L., Scovassi I., Izzo R., Menegazzi M., DePrati A.C., Orunesu M., Bertazzoni U. Changes in activity and mRNA levels of poly(ADP-ribose)polymerase during rat liver regeneration. Biochim. Biophys. Acta. 1990;1087:241–246. doi: 10.1016/0167-4781(90)90211-j. [DOI] [PubMed] [Google Scholar]
- Challiss R.A.J., Chilvers E.R., Willcocks A.L., Nahorski S.R. Heterogeneity of [3H]inositol 1,4,5-trisphosphate binding sites in adrenal-cortical membranes. Biochem. J. 1990;265:421–427. doi: 10.1042/bj2650421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Armon M., Sokolovsky M. Depolarization-induced changes in the muscarinic receptor in rat brain and heart are mediated by pertussis-toxin-sensitive G-proteins. J. Biol. Chem. 1991;266:2595–2605. [PubMed] [Google Scholar]
- Cohen-Armon M., Hammel I., Anis J., Homburg S., Dekel N. Evidence for endogenous ADP-ribosylation of GTP-binding proteins in neuronal cell nucleus. J. Biol. Chem. 1996;271:26200–26208. doi: 10.1074/jbc.271.42.26200. [DOI] [PubMed] [Google Scholar]
- D'Amours D., Desnoyers S., D'Silva I., Poirier G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z., Juan G. Selective extraction of fragmented DNA from apoptotic cells for analysis by gel electrophoresis and identification of apoptotic cells by flow cytometry. In: Henderson D.S., editor. DNA-repair Protocols, Eukaryotic Systems. Humana Press; New Jersey: 1999. pp. 599–602. [DOI] [PubMed] [Google Scholar]
- de Murcia G., Schreiber V., Molinete M., Saulier B., Poch O., Masson M., Niedergang C., Menissier-de Murcia J. Structure and function of poly(ADP-ribose)polymerase. Mol. Cell. Biochem. 1994;138:15–24. doi: 10.1007/BF00928438. [DOI] [PubMed] [Google Scholar]
- Desmarais Y., Menard L., Lagueux J., Poirier G.G. Enzymological properties of poly(ADP-ribose)polymerasecharacterization of automodification sites and NADase activity. Biochim. Biophys. Acta. 1991;1078:179–186. doi: 10.1016/0167-4838(91)99007-f. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. Oxidative damage to DNA mammalian chromatin. Mutat. Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- D'Mello S.R., Galli C., Ciotti T., Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassiuminhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich B.E. Functional properties of intracellular calcium-release channels. Curr. Opin. Neurobiol. 1995;5:304–309. doi: 10.1016/0959-4388(95)80042-5. [DOI] [PubMed] [Google Scholar]
- Ehrlich B.E., Kaftan E., Bezprozvannaya S., Bezprozvanny I. The pharmacology of intracellular Ca+2-release channels. Trends Pharmacol. Sci. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Exton J.H. Signaling through phosphatidylcholine breakdown. J. Biol. Chem. 1990;265:1–4. [PubMed] [Google Scholar]
- Ferris C.D., Snyder S.H. Inositol phosphate receptors and calcium disposition in the brain. J. Neurosci. 1992;12:1567–1574. doi: 10.1523/JNEUROSCI.12-05-01567.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro A.M., Olivera B.M. Poly(ADP-ribosylation) in vitro reaction parameters and enzyme mechanism. J. Biol. Chem. 1982;257:7808–7813. [PubMed] [Google Scholar]
- Ferro A.M., Olivera B.M. Poly(ADP-ribosylation) of DNA topoisomerase I from calf thymus. J. Biol. Chem. 1984;259:547–554. [PubMed] [Google Scholar]
- Ferro A.M., Higgins N.P., Olivera B.M. Poly(ADP-ribosylation) of a DNA topoisomerase. J. Biol Chem. 1983;258:6000–6003. [PubMed] [Google Scholar]
- Franklin J.L., Johnson E.M., Jr. Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- Friedberg E.C., Walker G.C., Siede W. DNA Repair and Mutagenesis 1995. American Society for Microbiology (ASM) Press; Washington DC: pp. 222–223 [Google Scholar]
- Fruman D.A., Meyers R.E., Cantley L.C. Phosphoinositide kinases. Annu. Rev. Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Galli C., Meucci O., Scorziello A., Werge T.M., Calissano P., Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-I through distinct mechanisms of actionthe involvement of intracellular calcium and RNA synthesis. J. Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko O.V., Gerasimenko J.V., Tepikin A.V., Petersen O.H. ATP-dependent accumulation and inositol trisphosphate- or cyclic ADP-ribose-mediated release of Ca2+ from the nuclear envelope. Cell. 1995;80:439–444. doi: 10.1016/0092-8674(95)90494-8. [DOI] [PubMed] [Google Scholar]
- Gurwitz D., Sokolovsky M. Dual pathways in muscarinic receptor stimulation of phosphoinositide hydrolysis. Biochemistry. 1987;26:633–638. doi: 10.1021/bi00376a039. [DOI] [PubMed] [Google Scholar]
- Gusovsky F., Hollingworth E.B., Daly J.W. Regulation of phosphatidyl-inositol turnover in brain synaptoneurosomesstimulatory effects of agents that enhance influx of sodium ions. Proc. Natl. Acad. Sci. USA. 1986;83:3003–3007. doi: 10.1073/pnas.83.9.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp technique for high-resolution current recording from cells and cell-free membrane patches. Eur. J. Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hanawalt P.C., Donahue B.A., Sweder K.S. Collision or collusion. Curr. Biol. 1994;4:518–521. doi: 10.1016/s0960-9822(00)00112-3. [DOI] [PubMed] [Google Scholar]
- Hardingham G.E., Chawla S., Johnson C.M., Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- Hennager D.J., Welsh M.J., DeLisle S. Changes in either cytosolic or nucleoplasmic inositol 1,4,5-trisphosphate levels can control nuclear Ca+2 concentration. J. Biol. Chem. 1995;270:4959–4962. doi: 10.1074/jbc.270.10.4959. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell culture. J. Mol. Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Huang Z.J., Kirkwood A., Pizzorusso T., Porciatti V., Morales B., Bear M.F., Maffei L., Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Jones D.P., McConkey D.J., Nicotera P., Orrenius S. Calcium-activated DNA fragmentation in rat liver nuclei. J. Biol. Chem. 1989;264:6398–6403. [PubMed] [Google Scholar]
- Kandel E.R. Genes, synapses and long-term memory. J. Cell Physiol. 1997;173:124–125. doi: 10.1002/(SICI)1097-4652(199711)173:2<124::AID-JCP6>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Kasid U.N., Halligan B., Liu L.F., Dritschilo A., Smulson M. Poly(ADP-ribose)-mediated post-translational modification of chromatin-associated human topoisomerase-Iinhibitory effects on catalytic activity. J. Biol. Chem. 1989;264:18687–18692. [PubMed] [Google Scholar]
- Kim H., Jacobson M.K., Rolli V., Menissier-de Murcia J., Reinbolt J., Simonin F., Ruf A., Schulz G., de Murcia G. Photoaffinity labeling of human poly(ADP-ribose)polymerase catalytic domain. Biochem. J. 1997;322:469–475. doi: 10.1042/bj3220469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupper J.H., de Murcia G., Burkle A. Inhibition of poly(ADP-ribosyl)ation by overexpressing the poly(ADP-ribose)polymerase DNA-binding domain in mammalian cells. J. Biol. Chem. 1990;265:18721–18724. [PubMed] [Google Scholar]
- Lamarre D., Talbot B., de Murcia G., Laplante C., Leduc Y., Mazen A., Poirier G.G. Structural and functional analysis of poly(ADP-ribose) polymerasean immunological study. Biochim. Biophys. Acta. 1988;950:147–160. doi: 10.1016/0167-4781(88)90007-3. [DOI] [PubMed] [Google Scholar]
- Lautier D., Lagueux J., Thibodeau J., Menard L., Poirier G.G. Molecular and biochemical features of poly(ADP-ribose) metabolism. Mol. Cell. Biochem. 1993;122:171–193. doi: 10.1007/BF01076101. [DOI] [PubMed] [Google Scholar]
- Lazebnik Y.A., Kaufmann S.H., Desnoyers S., Poirier G.G., Earnshaw W.C. Cleavage of poly(ADP-ribose)polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Li Oei S., Griesenbeck J., Scheiger M., Ziegler M. Regulation of RNA polymerase II-dependent transcription by poly(ADP-ribosyl)ation of transcription factors. J. Biol. Chem. 1998;273:31644–31647. doi: 10.1074/jbc.273.48.31644. [DOI] [PubMed] [Google Scholar]
- Lin W., Ame J.C., Aboul-Ela N., Jacobson E.L., Jacobson M.K. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose)-glycohydrolase. J. Biol. Chem. 1997;272:11895–11901. doi: 10.1074/jbc.272.18.11895. [DOI] [PubMed] [Google Scholar]
- Lindahl T., Satoh M.S., Poirier G.G., Klungland A. Post-translational modification of poly(ADP-ribose)polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–412. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- Liu L.F., Miller K.G. Eukaryotic DNA topoisomerasestwo forms of type I DNA topoisomerases from HeLa cell nuclei. Proc. Natl. Acad. Sci. USA. 1984;78:3487–3491. doi: 10.1073/pnas.78.6.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackrill J.J. Protein-protein interactions in intracellular Ca+2-release channel function. Biochem. J. 1999;337:345–361. [PMC free article] [PubMed] [Google Scholar]
- Malviya A.N., Rogue P.J. “Tell me where is calcium bred”clarifying the roles of nuclear calcium. Cell. 1998;92:17–23. doi: 10.1016/s0092-8674(00)80895-8. [DOI] [PubMed] [Google Scholar]
- Martinou J.C. ICE-like proteases execute the neuronal death program. Curr. Opin. Neurosci. 1996;6:609–614. doi: 10.1016/s0959-4388(96)80092-4. [DOI] [PubMed] [Google Scholar]
- Meisterernst M., Stelzer G., Roeder R.G. Poly(ADP-ribose)polymerase enhances activator-dependent transcription in vitro. Proc. Natl. Acad. Sci. USA. 1997;94:2261–2265. doi: 10.1073/pnas.94.6.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menissier-de Murcia J., Molinete M., Gradwohl G., Simonin F., de Murcia G. Zinc-binding domain of poly(ADP-ribose)polymerase participates in the recognition of single strand breaks on DNA. J. Mol. Biol. 1989;210:229–233. doi: 10.1016/0022-2836(89)90302-1. [DOI] [PubMed] [Google Scholar]
- Mignery G.A., Johnston P.A., Sudhof T.C. Mechanism of Ca+2 inhibition of inositol 1,4,5-trisphosphate (InsP3) binding to the cerebellar InsP3 receptor. J. Biol. Chem. 1992;267:7450–7455. [PubMed] [Google Scholar]
- Mikoshiba K. The InsP3 receptor and intracellular Ca+2 signaling. Curr. Opin. Neurobiol. 1997;7:339–345. doi: 10.1016/s0959-4388(97)80061-x. [DOI] [PubMed] [Google Scholar]
- Minta A., Kao J.P.Y., Tsien R.Y. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 1989;264:8171–8178. [PubMed] [Google Scholar]
- Miyakawa T., Maeda A., Yamazawa T., Hirose K., Kurosaki T., Lino M. Encoding of Ca+2 signals by differential expression of IP3 receptor subtypes. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1303–1308. doi: 10.1093/emboj/18.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler E.J., Greengard P. Dopamine and depolarizing agents regulate the state of phosphorylation of protein I in the mammalian superior cervical sympathetic ganglion. Proc. Natl. Acad. Sci. USA. 1980;77:7479–7483. doi: 10.1073/pnas.77.12.7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D.W., Ali A., Thornberry N.A., Vaillancourt J.P., Ding C.K., Gallant M., Gareau Y., Lazebnik Y.A., Raju S.M., Smulson M.E. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nicotera P., Orrenius S., Nilsson T., Berggren P.O. An inositol 1,4,5-trisphosphate sensitive Ca+2 pool in liver nuclei. Proc. Natl. Acad. Sci. USA. 1990;87:6858–6862. doi: 10.1073/pnas.87.17.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver F.J., de la Rubia G., Rolli V., Ruiz-Ruiz M.C., de Murcia G., Menissier-de Murcia J. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. J. Biol. Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- Olivera B.M., Miljanich G.P., Ramachandran J., Adams M.E. Calcium channel diversity and neurotransmitters releasethe ω-conotoxins and ω-agatoxins. Annu. Rev. Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991;14:453–454. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Park J.S., Wang M., Park S.J., Lee S.H. Zinc finger of replication protein A, a non-DNA binding element, regulates its DNA binding activity through redox potential. J. Biol. Chem. 1999;274:29075–29080. doi: 10.1074/jbc.274.41.29075. [DOI] [PubMed] [Google Scholar]
- Payne M.E., Fong Y.L., Ono T., Colbran R.J., Kemp B.E., Soderling T.R., Means A.R. Calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 1988;263:7190–7199. [PubMed] [Google Scholar]
- Peitsch M.C., Polzar B., Stephan H., Crompton T., MacDonald H.R., Mannherz H.G., Tschopp J. Characterization of the endogenous deoxyribonuclease involved in nuclear DNA degradation during apoptosis (programmed cell death) EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:371–377. doi: 10.1002/j.1460-2075.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitsch M.C., Mannherz H.G., Tschopp J. The apoptosis endonucleasescleaning up after cell death. Trend Cell Biol. 1994;4:37–41. doi: 10.1016/0962-8924(94)90002-7. [DOI] [PubMed] [Google Scholar]
- Rawling J.M., Alvarez-Gonzalez A. TFIIF, a basal eukaryotic transcription factor, is a substrate for poly(ADP-ribosyl)ation. Biochem. J. 1997;324:249–253. doi: 10.1042/bj3240249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C.A., Danoff S.K., Schell M.J., Snyder S.H., Ullrich A. Three additional inositol 1,4,5-trisphosphate receptorsmolecular cloning and differential localization in brain and peripheral tissues. Proc. Natl. Acad. Sci. USA. 1992;89:4265–4269. doi: 10.1073/pnas.89.10.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscetti T., Lehnert B.E., Halbrook J., Le Trong H., Hoekstra M.F., Chen D.J., Peterson S.R. Stimulation of the DNA-dependent proteins kinase by poly(ADP-ribose)polymerase. J. Biol. Chem. 1998;273:14461–14467. doi: 10.1074/jbc.273.23.14461. [DOI] [PubMed] [Google Scholar]
- Satoh M.S., Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- Satoh M.S., Poirier G.G., Lindahl T. Dual effect for poly(ADP-ribose) synthesis in response to DNA strand breakage. Biochemistry. 1994;33:7099–7106. doi: 10.1021/bi00189a012. [DOI] [PubMed] [Google Scholar]
- Schreiber V., Hunting D., Trucco C., Gowans B., Grunwald D., de Murcia G., Menissier-de Murcia J. A dominant-negative mutant of human poly(ADP-ribose)polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc. Natl. Acad. Sci. USA. 1995;92:4753–4757. doi: 10.1073/pnas.92.11.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selden J.R., Dolbeare F. A flow cytometric technique for detection of DNA repair in mammalian cells. Methods Cell Biol. 1994;42:1–19. doi: 10.1016/s0091-679x(08)61064-x. [DOI] [PubMed] [Google Scholar]
- Shah G.M., Kaufmann S.H., Poirier G.G. Detection of poly(ADP-ribose)-polymerase and its apoptosis-specific fragment by a nonisotopic activity-Western blot technique. Anal. Biochem. 1995;232:251–254. doi: 10.1006/abio.1995.0016. [DOI] [PubMed] [Google Scholar]
- Shah G.M., Poirier D., Desnoyers S., Saint-Martin S., Hoflack J.C., Rong P., ApSimon M., Kirkland J.B., Poirier G.G. Complete inhibition of poly(ADP-ribose)polymerase activity prevents the recovery of C3H10T1/2 cells from oxidative stress. Biochim. Biophys. Acta. 1996;1312:1–7. doi: 10.1016/0167-4889(96)00004-3. [DOI] [PubMed] [Google Scholar]
- Sharkey J., Ritchie I.M., Butcher S.P., Kelly J.S. Comparison of the patterns of altered cerebellar glucose utilization produced by competitive and non-competitive NMDA receptor antagonists. Brain Res. 1996;735:67–82. doi: 10.1016/0006-8993(96)00574-4. [DOI] [PubMed] [Google Scholar]
- Sierro C.D., Vitus J., Dunant Y. Effect of muscarinic agonists and depolarizing agents on inositol mono-phosphate accumulation in the rabbit vagus nerve. J. Neurochem. 1992;59:456–466. doi: 10.1111/j.1471-4159.1992.tb09392.x. [DOI] [PubMed] [Google Scholar]
- Simbulan C.-M.G., Suzuki M., Izuta S., Sakura T., Savoysky E., Kojima K., Miyahara K., Shizuta Y., Yoshida S. Poly(ADP-ribose)polymerase stimulates DNA polymerase alpha by physical association. J. Biol. Chem. 1993;268:93–99. [PubMed] [Google Scholar]
- Spitzer N.C. A developmental handshakeneuronal control of ionic currents and their control of neuronal differentiation. J. Neurobiol. 1991;22:659–673. doi: 10.1002/neu.480220702. [DOI] [PubMed] [Google Scholar]
- Sutherland B.M., Bennett P.V., Sutherland J.C. DNA damage quantitation by alkaline gel electrophoresis. In: Henderson D.S., editor. DNA Repair Protocols, Eukaryotic Systems. Humana Press; New Jersey: 1999. pp. 183–193. [DOI] [PubMed] [Google Scholar]
- Takemura H., Hughes A.R., Thastrup O., Putney J.W., Jr. Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. J. Biol. Chem. 1989;264:12266–12271. [PubMed] [Google Scholar]
- Toker A. The synthesis and cellular roles of phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Cell Biol. 1998;10:254–261. doi: 10.1016/s0955-0674(98)80148-8. [DOI] [PubMed] [Google Scholar]
- Trucco C., Oliver J.F., de Murcia G., Menissier-de Murcia J. DNA repair defect in poly(ADP-ribose)polymerase-deficient cell lines. Nucleic Acids Res. 1998;26:2644–2649. doi: 10.1093/nar/26.11.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y.J., Aoki T., Maruta H., Abe H., Sakagami H., Hatano T., Okuda T., Tanuma S. Mouse mammary tumor virus gene expression is suppressed by oligomeric ellagitannins, novel inhibitors of poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 1992;267:14436–14442. [PubMed] [Google Scholar]
- Udea K. Poly(ADP-ribose) synthetase. In: Moss J., Vaughan M., editors. ADP-ribosylating Toxins and G-proteinsInsight into Signal Transduction. American Society for Microbiology (ASM); Washington, DC: 1990. pp. 525–542. [Google Scholar]
- Wang J.C. DNA topoisomerases. Annu. Rev. Biochem. 1996;65:632–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- Wu X., Bishopric N.H., Discher D.J., Murphy B.J., Webster K.A. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol. Cell Biol. 1996;16:1035–1046. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchetti D., Clementi E., Fasolato C., Lorenzon P., Zottini M., Grohovaz F., Fumagalli G., Pozzan T., Meldolesi J. Intracellular Ca+2 pools in PC12 cells. J. Biol. Chem. 1991;266:20152–20158. [PubMed] [Google Scholar]
- Zhang J., Dawson V.L., Dawson T.M., Snyder S.H. Nitric oxide activation of poly(ADP-ribose)synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]