

Abstract
T cell reconstitution following lymphopenia from chemotherapy or stem cell transplant is often slow and incompetent, contributing to the development of infectious diseases, relapse, and graft-versus-host disease. This is due to the fact that de novo T cell production is impaired following cytoreductive regimens. T cells can be generated from two pathways: 1) thymus derived through active thymopoiesis and 2) peripherally expanded clones through homeostatic proliferation. In the development of lymphopenia, the thymic pathway is commonly compromised in adults and T cells rely upon peripheral expansion to recover T cell numbers. This homeostatic proliferation exploits the high cytokine levels following lymphopenia to rapidly generate T cells in the periphery. Moreover, this early peripheral expansion of T cells can also be driven by exogenous antigen. This results in loss of T cell repertoire diversity and may predispose to auto- or alloimmunity. Alternatively, the high homeostatic proliferation following lymphopenia may facilitate expansion of anti-tumor immunity. Murine and human studies have provided insight into the cytokine and cellular regulators of these two pathways of T cell generation and the disparate portraits of T cell immunity created through robust thymopoiesis or peripheral expansion following lymphopenia. This insight has permitted the manipulation of the immune system to maximize anti-tumor immunity through lymphopenia and led to an appreciation of mechanisms that underlie graft vs. host disease.
Keywords: Immune reconstitution, lymphopenia, thymopoiesis, thymus, homeostatic peripheral expansion, GVHD, immunotherapy
1. Introduction
Reconstitution of T cell dependent immunity is a critical issue for patients treated with lymphodepleting regimens. Chemotherapy or transplant preparative regimens result in a severe and protracted lymphopenia. The recovery of T cell populations is delayed compared to that of myeloid, NK or B cells [1]. Furthermore, T cell function often remains compromised even after normal lymphocyte numbers have recovered [2-4]. This prolonged period of T cell dysfunction may have serious clinical consequences. It may limit response to vaccines, reduce resistance to infection, permit tumor relapse, and contribute to the development of autoimmunity [3-6]. Yet at the same time, recent work has suggested that the immediate period of lymphopenia post cytoreduction provides a unique opportunity for effective anti-tumor immunotherapy [7-10]. An understanding of the processes of immune reconstitution that underlie this apparent paradox is critical, not only to address the deficits in immune competence, but also to manipulate the process of reconstitution to enhance immune therapy.
T cell immune reconstitution is dependent upon the contributions of two primary pathways: generation of new T cells from progenitors via thymopoiesis, and peripheral expansion of residual mature lymphocytes by antigenic stimulation and homeostatic cytokines [11]. The process of reconstitution involves a dynamic balance between the two pathways. Peripheral expansion pathways affect immediate reconstitution, while thymic dependent pathways may not fully impact on reconstitution for 1 – 2 years. The cumulative contribution from each pathway may vary depending upon host age, residual T cell subsets, homeostatic cytokine levels and endogenous antigenic stimulation. The sequelae of the two pathways are also quite disparate, resulting in either a diverse T cell repertoire through active thymopoiesis or a skewed, oligoclonal, peripherally-derived population. In the years since the two developmental pathways were described, significant progress has been made toward understanding the cellular and cytokine regulators of both renewed thymopoiesis and peripheral expansion. This appreciation of the mechanisms of T cell recovery has, in turn, suggested new strategies to manipulate these processes to improve immune therapy.
2. Thymic Dependent T Cell Reconstitution: Assessment of thymic contribution
The fundamental route for the generation of T cells is thymopoiesis. When peripheral T cell populations are severely depleted, a renewal of thymic activity can contribute to the reconstitution of these peripheral T cell populations by generating naïve T cells de novo. CD4 helper, CD8 cytotoxic effector and CD4+CD25++ regulatory T cells all mature in the thymus. During thymopoiesis, bone marrow-derived T progenitors traverse to the thymus, become committed to the T lineage, and undergo proliferative expansion and maturation. During thymopoiesis, the T cell receptor (TCR) is generated through recombinant rearrangement of Variable (V), Diversity (D) and Joiner (J) genes, resulting in a broadly diverse repertoire of T cell receptors (TCR). Through the positive and negative selection process, the thymocytes are negatively selected for reactivity to autologous antigen prior to emigration to the periphery as naïve cells.
Naïve T cells are severely reduced during cytoreductive regimens, whether through activation or cell loss [1, 12]. With renewal of thymopoiesis, the proportion of cells with a “naïve” phenotype increases in peripheral T cell populations [12-14]. In murine models these naïve cells were recognized as CD44low CD45RB; in man, CD45RA expression was initially used to monitor naïve T cell populations [12, 13]. The extent of the thymopoietic contribution was unclear, however, because of concerns that phenotypically “naïve” cells could expand in the periphery or that previously activated cells could revert to naïve phenotpe. This ambiguity has been resolved by use of additional phenotypic markers (CCR7+CD62L+), and by evidence that reversion of memory or activated cells does not contribute to populations with a true naïve phenotype [15].
Thymic productivity has been further quantified by measurement of T cell receptor rearrangement excision circles (TREC) [16]. TREC are the episomal DNA circles generated during the rearrangement of the VDJ genes of the TCR α and β chains. These circles are stably retained during cell division but do not replicate, hence becoming diluted among the daughter cells. The signal joint (sj) TREC, formed during rearrangement of the TCRα chain, is readily measurable by PCR assays in circulating peripheral T cells [17] and their quantification has allowed insights into thymus activity in unperturbed steady state settings. TREC frequencies are severely reduced by lymphodepletion, but recover with renewed thymopoiesis [16]. Elevated frequencies of TREC-bearing cells are consistent with export of large numbers of recent thymic emigrants (RTE) into the peripheral blood. In contrast, in the absence of robust thymopoiesis, peripheral TREC frequencies remain low.
Renewed thymopoiesis is also measurable by TCR repertoire diversity, as assessed by spectratyping, a PCR-based analysis of length variation in the complementarity-determining region 3 (CDR3) of the TCR β chain. The CDR3 forms the contact site for the binding of peptides and plays a critical role in antigen recognition. The enormous diversity of CDR3 results from the random insertion of nucleotides during the process of VDJ rearrangement; this random process results in a Gaussian distribution of CDR3 lengths. Severe depletion of T cells and antigen-driven clonal expansions result in an oligoclonal pattern of limited CDR3 lengths. Renewal of thymopoiesis results in the re-establishment of polyclonal Gaussian patterns in the CDR3 spectratypes, first among naïve cells and subsequently among memory T cells [14, 18].
3. Thymic Dependent Immune Reconstitution: Determinants and Consequences
Although the thymus involutes with age and injury, the thymus has a remarkable capacity for renewal. With active thymopoiesis, young mice and children rapidly recover naïve cells after high dose chemotherapy. In contrast, in thymectomized mice or individuals with inadequate thymopoiesis, naïve cell reconstitution is impaired [13, 14, 16, 19, 20]. In as few as 2 weeks post transplant in mice, thymic size and cellularity increase and maturing thymocyte subsets reappear [21]. In humans with thymic renewal capacity, following chemotherapy, transplant conditioning or HAART therapy for AIDS, the thymus similarly expands, even ‘rebounds’ to greater than normal size, and becomes densely cellular when viewed by computerized tomography (CT) [12, 14, 22-24]. This change in thymic size and radiodensity may represent an increase in thymic productivity, as demonstrated by an increase in the ratio of sjTREC:Dβ-Jβ TREC [25]. This expansion precedes and predicts an increase in export of naïve T cells [14]. Furthermore, this reflects enhanced thymic activity with increased TREC frequency in the peripheral blood [14, 16, 26].
Because thymic activity is dependent upon age, the ability of the thymus to recover is also significantly influenced by host age. The thymus is most productive during the first 6 months of life, but remains active throughout childhood. With time, the thymus dramatically involutes. During the involutional process the densely cellular cortical and medullary tissues of the neonatal thymus are reduced and the proliferative expansion of early thymocytes that underlies thymopoietic productivity declines [25, 27]. In the elderly, although the involuted thymus continues to generate new T cells, renewing the diversity of the TCR repertoire, this occurs at a much lower rate [27]. Age both affects the rate of recovery and diminishes the final extent of thymic renewal (Figure 1). Aged mice, when irradiated and given marrow, do not exhibit the degree of thymic rebound observed in young hosts [28]. In young children, robust renewal of thymopoiesis is evident as early as 6 months after chemotherapy, but is slower in adolescents [12, 29]. In adults, each successive decade reduces the incidence of renewal of thymopoiesis, limits the level of naive cells produced during this renewal and delays the time course of export of naïve T cells into the peripheral blood [14, 20, 22] (Figure 1, 3). Over age 45, the frequency of significant renewal of thymopoiesis is severely reduced. TREC-bearing and phenotypically naïve CD4 T cells exported into the periphery approach normal levels in adults only after 2 years [14, 16]. In many older adults recovery of naïve cells takes 3 – 5 years; in some, naïve cells remain below normal levels even after decades [30, 31].
Figure 1.
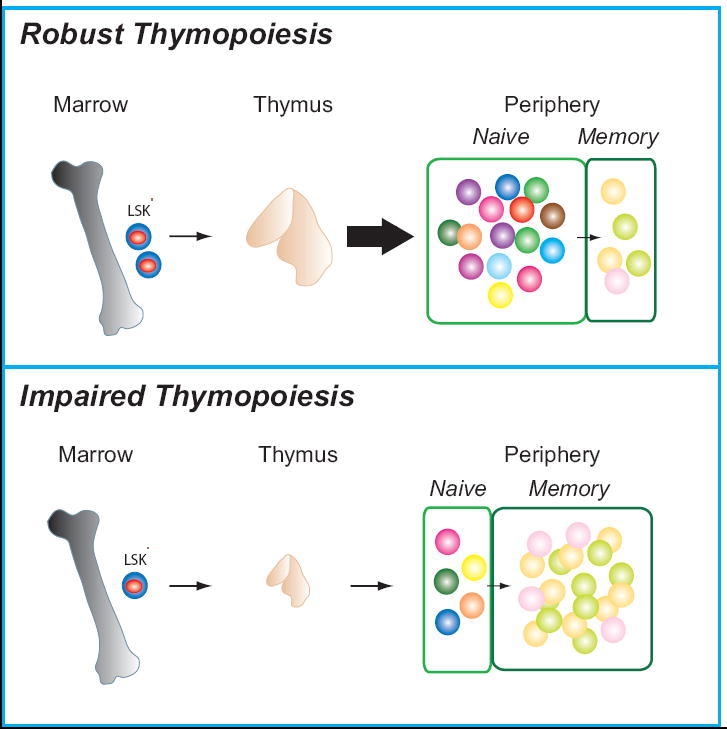
Diagrammatic representation of the consequences of robust versus impaired thymopoiesis. In young individuals without GVHD, thymic renewal rapidly ensues following transplant induced lymphopenia, with an increase in thymic size and a subsequent increase in thymic emigrants. The naïve pool is thus enlarged and enriched for newly derived thymic T cells and displays a diverse T cell repertoire, shown diagrammatically as multi-colored cells. In contrast, when thymopoiesis is impaired by age or GVHD, the thymus remains small and releases few new naïve T cells to the peripheral pool during immune reconstitution. The memory pool is expanded and there is less diversity within both the naïve and memory pool.
Figure 3.
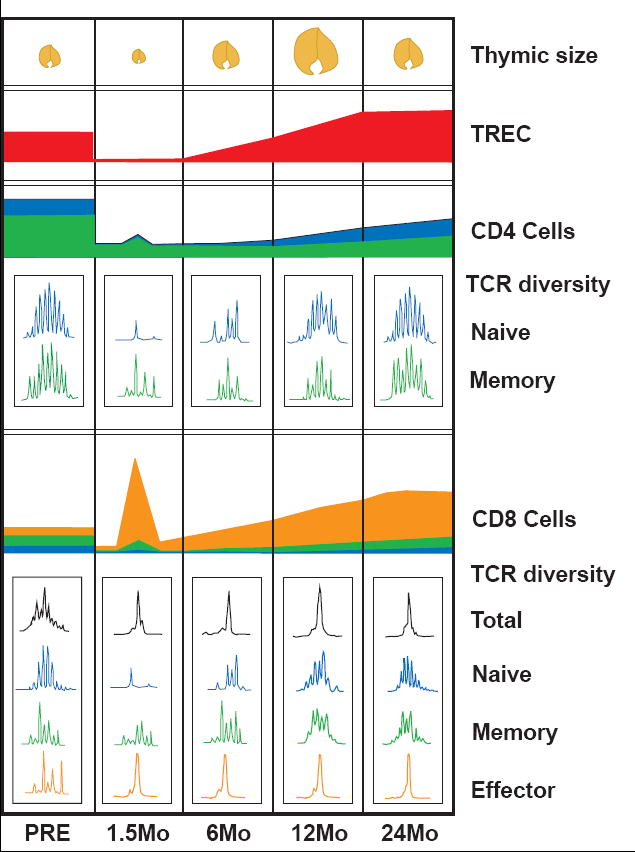
Diagrammatic representation of the timeline of immune reconstitution in a 40 year old CMV seropositive adult. Following cytoreductive therapy, the peripheral CD4 and CD8 T cell populations are severely depleted. The thymus is reduced to a small remnant (shown at 1.5 month post-transplant). CD4 and CD8 T cells immediately undergo a marked expansion in response to homeostatic cytokines and endogenous antigens, generating a population that is mainly composed of memory (green) and effector (orange) T cells, with few naïve (blue) or TREC-bearing cells (also represented at 1.5 month post-transplant). The CD4:CD8 ratio becomes inverted by the more rapid expansion of CD8+ T cells, demonstrated by the peak of (orange) effector CD8 cells as compared to the smaller (green) peak of CD4+ cells at 1.5 months. TCR repertoire diversity, that has been lost by lymphodepletion, is further skewed by oligoclonal expansion of the limited number of remaining cells. The expanded population of CD8 T cells persists and may continue to dominate the CD8 TCR repertoire as shown in subsequent months by the large effector (orange) CD8 population. Renewed thymopoiesis begins within the first 6 months, but the full contribution of naïve, TREC-bearing T cells with a diverse TCR repertoire may require 1 - 2 years to be evident.
While age has an immutable impact on thymic recovery, the contribution of thymopoiesis may also be altered by the availability of T progenitors, the extent of thymic damage from irradiation or chemotherapy agents, or the presence of post transplant factors that restrict thymopoiesis, such as immuno-suppressive drugs or GVHD (see the GVHD section below). Studies transplanting mixtures of congenic-labeled normal and recombinase activating gene knockout (RAG-/-)marrows (that cannot generate T cells) have demonstrated that thymic productivity is limited by the availability of functional marrow progenitors [32]. A decline in functionality in early thymocyte progenitors may underlie reduced thymic productivity in the aged [33, 34]. Conversely, increasing the availability of functional T progenitors may enhance thymopoiesis. Umbilical cord stem cells resulted in higher TREC frequencies than adult bone marrow stem cells [35], possibly due to a higher frequency of T progenitors in the younger marrows. When isolated lymphoid progenitors were infused into irradiated mice, the increased precursor doses enhanced thymocyte numbers and TREC [36]. Indeed when committed T progenitors were adoptively transplanted, donor-derived thymopoiesis was enhanced and donor T cells increased in the periphery [37].
In addition to a dependence upon functional T progenitors, thymic productivity is also influenced by cytokines, growth factors, and hormones. These impact on developing thymocytes or the thymic epithelial cells (TEC) that support them. Of the cytokines, the best characterized is Interleukin-7 (IL-7). IL-7 is an important survival factor for developing thymocytes that is produced constitutively by TEC. T cell maturation is severely reduced in IL-7-/- and IL-7Rα-/- mice [38], due to a IL-7 dependent block at the DN1-DN2 transition. IL-7 therapy in vivo and in vitro improved early DN2 survival, but did not enhance thymopoiesis or prevent age-associated involution [39, 40]. IL-7 administration in the post transplant period has been shown to significantly enhance donor-derived thymopoiesis [41-43], yet has had no impact on ongoing thymopoiesis in intact mice [44]. Since TEC production of IL-7 is reduced by radiation therapy [45], post-transplant IL-7 therapy may be effective because it restores depleted levels of the cytokine. In intact mice intrathymic IL-7 levels may have reached a cytokine plateau at which additional IL-7 produces no gain.
In contrast to IL-7, keratinocyte growth factor has shown promise to boost thymic productivity by expanding TEC both post transplant and in intact mice [46-48]. Whereas KGF was not required for initial thymic organogenesis and thymocyte maturation, KGF-/- mice were more susceptible to thymic damage on sublethal irradiation and KGF administration before transplant accelerated recovery of thymopoiesis [46]. In intact mice, KGF enhanced the level of thymopoiesis by expanding the TEC populations that supported T progenitor engraftment and thymocyte expansion [48].
Declines in the systemic levels of growth hormone and insulin-like growth factor - 1 (IGF-1) also contribute to reduction in thymopoietic productivity. Growth factor administration enhanced thymopoiesis in lymphopenic individuals [49-51]. In contrast to these agents that support the thymopoietic process, systemic androgen levels in adults reduce thymopoiesis through effects on the TEC [52]. Androgen withdrawal, by surgical or chemically induced blockade, has enhanced thymopoietic size and productivity in intact hosts and accelerated recovery of thymopoiesis after transplant therapy [34, 53, 54].
Renewal of thymopoiesis has profound consequences for recovery of immune function. The ability of the individual to recover normal CD4+ T cell number is dependent upon active thymopoiesis [14]. Without robust renewal of thymopoeisis, CD4+ T cell reconstitution is impaired indefinitely [12, 14, 16, 55]. Even 20 years after transplant, deficits in naïve CD4 numbers and total CD4 numbers persist in patients with inadequate renewal of thymopoiesis [31]. Additionally, severe lymphopenia depletes the diversity of the TCR repertoire; only renewal of thymopoiesis restores this diversity. Spectratyping demonstrates that TCR β-chain CDR3 diversity recovers in naïve populations only when thymopoiesis recovers [14, 56]. Only this recovery of a diverse collection of naïve T cells will lead to a diverse memory population with a normalization of oligoclonal and skewed TCR repertoires [14, 19, 29]. This thymic dependent T cell reconstitution has significant functional consequences. Without a diverse, thymic derived, naïve pool of T cells, vaccine responses and infectious disease clearance are impaired [20, 55, 56]. Furthermore, the recovery of repertoire diversity of naïve CD4+ cells can abate autoimmune disease [57].
4. T Cell Reconstitution by Homeostatic Proliferation
Although the thymic dependent pathway can provide stable, diverse T cell reconstitution, the contributions of this path to immune recovery are delayed by the period of thymic reconstruction. In contrast, mature T cells transferred into lymphopenic hosts immediately begin to proliferate and replenish the T cell compartment [13]. Adoptive transfer of syngeneic bone marrow (BM) cells and congenic lymph node cells to C57BL/6 irradiated, thymus-intact or thymectomized recipients resulted in sharply different patterns of T cell reconstitution. Thymus-bearing hosts largely reconstituted with syngeneic marrow-derived T cells, through a thymus-dependent mechanism. Thymectomized mice derived the majority of the peripheral T cells from the congenic lymph node innocula [13]. This process has been termed homeostatic peripheral expansion (HPE) [58] (Figure 2). HPE occurs in lymphopenic, not lympho-replete hosts, results in rapid significant expansion of the T cell pool, and is associated with a shift from naïve to memory/activated phenotype in the proliferating cells [58]. HPE does not depend upon renewal of thymopoiesis. Indeed, HPE makes a larger and more long lasting contribution to immune reconstitution in thymectomized hosts than in euthymic hosts [13]. HPE also is a distinct process from ‘homeostatic cycling’, whereby naïve and memory cells cycle in a lympho-replete host, without change in the cell phenotype or overall expansion in the T cell pool [58]. (See Figure 2).
Figure 2.
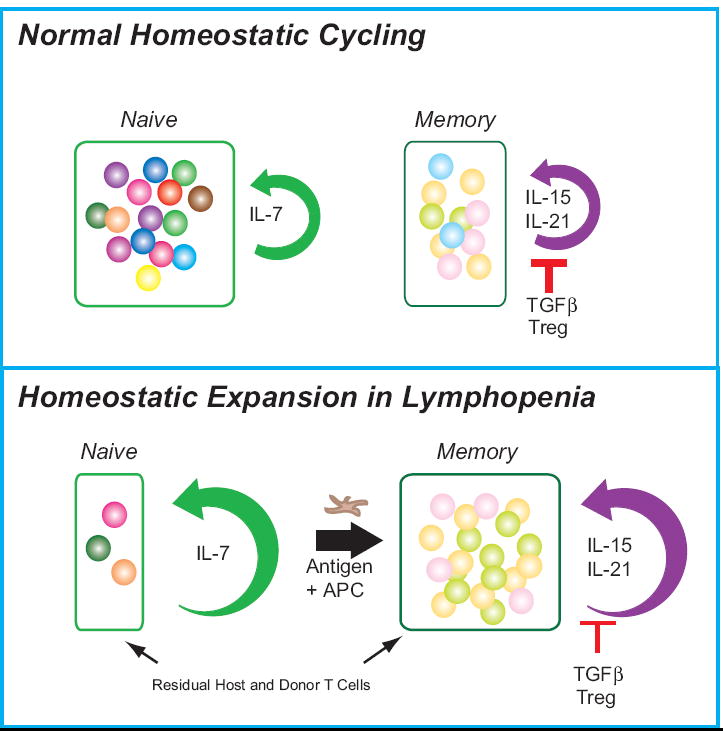
Diagrammatic representation of homeostatic cycling versus homeostatic peripheral expansion. In normal steady state homeostatic cycling, constitutive IL-7 production provides a survival signal to a diverse array of naïve T cells and maintains a low level of proliferation. Memory CD8+ compartment is similarly regulated by IL-15 production, IL-21 production, and the influence of TGFβ and Treg cells. The influence of these competing factors leads to a smaller and more diverse memory population. In contrast, following depletion of lymphocytes, residual mature T cells rapidly proliferate in response to homeostatic cytokines and antigenic stimulation by homeostatic peripheral expansion. Residual naive cells expand in response to the elevated levels IL-7. Antigen-driven stimulation results in a rapid activation of naïve cells and expansion of memory cells. Furthermore, elevated levels of IL-15, and possibly IL-21, contribute to an early increase in CD8+ memory T cells. Since there are low levels of Treg cells circulating, there is little regulation of this expansion by these cells. This results in a skewed naïve and memory T cell repertoires.
Following autologous or allogeneic transplantation in man, the residual T cells remaining in the host and any T cells contained in the stem cell graft begin to rapidly divide, consistent with HPE. The relative frequencies of the T subsets shift toward a preponderance of activated/memory T cells. Many of these T cells express HLA-DR [1]. All T subsets – naïve, memory and activated/effector – are in cycle [59]. The TREC frequency declines as TREC-bearing cells become diluted by proliferative expansion and lack of thymopoietic output of RTE [59]. T cells expand rapidly in the first weeks, but this expansion is often unstable, with longitudinal studies often noting a sharp decline in overall T cell numbers by 3 - 6 months [1]. Serial studies of TCR repertoire diversity have demonstrated that the early expansions are typically skewed and oligoclonal, with very limited repertoire diversity. These limited repertoires may persist unless supplemented either by an infusion of diverse repertoire mature cells via a donor lymphocyte infusion, or by the protracted process of renewal of thymopoiesis [14, 16, 19, 60, 61]. (See Figure 3).
HPE is dependent upon both the support of homeostatic cytokines and upon cognate antigen-driven interactions. In murine models, the influence of these two mechanisms is illustrated by studies of CFSE labeled cells administered into a lymphopenic host. CFSE is a fluorescent tracking molecule that binds to cell membranes; CFSE content is halved with each successive cell division. These CFSE dilution studies have permitted the discrimination of (relatively) slowly dividing cells, which seem to be dependent upon host cytokines, and rapidly dividing cells, that seem dependent on cognate antigenic activation of cells. Although these two mechanisms were defined independently, the process of HPE in lymphopenic hosts likely results from the interaction of both of these. Although cognate antigen-specific interactions may proceed more rapidly and skew the repertoire of T cell expansion, competition for limiting levels of homeostatic cytokines both fuels and determines the limits of HPE.
5. IL-7 tightly regulates the naïve T cell compartment in normal states and drives HPE following lymphopenia
IL-7 is a critical, non-redundant cytokine required for stimulating naïve T cell expansion during HPE and sustaining naïve T cell survival. Naïve CD4+ and CD8+ T cells undergo several rounds of CFSE dilution within a week after transfer into irradiated lymphopenic hosts. Naïve T cells adoptively transferred into IL-7-/- mice not only failed to undergo the HPE observed in wild type (WT) hosts, but also failed to survive [62, 63]. Administration of IL-7 led to a normalization of naïve T cell subsets [62, 63]. This IL-7 effect was maintained even in thymectomized hosts, implicating HPE not thymopoiesis in the early T cell recovery in lymphodepleted hosts [64]. In unmanipulated lympho-replete mice, moreover, exogenous IL-7 preferentially increased proliferation in peripheral naïve CD4+ and CD8+ T cells without altering thymopoiesis [44]. In nonhuman primates, IL-7 produced a rapid increase in the total number and in the percentage of cycling T cells in the peripheral blood, particularly within the naïve subset [65, 66]. This IL-7 treatment concurrently induced marked declines in the frequency of TREC-bearing cells in the peripheral blood, consistent with HPE [65]. Similarly, IL-7 administered to refractory melanoma patients produced a dose-dependent increase in circulating CD4+ and CD8+ T cell numbers [67]. Interleukin 7 treatment also influenced the trafficking of T cells, driving recent thymic emigrants from the spleen to the lymph nodes [44]. This trafficking effect may have contributed to the enhanced functional activity of T cells treated with IL-7 post transplant [64]. Finally, IL-7 provides a powerful anti-apoptotic signal to T cells, increasing expression of anti-apoptotic factors [68-71]. Thus, IL-7 profoundly influences the T cell compartment by enhancing proliferation, driving cells to lymph nodes, and providing an anti-apoptotic signal that enhances survival.
IL-7 is also a true regulator of the size of the naïve pool, driving proliferation of naïve cells in HPE and restricting expansion following T cell recovery Although the primary effect of IL7 is to enhance the size of the naïve pool, the presence of the receptor at the cellular level and the availability of this cytokine provide homeostatic control for naïve T cells. Resting naïve T cells express high levels of IL-7Rα. After activation or IL-7 signaling, proliferating cells downregulate the IL-7R [72], minimizing IL-7 responsiveness. Because IL-7 is produced constitutively at limiting levels, T cells ‘share’ the cytokine by downregulating the IL-7R after signaling. IL-7 acts through a consumptive mechanism as well. Thus, expansion of the naïve pool decreases cytokine access. Conversely, when lymphocyte numbers are depleted, availability of the cytokine increases. These elevated levels drive cells into proliferation until the numbers return to levels at which cytokine is once more a limiting factor. Human studies conclusively demonstrated this inverse correlation between serum levels of IL-7 and T cell reconstitution after lymphodepletion. High serum levels of IL-7 were initially observed in patients with severe lymphopenia from either chemical depletion or HIV infection, but these levels rapidly fell with lymphocyte recovery post-transplant [73, 74]. Thus, following lymphodepletion in mice and man, IL7 drives T cell recovery via peripheral expansion of resident naïve T cell clones, and then, with recovery and depletion of the cytokine, proliferation decreases to basal rates.
6. IL-15 and TGF beta Regulation of Memory CD8+ Cells Influences T cell Reconstitution after Lymphopenia
Unlike IL-7, IL-15 has little effect on naïve T cells, but is important in the expansion and survival of memory CD8+ T cells. Although memory CD8 T cells can be generated in IL-15-/- mice, they do not persist [75, 76]. Administration of IL-15 reversed this phenotype [75]. Bromodeoxyuridine (BRDU) analysis of IL15-/- and wildtype mice confirmed that IL-15 acts by inducing proliferation of CD8+ T cells [77]. Subsequent studies revealed that IL-15 provides a survival signal as well, upregulating Bcl-2 on activated CD8+ memory T cells [78]. Human work has substantiated a role for IL-15 in memory T cell homeostasis as well. IL-15 led to enhanced proliferation of CD8+ memory populations in vitro [79, 80]. Furthermore, IL-15 led to enhanced replicative potential in cell cultures, by maintaining telomere length through induction of telomerase [79]. Consistent with these data is the finding that CD122, a receptor for IL-15, is most highly expressed on CD44high CD8+ cells murine memory cells [63, 76]. Additionally, IL-15 is important for memory CD8+ functions. Diminished IL-15 leads to blunted memory CD8+ T cell response to bacterial and viral antigens. After varicella or Listeria inoculation, there were significantly fewer antigen specific T cells in the IL15-/- as compared to control mice [76, 78]. Thus, IL-15 supports the proliferation and survival of CD8+ memory T cells.
Recent evidence suggests that IL-15 as well as IL-7 may act as homeostatic cytokines, supporting HPE in lymphopenic hosts. Like IL-7, IL-15 is produced constitutively by many tissues although hematopoietic antigen presenting cells (APC) may be key producers; unlike IL-7, it is also subject to modulation by inflammation. When lymphocytes have been severely depleted, plasma IL-15 levels increase dramatically compared to normal levels [81, 82]. Concomitantly, in the early post transplant period, CD8 memory cells expand disproportionately, compared to CD4+ T cells [1, 83]. The predominant CD8+ subpopulations observed in the early expansion post transplant are the central memory (CM) (CD45RA-CCR7+), effector memory (EM) (CD45RA-CCR7-) and CD45RA+ effector (EMRA) (CD45RA+CCR7-) [84]. IL-15R expression and responsiveness is low in naïve CD8+ T cells and progressively increases in CM to EM to EMRA [85]. This differential expansion and receptor expression is consistent with a cytokine-driven HPE that is selective for CD8 T cells.
In addition to regulating CD8 homeostatic proliferation and enhancing antigenspecific CD8 responses, IL-15 also contributes to the expansion of a ‘bystander’ CD8 population. Administration of lipopolysaccharide (LPS) or polyinosinic: polycytidylic acid (poly I:C) to IL-15-/- or control hosts after adoptive transfer of WT CD8 memory T cells led to different profiles of T cell expansion. Both LPS and poly I:C stimulate T cells without engagement of the TCR. Proliferation of the WT T cells (enumerated by BRDU incorporation) was nearly absent in IL-15-/- mice in contrast to the rapidly dividing population in WT hosts despite the lack of TCR engagement [86]. Furthermore, the effect in control mice could be abrogated by the co-administration of anti-CD122 antibody or the adoptive transfer of CD122-/- memory T cells confirming that the bystander proliferative effect on CD8+ memory cells was due to IL-15 not simply the lack of innate immune cells in the IL-15-/- mouse [86].
While the greatest impact on memory T cell populations is mediated through IL-15, IL-21 may also contribute to the homeostatic proliferation of CD8+ T cells during immune reconstitution. Decreased CD 8+ vaccine responses were observed in an IL-21-/- mouse model as well [87]. IL-21 acted synergistically with IL-15 to increase murine CD8+ memory cell proliferation in vitro [87]. Increased CD8+ memory T cells were produced in response to IL-21 administration in vivo in a murine tumor model as well [87, 88]. Thus, IL-21 may also influence CD8+ memory expansion during HPE.
Since IL-15 is constitutively produced and could lead unchecked CD8+ memory expansion, it is balanced by factors negatively regulating CD8+ populations. Lucas showed that TGFß antagonizes the effect of IL-15 curtailing the proliferation and persistence of CD8 memory populations. When the activity of TGFß is abolished by a transgene encoding a dominant negative receptor (DNRII), a CD8 CD44high lymphoproliferative disorder and often leukemia ensues [89, 90]. This was shown to be a direct result of unchecked IL-15 influencing DNRII T cells. In vivo, crossing the DNRII host with an IL-15-/- ablated the CD8 expansion [91]. Administration of TGFβ to human and murine CD8+ memory cells in vitro decreases CD8+ proliferation and attenuates effector function [91, 92]. Thus, human in vitro and murine models of TGFß implicate this cytokine in the suppression of IL-15-driven memory CD8+ expansion.
7. Regulatory T Cells Constrain CD4+ and CD8+ T Cell Compartments, Influencing the Host-reactivity of T cells during Immune Reconstitution
In addition to the known homeostatic cytokine regulators of CD4 and CD8 T cells, peripheral expansion of both populations are influenced by regulatory T cells (Treg). Treg are CD127-CD25++ CD4+ T cells that function to control autoimmune responses [93, 94]. Characterized by the presence of the transcription factor FoxP3, Treg cells can mature in the thymus or can be generated in the periphery from CD25- CD4 cells [94]. Generation of Treg in the periphery is dependent upon the cytokine TGFβ, but expansion and survival of these cells is IL-2 dependent. Initial evidence for Treg regulation of CD8+ T cells, was indirectly shown by the significant increase in CD8 memory T cells after administration of IL-2 antibody [95]. This was corroborated by subsequent studies showing that IL-2-/- mice exhibited enhanced CD8 memory T cell proliferation as compared to wild type (WT) [96]. Even in the presence of Listeria antigen, depletion of Treg cells by anti-CD4+ antibody could diminish the directed CD8+ response [97]. Subsequent depletion of CD4+CD25+ cells led to a re-expansion of CD8 memory T cells, implicating Treg in the negative regulation of CD8 memory T cells [97]. Similarly, an uncontrolled expansion of CD4+ T cells in IL2-/- mice could be constrained by the administration of CD4+CD25+ Treg cells [98].
Studies have shown that Treg can modulate the peripheral expansion of T cells to low and high affinity antigen in lymphopenic hosts [99]. While regulatory T cells in mice and man display similar functional and phenotypic properties, the role of Treg in homeostatic expansion remains ill defined in man [100]. Treg levels appear to be low following transplant or chemotherapy but expand rapidly in the first month [101, 102]. As in mice, peripheral Treg levels in man are potentiated by IL-2 therapy,[102], but the time line for the generation of thymically derived Treg post transplant remains unknown.
8. Antigen-Driven Peripheral Expansion Independent of Cytokine Control
Antigen-induced T cell proliferation has long been identified as a key determinant in selective expansion of specific T cells; in 1996, Mackall et al. showed that peripheral expansion of TCR transgenic cells was exponentially increased when the cognate antigen was present [103]. Recently Min and Paul have explored a role for peripheral expansion that occurs independent of cytokines in lymphopenic conditions. Using a murine model, Min et al. demonstrated that adult congenic CD4+ T cells adoptively transferred into neonate recipients underwent a rapid expansion not observed in adult recipients [104]. The majority of these cells upregulated CD44 and adopted memory profiles of cytokine secretion [104]. The proportion of adoptively transferred cells undergoing proliferation was unaltered by the addition of anti-IL-7 or anti-IL-7Rα implying that it was an IL-7- independent expansion [104]. This pathway of T cell generation was also exacerbated by thymectomy of the neonatal mice, suggesting that this mechanism is influenced by the degree of lymphopenia [104]. In contrast to cytokine fueled expansions, MHC II interactions and CD28 ligation were shown to be critical to this process for naïve CD4 antigen-driven expansion [104]. This MHC II interaction in fact dictates the repertoire diversity of these antigen-expanded CD4 cells. Min et al. termed this endogenous proliferation to differentiate it from the homeostatic proliferation driven by cytokine signals [105]. In irradiated lymphopenic hosts, both processes could be demonstrated. Although the predominant contribution of peripheral expansion was homeostatic proliferation, driven by IL-7, there was a distinct population of rapidly proliferated cells unaffected by anti-IL-7 [105]. Finally, a similar mechanism was shown for a subset of CD8 T cells as well, implicating the antigen-driven endogenous proliferation in the development of memory CD4 and CD8 repertoires during immune reconstitution after lymphopenia [105].
Infectious antigens have been linked to the genesis of endogenous proliferation in the setting of lymphopenia. Gnotobiotic rearing of SCID hosts markedly reduced expansion of adoptively transferred cells compared to SCID hosts raised in usual specific pathogen free housing [106]. In human studies, the disproportionate expansion of CMV-reactive CD8+ T cells observed post transplant represents an example of antigen driven expansion [107-109]. CMV-reactive T cells, identified by binding tetramers of the dominant peptide of the CMV protein pp65, were found to expand following either autologous or allogeneic transplant [107-109]. The CMV reactive cells were CD45RA+, CCR7- CD27-, effector-memory/effector T cells [108], the phenotype of the terminally differentiated CD8+ T cells that dominate early post transplant expansions [83, 84]. These CMV-reactive populations constituted a significant percentage of the post transplant expansion, constituting more than 25% in some individuals which was disproportionately higher post-transplant than pre-transplant [107]. Furthermore, spectratyping demonstrated that the CMV-reactive population constituted an oligoclonal expansion of T cells [109]. Thus, endogenous proliferation likely contributes to the composition of the T cell repertoire generated following lymphopenia in mice and man. Homestatic proliferation permits the expansion of the T cell compartment as a whole; endogenous proliferation directs T cells to proliferate in response to specific antigen stimuli early in T cell recovery.
9. Peripheral Expansion after Lymphopenia Enhances Vaccine and Tumor Specific T cell Responses
The rapid cytokine-fueled, antigen-driven expansion of T cells transferred into a lymphopenic host lends this time frame to vaccine introduction or adoptive transfer of T cells to promote anti-tumor immunity. Several recent studies demonstrate that severe lymphodepletion can create optimal conditions to promote graft versus tumor responses. T cells undergoing peripheral expansion in sublethally irradiated lymphopenic mice developed CD8+ tumor-specific effectors against established tumors [8]. Indeed, the expansion of tumor-specific IFNγ producing T cells was significantly higher in the lymph nodes draining a tumor vaccination site in sublethally irradiated mice reconstituted with naïve T cells than in intact mice [110]. Two main mechanisms support targeted expansion of anti-tumor effectors after lymphodepletion: elevated availability of homeostatic cytokines and depletion of regulatory T cells that impede immune responses. IL-7 and IL-15 levels in the plasma are at the highest at the time of lymphodepletion, when the presence of lymphocytes competing for cytokines is the most reduced. Adoptive transfer of transgenic melanoma-antigen specific T cells blocked tumor growth better in irradiated hosts than in intact hosts [9]. The superior CD8 antitumor response after immune depletion was further enhanced by increasing the cytoreductive radiation dose in a syngeneic stem cell transplant model [10, 111]. This CD8+-mediated anti-tumor immunity was shown to be dependent on the presence of IL-7 and IL-15 using cytokine knockout mice, implicating these normal homeostasis mechanisms in the generation of anti-tumor responses by adoptive T cell transfer [9]. In addition to the contribution of HPE cytokines, lymphopenia also enhances directed anti-tumor immunity through depletion of T regulatory cells as a result of cytoreductive treatment [112]. Specific deletion of CD25+ CD4 Treg cells from a conventional CD25- CD4 T cell product improved the tumorcidal effect [112].
Maximal anti-tumor effects may be elicited with a combination of these effects: severe lymphopenia through syngeneic rescue, directed T cell responses through vaccines, and impaired Treg function. This was demonstrated in a B16F1 murine melanoma model using intact mice with a synergistic combination of tumor peptide vaccine with oligodeoxynucleotides (CPG ODN) and IL-2 [113]. Thymectomized mice that underwent lethal irradiation with syngeneic marrow rescue, followed by the same regimen of vaccine and adjuvant therapy were similarly capable of inhibiting melanoma growth and developing epitope-specific immunity in the post transplant period. Although IL-2 should diminish the CD8+ effector response through Treg expansion, the administration of CPG ODN has been shown to block Treg effects on conventional T cells [114]. This study thus highlights that vaccine strategies targeting multiple cell populations may best generate an antitumor response by enhancing tumor directed memory CD8+ effector populations through expansion during lymphopenia and diminishing the restraints of T regulatory cells.
The elevated levels of homeostatic cytokines may contribute to the persistence of the tumorcidal T cells as well as to their initial expansion. Exogenous administration of homeostatic cytokines has also been shown to strengthen the response and improve the lifespan of CD8+ tumor-specific immunity. IL-15 administration was shown to augment vaccine responses using a vaccine vector containing human IL-15. Mice treated with this vaccine elicited enhanced antigen-specific cytotoxic T lymphocytes generated against HIV and these persisted for up to 14 months [115]. Administration of IL-21 further improved tumor immunotherapy in murine vaccine models through the activation and prevention of cell death in memory CD8+ cells [87, 88, 116]. The majority of these IL-21 treated cells persisted for greater than 30 days after IL-21 therapy, far longer than IL-2 induced anti-tumor CD8+ T cells, further supporting the role for IL-21 in long-lived memory populations [88]. These studies have suggested that the elevated circulating cytokine milieu following lymphodepletion could benefit graft-versus-tumor reactions.
T cell immunotherapy studies in man have similarly demonstrated enhanced efficacy in the setting of lymphodepletion. Adoptive T cell transfers have had the greatest immunotherapy success in the setting of the highly immunogenic melanoma. Administration of ex vivo expanded melanoma-infiltrating lymphocytes along with high dose IL-2 following cytoreductive therapy has provided effective therapy against established tumors [7, 117, 118]. Over 50% of heavily pretreated patients with metastatic melanoma experienced significant tumor regression [117]. Tumor-specific clonotypes were found in responders at 1-2 months post-infusion, suggesting that oligoclonal expansion of tumor reactive T cells played a significant role in successful tumor clearance [118]. Furthermore, there was a significant correlation between tumor regression and the persistence of these clonotypes [118]. These studies have been limited by the ability to generate in vivo or ex vivo autologous T cells specific for melanoma that maintain proliferative and cytolytic potential. Loss of telomere length upon ex vivo expansion has limited the persistence of infused cells [119]. Use of IL-2 to support early expansion of the infused cells in vivo also has resulted in increasing regulatory T cells [120]. Finally, attack on normal melanocytes, resulting in vitilago or uveitis, has also been observed in patients with effective anti-melanoma responses [7]. Recent cutting edge work by Morgan et al. has demonstrated that T cells may be genetically engineered in vitro to target melanoma and that these cells can induce clinical responses after lymphodepletion [121].
Thus, these findings suggest that the early post-transplant or post-chemotherapy lymphopenic period is an optimal time to introduce T cell directed anti-tumor therapies. Furthermore, these studies highlight several critical components to the successful generation of antitumor immunity in the lymphopenic host. The timing of vaccines in the post-chemotherapy or post-autologous BMT host will be paramount to capitalize upon the proliferative cytokine milieu of elevated IL-7 and IL-15. Furthermore, it may be beneficial to increase these cytokine levels at given points during immune therapy when normal homeostatic mechanisms would normally lead to a depletion of these cytokines critical for persistence of expanded populations. For instance, future studies may administer exogenous IL-7 in the months following immune reconstitution to maximize naïve T cell expansion, or administer IL-15 to enhance T cell memory persistence at later time points. Finally, although immune responses may be elicited from a polyclonal T cell population, this may be hindered in older patients with a limited T cell repertoire or mechanisms inherent to tumors that effectively evade immune system recognition. Use of genetically engineered T cells to target tumor may address this. Thus, the novel approach of generating in vitro tumor specific T cells for infusion in the early after lymphopenia may push the horizon for T cell enhanced autologous tumor eradication in previously resistant malignancies.
10. Homeostatic Proliferation and Thymic Insufficiency Contribute to GVHD
The mechanisms that may enhance tumor immunity in autologous transplant can contribute to the development of acute and chronic graft versus host disease (GVHD) in the context of allogeneic transplant. Graft versus host disease has been linked to the development of donor anti-host T cells. Since the early post-transplant milieu favors proliferation of T cells within the graft rather than de novo T cell generation through thymopoiesis, alloimmunity can be exacerbated in the acute post-transplant setting. Furthermore, there is evidence that the initial expansions of CD8+ effector T cells are antigen-driven. Early experiments demonstrated antigen-driven expansion of mature T cells adoptively transferred into a thymectomized Ld+ host using host Ld-reactive transgenic T cells. The transferred T cells were potent donor anti-host effectors [103]. This was further substantiated using murine models in which the host expressed Mls antigens disparate from the donor. These endogenous viral epitopes act as superantigens, stimulating entire Vβ families of the TCR repertoire, the expansion of which can be monitored. Following infusion of donor cells, the stimulated Vβ families expanded disproportionately and rapidly dominated the total TCR repertoire [122-124]. Depletion of these Vβ families from the donor inoculum prior to infusion prevented or significantly reduced the morbidity of the GVHD [123, 124]. Adoptive transfer of T cells from these Vβ families from GVHD mice into sublethally irradiated secondary hosts led to GVHD in recipients, further implicating these peripheral T cell clones in the alloimmunity [124]. Much like the cells expanding in the periphery during autologous immune reconstitution, donor anti-host reactive T cells in murine GVHD models have a memory/activated effector phenotype, a limited TCR repertoire, expand quickly but then rapidly decline [122, 125].
Factors that support antigen-driven peripheral expansion can exacerbate GVHD. Enhancing naïve T cell proliferation through prolonged IL-7 administration aggravated GVHD in T cell replete transplants and decreased the T cell dose required to induce GVHD [126]. When shorter courses and lower doses of IL-7 were given, however, GVHD severity was unaffected [127, 128]. The key difference in these studies may have been the protracted maintenance of high IL-7 levels by exogenous administration after the period when consumption by proliferating cells would have depleted the cytokine to normal levels. Similarly, IL-15, the cytokine that expands and activates memory CD8+ cells, has been shown to play a critical role in the genesis of acute GVHD. Allogeneic transplants from IL-15-/- donors markedly reduced GVHD severity, whereas transplants from transgenic mice with elevated IL-15 production resulted in a more severe GVHD [129]. Similarly, exogenous IL-15 exacerbated GVHD in MHC-disparate and xenogeneic T cell-replete transplant models by increasing peripheral expansion of CD8 cells post transplant [130, 131]. In human studies, persistence of high IL-15 levels following allogeneic stem cell transplant was correlated with severe acute GVHD [82, 132]. The bystander effect of CD8+ proliferation has also led to GVHD flares in the setting of viral infections in murine models as well [133, 134].
In contrast, factors that reduce peripheral expansion can reduce GVHD. Regulatory T cells could control homeostatic expansion in lymphopenic mice [99]. Co-administration of CD25+CD4+ T regulatory cells along with the T replete donor inoculum prevented acute GHVD in murine models [135-137]. Similarly in clinical trials, the level of FoxP3+ T regulatory cells in the donor inoculum was inversely correlated with development of acute GVHD [101].
Rapid renewal of robust thymopoietic activity can offset the contribution of HPE post-transplant, minimizing the risk of GVHD. In part, this is because newly generated naïve cells may compete for homeostatic cytokines. This was first shown in the early studies of GVHD where transgenic T cells transferred into a mismatched host peripherally expanded maximally in thymectomzied hosts; thymus-intact mice had reduced HPE [13]. Additionally, newly generated thymic derived T cells have undergone negative selection, eradicating destructive host-reactive cells. In GVHD, however, Class II MHC expression in the thymus is reduced and negative selection of auto-reactive cells is impaired [138, 139].
Impaired thymic function is both caused by, and the result of, GVHD. The expansion of autoreactive clones target the thymus for graft vs. host attack, precluding thymic renewal [124, 140, 141]. Donor T cells infiltrate the host thymus and attack Class II MHC expressing TEC cells critical to thymic function [141]. Subsequent regeneration of normal thymic structural elements such as Hassall’s corpuscles is very slow [140]. Not only would this delay naïve T cell reconstiution, it may also impact on the generation of Treg cells that would control alloimmunity. Thymic maturation of CD4+CD25+ regulatory T cells is dependent upon interactions of these cells with dendritic cells in Hassall’s corpuscles [142]. Since MHC class II expressing host cells are a primary target of donor anti-host attack and since Hassall’s corpuscles are among the last structures to recover, [140, 141] GVHD may also reduce generation of regulatory T cells.
Overall thymic productivity is therefore reduced after GVHD. This finding from murine models has been substantiated in clinical studies; patients with chronic GVHD have reduced levels of naïve and TREC-bearing T cells compared to patients without GVHD or to those whose initial acute GVHD had resolved [20, 26, 143, 144]. Chronic GVHD has been associated with persistence of low TREC levels as well [20, 143, 145]. In the pediatric population, GVHD with low TREC levels and thus poor thymic function was associated with mortality [145]. Thus, in humans, TREC assays document the association between thymic dysfunction and persistent alloimmunity.
11. Conclusion and Trends for the Future: Manipulation of Immune Reconstitution to Enhance Anti-Tumor Effectors and Limit Immune Dysfunction
T cell recovery following lymphopenia is delayed and incompetent compared to other immune cells. This has profound consequences for the individual in terms of anti-tumor and overall immunity. Initial peripheral T cell proliferation, due to the prolific cytokine milieu for HPE and antigen-driven signals, leads to recovery of an oligoclonal, restricted repertoire of T cells. While this predisposes to alloimmunity and impairs clearance of infectious agents, it also permits a maximal expansion of anti-tumor clones, providing significant anti-tumor immunity. In patients with thymic renewal, de novo T cell production decreases peripheral expansion, restores repertoire diversity, and provides new receptors to recognize infectious diseases and tumor antigens. In the setting of allogeneic transplant, this has particular importance as thymic recovery may abate GVHD and restore functional T cell immunity.
Studies from mice and men have revealed regulators of thymopoiesis and peripheral expansion. In mice, recovery of thymopoiesis has been enhanced by infusion of committed T progenitors, administration of KGF, growth hormone, and withdrawal of androgens. In man, designing transplant regimens to minimize the toxicity to the thymus and maximize thymic recovery may permit the greatest possible graft vs. tumor effect while minimizing the risk of GVHD. Peripheral expansion is enhanced by cytokines and antigen-driven responses in the period following lymphopenia. Thus, in autologous transplant or chemotherapy settings, lymphopenia may provide the best opportunity for directing the immune system to target and delete residual disease. Vaccine and adoptive transfer of T cells may provide the most durable response if given in the immediate post-transplant period when cytokine levels are the highest and the antigen-driven response is maximal. Further administration of exogenous cytokines after the cytokine milieu has returned to normal levels may improve the persistence of anti-tumor clones. Therefore, an improved understanding of the cytokine and cellular influences that contribute to the genesis and persistence of aberrant T cell responses may have important implications for treating autoimmunity, GVHD, and malignant disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hakim FT, et al. Constraints on CD4 recovery postchemotherapy in adults: thymic insufficiency and apoptotic decline of expanded peripheral CD4 cells. Blood. 1997;90:3789–98. [PubMed] [Google Scholar]
- 2.Mackall CL, et al. Prolonged CD4 depletion after sequential autologous peripheral blood progenitor cell infusions in children and young adults. Blood. 2000;96:754–62. [PubMed] [Google Scholar]
- 3.Nordoy T, et al. Humoral immunity to viral and bacterial antigens in lymphoma patients 4-10 years after high-dose therapy with ABMT. Serological responses to revaccinations according to EBMT guidelines. Bone Marrow Transplant. 2001;28:681–7. doi: 10.1038/sj.bmt.1703228. [DOI] [PubMed] [Google Scholar]
- 4.Small TN, et al. Comparison of immune reconstitution after unrelated and related T-cell-depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood. 1999;93:467–80. [PubMed] [Google Scholar]
- 5.King C, et al. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–77. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 6.Parkman R, et al. Successful immune reconstitution decreases leukemic relapse and improves survival in recipients of unrelated cord blood transplantation. Biol Blood Marrow Transplant. 2006;12:919–27. doi: 10.1016/j.bbmt.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dummer W, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–92. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wrzesinski C, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest. 2007;117:492–501. doi: 10.1172/JCI30414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackall CL, Gress RE. Pathways of T-cell regeneration in mice and humans: implications for bone marrow transplantation and immunotherapy. Immunol Rev. 1997;157:61–72. doi: 10.1111/j.1600-065x.1997.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 12.Mackall CL, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med. 1995;332:143–9. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 13.Mackall CL, et al. T-cell regeneration after bone marrow transplantation: differential CD45 isoform expression on thymic-derived versus thymic-independent progeny. Blood. 1993;82:2585–94. [PubMed] [Google Scholar]
- 14.Hakim FT, et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005;115:930–9. doi: 10.1172/JCI22492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ge Q, et al. Different contributions of thymopoiesis and homeostasis-driven proliferation to the reconstitution of naive and memory T cell compartments. Proc Natl Acad Sci U S A. 2002;99:2989–94. doi: 10.1073/pnas.052714099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douek DC, et al. Assessment of thymic output in adults after haematopoietic stem-cell transplantation and prediction of T-cell reconstitution. Lancet. 2000;355:1875–81. doi: 10.1016/S0140-6736(00)02293-5. [DOI] [PubMed] [Google Scholar]
- 17.Douek DC, et al. Changes in thymic function with age and during the treatment of HIV infection. Nature. 1998;396:690–5. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 18.Wu CJ, et al. Reconstitution of T-cell receptor repertoire diversity following T-cell depleted allogeneic bone marrow transplantation is related to hematopoietic chimerism. Blood. 2000;95:352–9. [PubMed] [Google Scholar]
- 19.Klein AK, et al. T-Cell recovery in adults and children following umbilical cord blood transplantation. Biol Blood Marrow Transplant. 2001;7:454–66. doi: 10.1016/s1083-8791(01)80013-6. [DOI] [PubMed] [Google Scholar]
- 20.Lewin SR, et al. Direct evidence for new T-cell generation by patients after either T-cell-depleted or unmodified allogeneic hematopoietic stem cell transplantations. Blood. 2002;100:2235–42. [PubMed] [Google Scholar]
- 21.Fredrickson GG, Basch RS. Early thymic regeneration after irradiation. Dev Comp Immunol. 1994;18:251–63. doi: 10.1016/0145-305x(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 22.Sfikakis PP, et al. Age-related thymic activity in adults following chemotherapy-induced lymphopenia. Eur J Clin Invest. 2005;35:380–7. doi: 10.1111/j.1365-2362.2005.01499.x. [DOI] [PubMed] [Google Scholar]
- 23.Smith KY, et al. Thymic size and lymphocyte restoration in patients with human immunodeficiency virus infection after 48 weeks of zidovudine, lamivudine, and ritonavir therapy. J Infect Dis. 2000;181:141–7. doi: 10.1086/315169. [DOI] [PubMed] [Google Scholar]
- 24.Teixeira L, et al. Poor CD4 T cell restoration after suppression of HIV-1 replication may reflect lower thymic function. Aids. 2001;15:1749–56. doi: 10.1097/00002030-200109280-00002. [DOI] [PubMed] [Google Scholar]
- 25.Dion ML, et al. HIV infection rapidly induces and maintains a substantial suppression of thymocyte proliferation. Immunity. 2004;21:757–68. doi: 10.1016/j.immuni.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Fallen PR, et al. Factors affecting reconstitution of the T cell compartment in allogeneic haematopoietic cell transplant recipients. Bone Marrow Transplant. 2003;32:1001–14. doi: 10.1038/sj.bmt.1704235. [DOI] [PubMed] [Google Scholar]
- 27.Haynes BF, et al. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Annu Rev Immunol. 2000;18:529–60. doi: 10.1146/annurev.immunol.18.1.529. [DOI] [PubMed] [Google Scholar]
- 28.Mackall CL, et al. Thymic function in young/old chimeras: substantial thymic T cell regenerative capacity despite irreversible age-associated thymic involution. Eur J Immunol. 1998;28:1886–93. doi: 10.1002/(SICI)1521-4141(199806)28:06<1886::AID-IMMU1886>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 29.Sarzotti M, et al. T cell repertoire development in humans with SCID after nonablative allogeneic marrow transplantation. J Immunol. 2003;170:2711–8. doi: 10.4049/jimmunol.170.5.2711. [DOI] [PubMed] [Google Scholar]
- 30.Nordoy T, et al. Persistent changes in the immune system 4-10 years after ABMT. Bone Marrow Transplant. 1999;24:873–8. doi: 10.1038/sj.bmt.1702006. [DOI] [PubMed] [Google Scholar]
- 31.Storek J, et al. Immunity of patients surviving 20 to 30 years after allogeneic or syngeneic bone marrow transplantation. Blood. 2001;98:3505–12. doi: 10.1182/blood.v98.13.3505. [DOI] [PubMed] [Google Scholar]
- 32.Almeida AR, Borghans JA, Freitas AA. T cell homeostasis: thymus regeneration and peripheral T cell restoration in mice with a reduced fraction of competent precursors. J Exp Med. 2001;194:591–9. doi: 10.1084/jem.194.5.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min H, Montecino-Rodriguez E, Dorshkind K. Reduction in the developmental potential of intrathymic T cell progenitors with age. Journal of Immunology. 2004;173:245–250. doi: 10.4049/jimmunol.173.1.245. [DOI] [PubMed] [Google Scholar]
- 34.Heng TS, et al. Effects of castration on thymocyte development in two different models of thymic involution. J Immunol. 2005;175:2982–93. doi: 10.4049/jimmunol.175.5.2982. [DOI] [PubMed] [Google Scholar]
- 35.Talvensarri K, et al. A broad T-cell repertoire diversity and an efficient thymic function indicate a favorable long-term immune reconstitution after cord blood stem cell transplantation. Blood. 2002;99:1458–1464. doi: 10.1182/blood.v99.4.1458. [DOI] [PubMed] [Google Scholar]
- 36.Chen BJ, et al. Hematopoietic stem cell dose correlates with the speed of immune reconstitution after stem cell transplantation. Blood. 2004;103:4344–52. doi: 10.1182/blood-2003-07-2534. [DOI] [PubMed] [Google Scholar]
- 37.Zakrzewski JL, et al. Adoptive transfer of T-cell precursors enhances T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Nat Med. 2006;12:1039–47. doi: 10.1038/nm1463. [DOI] [PubMed] [Google Scholar]
- 38.Peschon JJ, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–60. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrew D, Aspinall R. Age-associated thymic atrophy is linked to a decline in IL-7 production. Experimental Gerontology. 2002;37:455–463. doi: 10.1016/s0531-5565(01)00213-3. [DOI] [PubMed] [Google Scholar]
- 40.Phillips JA, et al. IL-7 gene therapy in aging restores early thymopoiesis without reversing involution. Journal of Immunology. 2004;173:4867–4874. doi: 10.4049/jimmunol.173.8.4867. [DOI] [PubMed] [Google Scholar]
- 41.Bolotin E, et al. Enhancement of thymopoiesis after bone marrow transplant by in vivo interleukin-7. Blood. 1996;88:1887–94. [PubMed] [Google Scholar]
- 42.Mackall CL, et al. IL-7 increases both thymic-dependent and thymic-independent T-cell regeneration after bone marrow transplantation. Blood. 2001;97:1491–7. doi: 10.1182/blood.v97.5.1491. [DOI] [PubMed] [Google Scholar]
- 43.Li A, et al. Co-transplantation of bone marrow stromal cells transduced with IL-7 gene enhances immune reconstitution after allogeneic bone marrow transplantation in mice. Gene Ther. 2006;13:1178–87. doi: 10.1038/sj.gt.3302741. [DOI] [PubMed] [Google Scholar]
- 44.Chu YW, et al. Exogenous IL-7 increases recent thymic emigrants in peripheral lymphoid tissue without enhanced thymic function. Blood. 2004;104:1110–9. doi: 10.1182/blood-2003-10-3635. [DOI] [PubMed] [Google Scholar]
- 45.Adachi Y, et al. Semiquantitative detection of cytokine messages in X-irradiated and regenerating rat thymus. Radiat Res. 2005;163:400–7. doi: 10.1667/rr3331. [DOI] [PubMed] [Google Scholar]
- 46.Alpdogan O, et al. Keratinocyte growth factor (KGF) is required for postnatal thymic regeneration. Blood. 2006;107:2453–60. doi: 10.1182/blood-2005-07-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi S, et al. Keratinocyte growth factor preserves normal thymopoiesis and thymic microenvironment during experimental graft-versus-host disease. Blood. 2002;100:682–91. doi: 10.1182/blood.v100.2.682. [DOI] [PubMed] [Google Scholar]
- 48.Rossi SW, et al. Keratinocyte growth factor (KGF) enhances postnatal T-cell development via enhancements in proliferation and function of thymic epithelial cells. Blood. 2007;109 doi: 10.1182/blood-2006-10-049767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Napolitano LA, et al. Increased thymic mass and circulating naive CD4 T cells in HIV-1-infected adults treated with growth hormone. Aids. 2002;16:1103–11. doi: 10.1097/00002030-200205240-00003. [DOI] [PubMed] [Google Scholar]
- 50.Savino W, et al. The thymus gland: a target organ for growth hormone. Scand J Immunol. 2002;55:442–52. doi: 10.1046/j.1365-3083.2002.01077.x. [DOI] [PubMed] [Google Scholar]
- 51.Polgreen L, et al. Thymic hyperplasia in a child treated with growth hormone. Growth Horm IGF Res. 2006 doi: 10.1016/j.ghir.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 52.Olsen NJ, et al. Androgen receptors in thymic epithelium modulate thymus size and thymocyte development. Endocrinology. 2001;142:1278–83. doi: 10.1210/endo.142.3.8032. [DOI] [PubMed] [Google Scholar]
- 53.Sutherland JS, et al. Activation of thymic regeneration in mice and humans following androgen blockade. J Immunol. 2005;175:2741–53. doi: 10.4049/jimmunol.175.4.2741. [DOI] [PubMed] [Google Scholar]
- 54.Goldberg GL, et al. Sex steroid ablation enhances lymphoid recovery following autologous hematopoietic stem cell transplantation. Transplantation. 2005;80:1604–13. doi: 10.1097/01.tp.0000183962.64777.da. [DOI] [PubMed] [Google Scholar]
- 55.Roux E, et al. Recovery of immune reactivity after T-cell-depleted bone marrow transplantation depends on thymic activity. Blood. 2000;96:2299–303. [PubMed] [Google Scholar]
- 56.Dumont-Girard F, et al. Reconstitution of the T-cell compartment after bone marrow transplantation: restoration of the repertoire by thymic emigrants. Blood. 1998;92:4464–71. [PubMed] [Google Scholar]
- 57.Muraro PA, Douek DC. Renewing the T cell repertoire to arrest autoimmune aggression. Trends Immunol. 2006;27:61–7. doi: 10.1016/j.it.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 58.Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174:6571–6. doi: 10.4049/jimmunol.174.11.6571. [DOI] [PubMed] [Google Scholar]
- 59.Hazenberg MD, et al. T-cell receptor excision circle and T-cell dynamics after allogeneic stem cell transplantation are related to clinical events. Blood. 2002;99:3449–53. doi: 10.1182/blood.v99.9.3449. [DOI] [PubMed] [Google Scholar]
- 60.Bellucci R, et al. Immunologic effects of prophylactic donor lymphocyte infusion after allogeneic marrow transplantation for multiple myeloma. Blood. 2002;99:4610–7. doi: 10.1182/blood.v99.12.4610. [DOI] [PubMed] [Google Scholar]
- 61.Hochberg EP, et al. Quantitation of T-cell neogenesis in vivo after allogeneic bone marrow transplantation in adults. Blood. 2001;98:1116–21. doi: 10.1182/blood.v98.4.1116. [DOI] [PubMed] [Google Scholar]
- 62.Schluns KS, et al. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–32. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 63.Tan JT, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A. 2001;98:8732–7. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fry TJ, et al. Interleukin-7 restores immunity in athymic T-cell-depleted hosts. Blood. 2001;97:1525–33. doi: 10.1182/blood.v97.6.1525. [DOI] [PubMed] [Google Scholar]
- 65.Fry TJ, et al. IL-7 therapy dramatically alters peripheral T-cell homeostasis in normal and SIV-infected nonhuman primates. Blood. 2003;101:2294–9. doi: 10.1182/blood-2002-07-2297. [DOI] [PubMed] [Google Scholar]
- 66.Moniuszko M, et al. Recombinant interleukin-7 induces proliferation of naive macaque CD4+ and CD8+ T cells in vivo. J Virol. 2004;78:9740–9. doi: 10.1128/JVI.78.18.9740-9749.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg SA, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother. 2006;29:313–9. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li WQ, et al. Interleukin-7 inactivates the pro-apoptotic protein Bad promoting T cell survival. J Biol Chem. 2004;279:29160–6. doi: 10.1074/jbc.M401656200. [DOI] [PubMed] [Google Scholar]
- 69.Schober SL, et al. Expression of the transcription factor lung Kruppel-like factor is regulated by cytokines and correlates with survival of memory T cells in vitro and in vivo. J Immunol. 1999;163:3662–7. [PubMed] [Google Scholar]
- 70.Sasson SC, et al. Increased plasma interleukin-7 level correlates with decreased CD127 and Increased CD132 extracellular expression on T cell subsets in patients with HIV-1 infection. J Infect Dis. 2006;193:505–14. doi: 10.1086/499309. [DOI] [PubMed] [Google Scholar]
- 71.Vassena L, et al. Interleukin 7 reduces the levels of spontaneous apoptosis in CD4+ and CD8+ T cells from HIV-1-infected individuals. Proc Natl Acad Sci U S A. 2007;104:2355–60. doi: 10.1073/pnas.0610775104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park JH, et al. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21:289–302. doi: 10.1016/j.immuni.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 73.Bolotin E, et al. Serum levels of IL-7 in bone marrow transplant recipients: relationship to clinical characteristics and lymphocyte count. Bone Marrow Transplant. 1999;23:783–8. doi: 10.1038/sj.bmt.1701655. [DOI] [PubMed] [Google Scholar]
- 74.Fry TJ, et al. A potential role for interleukin-7 in T-cell homeostasis. Blood. 2001;97:2983–90. doi: 10.1182/blood.v97.10.2983. [DOI] [PubMed] [Google Scholar]
- 75.Kennedy MK, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191:771–80. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schluns KS, et al. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol. 2002;168:4827–31. doi: 10.4049/jimmunol.168.10.4827. [DOI] [PubMed] [Google Scholar]
- 77.Lodolce JP, et al. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–76. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- 78.Yajima T, et al. IL-15 regulates CD8+ T cell contraction during primary infection. J Immunol. 2006;176:507–15. doi: 10.4049/jimmunol.176.1.507. [DOI] [PubMed] [Google Scholar]
- 79.Li Y, et al. IL-15 activates telomerase and minimizes telomere loss and may preserve the replicative life span of memory CD8+ T cells in vitro. J Immunol. 2005;174:4019–24. doi: 10.4049/jimmunol.174.7.4019. [DOI] [PubMed] [Google Scholar]
- 80.Anichini A, et al. Differentiation of CD8+ T cells from tumor-invaded and tumor-free lymph nodes of melanoma patients: role of common gamma-chain cytokines. J Immunol. 2003;171:2134–41. doi: 10.4049/jimmunol.171.4.2134. [DOI] [PubMed] [Google Scholar]
- 81.Cooley S, et al. Adoptive Therapy with T Cells/NK Cells. Biol Blood Marrow Transplant. 2007;13(Suppl 1):33–42. [Google Scholar]
- 82.Chik KW, et al. Elevated serum interleukin-15 level in acute graft-versus-host disease after hematopoietic cell transplantation. J Pediatr Hematol Oncol. 2003;25:960–4. doi: 10.1097/00043426-200312000-00011. [DOI] [PubMed] [Google Scholar]
- 83.Mackall CL, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89:3700–7. [PubMed] [Google Scholar]
- 84.Ferrari V, et al. Distinct patterns of regeneration of central memory, effector memory and effector TCD8+ cell subsets after different hematopoietic cell transplant types: possible influence in the recovery of anti-cytomegalovirus immune response and risk for its reactivation. Clin Immunol. 2006;119:261–71. doi: 10.1016/j.clim.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 85.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260–6. doi: 10.1182/blood-2002-11-3577. [DOI] [PubMed] [Google Scholar]
- 86.Judge AD, et al. Interleukin 15 controls both proliferation and survival of a subset of memory-phenotype CD8(+) T cells. J Exp Med. 2002;196:935–46. doi: 10.1084/jem.20020772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zeng R, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201:139–48. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moroz A, et al. IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: comparative evaluation of IL-2, IL-15, and IL-21. J Immunol. 2004;173:900–9. doi: 10.4049/jimmunol.173.2.900. [DOI] [PubMed] [Google Scholar]
- 89.Lucas PJ, et al. Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J Exp Med. 2000;191:1187–96. doi: 10.1084/jem.191.7.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lucas PJ, et al. Transforming growth factor-beta pathway serves as a primary tumor suppressor in CD8+ T cell tumorigenesis. Cancer Res. 2004;64:6524–9. doi: 10.1158/0008-5472.CAN-04-0896. [DOI] [PubMed] [Google Scholar]
- 91.Lucas PJ, et al. Dysregulation of IL-15-mediated T-cell homeostasis in TGF-beta dominant-negative receptor transgenic mice. Blood. 2006;108:2789–95. doi: 10.1182/blood-2006-05-025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174:5215–23. doi: 10.4049/jimmunol.174.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu W, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–11. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakaguchi S, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 95.Ku CC, et al. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–8. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 96.Kamimura D, et al. Evidence of a novel IL-2/15R beta-targeted cytokine involved in homeostatic proliferation of memory CD8+ T cells. J Immunol. 2004;173:6041–9. doi: 10.4049/jimmunol.173.10.6041. [DOI] [PubMed] [Google Scholar]
- 97.Kursar M, et al. Regulatory CD4+CD25+ T cells restrict memory CD8+ T cell responses. J Exp Med. 2002;196:1585–92. doi: 10.1084/jem.20011347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Almeida AR, et al. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169:4850–60. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- 99.Shen S, et al. Control of homeostatic proliferation by regulatory T cells. J Clin Invest. 2005;115:3517–26. doi: 10.1172/JCI25463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nguyen VH, Zeiser R, Negrin RS. Role of naturally arising regulatory T cells in hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2006;12:995–1009. doi: 10.1016/j.bbmt.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 101.Rezvani K, et al. High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood. 2006;108:1291–7. doi: 10.1182/blood-2006-02-003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang H, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–43. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 103.Mackall CL, et al. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J Immunol. 1996;156:4609–16. [PubMed] [Google Scholar]
- 104.Min B, et al. Neonates support lymphopenia-induced proliferation. Immunity. 2003;18:131–40. doi: 10.1016/s1074-7613(02)00508-3. [DOI] [PubMed] [Google Scholar]
- 105.Min B, Paul WE. Endogenous proliferation: burst-like CD4 T cell proliferation in lymphopenic settings. Semin Immunol. 2005;17:201–7. doi: 10.1016/j.smim.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 106.Kieper WC, et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–63. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 107.Chalandon Y, et al. Pretransplantation CMV-specific T cells protect recipients of T-cell-depleted grafts against CMV-related complications. Blood. 2006;107:389–96. doi: 10.1182/blood-2005-07-2746. [DOI] [PubMed] [Google Scholar]
- 108.Ganepola S, et al. Patients at high risk for CMV infection and disease show delayed CD8+ T-cell immune recovery after allogeneic stem cell transplantation. Bone Marrow Transplant. 2007;39:293–9. doi: 10.1038/sj.bmt.1705585. [DOI] [PubMed] [Google Scholar]
- 109.Peggs KS, et al. Reconstitution of T-cell repertoire after autologous stem cell transplantation: influence of CD34 selection and cytomegalovirus infection. Biol Blood Marrow Transplant. 2003;9:198–205. doi: 10.1053/bbmt.2003.50010. [DOI] [PubMed] [Google Scholar]
- 110.Ma J, et al. Anti-tumor T cell response and protective immunity in mice that received sublethal irradiation and immune reconstitution. Eur J Immunol. 2003;33:2123–32. doi: 10.1002/eji.200324034. [DOI] [PubMed] [Google Scholar]
- 111.Borrello I, et al. Sustaining the graft-versus-tumor effect through posttransplant immunization with granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing tumor vaccines. Blood. 2000;95:3011–9. [PubMed] [Google Scholar]
- 112.Antony PA, et al. Interleukin-2-dependent mechanisms of tolerance and immunity in vivo. J Immunol. 2006;176:5255–66. doi: 10.4049/jimmunol.176.9.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kochenderfer JN, et al. Synergism between CpG-containing oligodeoxynucleotides and IL-2 causes dramatic enhancement of vaccine-elicited CD8+ T cell responses. J Immunol. 2006;177:8860–73. doi: 10.4049/jimmunol.177.12.8860. [DOI] [PubMed] [Google Scholar]
- 114.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 115.Oh S, et al. Coadministration of HIV vaccine vectors with vaccinia viruses expressing IL-15 but not IL-2 induces long-lasting cellular immunity. Proc Natl Acad Sci U S A. 2003;100:3392–7. doi: 10.1073/pnas.0630592100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Di Carlo E, et al. IL-21 induces tumor rejection by specific CTL and IFN-gamma-dependent CXC chemokines in syngeneic mice. J Immunol. 2004;172:1540–7. doi: 10.4049/jimmunol.172.3.1540. [DOI] [PubMed] [Google Scholar]
- 117.Dudley ME, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Robbins PF, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shen X, et al. Persistence of tumor infiltrating lymphocytes in adoptive immunotherapy correlates with telomere length. J Immunother. 2007;30:123–9. doi: 10.1097/01.cji.0000211321.07654.b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–14. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hakim FT, Mackall C. Immunopathogenesis of graft vs host disease. In: Ferrara J, Burakoff A, editors. Graft vs Host Disease. 2. 1996. [Google Scholar]
- 123.Muluk SC, Hakim FT, Shearer GM. Regulation of graft-versus-host-reaction by Mlsa-reactive donor T cells. Eur J Immunol. 1992;22:1967–73. doi: 10.1002/eji.1830220803. [DOI] [PubMed] [Google Scholar]
- 124.van den Brink MR, et al. Graft-versus-host-disease-associated thymic damage results in the appearance of T cell clones with anti-host reactivity. Transplantation. 2000;69:446–9. doi: 10.1097/00007890-200002150-00026. [DOI] [PubMed] [Google Scholar]
- 125.Hakim FT, et al. Repopulation of host lymphohematopoietic systems by donor cells during graft-versus-host reaction in unirradiated adult F1 mice injected with parental lymphocytes. J Immunol. 1991;146:2108–15. [PubMed] [Google Scholar]
- 126.Sinha ML, et al. Interleukin 7 worsens graft-versus-host disease. Blood. 2002;100:2642–9. doi: 10.1182/blood-2002-04-1082. [DOI] [PubMed] [Google Scholar]
- 127.Alpdogan O, et al. Administration of interleukin-7 after allogeneic bone marrow transplantation improves immune reconstitution without aggravating graft-versus-host disease. Blood. 2001;98:2256–65. doi: 10.1182/blood.v98.7.2256. [DOI] [PubMed] [Google Scholar]
- 128.Gendelman M, et al. Host conditioning is a primary determinant in modulating the effect of IL-7 on murine graft-versus-host disease. J Immunol. 2004;172:3328–36. doi: 10.4049/jimmunol.172.5.3328. [DOI] [PubMed] [Google Scholar]
- 129.Blaser BW, et al. Donor-derived IL-15 is critical for acute allogeneic graft-versus-host disease. Blood. 2005;105:894–901. doi: 10.1182/blood-2004-05-1687. [DOI] [PubMed] [Google Scholar]
- 130.Alpdogan O, et al. Interleukin-15 enhances immune reconstitution after allogeneic bone marrow transplantation. Blood. 2005;105:865–73. doi: 10.1182/blood-2003-09-3344. [DOI] [PubMed] [Google Scholar]
- 131.Roychowdhury S, et al. IL-15 but not IL-2 rapidly induces lethal xenogeneic graft-versus-host disease. Blood. 2005;106:2433–5. doi: 10.1182/blood-2005-04-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kumaki S, et al. Prolonged secretion of IL-15 in patients with severe forms of acute graft-versus-host disease after allogeneic bone marrow transplantation in children. Int J Hematol. 1998;67:307–12. doi: 10.1016/s0925-5710(97)00117-5. [DOI] [PubMed] [Google Scholar]
- 133.Via CS, Shanley JD, Shearer GM. Synergistic effect of murine cytomegalovirus on the induction of acute graft-vs-host disease involving MHC class I differences only. Analysis of in vitro T cell function. J Immunol. 1990;145:3283–9. [PubMed] [Google Scholar]
- 134.Levy RB, Jones M, Cray C. HSV-1 enhances GvHR-associated parent anti-F1 alloreactivity in vivo and in vitro. Cell Immunol. 1990;129:1–12. doi: 10.1016/0008-8749(90)90181-p. [DOI] [PubMed] [Google Scholar]
- 135.Trenado A, et al. Ex vivo-expanded CD4+CD25+ immunoregulatory T cells prevent graft-versus-host-disease by inhibiting activation/differentiation of pathogenic T cells. J Immunol. 2006;176:1266–73. doi: 10.4049/jimmunol.176.2.1266. [DOI] [PubMed] [Google Scholar]
- 136.Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. 2002;99:3493–9. doi: 10.1182/blood.v99.10.3493. [DOI] [PubMed] [Google Scholar]
- 137.Nguyen VH, et al. In vivo dynamics of regulatory T cell trafficking and survival predict effective strategies to control graft-versus-host disease following allogeneic transplantation. Blood. 2006 doi: 10.1182/blood-2006-08-044529. [DOI] [PubMed] [Google Scholar]
- 138.Hollander GA, Widmer B, Burakoff SJ. Loss of normal thymic repertoire selection and persistence of autoreactive T cells in graft vs host disease. J Immunol. 1994;152:1609–17. [PubMed] [Google Scholar]
- 139.Desbarats J, Lapp WS. Thymic selection and thymic major histocompatibility complex class II expression are abnormal in mice undergoing graft-versus-host reactions. J Exp Med. 1993;178:805–14. doi: 10.1084/jem.178.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ghayur T, et al. Complete sequential regeneration of graft-vs.-host-induced severely dysplastic thymuses. Implications for the pathogenesis of chronic graft-vs.-host disease. Am J Pathol. 1988;133:39–46. [PMC free article] [PubMed] [Google Scholar]
- 141.Hauri-Hohl MM, et al. Donor T-cell alloreactivity against host thymic epithelium limits T-cell development after bone marrow transplantation. Blood. 2007 doi: 10.1182/blood-2006-07-034157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Watanabe N, et al. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–5. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- 143.Jimenez M, et al. Clinical factors influencing T-cell receptor excision circle (TRECs) counts following allogeneic stem cell transplantation in adults. Transpl Immunol. 2006;16:52–9. doi: 10.1016/j.trim.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 144.Weinberg K, et al. Factors affecting thymic function after allogeneic hematopoietic stem cell transplantation. Blood. 2001;97:1458–66. doi: 10.1182/blood.v97.5.1458. [DOI] [PubMed] [Google Scholar]
- 145.Olkinuora H, et al. T cell regeneration in pediatric allogeneic stem cell transplantation. Bone Marrow Transplant. 2007;39:149–56. doi: 10.1038/sj.bmt.1705557. [DOI] [PubMed] [Google Scholar]