

Abstract
We combined behavioral testing with brain imaging using 99mTc-HMPAO (Amersham Health), to identify CNS structures reflecting alterations in pain perception in the streptozotocin (STZ) model of Type 1 diabetes. We induced diabetic hyperglycemia (blood glucose >300 mg/dl) by injecting male Sprague-Dawley rats with STZ (45 mg/kg i.p.). Four weeks after STZ, diabetic rats exhibited behaviors indicative of neuropathic pain (hypersensitivity thermal stimuli) and this hypersensitivity persisted for up to six weeks. Imaging data in STZ-diabetic rats revealed significant increases in the activation of brain regions involved in pain processing after six weeks duration of diabetes. These regions included secondary somatosensory cortex, ventrobasal thalamic nuclei and the basolateral amygdala. In contrast, the activation in habenular nuclei and the midbrain periaqueductal gray were markedly decreased in STZ rats. These data suggest that pain in diabetic neuropathy may be due in part to hyperactivity in somatosensory structures coupled with a concurrent deactivation of structures mediating antinociception.
Diabetes mellitus (DM) is one of the most common chronic medical problems affecting millions of people world-wide (Spruce, et al., 2003), and is the most prevalent cause of neuropathy in the United States, affecting more than 14 million persons (Mokdad, et al., 2000). A frequent complication of diabetes is unremitting pain and a reduced quality of life (Benbow, et al., 1994). Patients with DM may experience a variety of aberrant sensations including spontaneous pain and hypersensitivity to mechanical or thermal stimuli, followed by the long term paradoxical loss of stimulus-evoked sensation (Benbow, et al., 1994). Other symptoms encountered are an inability to detect heat and cold, cutaneous hyperaesthesia, loss of vibration sensation, and paradoxically, the loss of pain perception. Although numerous studies have indicated that DM produces various metabolic and morphological changes in the peripheral nervous system, and have suggested that these changes may be associated with neuropathic symptoms (Sima and Sugimoto, 1999), the pathophysiology of neuropathic pain in diabetes remains unclear.
Current evidence suggests multiple possibilities for the development of chronic neuropathic pain (CNP) as a consequence of diabetes. Hyperexcitability and altered discharge patterns in damaged peripheral axons (Burchiel, et al., 1985, Chen and Levine, 2003) has been suggested as one possible cause for pain in DM. However Calcutt (Calcutt, 2002) suggests that evidence for a purely peripheral mechanism of DM-induced neuropathic pain is weak. Evidence from both animal and human studies suggests that neuropathic pain in DM may also be the result of aberrant processing of sensory input at the spinal cord level. Electrophysiological evidence of spinal nerve conduction slowing in both rodents (Biessels, et al., 1999) and patients with DM (Brands, et al., 2004) has been described. There have also been reports of increased activity in spinal cord wide dynamic range neurons of diabetic rats in the absence of any peripheral input (Pertovaara, et al., 2001). Moreover, Calcutt et al. (Calcutt, et al., 2000) described protracted behavioral hyperalgesia in rats with DM following spinal delivery of substance P. This finding supports the suggestion that neuropathic pain likely involves aberrant spinal processing of sensory input.
There is a considerable evidence indicating that many neuropathic conditions, including injuries to peripheral nerves, produce dramatic changes in supraspinal processing and modulation of pain, and that these plastic changes may contribute significantly to the neuropathic pain symptoms (Willis, 1994). Although several reports describe cerebral effects of diabetes, these studies have focused primarily on the hippocampus and cognitive processing (Biessels, 1999, Biessels and Gispen, 2006). How plasticity in supraspinal central nervous system regions contributes to the development of neuropathic pain in DM has been relatively unexplored, probably because nerve dysfunction is most apparent in the periphery of diabetic patients. Our understanding of the mechanisms of neuropathic pain in diabetes is therefore sadly incomplete.
Previous imaging studies in rats with neuropathic pain due to experimental neuropathy (chronic constriction injury, CCI) have demonstrated significant maladaptive alterations in the activation of multiple forebrain structures involved in somatosensory processing (Morrow, et al., 2000, Paulson, et al., 2002). Accordingly, for the studies described here, we combined quantitative assessment of diabetes-induced changes in pain-related behaviors with brain imaging to identify potential CNS mechanisms of neuropathic pain using the streptozotocin (STZ)-induced diabetes model of Type I diabetes mellitus in rat.
RESEARCH DESIGN AND METHODS
We conducted all experiments in accordance with the NIH Guide for the care and use of laboratory animals (1996) and the IASP ethical guidelines for the use of awake animals in pain research (Zimmermann, 1983). All experimental procedures were approved by the Institutional Animal Care and Use Committees at the University of Michigan and the Ann Arbor Veterans Affairs Healthcare System.
Induction of Experimental Diabetes
Diabetes mellitus was induced by a single i.p. injection of streptozotocin (Sigma Chemical Co., St. Louis, MO) dissolved in a citrate buffer (pH 5.5) at a dose of 45 mg/kg body weight. Age-matched control rats were injected with vehicle only. Prior to injection, animals were fasted overnight to maximize the effectiveness of the streptozotocin (STZ) treatment. STZ-treated animals also received 10% sucrose water for 48 hours after injection to prevent transient hypoglycemia. Diabetic hyperglycemia was verified 72 hours after STZ injection by measuring blood glucose (Beyer Elite™ Glucometer). Fasting whole-blood glucose ≥ 300mg/dl (normal 80−135mg/dl) was our criteria for the presence of experimental diabetes mellitus. Thereafter, we measured fasting whole-blood glucose levels in all rats once each week until the end of the study. Animals that became diabetic after this dose of STZ typically had blood sugars in the range of 300−500 mg/dl and did not require insulin supplementation. Previous studies with this model indicate that rats with diabetes for 4−8 weeks demonstrate a variety of functional abnormalities in the peripheral nervous system (Srinivasan, et al., 2000), as well as somatosensory abnormalities (see Calcutt, 2002 for review).
Animal Subjects
Twenty male Sprague-Dawley (Charles River) rats weighing approximately 300 grams at the start of the experiment served as subjects for the studies described here. Because we wished to compare the responses of healthy age-matched controls to those animals with confirmed diabetes, four of ten STZ-injected rats were excluded from further testing because they failed to develop hyperglycemia. Experimental groups were therefore comprised of an STZ-diabetic group (n=6) and a saline vehicle-treated, healthy control group (n=10). All animals were housed in pairs and maintained on 12/12 light - dark cycle, with lights on at 0600 hr. Ambient temperature in the animal facility was kept at 22°C. Food and water were provided ad libitum except prior to testing of glucose levels.
Quantification of Behavior
All animals were acclimated to the colony room for one week before the start of the data acquisition. During this acclimation the animals were handled daily for 10 minutes and habituated to the behavioral testing apparatus. The details of the behavioral test for thermal sensitivity is described below. In addition, because rats were restrained during brain imaging, each animal was also acclimated to placement in a soft towel restraint for approximately 45 minutes. Prior to injection of experimental animals with STZ or vehicle, baseline behavioral responses to thermal and mechanical somatic stimuli were determined for all rats in two separate test sessions, one week apart. Thereafter, diabetic and control rats were evaluated for changes in behavioral responsiveness on a weekly basis until the terminal brain imaging experiment conducted at six weeks duration of diabetes.
Thermal Hypersensitivity
Hargreaves Plantar Test
To quantify thermal nociception, a commercially available device modeled after that described by Hargreaves (1988) was employed (UARDG, Dept. of Anesthesiology, University of California San Diego). Briefly, rats were placed in a clear a Plexiglas chamber (10 cm × 20 cm × 10 cm) located on an elevated floor of clear glass (2mm thick) and given 10 minutes to habituate to the testing environment. The glass floor was maintained at 30°C ± 1°C. A radiant heat source delivered a thermal stimulus to the plantar surface of the hind foot. The latency to foot withdrawal (escape) served as the behavioral measure of thermal nociception. If the foot was not withdrawn within 20 seconds, the stimulus was automatically terminated to avoid tissue damage. Each foot was tested 5 times with a minimum of 3 minutes between stimulations to avoid peripheral sensitization effects. The mean withdrawal latency for each foot was computed by averaging the 5 measurements. As compared to the baseline (control) latency, a significant decrease in the latency of foot withdrawal in response to the thermal stimulus was interpreted as indicating the presence of thermal hypersensitivity (Hargreaves, et al., 1988).
Brain Imaging
Imaging studies were performed in diabetic rats 6 weeks after induction of diabetes and in age matched vehicle injected controls. Neuronal activation in select brain regions was determined by autoradiographic measurement of regional cerebral blood flow (rCBF) using a technique described previously (Morrow, et al., 2000). Briefly, each rat was placed in a soft towel restraint and a flexible intravenous catheter is inserted into the tail vein. Subjects were then permitted to rest quietly in the restraint for approximately 40 minutes to recover from the stress of tail vein catheterization. For imaging, 10 mCi of [99m]Tc (technetium) exametazime was injected into the tail vein as a bolus over 10 to 15 seconds. Approximately 2 minutes following tracer injection, the rat was euthanized with chloral hydrate, removed from the restraint and decapitated. The brain was removed from the skull, quick frozen and 20μm coronal frozen sections were cut at −18°C. Four consecutive brain sections were taken starting at Bregma level −0.30 through level −6.30 at approximately 250 μm intervals. Brain sections were mounted on glass slides and desiccated on a 40°C slide warmer. Standard autoradiograms were generated by direct apposition of the mounted sections to the emulsion side of Kodak BioMax™ MR-1 Imaging film for 1−2 hours.
Region of Interest Analysis
Densitometric analysis of autoradiograms was performed using a microcomputer-assisted video densitometer system (MCID Elite™, Imaging Research). Autoradiograms were coded to prevent experimenter bias when sampling the images. Each brain section on film was digitized to produce a high resolution, 256 level grayscale image. Anatomic location of selected regions of interest (ROIs) was determined by overlaying transparent stereotaxic templates (adapted from the Paxinos and Watson (1998) stereotaxic atlas of the rat brain) on digitized brain images displayed on the video monitor. Select ROIs were then sampled by following the outline provided by the overlaying transparency using an MCID sample tool. When the radioactivity level of the slides returned to background (2.5 days), the slides were stained with cresyl violet and structural identifications confirmed by careful comparison of the digitized and cresyl violet stained sections to coronal plates from the Paxinos & Watson atlas (1998). We limited sampled ROIs to eighteen supraspinal structures including cortical and thalamic ROIs and the brainstem periaqueductal gray (see Table 1). As described previously (Morrow, et al., 2000), an index of activation (AI) was calculated for each sampled ROI using the following formula:
Table 1.
Anatomical abbreviations and comparison of regional differences in brain activation in control and diabetic rats expressed as the mean percent difference from the average whole brain activity, the activation index (AI).
|
Region of Interest (ROI) |
Abbrev. |
Control ± sem (N = 10) |
Diabetic ± sem (N = 6) |
p values |
|---|---|---|---|---|
| Sensory-Motor | ||||
| Primary Somatosensory Cortex (hindlimb area) | S I | 22.35 ± 3.16 | 22.24 ± 4.04 | .929 |
| Secondary Somatosensory Cortex | S II | 21.75 ± 1.69 | 33.30 ± 2.97 | .001 |
| Ventral Lateral nucleus | VL | −2.00 ± 1.81 | 7.66 ± 3.05 | .001 |
| Ventral Medial nucleus | VM | 2.39 ± 1.32 | 7.63 ± 4.687 | .092 |
| Ventral Posterior Lateral nucleus | VPL | −1.77 ± 2.79 | 13.65 ± 3.02 | .000 |
| Ventral Posterior Medial nucleus |
VPM |
11.15 ± 3.08 |
21.67 ± 2.54 |
.001 |
| Limbic | ||||
| Anterior dorsal thalamic nucleus | AD | 23.58 ± 2.640 | 25.88 ± 2.70 | .320 |
| Medial Thalamus | MT | 4.91 ± 2.03 | 7.54 ± 2.42 | .227 |
| Parafasicular nucleus (thalamus) | PF | 10.49 ± 3.87 | 8.41 ± 1.64 | .423 |
| Posterior Group (thalamus) | PO | −0.52 ± 2.30 | 1.47 ± 1.00 | .379 |
| Cingulate Cortex | CC | 23.90 ± 3.36 | 18.88 ± 2.88 | .123 |
| Retrosplenial Cortex | RS | 31.08 ± 2.41 | 32.59 ± 3.78 | .674 |
| Hippocampus (dorsal) | HIP | −5.39 ± 2.52 | −5.97 ± 2.39 | .752 |
| Basal Lateral Amygdala | BLA | −1.67 ± 2.10 | 4.02 ± 2.91 | .036 |
| Diagonal Band of Broca |
DB |
32.32 ± 3.51 |
26.91 ± 2.77 |
.116 |
| Antinociceptive | ||||
| Habenular Complex | HBC | 50.88 ± 3.51 | 41.74 ± 3.40 | .019 |
| Interpeduncular nucleus | IPN | 48.73 ± 4.86 | 32.39 ± 6.95 | .068 |
| Periaqueductal Gray | PAG | −2.34 ± 3.36 | −17.01 ± 5.02 | .025 |
Briefly, the densitometer system converted sampled ROI optical densities to apparent tissue radioactivity concentrations (nCi/mg) by comparison with the optical densities of [14C] standards also imaged on each film. The average total brain activity was estimated by sampling all pixels in each brain section and averaging the activity across all sections for a given animal. AI values were then calculated for each sampled ROI as a percent difference from the average total activity of the entire brain for each animal. A within subject mean AI value for each region of interest was computed and averaged across all subjects in each experimental group to compute group means for each ROI (Morrow, et al., 2000).
Statistical Analysis
Significant within-group changes in pre- versus post-injection behavioral responses over time was performed by repeated measures ANOVA (p ≤ 0.05). Significant differences in behavioral responsiveness between groups were determined by mixed-model repeated measures ANOVA (p ≤ 0.05) with Dunnetts T3 for post-hoc analysis of specific time points. Autoradiograms were analyzed for differences in relative optical density within the eighteen ROIs listed in table 1. Distinctly bilateral structures were examined for side to side differences in activation using a paired t-test (p ≤ 0.05). In addition, we tested for significant differences in AI for each ROI between control and diabetic groups using a mixed-model repeated measures ANOVA (p ≤ 0.05). All statistical analyses were performed using the software package, SPSS for Windows (SPSS Inc., Chicago, Illinois).
RESULTS
Streptozotocin (STZ)-Induced Diabetic Hyperglycemia
Six of ten rats injected with streptozotocin (STZ, 45 mg/kg i.p.) developed diabetic hyperglycemia (defined as glucose > 300 mg/dL) within less than 72 hours. Blood glucose levels remained elevated and relatively stable throughout the period of this study (Figure 1, panel A). As expected none of the vehicle treated controls showed an elevation in glucose levels. In addition to increased blood glucose, STZ-diabetic (STZ-D) animals presented clinical signs commonly seen in human diabetes, including polyuria, and low weight gain as compared to controls (Figure 1, panel B). However, none of the STZ-D rats showed symptoms of cachexia or weight loss and all rats exhibited grooming behaviors indistinguishable from age matched healthy controls.
Figure 1.
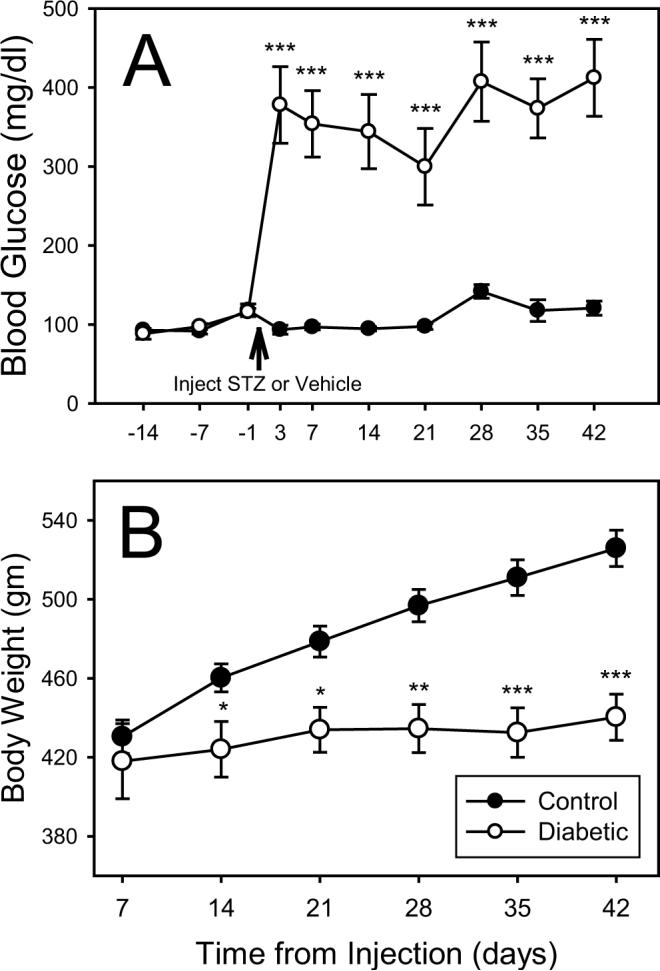
Graphs showing the blood glucose levels (A) and body weights (B) of control (black circles) and diabetic (open circles) rats. Differences in blood glucose (Beyer Elite™ Glucometer) before and after injection with vehicle (saline) or STZ are shown in Panel A. Note that diabetic rats developed a relatively stable level of hyperglycemia approximately 3 days after injection with STZ. Panel B shows the mean body weight for control and STZ-D groups over the six weeks following injection. STZ-D animals had a significantly lower weight gain than control animals, but did not lose weight. Asterisks indicate a significant difference between groups, determined using a repeated measures mixed model ANOVA (SPSS for Windows) with Dunnetts T3 for post-hoc analysis (* = p < .05; ** = p < .005; *** = p< .0001).
Behavioral Studies
Both the STZ-D and control groups showed a significant decrease in response latency to thermal stimuli over time in the Hargreaves plantar test and this decrease was significantly enhanced in the STZ-D group when compared to controls (Figure 2).
Figure 2.
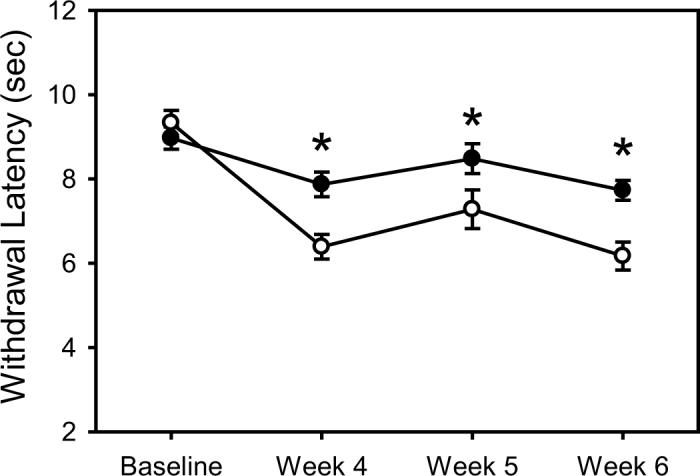
Graph showing changes in behavioral response to thermal stimuli applied to the plantar surface of the left hind foot in STZ-D (open circles) and control (black circles) rats. Differences in behavioral responsiveness to noxious heat between control and STZ injected rats developed by four weeks after induction of diabetes. All values are expressed as the group means at four time points: baseline, and 4, 5 and 6 weeks after confirmed diabetes. Asterisks indicate a significant difference between groups, determined using a repeated measures mixed model ANOVA (SPSS for Windows) with Dunnetts T3 for post-hoc analysis (* = p < .05).
Brain Activation: Imaging Regional Cerebral Blood Flow
Table 1 shows the basal activation index (AI value) for each ROI sampled in both the STZ-D rats with neuropathic pain and in the controls. There was no significant difference in total brain activity between the STZ-D and control group (17.43 ± 1.33 and 18.34 ± 0.82, respectively; p = 0.56). Animals of both experimental groups displayed a regional heterogeneity in the pattern of brain activation. In the absence of intentional somatic stimulation, neither control nor STZ-D rats showed significant side-to-side differences in the activation within any of the eighteen structures sampled (data not shown). Because neither experimental group showed significant lateralized differences in brain activation we averaged the AI values from both sides for each brain region with a bilateral representation for each animal before computing group means for graphical display purposes only. All statistics were performed on the un-pooled data as stated in the methods section.
Table 1 and Fig. 3 and 4 display the basal (unstimulated) level of activation within multiple forebrain regions and the midbrain periaqueductal gray (PAG) between control and STZ-D rats. The regions of interest in the table are divided into sensory-motor, limbic and antinociceptive regions. Seven of the eighteen (39%) sampled ROIs showed diabetes-induced changes, including both increases and decreases in activation when compared to age matched non-diabetic controls. STZ-D rats had significantly greater AI values in 7 of 18 brain regions sampled (Table 1 and Figure 3 and 4). These increases were found in 4 of 6 (67%) sampled regions involved in somatosensory or motor processing, including the secondary somatosensory cortex (SII), the ventral lateral (VL), ventral posterior lateral (VPL), and ventral posterior medial (VPM) nuclei of the thalamus. In contrast, STZ-D rats showed increased basal activation in only 1 of 9 (11%) sampled structures forming part of the limbic system, the basal lateral amygdala (BLA) when compared to controls.
Figure 3.

Sample colorized coronal sections showing differences in resting brain activation between non-diabetic control and STZ-D rats, at three A–P levels: −2.3, −3.3 and −6.3. Note: each image is taken from a single brain section in individual animals and therefore may not accurately depict group means presented in the table and graphs.
Figure 4.

The bar graphs compare the mean basal (unstimulated) bilateral level of activation (AI) in somatosensory and endogeneous antinociceptive ROIs for control (black circles) and diabetic experimental (open circles) groups six weeks after injection of vehicle or STZ. Data are shown only for ROIs showing a significant difference in activation between the two groups. Significant differences in AI between groups were determined using a mixed-model repeated measures ANOVA corrected for multiple comparisons (SPSS for Windows). Line drawings of sampled brain regions were modified from the stereotaxic atlas of the rat brain (Paxinos and Watson, 1998) to show the approximate Anterior-Posterior (AP) level where each brain region was sampled. Asterisks indicate: * = p < .05; ** = p < .005; *** = p< .0001.
In addition to the significant increases in AI found in the above sensory-motor and limbic structures, two of the eighteen sampled ROIs in diabetic animals showed a significant reduction in AI when compared to controls. These included the habenular complex (HBc) and the midbrain periaqueductal gray (PAG), structures known to participate in endogenous antinociceptive mechanisms. The PAG of diabetic animals exhibited a highly significant 6.26 fold reduction in basal activation when compared to controls.
DISCUSSION
In the present study we have shown that STZ-induced diabetic rats exhibit hypersensitivity to thermal stimuli when compared to similarly treated control rats. Using a well-validated test to assess nociceptive thermal sensitivity, we report that by four weeks after STZ injection, diabetic rats exhibit neuropathic pain as measured by a 20% or greater decrease in paw thermal withdrawal latencies. While we also found that control animals exhibited a decrease in withdrawal latency to thermal stimuli over time, the decreased latency in the STZ-D group was significantly greater. Reports of changes in thermal nociceptive thresholds have been variable, with hypersensitivity observed in some studies (Courteix, et al., 1993, Forman, et al., 1986, Lee and McCarty, 1992), others finding no alteration (Raz, et al., 1988), and others observing the loss of thermal sensation seen in the clinic. It is unclear why some laboratories report hypersensitivity in the STZ rat model and others do not. One possibility is the specific methodological details of the behavioral test employed. Indeed, we have found that differences in the location of the stimulus applied to the plantar surface of the foot can lead to vastly different results. Another possibility is that the disparity of data represents strain differences in the animal subjects (Paulson, et al., 2006).
It has been argued that STZ-treated rats show altered thresholds and withdrawal latencies in pain tests because they are ill and not because of the development of a painful peripheral neuropathy (Fox, et al., 1999). However, several factors suggest ill-health is not responsible for the abnormal behavioral responses we see here. The decrease in withdrawal latency in response to thermal stimuli shows that our STZ diabetic animals do detect, and are capable of, responding to external stimuli. Further, although our diabetic rats developed polyuria and exhibited a low weight gain, they otherwise appeared healthy, were well-groomed and did not lose weight.
We report here that STZ-D rats exhibiting signs of neuropathic pain show increased activation in the sensory-discriminative pain system, including secondary somatosensory cortex and the ventrobasal thalamic nuclei (VB). We have reported similar increases in these somatosensory structures following both chronic constriction injury (CCI) and spinal cord injury (SCI) in rats (Morrow, et al., 2000, Paulson, et al., 2001). Single neurons within VB and SI of rats with CCI are also especially susceptible to sensitization by noxious stimulation of the injured hindpaw (Guilbaud, et al., 1995). Guilbaud and colleagues (1995) described abnormal spontaneous “paroxysmal” discharges in S1 neurons occurring without stimulation and which lasted up to 5 minutes. In addition, noxious stimulation of the nerve damaged hindpaw also produced higher levels of discharge in S1, which lasted for extended periods following the termination of the stimulus (Guilbaud, et al., 1992). Furthermore, neuroimaging studies in patients with chronic low back pain, fibromyalgia and peripheral nerve injury have also reported increased activity in somatosensory cortices (Calcutt, 2002, Giesecke, et al., 2004).
In contrast, STZ-D rats did not show increased activation in select ROIs of the medial pain system, including the anterior cingulate (CC), retrosplenial cortices (RS), or medial thalamic nuclei, such as the anterior dorsal (AD), medial (MT) or posterior (PO) thalamic groups. The medial pain system is often referred to as the affective-motivational portion of the pain experience and is commonly activated in patients and animals experiencing chronic neuropathic pain (Calcutt, 2002, Paulson, et al., 2001). The lack of activation in these regions is in stark contrast to our previous reports using other neuropathic pain models (CCI and SCI) in rats (Morrow, et al., 2000, Paulson, et al., 2001). Perhaps it is the rapid-onset of the initial acute injury in the CCI and SCI models that induces a more robust activation of the medial pain system, in contrast to the more gradual onset of pain in diabetes.
Although we have shown diabetes-induced increases in activation within several somatosensory forebrain regions, it is not possible from these studies to conclude whether these enhanced CNS activations represent a primary generator for neuropathic pain or an increased response to altered PNS and spinal cord driven input. Several authors have described an association between neuropathic dysaesthetic pain and active nerve fiber degeneration or axonal atrophy (Britland, et al., 1992, Dyck, et al., 1976, Llewelyn, et al., 1991, Malik, et al., 2001), whereas others have failed to demonstrate any association between nerve pathology and pain (Kapur, 2003, Sorensen, et al., 2006) . Calcutt (Calcutt, 2002), however, has suggested that there is little evidence to support hyperexcitability of peripheral nerves or evidence of overt structural nerve damage at this early stage of diabetes. The abnormal pain sensations found in diabetic subjects may therefore be due in part to changes in the activation of CNS structures. Several evoked potential studies describe altered peripheral and central conduction times and amplitudes in somatosensory evoked potential studies in diabetic humans and STZ-diabetic rats (Biessels, et al., 1999, Comi, 1997, Gupta and Dorfman, 1981). One study also reports changes in the latency and duration of nociceptive laser evoked potentials in humans (Rossi, et al., 2002). Recent evidence from the Calcutt lab (Calcutt, 2002) suggests that facilitation or disinhibition in the spinal dorsal horn may be responsible, at least in part, for the tactile allodynia seen in the diabetic rats. Other studies in diabetic rats describe spontaneous spinal activity that is not driven by peripheral input (Calcutt, et al., 2000) and report protracted behavioral hyperalgesia following spinal delivery of substance P (Calcutt, et al., 1998). Such findings are consistent with spinal and/or supraspinal mechanisms that amplify sensory input in these animals, and thus support the contention that neuropathic pain may involve aberrant central processing of sensory input.
Besides amplification of sensory input, centrally-mediated hyper-responsiveness may occur secondary to changes in endogenous analgesia systems that modulate nociceptive processing. Spinal 5-HT release evoked by the stimulation of descending pathways is reduced in STZ diabetic rats despite normal tissue levels (Di Giulio, et al., 1989) and could contribute to spinally mediated hyperalgesia. Spinal levels of met-enkephalin have been reported as decreased (Mantyh, 1982) (Di Giulio, et al., 1989) by diabetes and descending noradrenergic inhibitory activity may also be depressed in diabetic animals (Britland, et al., 1992, Dyck, et al., 1976).
Finally, we report a robust decrease in activation of structures participating in endogeneous, descending modulation of pain, specifically in the habenular complex and the periaqueductal grey (PAG) (Klemm and Klemm, 2004, Millan, 2002). A considerable amount of evidence exists to establish that antinociception is mediated in part by descending pathways arising from the habenula and the periaqueductal gray (Klemm and Klemm, 2004, Millan, 2002). Early studies have shown that electrical stimulation or opioids microinjected into the habenula or PAG produced profound long-lasting antinociception (Klemm and Klemm, 2004, Millan, 2002). Decreases in PAG activation could result from a net decrease in input from forebrain structures that connect to the PAG, since neuroanatomical studies have revealed a major input from various limbic and somatosensory regions to the PAG (Millan, 2002). The periaqueductal gray and habenular complex might also show reduced activity if there was less nociceptive input ascending from the spinal dorsal horn. However, such an explanation seems unlikely, since our data shows that regions of somatosensory thalamus, as well as, SII cortex exhibit a significant increase in activation in diabetic rats as compared to controls. Alternatively, the decrease in activation in the PAG may be the result of neuronal loss within the PAG, as apoptosis has been reported in the brains of diabetic rats in the hippocampus and frontal cortex (Sima and Li, 2005). We are presently doing experiments to address this hypothesis.
There have been few reports of PET and SPET imaging of the CNS of type I and II diabetic patients. Two studies report a modest decrease in cerebral blood flow in cortical and cerebellar areas (MacLeod, et al., 1994, Quirce, et al., 1997); while there was no change in thalamic areas (MacLeod, et al., 1994). However, the duration of diabetes of the patients in these studies was six years (MacLeod, et al., 1994) and twenty-one years (Quirce, et al., 1997), respectively. Keymeulen and colleagues (Keymeulen, et al., 1996) found that alterations in cerebral blood flow were dependent on diabetes duration. Specifically, short term diabetic patients (< 5 years) showed increased blood flow in prefrontal and left frontal regions; while long term diabetic patients showed the opposite: reduced prefrontal uptake of Tc. Our imaging data presented in this report are not directly comparable to these clinical reports, primarily due to the extended duration of diabetes in the clinical subjects. While the report by Keymeulen et al may be more applicable, we did not sample the prefrontal regions that showed increased blood flow in this study (Keymeulen, et al., 1996). Ongoing experiments in our laboratory are currently assessing the affect of diabetes on frontal and prefrontal regions of cortex and their relationship to nociceptive processing.
In conclusion, this is the first study to use functional brain imaging to investigate the neural mechanisms underlying neuropathic pain in an animal model of experimentally induced type 1 diabetes. Our data shows that in the absence of overt somatic stimulation, the pattern of forebrain activation in STZ-diabetic rats with neuropathic pain exhibits not only similarities but also significant differences when compared to the patterns obtained during CCI-induced or SCI-induced nociception. These data provide strong and exciting evidence that neuropathic pain in diabetes mellitus is due not only to activation of supraspinal structures that participate in pain perception but also to deactivation of descending antinociceptive systems.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.NIH Guide for the care and use of laboratory animals. National Academy Press, Inc.; Washington D.C.: 1996. [Google Scholar]
- 2.Benbow J, Chan AW, Bowsher D, MacFarlane IA, Williams G. A prospective study of painful symptoms, small-fibre function and peripheral vascular disease in chronic painful diabetic neuropathy. Diabet.Med. 1994:17. doi: 10.1111/j.1464-5491.1994.tb00223.x. [DOI] [PubMed] [Google Scholar]
- 3.Biessels GJ. Cerebral complications of diabetes: clinical findings and pathogenetic mechanisms. Netherlands Journal of Medicine. 1999;54:35. doi: 10.1016/s0300-2977(98)00134-x. [Review] [162 refs] [DOI] [PubMed] [Google Scholar]
- 4.Biessels GJ, Cristino NA, Rutten GJ, Hamers FP, Erkelens DW, Gispen WH. Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats. Course of development and effects of insulin treatment. Brain. 1999;122:757. doi: 10.1093/brain/122.4.757. [DOI] [PubMed] [Google Scholar]
- 5.Biessels GJ, Cristino NA, Rutten GJ, Hamers FPT, Erkelens DW, Gispen WH. Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats - Course of development and effects of insulin treatment. Brain. 1999;122:757. doi: 10.1093/brain/122.4.757. [DOI] [PubMed] [Google Scholar]
- 6.Biessels GJ, Gispen WH. The impact of diabetes on cognition: What can be learned for rodent models? Neurobiology of Aging. 2006;265:S36–S41. doi: 10.1016/j.neurobiolaging.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Brands AM, Kessels RP, de Haan EH, Kappelle LJ, Biessels GJ. Cerebral dysfunction in type 1 diabetes: effects of insulin, vascular risk factors and blood-glucose levels. European Journal of Pharmacology. 2004;490:159. doi: 10.1016/j.ejphar.2004.02.053. [Review] [115 refs] [DOI] [PubMed] [Google Scholar]
- 8.Britland ST, Young RJ, Sharma AK, Clarke BF. Acute and remitting painful diabetic polyneuropathy: a comparison of peripheral nerve fibre pathology. Pain. 1992;48:361–370. doi: 10.1016/0304-3959(92)90085-P. [DOI] [PubMed] [Google Scholar]
- 9.Burchiel KJ, Russell LC, Lee RP, Sima AA. Spontaneous activity of primary afferent neurons in diabetic BB/Wistar rats. A possible mechanism of chronic diabetic neuropathic pain. Diabetes. 1985;34:1210. doi: 10.2337/diab.34.11.1210. [DOI] [PubMed] [Google Scholar]
- 10.Calcutt NA. Potential mechanisms of neuropathic pain in diabetes. Int.Rev.Neurobiol. 2002;50:205. doi: 10.1016/s0074-7742(02)50078-7. [DOI] [PubMed] [Google Scholar]
- 11.Calcutt NA. Potential mechanisms of neuropathic pain in diabetes. International Review of Neurobiology. 2002;50:205. doi: 10.1016/s0074-7742(02)50078-7. [Review] [149 refs] [DOI] [PubMed] [Google Scholar]
- 12.Calcutt NA, Chen P, Hua XY. Effects of diabetes on tissue content and evoked release of calcitonin gene-related peptide-like immunoreactivity from rat sensory nerves. Neurosci.Lett. 1998;254:129. doi: 10.1016/s0304-3940(98)00692-2. [DOI] [PubMed] [Google Scholar]
- 13.Calcutt NA, Freshwater JD, O'Brien JS. Protection of sensory function and antihyperalgesic properties of a prosaposin-derived peptide in diabetic rats. Anesthesiology. 2000;93:1271. doi: 10.1097/00000542-200011000-00021. [DOI] [PubMed] [Google Scholar]
- 14.Calcutt NA, Stiller CO, Gustafsson H, Malmberg AB. Elevated substance-P-like immunoreactivity levels in spinal dialysates during the formalin test in normal and diabetic rats. Brain Research. 2000;856:20. doi: 10.1016/s0006-8993(99)02345-8. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Levine JD. Altered temporal pattern of mechanically evoked C-fiber activity in a model of diabetic neuropathy in the rat. Neuroscience. 2003;121:1007. doi: 10.1016/s0306-4522(03)00486-x. [DOI] [PubMed] [Google Scholar]
- 16.Comi G. Evoked potentials in diabetes mellitus. Clinical Neuroscience. 1997;4:374. [Review] [59 refs] [PubMed] [Google Scholar]
- 17.Courteix C, Eschalier A, Lavarenne J. Streptozocin-induced diabetic rats: behavioural evidence for a model of chronic pain. Pain. 1993;53:81. doi: 10.1016/0304-3959(93)90059-X. [DOI] [PubMed] [Google Scholar]
- 18.Di Giulio AM, Tenconi B, La Croix R, Mantegazza P, Abbracchio MP, Cattabeni F, Gorio A. Denervation and hyperinnervation in the nervous system of diabetic animals. II. Monoaminergic and peptidergic alterations in the diabetic encephalopathy. J Neurosci Res. 1989;24:362. doi: 10.1002/jnr.490240304. [DOI] [PubMed] [Google Scholar]
- 19.Di Giulio AM, Tenconi B, La Croix R, Mantegazza P, Cattabeni F, Gorio A. Denervation and hyperinnervation in the nervous system of diabetic animals. I. The autonomic neuronal dystrophy of the gut. J Neurosci Res. 1989;24:355. doi: 10.1002/jnr.490240303. [DOI] [PubMed] [Google Scholar]
- 20.Dyck PJ, Lambert EH, O'Brien PC. Pain in peripheral neuropathy related to rate and kind of fiber degeneration. Neurology. 1976;26:466–471. doi: 10.1212/wnl.26.5.466. [DOI] [PubMed] [Google Scholar]
- 21.Forman LJ, Estilow S, Lewis M, Vasilenko P. Streptozocin diabetes alters immunoreactive beta-endorphin levels and pain perception after 8 wk in female rats. Diabetes. 1986;35:1309. doi: 10.2337/diab.35.12.1309. [DOI] [PubMed] [Google Scholar]
- 22.Fox A, Eastwood C, Gentry C, Manning D, Urban L. Critical evaluation of the streptozotocin model of painful diabetic neuropathy in the rat. Pain. 1999;81:307. doi: 10.1016/S0304-3959(99)00024-X. [DOI] [PubMed] [Google Scholar]
- 23.Giesecke T, Gracely RH, Grant MA, Nachemson A, Petzke F, Williams DA, Clauw DJ. Evidence of augmented central pain processing in idiopathic chronic low back pain. Arthritis Rheum. 2004;50:613. doi: 10.1002/art.20063. [DOI] [PubMed] [Google Scholar]
- 24.Guilbaud G, Benoist JM, Gautron M. Contribution of the sciatic and saphenous nerve to the ventrobasal thalamic neuronal responses to pinch in rats with a chronic sciatic nerve constriction: A study using anesthetic blocks and nerve section. Neurosci.Lett. 1995;187:197. doi: 10.1016/0304-3940(95)11375-7. [DOI] [PubMed] [Google Scholar]
- 25.Guilbaud G, Benoist JM, Levante A, Gautron M, Willer JC. Primary somatosensory cortex in rats with pain-related behaviours due to a peripheral mononeuropathy after moderate ligation of one sciatic nerve: Neuronal responsivity to somatic stimulation. Exp.Brain Res. 1992;92:227. doi: 10.1007/BF00227967. [DOI] [PubMed] [Google Scholar]
- 26.Gupta PR, Dorfman LJ. Spinal somatosensory conduction in diabetes. Neurology. 1981;31:841. doi: 10.1212/wnl.31.7.841. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 28.Kapur D. Neuropathic pain and diabetes. Diabetes/Metabolism Research Reviews. 2003;19(Suppl 1):S9. doi: 10.1002/dmrr.359. [Review] [45 refs] [DOI] [PubMed] [Google Scholar]
- 29.Keymeulen B, de Metz K, Cluydts R, Bossuyt A, Somers G. Technetium-99m hexamethylpropylene amine oxime single-photon emission tomography of regional cerebral blood flow in insulin-dependent diabetes. Eur J Nucl Med. 1996;23:163–168. doi: 10.1007/BF01731840. [DOI] [PubMed] [Google Scholar]
- 30.Klemm WR, Klemm WR. Habenular and interpeduncularis nuclei: shared components in multiple-function networks. Medical Science Monitor. 2004;10:RA261–273. [PubMed] [Google Scholar]
- 31.Lee JH, McCarty R. Pain threshold in diabetic rats: effects of good versus poor diabetic control. Pain. 1992;50:231. doi: 10.1016/0304-3959(92)90167-A. [DOI] [PubMed] [Google Scholar]
- 32.Llewelyn JG, Gilbey SG, Thomas PK, King RH, Muddle JR, Watkins PJ. Sural nerve morphometry in diabetic autonomic and painful sensory neuropathy. A clinicopathological study. Brain. 1991;114:867. doi: 10.1093/brain/114.2.867. [DOI] [PubMed] [Google Scholar]
- 33.MacLeod KM, Hepburn DA, Deary IJ, Goodwin GM, Dougall N, Ebmeier KP, Frier BM. Regional cerebral blood flow in IDDM patients: effects of diabetes and of recurrent severe hypoglycaemia. Diabetologia. 1994;37:257–263. doi: 10.1007/BF00398052. [DOI] [PubMed] [Google Scholar]
- 34.Malik RA, Veves A, Walker D, Siddique I, Lye RH, Schady W, Boulton AJM. Sural nerve fibre pathology in diabetic patients with mild neuropathy: relationship to pain, quantitative sensory testing and peripheral nerve electrophysiology. Acta Neuropathologica. 2001;101:367. doi: 10.1007/s004010000287. [DOI] [PubMed] [Google Scholar]
- 35.Mantyh PW. Forebrain projections to the periaqueductal gray in the monkey, with observations in the cat and rat. J Comp Neurol. 1982;206:146–158. doi: 10.1002/cne.902060205. [DOI] [PubMed] [Google Scholar]
- 36.Millan MJ. Descending control of pain. Progress in Neurobiology. 2002;66:355. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 37.Mokdad AH, Ford ES, Bowman BA, Nelson DE, Engelgau MM, Vinicor F, Marks JS. Diabetes trends in the U.S.: 1990−1998. Diabetes Care. 2000;23:1278–1283. doi: 10.2337/diacare.23.9.1278. [DOI] [PubMed] [Google Scholar]
- 38.Morrow TJ, Paulson PE, Brewer KL, Yezierski RP, Casey KL. Chronic, selective forebrain responses to excitotoxic dorsal horn injury. Experimental Neurology. 2000;161:220–226. doi: 10.1006/exnr.1999.7246. [DOI] [PubMed] [Google Scholar]
- 39.Paulson PE, Casey KL, Morrow TJ. Long Term Changes in Behavior and Regional Cerebral Blood Flow Associated with Painful Peripheral Mononeuropathy in the Rat. Pain. 2002;95:31. doi: 10.1016/s0304-3959(01)00370-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulson PE, Gorman AL, Yerzierski RP, Casey KL, Morrow TJ. Differences in forebrain activation in two strains of rat at rest and after spinal cord injury. Experimental Neurology. 2006;196:413–421. doi: 10.1016/j.expneurol.2005.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulson PE, Morrow TJ, Casey KL. Bilateral behavioral and regional cerebral blood flow changes during painful peripheral mononeuropathy in the rat. Pain. 2001;84:233–245. doi: 10.1016/s0304-3959(99)00216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1998. [DOI] [PubMed] [Google Scholar]
- 43.Pertovaara A, Wei H, Kalmari J, Ruotsalainen M. Pain behavior and response properties of spinal dorsal horn neurons following experimental diabetic neuropathy in the rat: Modulation by nitecapone, a COMT inhibitor with antioxidant properties. Experimental Neurology. 2001;167:425. doi: 10.1006/exnr.2000.7574. [DOI] [PubMed] [Google Scholar]
- 44.Quirce R, Carril JM, Jimenez-Bonilla JF, Amado JA, Gutierrez-Mendiguchia C, Banzo I, Blanco I, Uriarte I, Montero A. Semi-quantitative assessment of cerebral blood flow with 99mTc-HMPAO SPET in type I diabetic patients with no clinical history of cerebrovascular disease. Eur J Nucl Med. 1997;24:1507–1513. doi: 10.1007/s002590050181. [DOI] [PubMed] [Google Scholar]
- 45.Raz I, Hasdai D, Seltzer Z, Melmed RN. Effect of hyperglycemia on pain perception and on efficacy of morphine analgesia in rats. Diabetes. 1988;37:1253. doi: 10.2337/diab.37.9.1253. [DOI] [PubMed] [Google Scholar]
- 46.Rossi P, Morano S, Serrao M, Gabriele A, Di Mario U, Morocutti C, Pozzessere G. Pre-perceptual pain sensory responses (N1 component) in type 1 diabetes mellitus. NeuroReport. 2002;13:1009. doi: 10.1097/00001756-200206120-00005. [DOI] [PubMed] [Google Scholar]
- 47.Sima AA, Sugimoto K. Experimental diabetic neuropathy: an update. Diabetologia. 1999;42:773. doi: 10.1007/s001250051227. [DOI] [PubMed] [Google Scholar]
- 48.Sima AAF, Li Z.-g. The Effect of C-Peptide on Cognitive Dysfunction and Hippocampal Apoptosis in Type 1 Diabetic Rats. 2005:1497–1505. doi: 10.2337/diabetes.54.5.1497. [DOI] [PubMed] [Google Scholar]
- 49.Sorensen L, Molyneaux L, Yue DK, Sorensen L, Molyneaux L, Yue DK. The level of small nerve fiber dysfunction does not predict pain in diabetic Neuropathy: a study using quantitative sensory testing. Clinical Journal of Pain. 2006;22:261–265. doi: 10.1097/01.ajp.0000169670.47653.fb. [DOI] [PubMed] [Google Scholar]
- 50.Spruce MC, Potter J, Coppini DV. The pathogenesis and management of painful diabetic neuropathy: a review. Diabetic Medicine. 2003;20:88–98. doi: 10.1046/j.1464-5491.2003.00852.x. [DOI] [PubMed] [Google Scholar]
- 51.Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy - Evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932. doi: 10.2337/diabetes.49.11.1932. [DOI] [PubMed] [Google Scholar]
- 52.Willis WD. Central plastic responses to pain. Prog.Pain Res.Manage. 1994:301. [Google Scholar]
- 53.Zimmermann M. Ethical guidlines for investigations of experimental pain in conscious animals. Pain. 1983;16:109. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]