

SUMMARY
The human cytosolic thymidine kinase (TK) and structurally related TKs in prokaryotes play a crucial role in the synthesis and regulation of the cellular thymidine triphosphate pool. We are now reporting the crystal structures of the TK homotetramer from Thermotoga maritima in four different states, its apo-form, a binary complex with thymidine, as well as the ternary structures with the two substrates (thymidine/AppNHp) and the reaction products (TMP/ADP). In combination with fluorescence spectroscopy and mutagenesis experiments, our results demonstrate that ATP binding is linked to a substantial reorganization of the enzyme quaternary structure, leading to a transition from a closed, inactive conformation to an open, catalytic state. We hypothesize that these structure changes are relevant to enzyme function in situ as part of the catalytic cycle and serve an important role in regulating enzyme activity by amplifying the effects of feedback inhibitor binding.
INTRODUCTION
Thymidine kinases (TKs) catalyze the first phosphorylation step in the salvage pathway of thymidine triphosphate (TTP) synthesis by transferring the γ-phosphate group from an ATP molecule to the 5′-hydroxyl group of thymidine. Mammals contain two structurally distinct thymidine kinases referred to as TK1 and TK2. The latter is localized to the mitochondria and is constitutively expressed. In contrast, TK1, the focus of this report, is expressed in the cytoplasm and is subject to strict cell cycle regulation. TK1 is only transcribed during the S-phase of the cell cycle (Coppock and Pardee, 1987), and carries regulatory motifs that promote its degradation in the proteasome after mitosis exit (Ke and Chang, 2004). The strict control of TK1 activity highlights the enzyme’s importance in regulating the intracellular TTP levels. This has been confirmed by experiments in which disruption of human TK1 (hTK1) proteolysis leads to a dramatic increase in the intracellular TTP levels, resulting in growth retardation and increased gene mutation rate (Ke et al., 2005). Conversely, TK1-knockout mice exhibit a dramatic shortening of lifespan, kidney failure and compromised immune system, suggesting an indispensable role of the salvage pathway in vivo (Dobrovolsky et al., 2003). The phosphoryl donor ATP has also been shown to affect TK1 activity (Munch-Petersen et al., 1993). Here we report how ATP modulates the TK1 quaternary conformation from a closed to an open state, and provide experimental evidence for the essentiality of the open state for enzymatic activity.
Recently, the crystal structures of hTK1 and several TK1-like enzymes were solved and revealed a topology distinct from previously reported deoxynucleoside kinases (Birringer et al., 2005; Welin et al., 2004). Members of the TK1 family assemble into homotetramers with D2-symmetry, indicating a dimer-of-dimers with two distinct protein-protein interfaces. The first boundary, referred to as the strong dimer interface, is distal from the active site and is formed by aligning the β-sheets of the two flanking subunits in antiparallel fashion. The contact area is made up of side chains from strand β6 and helix α4 in both subunits. In contrast, the second boundary, called the weak dimer interface, is proximal to the ATP binding site and involves mostly interactions between the helix α1 of both subunits (Segura-Pena et al., 2007).
The individual subunits consist of a large amino terminal domain with a RecA fold (Welin et al., 2004) and a relatively small carboxy-terminal region. The latter contains a zinc binding site and an extended loop structure, called the “lasso” region. Thymidine binding is facilitated through specific hydrogen-bonding interactions between its pyrimidine moiety and the lasso region. At the other end of the active site, ATP binding is assisted by the enzyme’s P-loop region and a magnesium ion. Consistent with reports of broad phosphoryl donor specificity for TK1-like enzymes (Lutz et al., 2007; Segura-Pena et al., 2007), few specific interactions with the nucleosidyl portion of ATP were observed in the crystal structures. The structural analysis placed ATP’s adenosyl moiety at the weak dimer interface of the tetramer. In fact, earlier data for hTK1 and Thermotoga maritima TK (TmTK), co-crystallized with the bisubstrate inhibitor TP4A, suggest that ATP binding induced a conformational change on the quaternary structure level, consequently expanding the tetrameric complex along the weak dimer interface. The expanded conformation is critical for proper orientation of ATP and is stabilized by a hydrogen-bonding network involving protein side chains, the ribose portion of ATP, and numerous water molecules (Segura-Pena et al., 2007). Separate kinetic and spectroscopic experiments support the proposed quaternary structure change in TK1 and its relevance to enzyme function (Lutz et al., 2007).
From these data, a functional model for TK1-like enzymes has emerged. In the absence of a substrate in the phosphoryl donor binding site, the homotetramer preferentially exist in a closed, inactive state as observed in the structures of hTK1 (Birringer et al., 2005; Segura-Pena et al., 2007; Welin et al., 2004), Ureaplasma urealyticum TK (Welin et al., 2004), as well as Bacillus cereus TK (Kosinska et al., 2007). Upon nucleotide binding at the donor site, the enzyme complex undergoes conformational changes that lead to an open, catalytically competent conformation as seen for the TKs from T. maritima (Segura-Pena et al., 2007), Clostridium acetobutylicum (CaTK) (Kuzin et al., 2004), and Bacillus anthracis (BaTK) (Kosinska et al., 2007). These conformational changes concentrate on the weak dimer interface and might involve neighboring loops and domains of the individual subunits.
In the above model, ambiguity arises from the fact that the comparison of the closed and open tetrameric states was carried out with enzymes from different sources. One could not discount the possibility that the former enzymes maintain their closed state upon ATP binding, or that the latter remain in the open state in the absence of nucleotide at the donor site. To address this question, we have elucidated the crystal structures of the same TK ortholog in different substrate-bound states. The TK from T. maritima formed diffraction quality crystals in the nucleotide-free (apo) form, with thymidine (binary complex), and with thymidine and the non-hydrolyzable ATP analog AppNHp (ternary complex). Comparison of these structures enabled us to evaluate the effect of substrate binding on the tertiary and quaternary structure of TmTK. In parallel, we probed the functional importance of conformational changes at the protein-protein interface in solution, using site-directed mutagenesis and fluorescence spectroscopy. In the absence of tryptophan residues in the wild type TmTK, we introduced single tryptophan mutations in various locations throughout the protein structure to allow detection of conformational change as a function of substrate binding. Separately, we designed a double mutant carrying two cysteines at the critical weak dimer interface. Under oxidizing conditions, disulfide bond formation across this interface locks the tetramer in the closed, inactive state. The process is reversible upon addition of a strong reducing agent, yielding subunits that can oscillate between the closed and open state and thus regain TK activity.
RESULTS
Structural analysis of TmTK in three complexation states
The three TmTK structures presented here were obtained from crystals that diffract to high resolution: 1.95 Å for the apo form and the complex with thymidine, and to 1.5 Å for the structure of the ternary complex (Table 1). The structures were solved by the molecular replacement method, using the TmTK structure in complex with the bisubstrate inhibitor TP4A (PDB: 2ORW (Segura-Pena et al., 2007)) as the search model. Notably, this is the first time that an apo state and a ternary complex state are reported for a TK1-like enzyme. Crystals of apo TmTK had a complete tetramer as the asymmetric unit. A sulfate ion was observed bound to the phosphate binding loop (P-loop) of each subunit. This is analogous to the reported structure of the nucleotide-free form of uridylate (Segura-Pena et al., 2004) and adenylate kinase (Dreusicke et al., 1988) in which a sulfate group was located at the P-loop. The sulfate anion mimics the beta phosphate group of the nucleotide triphosphate, but is not able to elicit the conformational changes that occur upon ATP binding. Therefore, despite the presence of the sulfate group, we interpret the TmTK-sulfate complex as representing the apo form of the enzyme.
Table 1.
Data collection and refinement statistics
| TmTK Apo | TmTK Thy | TmTK Thy+AppNhp | |
|---|---|---|---|
| Wavelength (Å) /beam | 1.54 / in-house | 1.0 / SERCAT | 1.0 / SERCAT |
| Space group | P21 | C2 | C2 |
| Unit Cell (Å, °) |
a=52.6 b=113.5 c=54.8
α=90 β=109.2 γ=90 |
a=111.6 b=52.8 c=59.31
α=90 β=107.9 γ=90 |
a=102.2 b=59.3 c=61.3
α=90 β=103.2 γ=90 |
| Content of asymmetric unit | tetramer | dimer | dimer |
| Resolution (Å) | 1.95 | 1.95 | 1.5 |
| Rsyma,b (%) | 6.5 (22.5) | 7.6 (33.4) | 5.8 (25.8) |
| I/σIa | 13.8 (4.4) | 13.1 (5.8) | 12.7 (4.5) |
| Completenessa | 98.0 % (75.2) | 98.5% (95.5) | 95.3 (87.5) |
| No of observed reflections | 162886 | 85927 | 199812 |
| Redundancy | 3.8 | 3.6 | 3.7 |
| Rcrsytd (%) | 20.0 | 23.5 | 17.1 |
| Rfreed (%) | 25.8 | 28.5 | 21.6 |
| Rmsd bond lengthe (Å) | 0.015 | 0.017 | 0.014 |
| Rmsd bond anglee (°) | 1.6 | 1.6 | 1.7 |
| Number of atoms | 4928 | 2589 | 3142 |
| Protein | 4730 | 2504 | 2659 |
| Nucleotide | 0 | 4 (34 atoms) | 8 (96 atoms) |
| SO4 | 4 (20 atoms) | 0 | 0 |
| Water | 174 | 49 | 383 |
| Zinc | 4 | 2 | 2 |
| Mean B-factors (Å2) | |||
| Protein Atoms | 23.5 | 27.6 | 19.2 |
| Nucleotides | 24.7 | 18.2 | |
| Sulfate | 26.0 | ||
| Water molecules | 28.0 | 30.4 | 41.9 |
| Zinc atoms | 23.2 | 28.4 | 19.8 |
| Ramachandran plot (%) | |||
| Most allowed | 95.5 | 91.5 | 95.9 |
| Additional allowed | 4.5 | 7.0 | 4.1 |
| Generously allowed | 0 | 1.4 | 0 |
| Disallowed | 0 | 0 | 0 |
Data for the highest resolution shell are given in parenthesis
Rsym = Σ|Ii−<Ii>|/Σ<Ii>
Rcryst = Σ||Fobs|−|Fcalc||/Σ|Fobs|. Rfree is for 10% of the reflections excluded from the refinement.
Root mean square deviations (rmsd) from ideal values calculated with Refmac
For the binary and ternary complexes, a dimer was present in the asymmetric unit and a two-fold crystallographic symmetry operator recreated the biological tetramer. Surprisingly, in the ternary complex that was crystallized in the presence of thymidine and AppNHp we observe different nucleotides in the two monomers. While the electron density in the substrate binding sites of one monomer fits the expected thymidine and AppNHp, the second monomer shows electron density that corresponds to the presence of the reaction products TMP and ADP (Fig. S1). This observation suggests that the enzyme catalyzed phosphoryl transfer despite the more stable NH group that bridges the β and γ phosphates in AppNHp. This also implies that the enzyme was active in the conditions used for crystallization.
The overall fold for the three crystal structures is very similar (Fig. 1a). Overlay of the apo protein with the ternary complex produced a root mean square deviation (rmsd) of 0.4 Å for 144 Cα atoms (out of 185 residues). Similarly, the overlay of the binary and ternary complexes gave an rmsd of 0.4 Å over 162 Cα atoms. The analysis suggests that the three dimensional structure of TmTK remains largely unaltered over the course of the phosphoryl transfer reaction. However, there are two regions in TmTK that change conformation in response to substrate binding. The first region is the lasso loop, sensitive to the presence of substrate in the phosphoryl acceptor-binding site (shown in red, Fig. 1b). The second region includes residues 40 through 58, a flexible loop that only assumes an ordered hairpin-like structure upon binding of a nucleotide in the phosphoryl donor-binding site (shown in blue Fig. 1b).
Figure 1.

The TmTK three-dimensional structure remains largely unchanged upon substrate binding. (a) Stereo-view of an overlay of three TmTK backbones in different complexation states. Apo-form in cyan, in complex with thymidine in yellow and the ternary complex in magenta. Purple spheres represent the Mg ion in the active site. (b) Ribbon representation of the three individual TmTK structures, using the same color code as in panel a. Yellow spheres mark the position of the structural zinc atom. The two regions of the protein that undergo a conformational change upon substrate binding are the lasso loop (red lines) and the β hairpin loop (blue lines). The dashed lines represent parts of the flexible loops that could not be modeled due to lack of electron density. Most of the lasso loop, which is involved in thymidine binding was disorganized in the apo-form, yet became almost fully defined in the complex with thymidine and could be completely modeled in the ground state complex (thymidine + AppNHp). The β hairpin loop composed of βc1/β3 is sensitive to the presence of phosphoryl donor. Only in the presence of AppNHp, the electron density for βc1 was present.
The lasso region forms part of the C-terminal domain of TmTK and interacts with enzyme-bound thymidine via several main-chain atoms. In the apo protein (Fig. 1b, cyan), the lasso region is devoid of interpretable electron density but becomes mostly organized upon thymidine binding, as seen in the binary complex (Fig. 1b, yellow). In the binary complex only two residues in the lasso region were lacking electron density in the first monomer of the asymmetric unit, while five amino acids in the same location of the second monomer could not be assigned. The reason for this partial disorganization of the lasso is not clear, since previous structures with an occupied phosphoryl acceptor-binding site had electron density for the entire region (Birringer et al., 2005; Kosinska et al., 2005; Welin et al., 2004). Only upon ATP binding in our ternary complex (Fig. 1b, magenta) did the lasso region become completely organized as reflected in a clear continuous electron density and lower B-factor values for the entire sequence. The difference in apparent rigidity of the lasso region between the binary and the ternary complex cannot be attributed to differences in the crystallization conditions since both structures were crystallized under the same conditions. Moreover, inspection of the crystal packing of the TmTK-thymidine complex suggests ample space to accommodate the loop. Instead, our findings are consistent with ATP functioning as a modulator of the lasso loop flexibility and consequently the binding affinity for thymidine, an effect that was observed experimentally in hTK1 (Munch-Petersen et al., 1993).
The second region in TmTK that undergoes significant structural reorganization upon substrate binding is the flexible loop made up of residues 40 through 58. In the apo structure and binary complex, the region cannot be traced (shown as blue dotted lines in Fig. 1b) but becomes partially ordered in the presence of a nucleotide at the ATP binding site. In the ternary TmTK complex (Fig. 1b, magenta), we note that the previously disordered residues now form a β-hairpin structure, consistent of a short β-strand that we designate βc1 and β3. A similar conformation has been observed in the binary complexes of CaTK with ADP and BaTK with TTP bound at the ATP binding site. Thus, the reorganization of this second region is independent of the substrate in the phosphoryl acceptor site but responds to phosphoryl donor binding. Our earlier work (Segura-Pena et al., 2007) revealed that the highly conserved histidine in position 53 (TmTK numbering), located at the tip of the β-hairpin, could form hydrogen bonds to the phosphates of AppNHp, suggesting a possible role for the hairpin in catalysis (Fig. S1). To test this hypothesis, we introduced a H53A mutation but found that neither the enzymatic turnover nor the affinity for ATP and thymidine are affected (Table 2). Alternatively, the loop could function as a sensor for NTP binding, triggering more extensive conformational changes at the quaternary structure level of the homotetramer (see below). Evidence linking the conformational change in the β-hairpin region to ATP binding was obtained by fluorescence spectroscopy.
Table 2.
| thymidine | ATP | |||||
|---|---|---|---|---|---|---|
| KM (μM) | kcat (s−1) | kcat/KM (×105 M−1s−1) | KM (μM) | kcat (s−1) | kcat/KM (×104 M−1s−1) | |
| wild type | 0.5 ± 0.2 | 0.3 ± 0.08 | 6.0 | 40 ± 12 | 0.4 ± 0.04 | 1.0 |
| H53A | 1.2 ± 0.2 | 0.51 ± 0.01 | 4.3 | 86 ± 7 | 0.52 ± .01 | 0.6 |
| V34W | 0.9 ± 0.2 | 0.59 ± 0.02 | 6.3 | 23 ± 4 | 0.35 ± 0.01 | 1.5 |
| V51W | 0.7 ± 0.1 | 0.25 ± 0.01 | 3.4 | 35 ± 5 | 0.20 ± 0.005 | 0.6 |
| G55W | 0.8 ± 0.1 | 0.62 ± 0.01 | 7.7 | 112 ± 10 | 0.45 ± 0.005 | 0.4 |
| V58W | 0.5 ± 0.1 | 0.47 ± 0.01 | 9.6 | 29 ± 3 | 0.38 ± 0.008 | 1.3 |
| L129W | 3.0 ± 0.3 | 1.91 ± 0.05 | 6.4 | 46 ± 5 | 1.49 ± 0.02 | 3.2 |
Tryptophan fluorescence to monitor β-hairpin movement
Independent confirmation of the proposed conformational change in the β hairpin region of TmTK was sought by environmentally sensitive intrinsic tryptophan fluorescence. In the absence of native tryptophans in TmTK, we introduced five single mutations: three in the βc1/β3 region itself (V51W, G55W, V58W), one (V34W) proximal to the hairpin that provides an indirect probe for monitoring conformational changes, and one (L129W) which is located at the distant strong dimer interface and serves as a control (Fig. 2). Following overexpression and purification, the kinetic characterization of these mutants showed very few changes in the catalytic properties compared to wild type enzyme (Table 2). Noticeable, a 6-fold increase in KM for thymidine in L129W can be rationalized by its relative proximity to the thymidine binding site. In G55W, the ~3-fold raise in the apparent ATP binding constant can be explained by its proximity to the phosphoryl donor binding site.
Figure 2.
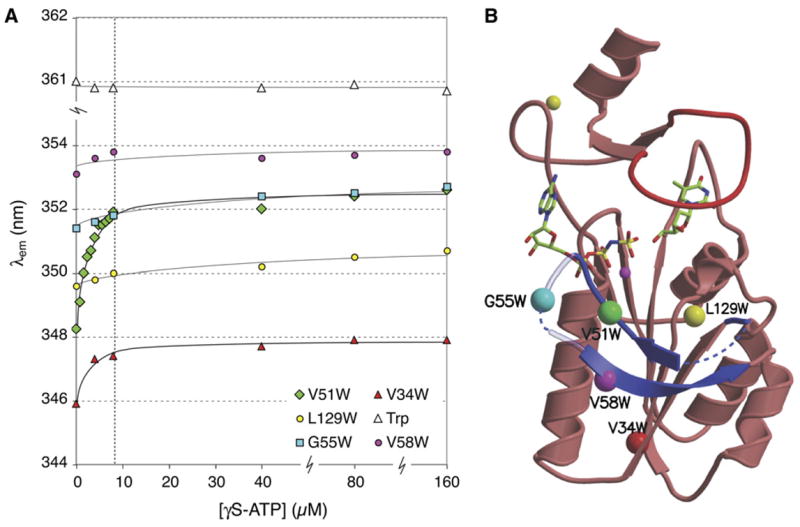
Conformational changes in the β hairpin loop of TmTK upon ATP binding. (a) Changes in the emission wavelength of single tryptophan residues during titration with γS-ATP were monitored via intrinsic tryptophan fluorescence. Tryptophans were introduced by site-directed mutagenesis to probe for conformational changes in the β hairpin loop region directly (V51W, G55W, V58W) or indirectly (V34W). The L129W mutant and tryptophan in solution served as controls. The enzyme concentration was kept constant at 8 μM and is marked with a vertical dashed line. (b). Ribbon diagram of a TmTK monomer in the ternary complex, indicating the position of the single tryptophan mutations with colored spheres.
The five tryptophan mutants were used to detect conformational changes upon ATP binding. The proteins’ intrinsic tryptophan fluorescence was monitored upon titration with non-hydrolyzable γS-ATP in the absence or after preincubation with thymidine. The presence of thymidine did not affect our fluorescence measurements (data not shown), consistent with the earlier observation that binding of the phosphoryl acceptor does not induce significant conformational changes in the βc1/β3 region of our binary complex. Possible unspecific changes in the fluorescence signal upon γS-ATP addition were accounted for in control experiments with free tryptophan in the same buffer.
All five mutants show distinct tryptophan emission spectra with λmax ranging from 346 to 354 nm (Fig. 2). The blue shift compared to free tryptophan at 361 nm is indicative of a more hydrophobic environment of the indole moieties in the proteins. Upon titration with the ATP analog, the V34W and V51W variants show clear red shifts in their emission spectra, ranging from 2 to 4 nm respectively. Furthermore, the titration curves reached saturation at equimolar concentrations of TmTK and the ATP analog. The data suggests a significant change in the residues’ local environment, consistent with a conformational change in TmTK’s βc1/β3 region upon ATP binding. For the V34W protein, substrate binding causes the upward movement of the βc1/β3 loop, thereby exposing the underlying position 34 to a more hydrophilic environment. In the case of V51W, the loop relocation could cause a λmax shift as it induces a rotation of the amino acid side chain from a shielded interior position to the protein surface. The absence of a similar wavelength shift in G55W and V58W, which are also located in the βc1/β3 region, can be rationalized with little change in the residues’ local environments upon organization of the hairpin structure. Consistent with this interpretation, the high λem for those two residues suggests that they are surface exposed, making them less susceptible to conformational changes in the protein. In both cases, the slight changes in λmax are comparable to the shifts observed for our control L129W. In summary, our fluorescence data corroborate our hypothesis of ATP-dependent conformational changes for the βc1/β3 region.
ATP binding induces a conformational change in the quaternary structure
In addition to the conformational changes in the tertiary structure of TmTK, significant changes in the homotetramer’s quaternary structure can be observed upon ATP binding. Among the tetrameric forms of our three TK structures, the apo protein and the TmTK-thymidine complex show nearly identical dimensions across the weak and strong dimer interfaces (Fig. 3a) and overlay very well in all four subunits (Fig. 3b). In contrast, the tetramer undergoes significant conformational changes upon ATP binding as seen in the ternary complex. While the dimensions and orientation of the two subunits connected by the strong dimer interface are unchanged, the distance across the weak dimer interface increases by almost 10% from ~51 Å in the apo and thymidine-bound structure to ~56 Å in the complex with thymidine and AppNHp (Fig. 3a). Consistent with that observation, the superposition of the binary and ternary complexes shows that only two of the four subunits overlay well (Fig. 3c). The overlay further indicates that the distance change is accompanied by an ~11° rotation between the subunits across the weak dimer interface.
Figure 3.
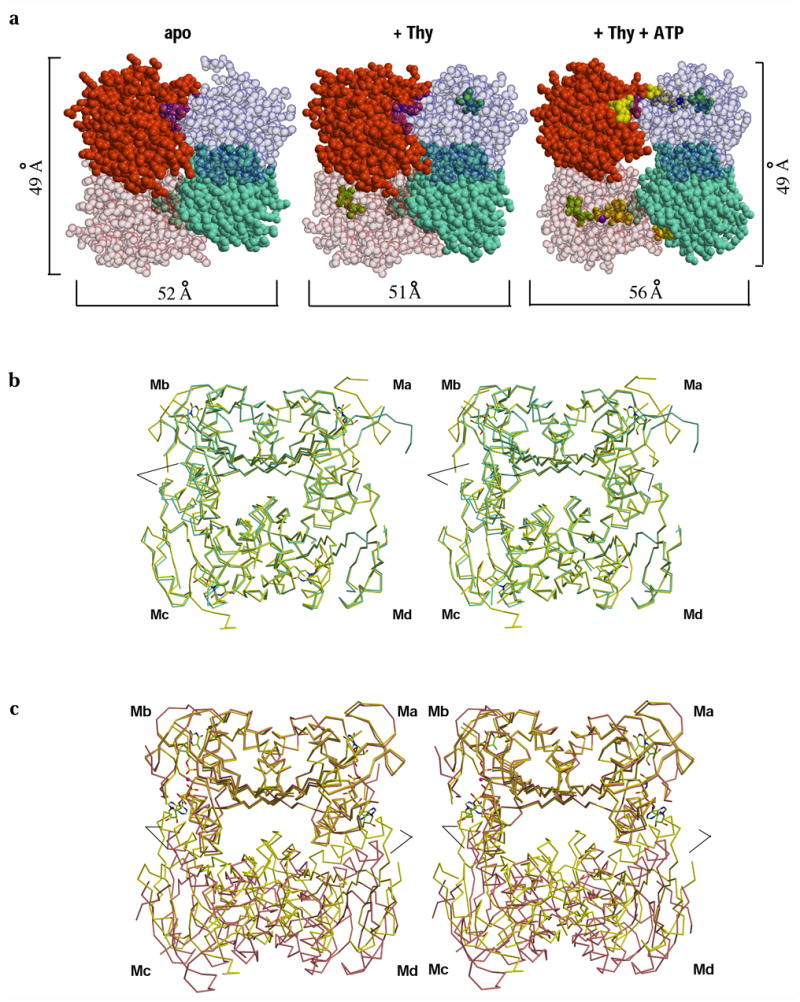
ATP induces a quaternary structural rearrangement in TmTK. (a) Sphere representation of TmTK homotetramer in the three substrate-binding states. Individual monomers are shown in different colors. Note that upon ATP binding the tetramer expands due to an increase in the separation between the monomers that make the interface to which the adenosine moiety of ATP is bound (weak dimer interface, horizontal brackets). In contrast the second type of monomer-monomer interface remains unchanged (vertical brackets) (b) Stereoview overlay of the TmTK apo-tetramer (cyan) with the tetramer in complex with thymidine (yellow). The overlay was done on the monomer A (Ma). There is an excellent superposition of the two tetrameric structures, indicating the same subunit organization for the two tetrameric structures (c) Analogous stereoview overlay between the TmTK binary complex (with thymidine) and the ternary complex (magenta color). Subunits across the strong dimer interface show an excellent overlay (Ma and Mb). In constrast, the relative orientation of the remaining two monomers is changed. Note the change of orientation between monomers across the weak interface (Md with respect to Ma and Mc with respect to Mb). Black lines mark the positions of helix α1 in each tetramer. In the close state of the tetramer (binary complex in yellow), helix α1 would clash with the adenosine moiety of ATP.
As a result of the quaternary structure expansion and rotation, there are substantial differences in the nature of the subunit contacts at the weak interface between the collapsed (closed) and the expanded (open) state of the tetramer (Fig. 4). In the closed state, interactions between monomers that form the weak dimer involve mostly side chains of residues in helix α1. There are 19 Van der Waals (VDW) contacts (≤ 3.8 Å), 3 salt bridges (≤ 3.0 Å) and 3 polar interactions (≤ 3.0 Å). In the open conformation, the same distance criterion identifies 31 VDW, 2 salt bridges and 6 polar interactions. The critical role of ATP as a bridging element between subunits across the weak dimer interface is apparent from the fact that 20 out of the 31 VDW interactions involve the phosphoryl donor and all the polar interactions are established with the NTP. An analysis of the buried surface area due to monomer-monomer interactions help to further illustrate the changes that take place during the transition from a closed to open state. In the closed state, the occluded surface area between subunits at the weak dimer interface is approximately 900 Å2 in the apo structure and 1030 Å2 for the thymidine complex. On the other hand, the ternary complex, representing the open state, has only 370 Å2 of protein-protein surface contact area. When taking into consideration the additional interactions contributed by ATP, the occluded surface area increases by an additional 260 Å2 to 630 Å2. Significantly, the same calculations at the strong dimer interface show no significant change among the three structures: 1070 Å2 (apo), 1130 Å2 (binary complex), and 1050 Å2 (ternary complex).
Figure 4.
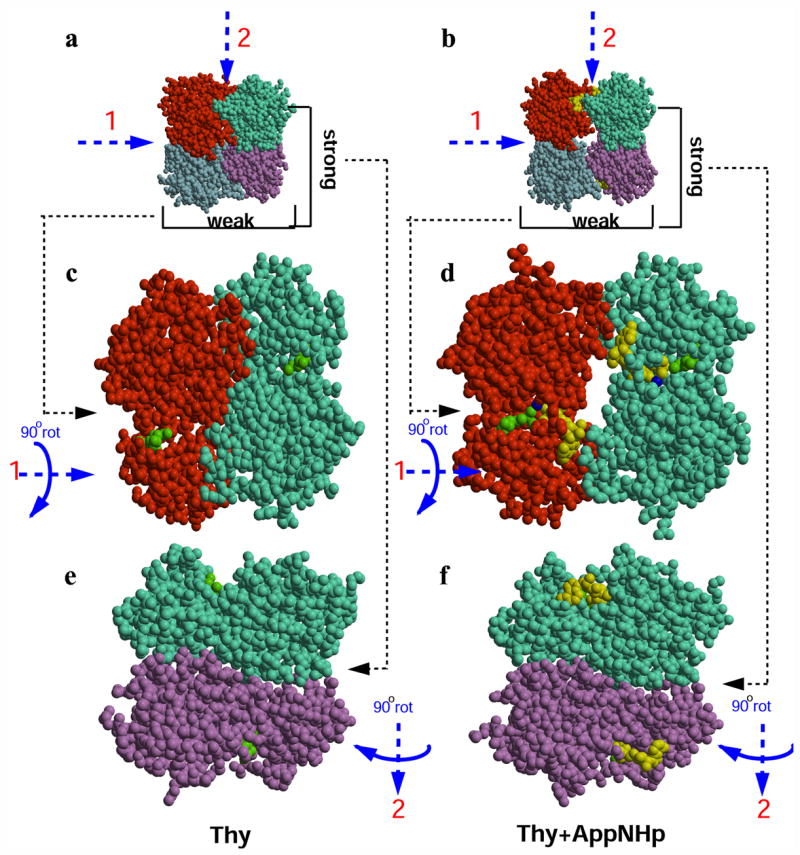
Frontal view of the two types of monomer-monomer interfaces present in TmTK tetramer (colored in red, cyan, purple and marine). AppNHp is showed in yellow and thymidine is labeled in green. (a) Top view of the tetrameric TmTK in complex with thymidine (closed conformation). (b) Top view of the tetramer in complex with AppNhp and thymidine (open conformation). (c) Frontal view of the monomers that make the weak dimer interface for the binary complex with thymidine (closed conformation), after 90° rotation around axis 1. In the absence of ATP, the two monomers are in close contact. (d) Frontal view for the ternary complex. Upon ATP binding, the two subunits that make the weak dimer interface separate from each other by ~5 Å. The water network formed at the weak interfaces of the open tetramer is not shown for the purpose of clarity. (e) Frontal view of the monomers that make the strong dimer interface for the enzyme in complex with thymidine (closed conformation), after 90° rotation around axis 2. (f) View for the ternary complex. Note that the strong dimer interface remains unchanged upon ATP binding.
The conclusion from this analysis is that in the absence of ATP, the closed conformation is more stable. In addition to having more protein-protein interactions in the closed state, the adoption of the closed tetrameric state might be favored by the increase of entropy generated by the exclusion of the water network that otherwise forms between the subunits in the open state. Consistent with this idea, a significant decrease in entropy due to γS-ATP binding (and hence, closed-to-open transition) to TmTK has been detected by isothermal titration calorimetry (Fig. S2).
While the closed tetramer seems to be more stable (especially in the absence of ATP), our previous structures of hTK1 with TP4A indicated that this state is unable to accommodate the phosphoryl donor in a catalytically productive orientation (Segura-Pena et al., 2007). To experimentally validate that the enzyme adopts the closed tetrameric state in solution, and that this closed state is not (or only weakly) enzymatically active, in contrast to the active open state, we created a double-cysteine mutant to reversibly lock the homotetramer in its closed conformation via disulfide bridges.
Trapping the closed conformation by disulfide linkage
The TmTK crystal structures in the closed state suggest that thiol side chains of cysteines in position 18 and 22 of helix α1 would be in close proximity to the corresponding residues of the neighboring subunit across the weak dimer interface (Fig. 5). Under oxidizing conditions, we predicted that these thiols could form two disulfide bridges, covalently linking the two subunits, thus locking them in the closed conformation. While the homotetramer in such a locked conformation is expected to have lower affinity for ATP and significantly lower catalytic efficiency, addition of a reducing agent should reestablish phosphoryl transfer activity.
Figure 5.
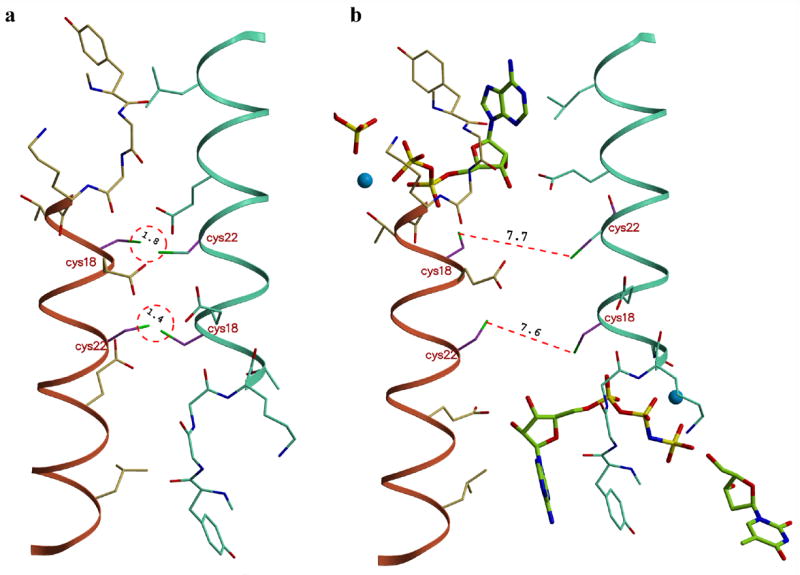
Modeling of the disulfide bond formation. (a) The close proximity of the helix α1 regions in the closed tetrameric conformation properly positions the side chains of cysteines 18 and 22 for disulfide bond formation across the weak dimer interface. (b) In the open state, the ~6 Å increase in distance between neighboring subunits prevents the covalent linkage. The formation of intra-subunits disulfide bonds is not possible due to steric constraints. Models were generated in the program ‘O’.
These predictions were tested with the double-cysteine TmTK mutant T18C/S22C. Wild type enzyme and T18A/S22A, a double-alanine mutant to account for possible effects of mutagenesis on activity, were used as controls. Following protein overexpression, the three enzymes were purified by affinity chromatography using Ni-NTA (Lutz et al., 2007). Unspecific thiol derivatization during purification was prevented by addition of 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) to all buffer solutions. The reducing agent was subsequently removed via dialysis, yielding fully active enzyme in the case of wild type TmTK and T18A/S22A (Table 3). In contrast, the double-cysteine mutant protein showed only ~3.7% of wild type activity. Analysis by non-reductive SDS-PAGE indicated that the majority of T18C/S22C mutant migrates as a dimer under these conditions (Fig. 6). In contrast, the wild type enzyme and the T18A/S22A migrate as monomers in the polyacrylamide gel. To eliminate the possibility for protein agglomeration of the double-cysteine mutant, we confirmed its quaternary structure by gel filtration chromatography. The mutant protein shows an identical elution profile with wild type TmTK (data not shown).
Table 3.
| enzyme | protein treatment | v (nmol mg−1 min−1) | relative activity (%) |
|---|---|---|---|
| wt TmTK | w/ TCEP & dialysis | 674 ± 33 | 100 |
| T18A/S22A | w/ TCEP & dialysis | 622 ± 28 | 92 |
| T18C/S22C | w/ TCEP & dialysis | 25 ± 0.1 | 3.7 |
| wt TmTK | post-dialysis; 0 mM TCEP | 668 ± 18 | 99 |
| post-dialysis; 8 mM TCEP | 351 ± 25 | 52 | |
| post-dialysis; 16 mM TCEP | 76 ± 0.5 | 11 | |
| T18C/S22C | post-dialysis; 0 mM TCEP | 7.6 ± 2.3 | 1.1 |
| post-dialysis; 8 mM TCEP | 222 ± 5 | 33 | |
| post-dialysis; 16 mM TCEP | 86 ± 2 | 13 |
Figure 6.

SDS-PAGE analysis of the oxidized and reduced forms of wild type and double-cysteine mutant TmTK. Incubation of oxidized protein samples at various TCEP concentrations leaves the monomeric wild type enzyme unchanged. Similar conditions result in the [TCEP]-dependent reduction of the disulfide linkage in the mutant TmTK, converting the inactive enzyme (disulfide-locked in the closed conformation) dimer into its catalytically competent monomeric form. Higher order bands in the TmTK (T18C/S22C) sample at 0 mM TCEP suggest formation of oligomeric structures due to unspecific disulfide linkage. Incomplete denaturation of these highly thermostable proteins prior to gel analysis is responsible for the observed minor bands in some sample lanes.
The reversibility of the T18C/S22C inactivation was demonstrated by adding various amounts of TCEP. The double-cysteine mutant and, for control purposes, wild type TmTK were incubated with either 0, 8, or 16 mM TCEP for 3 days at ambient temperature. Protein samples were then analyzed by SDS-PAGE and kinase activity assay. While the wild type enzyme’s gel mobility remains unchanged at all conditions, T18C/S22C showed approximately 70% and 90% monomeric bands at 8 mM and 16 mM TCEP respectively (Fig. 6). More importantly, the reduction of the disulfide bonds in T18C/S22C resulted in an up to 30-fold higher kinase activity compared to double mutant incubated without TCEP (Table 3). In fact, the recovered activity in T18C/S22C (222 nmol mg−1 min−1) in the presence of 8 mM TCEP was comparable to similarly treated wild type TmTK (351 nmol mg−1 min−1), especially when one considers that only ~70% of the double-cysteine mutant were reduced (Fig. 6) allowing the enzyme to adopt the catalytically active tetramer upon ATP binding. Our results also suggest that prolonged incubation with TCEP does interfere with enzyme activity. Both the mutant and wild type enzymes show a TCEP-dependent drop in activity, which we attribute to interference of the reducing agent with the native cysteines of the zinc binding site. In summary, the SDS-PAGE analysis and kinetic data confirm that the double cysteine mutant T18C/S22C can be locked in the catalytically incompetent, closed conformation under oxidative conditions, a process that is reversible upon addition of a reducing agent.
DISCUSSION
Our study presents the first collection of high-resolution structures of a single member of the TK1-like enzyme family in various stages along the reaction coordinate. The crystal structures of TmTK in the apo form, binary complex with thymidine, as well as its ternary complex with thymidine/AppNHp and the product complex with TMP/ADP strongly supports the hypothesis that the enzyme’s conformational changes are part of its catalytic cycle and are not due to species variation. The combination of crystallographic data with fluorescence and mutagenesis experiments further confirms that substrate binding to the homotetramer results in a more ordered tertiary structure and induces conformational changes at the quaternary structure level that are critical for catalytic function. While thymidine binding in the phosphoryl acceptor site organizes the lasso region, ATP binding in the phosphoryl donor site coincides with the folding of the βc1/β3-loop region into a defined β-hairpin structure and the elongation of the tetramer’s weak dimer interface by 4–5 Å. We believe that this elongation step of the enzyme’s quaternary structure is critical for proper ATP binding, eliminating steric clashes that prevent phosphoryl donor binding in the tetramer’s closed state. Consistent with that observation, our TmTK double-cysteine mutant, that under oxidative conditions locks the tetramer in its closed conformation via two disulfide bridges, is catalytically inactive.
These observations raise the question on the order of events leading from the apo-tetramer in its closed conformation to the ATP-bound enzyme complex in the open state. In an early model, we hypothesized that the β and γ-phosphates of ATP establishes initial contacts with the P-loop region as seen in the hTK1 structure with T4PA (Segura-Pena et al., 2007). Following this pre-positioning, ATP could either take advantage of the enzyme’s spontaneous oscillation between the closed and open conformation to trap the complex in the open state or, alternatively, actively facilitate the conformational changes in the β-hairpin region and at the dimer interface, forcing the enzyme into the open conformation. However, the model was inconsistent with subsequent kinetic experiments with sodium triphosphate (TP) as donor that showed no phosphoryl transfer activity. More importantly, competition experiments with ATP showed that TP had no measurable inhibitory effect on catalytic performance (Lutz, unpublished data). Our second-generation model now assumes sporatic oscillation of the enzyme between the two conformational states, enabling ATP to efficiently bind in the pre-organized phosphoryl donor site of the open form and trapping the homotetramer in the catalytically relevant conformation.
Our model of the observed quaternary structure changes in connection with ATP binding can also explain the reported allosterism. Human TK1 and TmTK both show positive cooperativity for ATP with a Hill coefficient of ~2 (Lutz et al., 2007; Munch-Petersen, 1984; Segura-Pena et al., 2007). This functional codependency can be rationalized in light of the two proposed conformational states. The closed state, equivalent to the tense T state in the Monod, Wyman and Changeux (MWC) model (Monod et al., 1965) shows low substrate affinity due to the lack of space to properly accommodate the adenosine moiety of ATP. Upon binding of phosphoryl donor to the first subunit, the entire tetramer is transformed into the open conformation, representing the R state in the MWC model. As a consequence, the remaining ATP binding sites will show increased affinity due to pre-organization of the binding pockets.
Beyond our laboratory experiments, we have been wondering about the functional relevance of our two-state model under physiological conditions. A critical question in that regard is the timing of catalysis in the enzyme complex. It is currently unclear whether the four reaction sites of the enzyme complex operate in a coordinated fashion in performing the actual phosphoryl transfer step. The tetramer could require all four sites to be charged prior to catalysis but it is equally possible for the four reaction centers to perform phosphoryl transfer independent of one another. The former case of synchronized substrate turnover could result in an apo-protein complex, giving the closed conformation relevance as part of the reaction cycle. In contrast, random catalysis would significantly lower the probablility for all four sites to simultaneously exist in the ATP-free state, making the closed state a rare occurance. Furthermore, the cell’s ATP concentration in the millimolar range and the observed allosterism raises the question whether the enzyme complex ever exist in its ATP-free, closed conformation in situ.
Besides playing a possible role in the enzyme’s reaction cycle, the closed conformation could be relevant in a regulatory function. Evidence in support of this hypothesis comes from earlier crystal structures of TK1-like family members. Despite attempts to isolate the apo-form of the enzyme, these experiments consistently found the known feedback inhibitor TTP in the phosphoryl acceptor binding site of the tetramer in the closed conformation. In addition to blocking the thymidine binding site, the inhibitor also occupies part of the active site that holds the β and γ-phosphates from the phosphoryl donor. Separately, kinetic data have shown that TTP inhibits ATP competively with a Ki value of 0.6 μM (Lee and Cheng, 1976). As the TTP concentration reaches up to 20 μM at the end of the S-phase (Hu and Chang, 2007; Spyrou and Reichard, 1988), the KM value for ATP of the TK1-like enzyme increases by 100 to 1000-times. The magnitude of the response to feedback inhibitor binding is rather surprising as TTP and ATP occupy distinct binding sites which overlap by only two phosphate groups. Beyond simple competitive binding, the dramatic effect of TTP on down-regulating thymidine phosphorylation could be explained by a TTP-induced conformational change into the closed state of the tetramer. Such a hypothesis raises the interesting possibility of a cellular mechanism to rapidly turn off TK1 activity, prior to enzyme degradation by the proteasome during mitosis. Upon TTP binding, the inhibitor not only occupies parts of the active site but forces the enzyme complex into the inactive, closed tetrameric conformation which dismantles the ATP binding site and further reduces the enzyme’s affinity for phosphoryl donor and consequently catalysis.
In summary, we have presented conclusive evidence for the existence of two distinct quaternary structures in TmTK, an open and closed tetrameric state. The closed state is inactive and the open state is the catalytically active one. The transition between the two conformations is physiologically relevant as they might be part of the enzyme’s reaction cycle but more likely is involved in regulating enzyme activity. Taking into consideration the high degree of structural conservation between TmTK and its orthologs (Birringer et al., 2005; Segura-Pena et al., 2007; Welin et al., 2004), the conclusions derived from the results presented here can be generalized to the human TK1 and the rest of TK1-like enzyme members.
EXPERIMENTAL PROCEDURES
Crystallization
Purified TmTK (Lutz et al., 2007) at a concentration of 5 mg/ml was used for crystallization. Crystals were grown at 22 °C in hanging drops by mixing 1 μl of purified TmTK with 1 μl of the precipitant. The TmTK in the apo form was crystallized with 20% PEG3350 + 200 mM NaSO4, Before flash freezing in liquid nitrogen the TmTK apo crystals were transferred to a cryoprotectant solution containing 20% PEG3350 + NaSO4 0.2 M + 20% of PEG400. The crystals of TmTK binary and ternary complexes were obtained with 60%–65% of 2-methyl-2,4-pentanediol (MPD) and 0.1 M Na OAc (pH 5.0) . The above precipitant solution also acted as a cryoprotectant.
Structure determination
The X-ray diffraction data was reduced with the program XDS (Kabsch, 1993). The structures were solved by the molecular replacement method using the CaTK (Kuzin et al., 2004) or the previously reported structure of TmTK in complex with the substrate inhibitor TP4A (Segura-Pena et al., 2007) as search models. The programs molrep (Vagin and Teplyakov, 2000) and phaser (McCoy et al., 2005) were used to obtain the initial phases. Model building was done with the program ‘O’ (Jones et al., 1991) and the models were refined with CNS and REFMAC (Brunger et al., 1998; Murshudov et al., 1997). The structure figures were generated using the programs Molscript (Kraulis, 1991) and Bobscript (Esnouf, 1997) and rendered with Raster3D (Merritt and Murphy, 1994).
Site-directed mutagenesis
All mutants of TmTK were prepared via primer overlap extension PCR using wild-type tmtk as template. Following restriction endonuclease digestion, the PCR products were ligated into pET14b and transformed into bacterial host cells. All clones were confirmed by DNA sequencing. Protein overexpression in E. coli BL21(DE3) and isolation via metal affinity chromatography was performed as previously described (Lutz et al., 2007). Where indicated, the cell lysis buffer (50 mM Tris-HCl pH 8, 0.5M NaCl, 2.5 mM MgCl2) was supplemented with 5 mM TCEP.
Disulfide experiment
For disulfide bond formation, TCEP was removed from double-cysteine mutant after protein purification via dialysis. Sample (~2 mg/ml) was loaded into Slide-A-Lyzers (MWCO: 10 kDa; Pierce, Rockford IL) and dialyzed against 3 Lt of cell lysis buffer at 4 °C for 2 days. Buffer was exchanged once after 24 h. To reduce the disulfide linker, TCEP was added back to protein solution and sample was incubated at ambient temperature for 3 days.
Enzyme kinetics
The kinetic constants for all enzymes were determined by the spectrophotometric coupled enzyme assay at 37 °C (Schelling et al., 2001). Experiments were performed in triplicates and data were fit to the Michaelis-Menten equation, using Origin software (OriginLab, Northhampton, MA).
Fluorescence spectroscopy
Tryptophan mutants (~8 μM) in assay buffer (50 mM Tris-HCl pH 8, 2.5 mM MgCl2) were excited at 295 nm in a 1 cm path length cuvette. Emission data were collected at 10 °C from 310 – 390 nm in 0.2 nm increments. Each spectrum represents the mean of three scans. For titration experiments with γ-thio ATP stock solution (45 mM in assay buffer), samples were mixed and incubated for 2 minutes prior to spectrum acquisition. All measurements were performed in triplicates and data were analyzed with Origin software.
Coordinates
Coordinates for the TK from Thermotoga maritima in its apo-form (1.95 Å), the binary complex with thymidine (1.95 Å), and the ternary complex with thymidine and AppNHp (1.5 Å) were deposited in the Protein Data Bank (accession codes 2QPO, 2QQ0, and 2QQE respectivley).
Supplementary Material
Acknowledgments
We are grateful to the staff of SERCAT for help in data collection. This work was supported in part by NIH grant AI046943 (to D.S.-P., A.L., and M.K.), as well as GM69958 (to J.L., M.T., and S.L.) and by a grant to the Emory Center for AIDS Research (AI050409) from the NIH and by institutional funding from the Emory University HSC. M.K. was also supported by the Deutsche Forschungsgemeinschaft and the Max-Planck-Society.
Abbreviations used
- hTK1
human thymidine kinase 1
- TmTK
Thermotoga maritima thymidine kinase
- TP4A
P1-(5′-adenosyl)P4-(5′-(2′-deoxy-thymidyl)) tetraphosphate
- TTP
thymidine triphosphate
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- AppNHp
Adenosine-5′-[(β,γ)-imido]triphosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Birringer MS, Claus MT, Folkers G, Kloer DP, Schulz GE, Scapozza L. Structure of a type II thymidine kinase with bound dTTP. FEBS Lett. 2005;579:1376–1382. doi: 10.1016/j.febslet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Coppock DL, Pardee AB. Control of thymidine kinase mRNA during the cell cycle. Mol Cell Biol. 1987;7:2925–2932. doi: 10.1128/mcb.7.8.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrovolsky VN, Bucci T, Heflich RH, Desjardins J, Richardson FC. Mice deficient for cytosolic thymidine kinase gene develop fatal kidney disease. Mol Genet Metab. 2003;78:1–10. doi: 10.1016/s1096-7192(02)00224-x. [DOI] [PubMed] [Google Scholar]
- Dreusicke D, Karplus PA, Schulz GE. Refined structure of porcine cytosolic adenylate kinase at 2.1 Å resolution. J Mol Biol. 1988;199:359–371. doi: 10.1016/0022-2836(88)90319-1. [DOI] [PubMed] [Google Scholar]
- Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model. 1997;15:132–134. 112–133. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- Hu CM, Chang ZF. Mitotic control of dTTP pool: a necessity or coincidence? J Biomed Sci. 2007;14:491–497. doi: 10.1007/s11373-007-9175-1. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Automatic Processing of Rotation Diffraction Data from Crystals of Initially Unknown Symmetry and Cell Constants. J Appl Crystal. 1993;26:795–800. [Google Scholar]
- Ke PY, Chang ZF. Mitotic degradation of human thymidine kinase 1 is dependent on the anaphase-promoting complex/cyclosome-CDH1-mediated pathway. Mol Cell Biol. 2004;24:514–526. doi: 10.1128/MCB.24.2.514-526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke PY, Kuo YY, Hu CM, Chang ZF. Control of dTTP pool size by anaphase promoting complex/cyclosome is essential for the maintenance of genetic stability. Genes Dev. 2005;19:1920–1933. doi: 10.1101/gad.1322905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinska U, Carnrot C, Eriksson S, Wang L, Eklund H. Structure of the substrate complex of thymidine kinase from Ureaplasma urealyticum and investigations of possible drug targets for the enzyme. FEBS J. 2005;272:6365–6372. doi: 10.1111/j.1742-4658.2005.05030.x. [DOI] [PubMed] [Google Scholar]
- Kosinska U, Carnrot C, Sandrini MP, Clausen AR, Wang L, Piskur J, Eriksson S, Eklund H. Structural studies of thymidine kinases from Bacillus anthracis and Bacillus cereus provide insights into quaternary structure and conformational changes upon substrate binding. FEBS J. 2007;274:727–737. doi: 10.1111/j.1742-4658.2006.05617.x. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystal. 1991;24:946–950. [Google Scholar]
- Kuzin AP, Abashidze M, Forouhar F, Vorobiev SM, Acton TB, Ma LC, Xiao R, Montelione GT, Tong L, Hunt JF. X-Ray Structure of Clostridium Acetobutylicum Thymidine Kinase with ADP. Northeast Structural Genomics Target Car26. 2004 to be published. [Google Scholar]
- Lee LS, Cheng Y. Human deoxythymidine kinase II: substrate specificity and kinetic behavior of the cytoplasmic and mitochondrial isozymes derived from blast cells of acute myelocytic leukemia. Biochemistry. 1976;15:3686–3690. doi: 10.1021/bi00662a007. [DOI] [PubMed] [Google Scholar]
- Lutz S, Lichter J, Liu L. Exploiting Temperature-Dependent Substrate Promiscuity for Nucleoside Analogue Activation by Thymidine Kinase from Thermotoga maritima. J Am Chem Soc. 2007;129:8714–8715. doi: 10.1021/ja0734391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Murphy ME. Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: a Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Munch-Petersen B. Differences in the kinetic properties of thymidine kinase isoenzymes in unstimulated and phytohemagglutinin-stimulated human lymphocytes. Mol Cell Biochem. 1984;64:173–185. doi: 10.1007/BF00224774. [DOI] [PubMed] [Google Scholar]
- Munch-Petersen B, Tyrsted G, Cloos L. Reversible ATP-dependent transition between two forms of human cytosolic thymidine kinase with different enzymatic properties. J Biol Chem. 1993;268:15621–15625. [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Schelling P, Folkers G, Scapozza L. A spectrophotometric assay for quantitative determination of kcat of herpes simplex virus type 1 thymidine kinase substrates. Anal Biochem. 2001;295:82–87. doi: 10.1006/abio.2001.5191. [DOI] [PubMed] [Google Scholar]
- Segura-Pena D, Lutz S, Monnerjahn C, Konrad M, Lavie A. Binding of ATP to TK1-like Enzymes Is Associated with a Conformational Change in the Quaternary Structure. J Mol Biol. 2007;369:129–141. doi: 10.1016/j.jmb.2007.02.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura-Pena D, Sekulic N, Ort S, Konrad M, Lavie A. Substrate-induced conformational changes in human UMP/CMP kinase. J Biol Chem. 2004;279:33882–33889. doi: 10.1074/jbc.M401989200. [DOI] [PubMed] [Google Scholar]
- Spyrou G, Reichard P. Dynamics of the thymidine triphosphate pool during the cell cycle of synchronized 3T3 mouse fibroblasts. Mut Res. 1988;200:37–43. doi: 10.1016/0027-5107(88)90069-3. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- Welin M, Kosinska U, Mikkelsen NE, Carnrot C, Zhu C, Wang L, Eriksson S, Munch-Petersen B, Eklund H. Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc Natl Acad Sci U S A. 2004;101:17970–17975. doi: 10.1073/pnas.0406332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.