

Abstract
The cyclobutane thymine dimer is the major DNA lesion induced in human skin by sunlight and is a primary cause of skin cancer, the most prevalent form of cancer in the Northern Hemisphere. In humans, the only known cellular repair mechanism for eliminating the dimer from DNA is nucleotide excision repair. Yet the mechanism by which the dimer is recognized and removed by this repair system is not known. Here we demonstrate that the six-factor human excision nuclease recognizes and removes the dimer at a rate consistent with the in vivo rate of removal of this lesion, even though none of the six factors alone is capable of efficiently discriminating the dimer from undamaged DNA. We propose a recognition mechanism by which the low-specificity recognition factors, RPA, XPA, and XPC, act in a cooperative manner to locate the lesion and, aided by the kinetic proofreading provided by TFIIH, form a high-specificity complex at the damage site that initiates removal of thymine dimers at a physiologically relevant rate and specificity.
Keywords: Damage recognition, random assembly, cooperative binding, kinetic proofreading, xeroderma pigmentosum
Nucleotide excision repair (excision repair) is the removal of DNA lesions by dual incisions bracketing the damaged base(s) followed by filling in of the resulting gap and ligation. Excision repair was first discovered in Escherichia coli as a repair system that removes ultraviolet (UV)-induced cyclobutane thymine dimers (T<>T) from DNA (Boyce and Howard-Flanders 1964; Setlow and Carrier 1964). Subsequently, a similar repair system was discovered in humans, and it was found that patients who suffer from the disease xeroderma pigmentosum (XP), which is characterized by extreme photosensitivity and a high incidence of skin cancer, are defective in excision repair of UV damage (Cleaver and Kraemer 1995). Further work showed that this repair system removes not only T<>T but also the second most abundant UV photoproduct, the (6–4) photoproduct, and other bulky DNA lesions caused by a variety of chemical and physical agents.
In recent years, the human excision repair system has been characterized in some detail (Sancar 1996; Wood 1997). The damaged base(s) is removed from the duplex in the form of a 24–32-nt-long oligomer by dual incisions at the 6th ± 3 phosphodiester bond 3′ and the 20th ± 5 phosphodiester bond 5′ to the lesion (Huang et al. 1992). Six repair factors—RPA, XPA, XPC, TFIIH, XPG, and XPF · ERCC1—are necessary and sufficient to carry out the dual incisions and release the excised oligomer (Mu et al. 1995, 1996; Evans et al. 1997). We refer to the joint activity of these six factors as the six-factor excision nuclease. The six-factor excision nuclease system has been reconstituted in Saccharomyces cerevisiae as well (Guzder et al. 1995; Prakash and Prakash 2000) and appears to be universal in eukaryotes. The basic features of excision repair are relatively well-understood: RPA, XPA, and XPC recognize damage and recruit TFIIH (composed of six polypeptides including the XPB and XPD helicases) to the damage site. TFIIH unwinds ∼20 bp around the lesion (Evans et al. 1997; Mu et al. 1997), enabling the XPG and XPF · ERCC1 nucleases to make the 3′ and 5′ incisions, respectively, and the excised oligomer is released from the duplex.
A fundamental step in the overall reaction, the recognition of DNA damage, remains poorly defined, however. Using substrates containing an acetylaminofluorene-guanine adduct (AAF-G) or the (6–4) photoproduct in DNA-binding assays and in order-of-addition assays for repair, two general models have been proposed for the assembly of the enzyme system: “RPA/XPA First” or “XPC First.” In the RPA/XPA First model (Mu et al. 1996; Wakasugi and Sancar 1999; Missura et al. 2001), it is proposed that either RPA or XPA or the RPA/XPA complex (He et al. 1995; Li et al. 1995; Park et al. 1995) recognizes the damage first and then recruits the TFIIH · XPC complex followed by the other repair factors. In the XPC First model, this protein binds to the damage first and then recruits TFIIH and other repair factors (Sugasawa et al. 1998, 2001). The latter model appears more attractive because XPC has higher affinity for DNA than does RPA or XPA (Reardon et al. 1996). However, even though XPC can discriminate between a DNA fragment containing a (6–4) photoproduct or AAF-G and an undamaged fragment, it was reported to be incapable of discriminating T<>T-containing DNA from undamaged DNA (Batty et al. 2000; Sugasawa et al. 2001) and that, in fact, the human excision nuclease was incapable of repairing T<>T in vitro (Szymkowski et al. 1993; Sugasawa et al. 2001). Thus, it was suggested that in the case of the cyclobutane thymine dimer, the XPC First model does not apply, and it was proposed that there must be another protein that functions as the initiator of global genomic repair of T<>T (Sugasawa et al. 2001).
A reasonable candidate for the initiator of global genomic repair of T<>T is DDB (Hanawalt 2002; Tang and Chu 2002). DDB is a heterodimer of p125 (Ddb1) · p48 (Ddb2) polypeptides (Keeney et al. 1993) whose small subunit is encoded by the XPE gene (Nichols et al. 1996). DDB binds to damaged DNA with high affinity; however, it is unclear whether it plays a role in excision repair (see Hanawalt 2002). It is generally agreed that DDB is not required for the removal of many bulky lesions including the (6–4) photoproduct (Kazantsev et al. 1996; Mu et al. 1996; Rapic-Otrin et al. 1998), but it appeared to be a good candidate for the initial sensor of T<>T for several reasons. First, in contrast to XPC, it was reported that DDB specifically binds T<>T and bends the DNA around the lesion so that XPC can now bind to it (Fujiwara et al. 1999). Second, it was reported that although XP-E cells have essentially normal transcription-coupled repair, they are severely compromised in global genomic repair of T<>T (Hwang et al. 1999). Third, Chinese hamster ovary (CHO) cells, which lack DDB activity because of transcriptional silencing of DDB2 (Hwang et al. 1998), are inefficient in pyrimidine dimer (Pyr<>Pyr) repair relative to human cells (Mitchell 1988), and this deficiency can be corrected by the ectopic expression of DDB2 (Tang et al. 2000). [It must be noted, however, that, even though nearly all tested rodent cell lines repair T<>T at a slower rate than human cell lines, only CHO cells lack DDB. For example, two mouse cell lines that express DDB are equally inefficient in T<>T repair in vivo (Zolezzi and Linn 2000).] Finally, it was found that under certain conditions DDB stimulated the excision of T<>T by CHO cell-free extracts 5- to 15-fold and by the six-factor excision nuclease fivefold (Wakasugi et al. 2001, 2002). Based on these findings, a widely accepted model for repair of UV photoproducts by human nucleotide excision repair is as follows (see Hanawalt 2002). The (6–4) photoproduct is directly recognized by XPC, which binds tightly to this lesion and recruits TFIIH to the damage site; TFIIH unwinds the duplex around the lesion, enabling RPA and XPA to bind; and, finally, XPG and XPF · ERCC1 are recruited to make the dual incisions. In the case of T<>T, the damage is first recognized by DDB, which, by bending the DNA at the damage site, facilitates the binding of XPC and the cascade of subsequent reactions leading to excision. However, this model is inconsistent with certain facts, some of which were alluded to above.
To address these three interrelated issues: (1) the recognition of T<>T in human cells, (2) the role of DDB in recognition and repair of T<>T, and (3) the order of assembly of the human excision nuclease at damage sites, we conducted a systematic study of damage recognition factor binding to T<>T and (6–4) photoproducts and removal of these lesions by mammalian cell-free extracts and the human six-factor excision nuclease. Our data show that the six-factor excision nuclease is necessary and sufficient for removing T<>T at a physiologically relevant rate. We propose a model whereby the three damage recognition factors, RPA, XPA, and XPC, assemble at a damage site by a random mechanism. The low discrimination for T<>T afforded by the three damage recognition factors becomes amplified by (1) cooperative interactions among the three factors aided by (2) the kinetic proofreading function of TFIIH, which favors a four-factor (RPA, XPA, XPC, TFIIH) assembly at the damage site and disassembly from undamaged DNA.
Results
Efficient excision of thymine dimers in vitro
It has been reported that T<>T is not removed by the human excision nuclease in vitro as measured by the repair synthesis assay, which measures incorporation of radiolabel at sites of damage (Szymkowski et al. 1993), or the excision assay, which measures release of T<>T as a radiolabeled oligomer (Sugasawa et al. 2001). The lack of excision by the human excision nuclease was attributed to the inability of XPC to recognize T<>T and led to the suggestion that global T<>T repair is initiated by DDB (Hwang et al. 1999; Sugasawa et al. 2001). In support of this model, an in vitro study suggested that T<>T excision above background level was dependent on DDB (Wakasugi et al. 2001).
To test the hypothesis that DDB is the initiator of global genomic repair of T<>T, we conducted excision assays with CHO cell extracts that lack DDB and with the six-factor excision nuclease. We used undamaged DNA and a (6–4) photoproduct-containing duplex as reference substrates for background excision (gratuitous repair) and for an efficiently excised substrate, respectively. As seen in Figure 1A, T<>T was excised by CHO cell extracts at ∼15% the rate of (6–4) photoproduct. In agreement with earlier observations (Branum et al. 2001), even undamaged DNA was attacked by the excision nuclease in the cell extract at an uncorrected rate of ∼10% of T<>T substrate. Importantly, the ratio of [rate of T<>T excision]/[rate of (6–4) photoproduct excision] is comparable to the in vivo ratio observed in human cells (Mitchell 1988) and suggests that the lack of DDB in CHO cells does not adversely affect the repair of T<>T. Moreover, when the substrate was treated with the six-factor excision nuclease, essentially identical results were obtained, thus eliminating the possibility of participation of an unknown cell extract protein in T<>T recognition (Fig. 1B).
Figure 1.

Removal of UV photoproducts by mammalian excision nuclease. (A) Excision by CHO cell-free extract (CFE). Duplexes of 136–140 bp (20 fmole) with no damage (UM, unmodified), with a cyclobutane thymine dimer (T<>T), or with a (6–4) photoproduct [(6–4)PP] were incubated with CHO CFE, and excision products were separated in 10% denaturing polyacrylamide gels. (Top) Autoradiogram of a representative assay; excision products are indicated by brackets to the right; numbers to the left indicate positions of DNA size markers. The reaction time was 60 min, and the excision levels as percent of substrate were 0.3% for UM DNA (lane 2), 1.8% for T<>T (lane 4), and 9.6% for (6–4)PP (lane 6). (Bottom) Excision kinetics. The averages of two experiments are plotted. (×) UM DNA; (▵) T<>T; (○) (6–4) photoproduct. (B) Excision with reconstituted human excision nuclease, RFI–VI (repair factors RPA, XPA, XPC, TFIIH, XPG, and XPF · ERCC1). (Top) Autoradiogram of a representative gel. Excision levels after 90 min of incubation were 0.1% for UM DNA (lane 2), 2.2% for T<>T (lane 4), and 16.4% for (6–4)PP (lane 6). (Bottom) Excision kinetics with RFI–VI. The averages of two to four experiments are plotted. (×) UM DNA; (▵) T<>T; (○) (6–4) photoproduct.
Because of the sharp contrast between our results and a previous report (Sugasawa et al. 2001) suggesting that the (6–4) photoproduct, but not T<>T, is excised by the human excision nuclease, we were concerned that the T<>T-containing 8-mer used to construct our 136-bp substrate might be contaminated with (6–4) photoproduct. Even a 10%–20% level of contamination of T<>T oligomer with the (6–4) photoproduct 8-mer could explain the relative rates we obtain with nominally pure T<>T and (6–4) photoproducts. We addressed this issue by treating the T<>T substrate with the E. coli photolyase prior to using it in an excision assay. As seen in Figure 2, enzymatic photoreactivation reduced the level of excision to that observed with undamaged DNA. Thus, we conclude that all of the excision we observe from the T<>T substrate above the undamaged DNA level is exclusively due to excision of T<>T. This finding, in turn, raises the question of T<>T recognition. If this lesion is not recognized by either XPC or DDB, then what is the initiator of thymine dimer repair in mammalian cells? In an attempt to answer this question, we performed DNA-binding studies with all four factors implicated in damage recognition.
Figure 2.
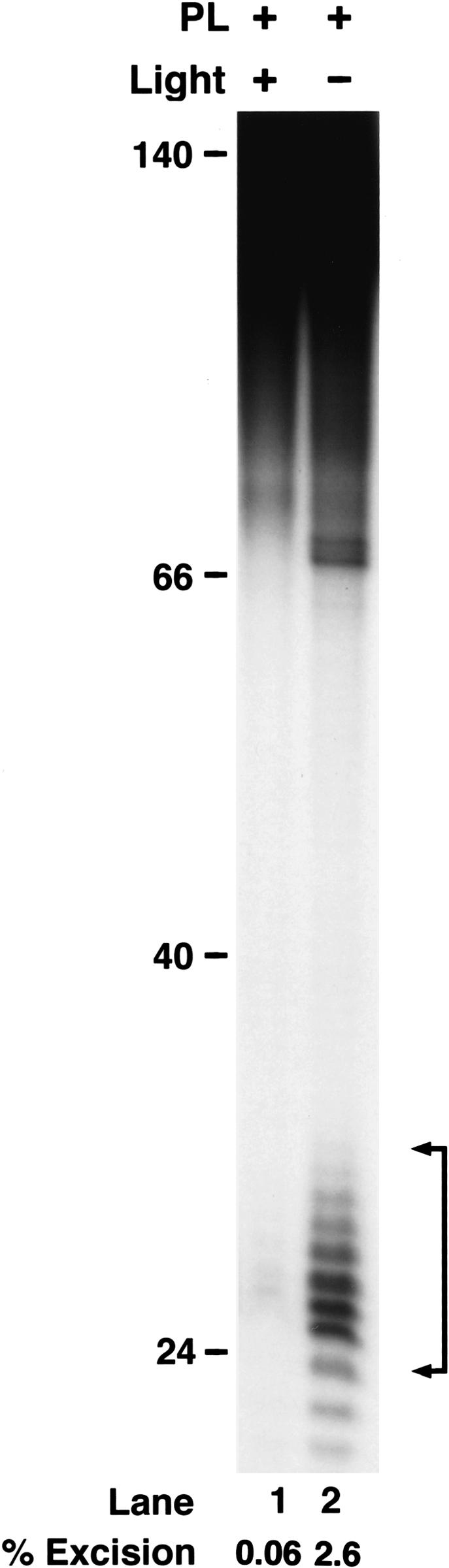
Effect of photoreactivation on excision from the T<>T substrate. The substrate was mixed with photolyase (PL) and either kept in the dark or exposed to photoreactivating light as indicated. Then the DNA was extracted with phenol/chloroform, used in excision assays with CHO cell extracts, and separated in a 10% denaturing polyacrylamide gel. The numbers below the lanes indicate the average excision from two independent experiments.
Binding of repair factors to UV photoproducts
The binding of RPA, XPA, XPC, and DDB to a 50-bp duplex with no damage, with a T<>T, or with a (6–4) photoproduct was investigated using electrophoretic mobility shift assays. The binding experiments were performed with a 50-bp duplex rather than the 136-bp substrate we routinely use in our repair studies because with the longer substrate no discrimination can be detected between undamaged duplex and a duplex with a (6–4) photoproduct (Wakasugi and Sancar 1998, 1999). In agreement with previous reports, only the (6–4) photoproduct was bound preferentially by repair factors (Fig. 3; Table 1). The best discrimination between an undamaged probe and the (6–4) photoproduct-containing duplex was achieved by DDB. The other three factors, RPA, XPA, and XPC, exhibited similar preferences for the (6–4) photoproduct relative to undamaged DNA as reported previously (Wakasugi and Sancar 1999). Importantly, this analysis failed to reveal a clear damage recognition factor for the T<>T substrate.
Figure 3.

Binding of human damage recognition proteins to UV photoproducts. Electrophoretic mobility shift assays were done with the indicated proteins and 50-bp duplexes (0.4 nM) with no lesions (UM), with a T<>T, or with a (6–4) photoproduct as indicated. The lowest and highest protein concentrations in the assays are indicated by triangles above the autoradiograms, and the positions of free DNA are indicated to the left of each panel. At the right of each panel, binding isotherms generated from average values for three to five experiments are shown. (×) UM DNA; (▵) T<>T; (○) (6–4) photoproduct. For simplicity, only curves for UM and (6–4)PP DNA are shown.
Table 1.
Affinities of human DNA damage binding proteins to UV photoproducts
| KNSa | KT<>Ta | K(6—4)a | |
|---|---|---|---|
| RPA | 5.2 × 10-7 | 6.3 × 10-7 | 2.2 × 10-7 |
| XPA | 2.2 × 10-7 | 2.1 × 10-7 | 1.5 × 10-7 |
| XPC | 3.9 × 10-8 | 3.8 × 10-8 | 2.6 × 10-8 |
| DDB | 4.0 × 10-9 | 3.1 × 10-9 | 8.0 × 10-10 |
KNS, KT<>T, and K(6—4) are the molar concentrations of repair factors at which 50% unmodified, T<>T-containing, or (6—4) photoproduct-containing 50-nt DNA (0.4 nM), respectively, were bound under conditions of excess repair factors
Because damage is recognized in vivo in the presence of excess undamaged DNA, we investigated the effect of poly(dI · dC) on the binding of repair factors with the expectation that under these conditions we might uncover a factor with a significant preference for T<>T. In many cases of specific DNA–protein interactions, inclusion of poly(dI · dC) in the reaction mixture improves the specificity of binding of the protein to its target sequence. This was, indeed, observed with DDB and the (6–4) photoproduct. Concentrations of poly(dI · dC) that reduced nonspecific binding of DDB to nearly background levels had essentially no effect on its binding to the (6–4) photoproduct in agreement with a previous report (Batty et al. 2000; data not shown). No such drastic enhancement in specificity was seen with any of the three factors, RPA, XPA, and XPC, known to be essential for excision nuclease function (data not shown). Importantly, in the presence of poly(dI · dC), the specificity for T<>T did not measurably change for RPA or XPC, and only slightly improved for XPA and DDB (Fig. 4).
Figure 4.

Binding of human damage recognition proteins to T<>T in the presence of competitive undamaged DNA. DNA substrates (5 fmole of 50-bp duplex) were mixed with indicated amounts of poly(dI · dC) and incubated with damage recognition proteins (concentrations in parentheses); then the fractions of bound radiolabeled probe were measured by electrophoretic mobility shift assays. The fractions bound in the absence of competitor were as follows. RPA (500 nM): 46.8% UM and 49.2% T<>T; XPA (200 nM): 58.1% UM and 73.2% T<>T; XPC (35 nM): 43.8% UM and 44.7% T<>T; DDB (3 nM): 42.9% UM and 70.1% T<>T. The average values for two to four independent experiments are plotted: (○) undamaged DNA; (▵) T<>T substrate.
Because these experiments did not reveal a clear damage sensor of T<>T, we were concerned that the lack of discrimination might have been due to the small size of the duplex used in the binding assays. It is conceivable that the 50-mer was too small to act as a substrate and that we were measuring mostly nonspecific (end) binding. To address this concern, we carried out excision assays with the 50-mers used in the binding assays. As shown in Figure 5, the 50-bp duplex is a substrate for the excision assay. Hence, the discrimination or lack thereof observed with the 50-mer containing a T<>T and control substrates is a valid reflection of repair factor–substrate interactions, and efficient excision of T<>T does occur even in the absence of an obvious “T<>T sensor” detectable by DNA-binding assays.
Figure 5.

Excision of DNA damage from 50-bp duplex. Substrate DNA (20 fmole), with no lesion (UM), with a T<>T, or with a (6–4) photoproduct [(6–4)PP], was incubated with CHO cell extracts for 60 min, and products were separated in a 12% denaturing gel. Excision products are indicated by brackets to the right, and numbers below the lanes indicate excision levels as a percent of substrate DNA. With the 50-mer undamaged substrate no background excision was detected (lane 2).
Effect of order of addition of repair factors on excision of T<>T
Previous experiments to determine the order of assembly of the human excision nuclease were performed with AAF-G or (6–4) photoproduct substrates, and contradictory results were reported with respect to whether preincubation of substrate with XPC or RPA and XPA led to faster rates of excision (Sugasawa et al. 1998; Wakasugi and Sancar 1999). We wished to conduct a similar type of experiment to find out if a T<>T sensor and hence an order of assembly could be identified by this approach. The results of such an order-of-addition experiment are shown in Figure 6. It appears that preincubation with XPA or RPA conferred a slight advantage at early time points relative to preincubation with XPC. The minor differences were reproducible, but were not statistically significant, and thus it is not possible to conclude from these data that a particular protein functions as the sensor of damage and initiator of global genomic repair of cyclobutane pyrimidine dimers. Clearly, order-of-addition experiments do not have the resolution to determine the order of assembly of the human excision nuclease.
Figure 6.

Effect of order-of-addition of repair factors on excision of T<>T. (A) Excision assays. Substrate (20 fmole) was preincubated with the indicated repair factor at 30°C for 10 min, then the remaining repair factors were added and incubation continued for the indicated times, and reaction products were separated in 10% denaturing polyacrylamide gels. The bracket indicates excision products. (B) Quantitative analysis of T<>T excision. The data points are averages for two to four independent experiments; for clarity, curve and error bars are shown only for the “no preincubation” reaction condition.
Effect of DDB on excision
The data presented so far indicate that T<>T can be excised relatively efficiently by the human excision nuclease in the absence of DDB. However, these data do not eliminate the possibility that DDB is the actual sensor of T<>T and that, in the absence of DDB, the photodimer is excised by an alternative and lower-efficiency pathway. We investigated the effect of DDB on excision of T<>T and (6–4) photoproduct with CHO cell-free extracts that lack DDB and with the reconstituted human excision nuclease. As seen in Figures 7 and 8, at low concentrations, DDB had a marginal (but not statistically significant) stimulatory effect on T<>T excision in both systems and had no effect at higher concentrations. At subnanomolar concentrations, DDB did not affect (6–4) photoproduct excision, and at higher concentrations it inhibited repair. We considered the possibility that the stimulatory effect of DDB may be observable only under conditions of limiting concentrations of the other damage sensors, a situation that might more closely approximate in vivo conditions. Using the reconstituted system, we empirically determined limiting concentrations of RPA, XPA, XPC, and TFIIH in our assay system and tested for a DDB effect under these conditions. The results were essentially identical to those under our optimal assay system (Fig. 8), suggesting no direct role for DDB in the repair of UV photoproducts.
Figure 7.

Effect of DDB on excision of UV photoproducts by CHO cell-free extract. Substrates with a T<>T or (6–4) photoproduct (20 and 8 fmole, respectively) were incubated with the indicated amounts of DDB, and then CHO CFE with or without pBR322 was added and incubation continued for 90 min. (A) T<>T substrate. (Top) Excision gel (-pBR322) showing only the region encompassing the excision products; excision gels in the presence of pBR322 are not shown. (Bottom) Quantitative analysis of the data: (open bar) excision in the absence of pBR322; (closed bar) excision in the presence of pBR322. In the absence of DDB, the average excision in the absence or presence of pBR322 was 0.9% and 2.3%, respectively. Other values are expressed relative to these two values within each set. (B) The (6–4) photoproduct substrate. The gel and plot are as described in A. Without DDB, the average excision of input substrate in the absence and presence of pBR322 was 9.6% and 16.5%, respectively.
Figure 8.

Effect of DDB on excision of UV photoproducts by the six-factor human excision nuclease. T<>T and (6–4) photoproduct substrates (20 and 10 fmole, respectively) were incubated with DDB as indicated, and then RFI–VI were added and incubation was continued for another 90 min. (A) T<>T substrate. (Top) Excision gel showing only the region encompassing the excision products. The reactions in lanes 1–4 were carried out with optimal concentrations of RPA, XPA, XPC, and TFIIH; those in lanes 5–8 were performed with limiting concentrations of these repair factors. In the absence of DDB, the average excision values for optimal and limiting concentrations were 2.3% and 0.6%, respectively. (Bottom) Quantitative analysis of excision data from two to three experiments. (Closed bars) Optimal reaction conditions; (open bars) limiting recognition factors. Values are relative to the reaction with no DDB within each set, and standard errors are indicated. (B) The (6–4) photoproduct substrate. (Top) Excision gels; the average percent excision in the absence of DDB under optimal reaction conditions was 28.6% and under conditions of limiting recognition factors was 5.7%. (Bottom) Bar graph of averages for two to three experiments. (Closed bars) Optimal reaction conditions; (open bars) limiting recognition factors. Values are relative to the reaction with no DDB within each set, and standard errors are indicated.
Finally, we considered the possibility that photoproduct density may affect the outcome of the excision assay. In our standard assay, the photoproduct density is relatively high (one lesion in 136 bp). We reasoned that the stimulatory effect of DDB might become apparent only under conditions approximating in vivo lesion density. Therefore, we conducted excision assays in which the photoproduct density was one lesion/3000 bp, a Pyr<>Pyr density that would be expected when cells are irradiated with 25 J/m2 of 254-nm light. As seen in Figure 7, no stimulatory effect of DDB was seen under this condition either. Thus, our data on the whole show that the six-factor excision nuclease is necessary and sufficient for carrying out excision of cyclobutane pyrimidine dimers at a physiologically relevant rate, and that the damage recognition activity of the DDB protein is not germane to the reaction mechanism of the human excision nuclease.
Discussion
We find that the six-factor human excision nuclease is capable of recognizing and repairing Pyr<>Pyr at ∼10%–20% the rate of repair of (6–4) photoproduct. This is in contrast to reports suggesting that T<>T cannot be repaired by human excision nuclease at a detectable rate in vitro (Szymkowski et al. 1993; Sugasawa et al. 2001). We have no explanation for this discrepancy. Suffice it to say that we consistently observe T<>T excision at ∼10%–20% the rate of the (6–4) photoproduct with linear DNA fragments in the 50-bp to 4-kb range and with 4–5-kb plasmids containing a single T<>T (Mu et al. 1996; Selby et al. 1997). Because the relative rates of excision for the two photoproducts that we find in vitro are comparable to those observed in human cells in vivo (Mitchell 1988), we conclude that the six-factor excision nuclease is sufficient for repairing Pyr<>Pyr in vivo without the aid of additional damage recognition factors. Our findings necessitate a reevaluation of the role of DDB in repair and of models for damage recognition by and assembly of the human excision nuclease.
Role of DDB in excision repair
In electrophoretic mobility shift assays with damaged DNA and human nuclear extracts, the most prominent DNA–protein band is that corresponding to the DDB–DNA complex (Chu and Chang 1988). Indeed, when purified DDB is compared with excision repair proteins implicated in damage recognition, it is found that DDB has the highest selectivity for both T<>T and (6–4) photoproducts (Reardon et al. 1993; Fujiwara et al. 1999; Batty et al. 2000). It is, therefore, somewhat ironic that this protein with the best discriminatory power between damaged and undamaged DNA plays either no direct role or only a minor role in nucleotide excision repair. Our results with CHO extracts and reconstituted human excision nuclease make the model of DDB-facilitated binding of XPC (or of any other repair factor) to damaged DNA quite unlikely. It has been speculated and recently indirect evidence was obtained that DDB may recognize DNA lesions in chromatin and, as part of a multiprotein complex, perform some chromatin remodeling that facilitates repair (Brand et al. 2001; Datta et al. 2001; Martinez et al. 2001). We do not find any stimulatory effect of DDB on in vitro excision from nucleosomal DNA (Hara et al. 2000). However, it is conceivable that the recently identified DDB/Cullin 4A/COP9 signalosome complex may play a role by chromatin modification and remodeling and thus facilitate repair in vivo (Shiyanov et al. 1999; Groisman et al. 2003; Liu et al. 2003). It must be noted, however, that DDB is not required for Pyr<>Pyr recognition and repair in vitro and that at least one XP-E cell line appears to carry out Pyr<>Pyr excision at a normal rate in vivo (Itoh et al. 1999). DDB seems to be involved in many other cellular processes, including transcription through its interaction with E2F1 (Hayes et al. 1998) and DNA damage checkpoint through a regulatory circuit involving p53 (Itoh et al. 2003), and it is likely that DDB influences cellular responses to DNA damage through these pathways.
In vivo studies on the order of assembly
An elegant method called focal UV irradiation has been developed to investigate redistribution of repair proteins in the cell following UV irradiation (Katsumi et al. 2001; Moné et al. 2001). Using this method, it was reported that XPC accumulated in UV foci in both wild-type and XP-A cells but that XPA did not accumulate in these foci in XP-C cells; this was taken as evidence for XPC being the damage sensor in global genomic repair (Volker et al. 2001). A separate study found that DDB accumulated very rapidly in UV foci in wild-type and XP-C cells, indicating that DDB was capable of damage recognition in the absence of XPC (Wakasugi et al. 2002); this is consistent with the model in which DDB is responsible for the primary recognition of at least some Pyr<>Pyr lesions and therefore may be the initiator of global Pyr<>Pyr repair. Finally, a third study found p53 colocalized with excision repair proteins in UV foci and that this colocalization of p53 was essential for excision repair (Rubbi and Milner 2003).
An exhaustive analysis of these studies and of the power and limitations of the focal-UV-irradiation method is beyond the scope of this discussion. The following points, however, are pertinent to the question of the order of assembly. First, TFIIH · XPC remains in UV foci that are not expected to contain (6–4) photoproducts (Volker et al. 2001); yet XPC cannot discriminate Pyr<>Pyr from undamaged DNA in any measurable way. Second, DDB accumulates in UV foci with identical kinetics in wild-type and XP-C cells (Wakasugi et al. 2002), consistent with its high affinity for damaged DNA; yet both Pyr<>Pyr and the (6–4) photoproduct are repaired at essentially the same relative rates in vitro in the absence or presence of DDB. According to at least one report, the same is also true in vivo—at best, DDB has only a modest effect on the rate of repair (Itoh et al. 1999). Finally, the accumulation of p53 in UV foci is most likely related to its checkpoint or chromatin remodeling functions, as repeated attempts to detect an effect of p53 on excision repair have shown neither stimulation nor inhibition (Sancar 1995; Leveillard et al. 1996).
Model for human excision nuclease: damage recognition by cooperativity and kinetic proofreading
At present there are two models for the assembly of the human excision nuclease: “RPA/XPA First” or “XPC First” and its variant “DDB-aided XPC First” (see Hanawalt 2002; Tang and Chu 2002). The data presented in this paper show that, in the case of Pyr<>Pyr, arguably the physiologically most relevant substrate for human nucleotide excision repair (Brash 1997), the two criteria (damage-binding specificity and effect of order of addition on repair rate) used to elaborate the two order-of-assembly models do not apply. Hence, an alternative model, within the context of the general problem of specific DNA–protein interactions, is presented below.
For proper execution of their specific functions, DNA-binding proteins must be able to recognize their targets (a unique sequence, structure, or lesion) with high affinities, but very high specific affinities are invariably associated with high nonspecific affinities as well. Within a cell, a protein possessing such a property may be mostly sequestered at nonspecific sites. A classic example of this problem is the XL186 mutant of the Lac repressor, which has higher affinities than the wild-type repressor for both the lac operator and nonoperator DNA. In cells carrying this mutation, repression occurs in the presence of lactose (or IPTG), which normally causes derepression. This abnormal behavior is explained as follows (Ptashne and Gann 2002). In an uninduced cell, the mutant repressor is sequestered on nonspecific sites because of its increased overall DNA affinity and low concentration in the cell. In the presence of IPTG, the specific and nonspecific affinities are lowered to a level comparable to those of the wild-type repressor, resulting in repression rather than induction by IPTG. Hence, high specificity, in particular in mammalian cells, cannot be achieved by increasing the affinity of a single protein to its target. The specificity is achieved either by cooperativity (Ptashne and Gann 2002) or by kinetic proofreading (Hopfield 1974), or, as in the case of human excision repair, by both. A model that incorporates both concepts for the human excision nuclease is presented in Figure 9.
Figure 9.

Model for human excision nuclease. Damage is recognized by RPA, XPA, and XPC in a “cooperative” manner, and the order of binding of these factors is “random.” Recruitment of TFIIH to form the four-factor preincision complex PIC1 and the ensuing helix unwinding provide an additional degree of specificity by “kinetic proofreading.” Further specificity is conferred by the preferential binding of XPG to the unwound DNA (PIC2). PIC3, a short-lived intermediate, is formed upon entry of XPF · ERCC1 and leads to dual incisions/excision.
In cooperative binding, specificity is achieved by binding of two or more interacting proteins to adjacent or overlapping sites on DNA such that occupancy of one site by a protein facilitates occupancy of adjacent sites by other proteins by increasing the local concentration of and imposing proper orientation on other proteins through protein–protein interactions. In human excision repair, damage may initially be detected by RPA, XPA, or XPC (“random” rather than “ordered” assembly). The cooperative actions of XPA (which binds RPA and TFIIH), RPA, and XPC (which binds TFIIH; Drapkin et al. 1994) leads to formation of the four-factor preincision complex 1 (PIC1) regardless of the order of initial contact. Within PIC1, ∼20 bp around the lesion are unwound by the TFIIH helicase, leading to more intimate contacts between repair factors and DNA (Reardon and Sancar 2002). The moderate specificity provided by cooperative binding of RPA, XPA, and XPC is amplified by the kinetic proofreading function of TFIIH.
In kinetic proofreading, one or more high-energy intermediates are placed in the reaction pathway such that nonspecific (or erroneous) products can be aborted at any step along the reaction pathway without seriously compromising the rate of the specific reaction by overspecification at the initial binding step (Hopfield 1974). In excision repair, once TFIIH is recruited by the three damage recognition factors to form PIC1, the DNA is unwound by ∼20 bp at the assembly site by the helicase activities of XPB and XPD. If the assembly is at a nondamage site, ATP hydrolysis by TFIIH aborts the reaction (kinetic proofreading). In contrast, PIC1 formed at a damage site is more stable, and the unwound DNA constitutes a high-affinity binding site for XPG (Hohl et al. 2003). The binding of XPG, concurrent with dissociation of XPC from the complex (Wakasugi and Sancar 1998), leads to formation of PIC2, which by virtue of XPG entry and XPC exit provides an additional layer of specificity. Finally, entry of XPF · ERCC1 into the complex to form PIC3 provides an additional proofreading opportunity because even at this stage the reaction can be aborted (Wakasugi et al. 1997).
To summarize, the human excision nuclease system uses cooperative binding and kinetic proofreading to achieve biologically acceptable specificity at a physiologically relevant rate. It must be noted, however, that despite all these thermodynamic and kinetic safeguards aimed at ensuring excision of only damaged nucleotides, the discrimination of the human excision nuclease between undamaged and damaged DNA is not absolute, and the enzyme excises oligomers from undamaged DNA (gratuitous repair) at a significant and potentially mutagenic rate (Branum et al. 2001).
Materials and methods
Repair factors
CHO AA8 cells were grown in 10-L suspension cultures (Eagle's minimal essential medium + 10% FCS) and harvested while in log phase. Cell-free extracts (CFE) were prepared and stored as described (Reardon et al. 1997). The his-tagged XPF · ERCC1 heterodimer was expressed in SF21 cells and purified by sequential chromatography on SP Sepharose and Ni-NTA columns (Amersham Biosciences; QIAGEN). Other essential repair factors (XPA, RPA, TFIIH, XPC · hR23B, and XPG), DDB (Ddb1 · Ddb2 heterodimer), and E. coli photolyase were purified using schemes similar to those described previously (Sancar et al. 1984; Keeney et al. 1993; Henricksen et al. 1994; Mu et al. 1995, 1996; Matsunaga et al. 1996; Reardon et al. 1996). Although referred to as XPC in the text, the XPC · hR23B heterodimer was used for experiments described in this work. TFIIH was purified from HeLa cells, tested by ECL detection of Western blots (Amersham Biosciences), and found to contain <1 ng of Ddb1 in 150 ng of TFIIH (data not shown).
DNA substrates
For most excision assays, the substrates were internally radiolabeled 136-bp or 140-bp duplexes prepared as described (Huang et al. 1994). Unmodified oligomers were from Operon Technologies and the 8-mers containing UV-induced damage were prepared and isolated as described (Zhao et al. 1995). Substrates containing UV-induced lesions, the cis–syn cyclobutane thymine–thymine dimer (T<>T), and the (6–4) photoproduct (6–4PP) were 136 bp in length, and the unmodified (UM) substrate was a 140-bp duplex. Enzymatic photoreactivation of the T<>T lesion was accomplished by irradiating photolyase–DNA complexes with 366-nm light (Sancar et al. 1984). For electrophoretic mobility shift assays, and some excision assays, internally radiolabeled 50-bp duplexes were prepared in a similar manner using four oligomers. The sequences of these 50-bp duplexes correspond to nucleotide positions 44–93 of the 136-bp substrate (Hara et al. 2000) and nucleotide positions 46–95 of the 140-bp substrate (Reardon and Sancar 2002).
Excision assay
Assays with CHO CFE were conducted in 25-μL reaction mixtures as described (Reardon et al. 1997). Unless indicated otherwise, the reaction mixtures included 75 μg of CFE, 20 fmole of substrate DNA, and 35 ng of pBR322; incubation was at 30°C for 60–90 min. For kinetic analyses, aliquots were removed at the indicated time points and processed as described below. For experiments with DDB, there was a 10-min preincubation of the substrate with DDB at 30°C, and then 75 μg of CFE was added; incubation continued at 30°C for 60–90 min. Excision assays reconstituted with purified repair factors (RFI–VI) were performed using reaction conditions as described (Reardon and Sancar 2002) with 50 nM XPA, 200 nM RPA, 12.5 nM TFIIH, 2.5 nM XPC · hR23B, 4.5 nM XPF · ERCC1, and 3 nM XPG. Unless indicated otherwise, incubations were with these protein concentrations at 30°C for 90 min, and reaction mixtures did not contain pBR322. The 25-μL reactions typically contained 8–20 fmole of (6–4)PP or 20 fmole of other substrates. In order-of-addition experiments, DNA was preincubated with damage recognition factors at 30°C for 10 min, and then the missing repair factors were added and incubation was continued at 30°C for the indicated times. For suboptimal excision experiments, we used limiting concentrations of repair factors: 25 nM XPA, 33 nM RPA, 6 nM TFIIH, 0.6 nM XPC · hR23B, 4.5 nM XPF · ERCC1, and 3 nM XPG. To detect and quantify excision products, deproteinized DNA was resolved in 10%–12% denaturing (7.7 M urea) polyacrylamide gels with radiolabeled HinfI-digested ΦX 174 DNA (Promega) as size markers, visualized by autoradiography, and quantitated using the Storm 860 system and ImageQuant 5.2 software (Amersham Biosciences).
Electrophoretic mobility shift assay
Repair factors and 5 fmole of 50-bp duplexes were incubated in 12.5-μL reaction mixtures under reconstitution assay reaction conditions at 30°C for 30 min, except ATP was omitted. Glycerol was added to ∼8.5%, and samples were resolved in 5% nondenaturing polyacrylamide gels, with electrophoresis at room temperature and a constant current of 25 mA. Visualization and quantitation were as described above, and binding isotherms were generated from these data.
Acknowledgments
We thank R. Hara for the UV-damaged 8-mers, T. Matsunaga and M. Wakasugi (Kanazawa University) for his-tagged XPF and ERCC1 baculovirus stocks, K. Ünsal-Kaçmaz (University of North Carolina, Chapel Hill) for purified RPA, and P. Raychaudhuri (University of Illinois, Chicago) for DDB antibodies. We thank T. Itoh for a crucial role in initiating this work and T. Itoh and S. Linn (University of California, Berkeley) for communicating data prior to publication. This work was supported by NIH grant GM32833.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1131003.
References
- Batty D., Rapic-Otrin, V., Levine, A.S., and Wood, R.D. 2000. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J. Mol. Biol. 300: 275-290. [DOI] [PubMed] [Google Scholar]
- Boyce R.P. and Howard-Flanders, P. 1964. Release of ultraviolet light-induced thymine dimers from DNA in E. coli K-12. Proc. Natl. Acad. Sci. 51: 293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M., Moggs, J.G., Oulad-Abdelghani, M., Lejeune, F., Dilworth, F.J., Stevenin, J., Almouzni, G., and Tora, L. 2001. UV-damaged DNA-binding protein in the TFTC complex links DNA damage recognition to nucleosome acetylation. EMBO J. 20: 3187-3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branum M.E., Reardon, J.T., and Sancar, A. 2001. DNA repair excision nuclease attacks undamaged DNA. J. Biol. Chem. 276: 25421-25426. [DOI] [PubMed] [Google Scholar]
- Brash D.E. 1997. Sunlight and the onset of skin cancer. Trends Genet. 13: 410-414. [DOI] [PubMed] [Google Scholar]
- Chu G. and Chang, E. 1988. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 242: 564-567. [DOI] [PubMed] [Google Scholar]
- Cleaver J.E. and Kraemer, K.H. 1995. Xeroderma pigmentosum and Cockayne syndrome. In The metabolic basis of inherited disease, 7th ed. (eds. C.R. Scriver et al.), Vol. 3, pp. 4393-4419. McGraw-Hill, New York. [Google Scholar]
- Datta A., Bagchi, S., Nag, A., Shiyanov, P., Adami, G.R., Yoon, T., and Raychaudhuri, P. 2001. The p48 subunit of the damaged-DNA binding protein DDB associates with the CBP/p300 family of histone acetyltransferase. Mutation Res. 486: 89-97. [DOI] [PubMed] [Google Scholar]
- Drapkin R., Reardon, J.T., Ansari, A., Huang, J.-C., Zawel, L., Ahn, K., Sancar, A., and Reinberg, D. 1994. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature 368: 769-772. [DOI] [PubMed] [Google Scholar]
- Evans E., Moggs, J.G., Hwang, J.R., Egly, J.-M., and Wood, R.D. 1997. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 16: 6559-6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y., Masutani, C., Mizukoshi, T., Kondo, J., Hanaoka, F., and Iwai, S. 1999. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J. Biol. Chem. 274: 20027-20033. [DOI] [PubMed] [Google Scholar]
- Groisman R., Polanowska, J., Kuraoka, I., Sawada, J., Saijo, M., Drapkin, R., Kisselev, A.F., Tanaka, K., and Nakatani, Y. 2003. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113: 357-367. [DOI] [PubMed] [Google Scholar]
- Guzder S.N., Habraken, Y., Sung, P., Prakash, L., and Prakash, S. 1995. Reconstitution of yeast nucleotide excision repair with purified Rad proteins, replication protein A, and transcription factor TFIIH. J. Biol. Chem. 270: 12973-12976. [DOI] [PubMed] [Google Scholar]
- Hanawalt P.C. 2002. Subpathways of nucleotide excision repair and their regulation. Oncogene 21: 8949-8956. [DOI] [PubMed] [Google Scholar]
- Hara R., Mo, J., and Sancar, A. 2000. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 20: 9173-9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes S., Shiyanov, P., Chen, X., and Raychaudhuri, P. 1998. DDB, a putative DNA repair protein, can function as a transcriptional partner of E2F1. Mol. Cell. Biol. 18: 240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., Henricksen, L.A., Wold, M.S., and Ingles, C.J. 1995. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 374: 566-569. [DOI] [PubMed] [Google Scholar]
- Henricksen L.A., Umbricht, C.B., and Wold, M.S. 1994. Recombinant replication protein A: Expression, complex formation, and functional characterization. J. Biol. Chem. 269: 11121-11132. [PubMed] [Google Scholar]
- Hohl M., Thorel, F., Clarkson, S.G., and Schärer, O.D. 2003. Structural determinants for substrate binding and catalysis by the structure-specific endonuclease XPG. J. Biol. Chem. 278: 19500-19508. [DOI] [PubMed] [Google Scholar]
- Hopfield J.J. 1974. Kinetic proofreading: A new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. 71: 4135-4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.-C., Svoboda, D.L., Reardon, J.T., and Sancar, A. 1992. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. 89: 3664-3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.-C., Hsu, D.S., Kazantsev, A., and Sancar, A. 1994. Substrate spectrum of human excinuclease: Repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc. Natl. Acad. Sci. 91: 12213-12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B.J., Toering, S., Francke, U., and Chu, G. 1998. p48 activates a UV-damaged-DNA binding factor and is defective in xeroderma pigmentosum group E cells that lack binding activity. Mol. Cell. Biol. 18: 4391-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang B.J., Ford, J.M., Hanawalt, P.C., and Chu, G. 1999. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. 96: 424-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T., Mori, T., Ohkubo, H., and Yamaizumi, M. 1999. A newly identified patient with clinical xeroderma pigmentosum phenotype has a non-sense mutation in the DDB2 gene and incomplete repair in (6–4) photoproducts. J. Invest. Dermatol. 113: 251-257. [DOI] [PubMed] [Google Scholar]
- Itoh T., O'Shea, C., and Linn, S. 2003. Impaired regulation of tumor suppressor p53 caused by mutations in the xeroderma pigmentosum DDB2 gene: A regulatory interaction between p53 and p48DDB2. Mol. Cell. Biol. (in press). [DOI] [PMC free article] [PubMed]
- Katsumi S., Kobayashi, N., Imoto, K., Nakagawa, A., Yamashina, Y., Muramatsu, T., Shirai, T., Miyagawa, S., Sugiura, S., Hanaoka, F., et al. 2001. In situ visualization of ultraviolet-light-induced DNA damage repair in locally irradiated human fibroblasts. J. Invest. Dermatol. 117: 1156-1161. [DOI] [PubMed] [Google Scholar]
- Kazantsev A., Mu, D., Nichols, A.F., Zhao, X., Linn, S., and Sancar, A. 1996. Functional complementation of xeroderma pigmentosum complementation group E by replication protein A in an in vitro system. Proc. Natl. Acad. Sci. 93: 5014-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S., Chang, G.J., and Linn, S. 1993. Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J. Biol. Chem. 268: 21293-21300. [PubMed] [Google Scholar]
- Leveillard T., Andera, L., Bissonnette, N., Schaeffer, L., Bracco, L., Egly, J.M., and Wasylyk, B. 1996. Functional interactions between p53 and the TFIIH complex are affected by tumour-associated mutations. EMBO J. 15: 1615-1624. [PMC free article] [PubMed] [Google Scholar]
- Li L., Lu, X., Peterson, C.A., and Legerski, R.J. 1995. An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol. Cell. Biol. 15: 5396-5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Powell, K.A., Mundt, K., Wu, L., Carr, A.M., and Caspari, T. 2003. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes & Dev. 17: 1130-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E., Palhan, V.B., Tjernberg, A., Lymar, E.S., Gamper, A.M., Kundu, T.K., Chait, B.T., and Roeder, R.G. 2001. Human STAGA complex is a chromatin-acetylating transcription coactivator that interacts with pre-mRNA splicing and DNA damage-binding factors in vivo. Mol. Cell. Biol. 21: 6782-6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga T., Park, C.-H., Bessho, T., Mu, D., and Sancar, A. 1996. Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J. Biol. Chem. 271: 11047-11050. [DOI] [PubMed] [Google Scholar]
- Missura M., Buterin, T., Hindges, R., Hübscher, U., Kaspárková, J., Brabec, V., and Naegeli, H. 2001. Double-check probing of DNA bending and unwinding by XPA-RPA: An architectural function in DNA repair. EMBO J. 20: 3554-3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D.L. 1988. The relative cytotoxicity of (6–4) photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol. 48: 51-57. [DOI] [PubMed] [Google Scholar]
- Moné M.J., Volker, M., Nikaido, O., Mullenders, L.H.F., van Zeeland, A.A., Verschure, P.J., Manders, E.M.M., and van Driel, R. 2001. Local UV-induced DNA damage in cell nuclei results in local transcription inhibition. EMBO Rep. 2: 1013-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D., Park, C.-H., Matsunaga, T., Hsu, D.S., Reardon, J.T., and Sancar, A. 1995. Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270: 2415-2418. [DOI] [PubMed] [Google Scholar]
- Mu D., Hsu, D.S., and Sancar, A. 1996. Reaction mechanism of human DNA repair excision nuclease. J. Biol. Chem. 271: 8285-8294. [DOI] [PubMed] [Google Scholar]
- Mu D., Wakasugi, M., Hsu, D.S., and Sancar, A. 1997. Characterization of reaction intermediates of human excision repair nuclease. J. Biol. Chem. 272: 28971-28979. [DOI] [PubMed] [Google Scholar]
- Nichols A.F., Ong, P., and Linn, S. 1996. Mutations specific to the xeroderma pigmentosum group E Ddb- phenotype. J. Biol. Chem. 271: 24317-24320. [DOI] [PubMed] [Google Scholar]
- Park C.-H., Mu, D., Reardon, J.T., and Sancar, A. 1995. The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J. Biol. Chem. 270: 4896-4902. [DOI] [PubMed] [Google Scholar]
- Prakash S. and Prakash, L. 2000. Nucleotide excision repair in yeast. Mutation Res. 451: 13-24. [DOI] [PubMed] [Google Scholar]
- Ptashne M. and Gann, A. 2002. Genes & signals. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rapic-Otrin V., Kuraoka, I., Nardo, T., McLenigan, M., Eker, A.P.M., Stefanini, M., Levine, A.S., and Wood, R.D. 1998. Relationship of the xeroderma pigmentosum group E DNA repair defect to the chromatin and DNA binding proteins UV-DDB and replication protein A. Mol. Cell. Biol. 18: 3182-3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon J.T. and Sancar, A. 2002. Molecular anatomy of the human excision nuclease assembled at sites of DNA damage. Mol. Cell. Biol. 22: 5938-5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon J.T., Nichols, A.F., Keeney, S., Smith, C.A., Taylor, J.-S., Linn, S., and Sancar, A. 1993. Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6–4]T, and T[Dewar]T. J. Biol. Chem. 268: 21301-21308. [PubMed] [Google Scholar]
- Reardon J.T., Mu, D., and Sancar, A. 1996. Overproduction, purification, and characterization of the XPC subunit of the human DNA repair excision nuclease. J. Biol. Chem. 271: 19451-19456. [DOI] [PubMed] [Google Scholar]
- Reardon J.T., Thompson, L.H., and Sancar, A. 1997. Rodent UV-sensitive mutant cell lines in complementation groups 6–10 have normal general excision repair activity. Nucleic Acids Res. 25: 1015-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi C.P. and Milner, J. 2003. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 22: 975-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A. 1995. Excision repair in mammalian cells. J. Biol. Chem. 270: 15915-15918. [DOI] [PubMed] [Google Scholar]
- ____. 1996. DNA excision repair. Annu. Rev. Biochem. 65: 43-81. [DOI] [PubMed] [Google Scholar]
- Sancar A., Smith, F.W., and Sancar, G.B. 1984. Purification of Escherichia coli DNA photolyase. J. Biol. Chem. 259: 6028-6032. [PubMed] [Google Scholar]
- Selby C.P., Drapkin, R., Reinberg, D., and Sancar, A. 1997. RNA polymerase II stalled at a thymine dimer: Footprint and effect on excision repair. Nucleic Acids Res. 25: 787-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlow R.B. and Carrier, W.L. 1964. The disappearance of thymine dimers from DNA: An error-correcting mechanism. Proc. Natl. Acad. Sci. 51: 226-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiyanov P., Nag, A., and Raychaudhuri, P. 1999. Cullin 4A associates with the UV-damaged DNA-binding protein DDB. J. Biol. Chem. 274: 35309-35312. [DOI] [PubMed] [Google Scholar]
- Sugasawa K., Ng, J.M.Y., Masutani, C., Iwai, S., van der Spek, P.J., Eker, A.P.M., Hanaoka, F., Bootsma, D., and Hoeijmakers, J.H.J. 1998. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2: 223-232. [DOI] [PubMed] [Google Scholar]
- Sugasawa K., Okamoto, T., Shimizu, Y., Masutani, C., Iwai, S., and Hanaoka, F. 2001. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes & Dev. 15: 507-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymkowski D.E., Lawrence, C.W., and Wood, R.D. 1993. Repair by human cell extracts of single (6–4) and cyclobutane thymine–thymine photoproducts in DNA. Proc. Natl. Acad. Sci. 90: 9823-9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. and Chu, G. 2002. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair 1: 601-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.Y., Hwang, B.J., Ford, J.M., Hanawalt, P.C., and Chu, G. 2000. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell 5: 737-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker M., Moné, M.J., Karmakar, P., van Hoffen, A., Schul, W., Vermeulen, W., Hoeijmakers, J.H.J., van Driel, R., van Zeeland, A.A., and Mullenders, L.H.F. 2001. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8: 213-224. [DOI] [PubMed] [Google Scholar]
- Wakasugi M. and Sancar, A. 1998. Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc. Natl. Acad. Sci. 95: 6669-6674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 1999. Order of assembly of human DNA repair excision nuclease. J. Biol. Chem. 274: 18759-18768. [DOI] [PubMed] [Google Scholar]
- Wakasugi M., Reardon, J.T., and Sancar, A. 1997. The noncatalytic function of XPG protein during dual incision in human nucleotide excision repair. J. Biol. Chem. 272: 16030-16034. [DOI] [PubMed] [Google Scholar]
- Wakasugi M., Shimizu, M., Morioka, H., Linn, S., Nikaido, O., and Matsunaga, T. 2001. Damaged DNA-binding protein DDB stimulates the excision of cyclobutane pyrimidine dimers in vitro in concert with XPA and replication protein A. J. Biol. Chem. 276: 15434-15440. [DOI] [PubMed] [Google Scholar]
- Wakasugi M., Kawashima, A., Morioka, H., Linn, S., Sancar, A., Mori, T., Nikaido, O., and Matsunaga, T. 2002. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J. Biol. Chem. 277: 1637-1640. [DOI] [PubMed] [Google Scholar]
- Wood R.D. 1997. Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272: 23465-23468. [DOI] [PubMed] [Google Scholar]
- Zhao X., Kao, J.L.-F., and Taylor, J.-S. 1995. Preparation and characterization of a deoxyoligonucleotide 49-mer containing a site-specific thymidylyl-(3′,5′)-deoxyadenosine photoproduct. Biochemistry 34: 1386-1392. [DOI] [PubMed] [Google Scholar]
- Zolezzi F. and Linn, S. 2000. Studies of the murine DDB1 and DDB2 genes. Gene 245: 151-159. [DOI] [PubMed] [Google Scholar]