

Abstract
Transcription by σ54 RNA polymerase depends on activators that contain ATPase domains of the AAA+ class. These activators, which are often response regulators of two-component signal transduction systems, remodel the polymerase so that it can form open complexes at promoters. Here, we report the first crystal structures of the ATPase domain of an activator, the NtrC1 protein from the extreme thermophile Aquifex aeolicus. This domain alone, which is active, crystallized as a ring-shaped heptamer. The protein carrying both the ATPase and adjacent receiver domains, which is inactive, crystallized as a dimer. In the inactive dimer, one residue needed for catalysis is far from the active site, and extensive contacts among the domains prevent oligomerization of the ATPase domain. Oligomerization, which completes the active site, depends on surfaces that are buried in the dimer, and hence, on a rearrangement of the receiver domains upon phosphorylation. A motif in the ATPase domain known to be critical for coupling energy to remodeling of polymerase forms a novel loop that projects from the middle of an α helix. The extended, structured loops from the subunits of the heptamer localize to a pore in the center of the ring and form a surface that could contact σ54.
Keywords: AAA+ ATPase, crystal structure, σ54, transcriptional activator, response regulator, two-component system
Activators of the alternative σ54-holoenzyme form of RNA polymerase catalyze an ATP-dependent conformational change in the polymerase that allows it to form transcriptionally productive open complexes at promoters (Rombel et al. 1998; Buck et al. 2000; Zhang et al. 2002). Remodeling depends on previous binding of the polymerase to a promoter in a closed complex. The activators, which are usually composed of regulatory, oligomerization/ATPase, and DNA-binding domains, respond to different environmental signals and are unusual in binding to DNA enhancer-like sequences between 100 and 150 bp upstream of promoters. In many cases, their activity is controlled by phosphorylation of an aspartate residue in the regulatory domain, which induces a conformational change that allows the central ATPase domain to oligomerize. Oligomerization is essential for ATP hydrolysis and productive contact with σ54-holoenzyme. Although the structures of isolated regulatory and DNA-binding domains have been solved (Kern et al. 1999; Meyer et al. 2001; Park et al. 2002a), there is not yet a structure for the oligomerization/ATPase domain of an activator of σ54-holoenzyme, and hence, many questions remain unanswered. What are the molecular bases for regulation of the central ATPase domain by the regulatory domain? How do the activators, which are dimers in their inactive state, form active oligomers, which appear to be hexamers or octamers? How is ATP hydrolysis by the activators coupled to the conformational change in σ54-holoenzyme that allows DNA melting at promoters? We have begun structural studies of NtrC1 (GI: 2983588) from the hyperthermophilic bacterium Aquifex aeolicus to try to answer some of these questions. Although this protein was classified as a nitrogen regulatory protein C (NtrC) family member on the basis of its high amino acid similarity (59%) to the well-studied NtrC from Salmonella enterica serovar typhimurium, the genes regulated by NtrC1 and the environmental signals controlling its function have not been identified (Deckert et al. 1998). Here, we describe in both its inactive and active states, the structure of the ATPase domain of NtrC1. We compare the structure of the active ATPase with those of other ATPases associated with various cellular activities (AAA+) proteins, namely, N-ethylmaleimide sensitive factor (NSF), which dissociates a complex of SNAREs [soluble NSF attachment protein (SNAP) receptors] to allow further cycles of membrane fusion (Lenzen et al. 1998; Yu et al. 1998); p97, which is thought to have an analogous role in disassembling SNARE complexes;heat-shock locus protein U (HslU), which completes the unfolding of partially unfolded proteins and transfers them to the protease HslV (Bochtler et al. 2000); RuvB, which promotes DNA branch migration at Holliday junctions during genetic recombination; and the clamp loader γ complex, which opens the β-sliding clamp ring and loads it onto DNA to mediate processive movement of DNA polymerase (Neuwald et al. 1999; Vale 2000; Jeruzalmi et al. 2001).
Results
Overall architecture of the protein structures
The structure of the joined N-terminal regulatory and central ATPase domains (NtrC1RC) of NtrC1 from Aquifex aeolicus and the structure of the isolated central domain (NtrC1C) were determined to resolutions of 2.4 Å and 3.1 Å, respectively, by X-ray crystallography (Table 1). Both were in their ADP-bound form. NtrC1RC crystallized with a dimer in the asymmetric unit, whereas NtrC1C crystallized with two heptamers (Fig. 1). This change allows us to infer the fundamental process by which the protein is activated, the details of which are discussed below.
Table 1.
Data collection, phasing, refinement statistics
| Crystal | NtrClRC(dimer) | NtrClC(heptamer) |
| Space group | P3221 | P1 |
| Cell parameters (Å) | a = b = 94.76, c = 195.01 | a = 106.79, b = 108.26, c = 110.02 |
| α = 70.25, β = 85.90, γ = 73.27 | ||
| Source | ALS 5.0.2 | ALS 8.3.1 |
| Data and Phasing | ||
| Resolution (Å) | 50.0-2.4 | 62.02-3.1 |
| Wavelength (Å) | 0.9793 | 1.0 |
| Completeness (%) | 99.4 (83.4) | 98.0 (97.1) |
| Rsym (%)a | 8.0 (41.5) | 6.1 (42.1) |
| Rcullis (ano)b | 0.38 | — |
| Figure of meritc | 0.52 | — |
| Refinement Statistics | ||
| Resolution (Å) | 42.6-2.4 | 20.0-3.1 |
| No. of reflections | 36719 | 78375 |
| No. of atoms | Protein/ADP/solvent 6192/269/54 | 27677/378/0 |
| No. of molecules/AU | 2 | 14 |
| R factors (%)d | Rwork/Rfree, 21.3/25.7 | 26.6/32.9 |
| R.m.s.d of bondse | Length/angle, 0.007 Å/1.4° | 0.01 Å/1.6° |
Rsym = ∑|Ii - <Ii>|/∑Ii, where <Ii> is the average intensity of symmetry equivalent reflections. Numbers in parenthesis are statistics for the highest resolution bin. Same for completeness
Rcullis (ano) = ∑|Δano(obs) - Δano(cal)|/Δano(obs) for acentric reflections, where Δano(obs) is the anomalous difference
Both figure of merit and Rcullis were calculated with reflections up to 3.0 Å
R factor = ∑|F(obs) - F(calc)|/∑ F(obs)
R.m.s.d = root-mean-square deviations
Figure 1.

Overall structures of NtrC1RC and NtrC1C. (A) Structure of NtrC1RC. NtrC1RC forms a dimer in the asymmetric unit. The monomers are colored gray and gold. The GAFTGA and β-hairpin insertions into the central ATPase domain are colored blue and green, respectively. ADP and the catalytic arginine residue (R293) are shown using a ball-and-stick representation. R293 cannot contact the nucleotide in the inactive dimer. (B) Structure of the NtrC1C heptamer. At left is a top view illustrating how the protomers pack in the context of the heptamer. Each protomer is labeled with a letter, A through G. The GAFTGA insertion (blue) and β-hairpin (green) comprise the pore in the heptamer. The Sensor II helix is colored cyan. At right is a side view.
Structure of the inactive dimer
NtrC1RC is a dimer in which the regulatory domain of each monomer is adjacent to the central domain of its dimeric partner (Fig. 1A). The regulatory domain, often called a receiver domain in the context of two-component signal transduction (Nixon et al. 1986), has the expected (α/β)5-fold and contains the conserved active site residues grouped together as they were in many previous structures of homologs. However, NtrC1R has a substantial extension of C-terminal helix 5 (to residue 135, ∼10 residues longer than the equivalent helix in S. typhimurium NtrC), and the extensions from the two monomers form a pair of crossed helices that are major dimerization determinants of NtrC1RC. The dimerization interface between the two receiver domains and the extended helices is very similar to that seen for a homologous σ54 activator, dicarboxylate transport regulator D (DctD) of Sinorhizobium meliloti (Meyer et al. 2001; Park et al. 2002a).
The central ATPase domain (residues 143-387) of NtrC1 is composed of an α/β subdomain that is typical of P-loop NTPases and an α-helical subdomain that is a distinctive feature of AAA+ ATPases (Neuwald et al. 1999). The five β-strands in the central ATPase domain of NtrC1RC could be superimposed on those of NSF domain 2 (D2), considered a prototype structure of a AAA+ ATPase (Neuwald et al. 1999), with an r.m.s.d of 1.0 Å between the aligned segments (proteins displayed side-by-side, Fig. 2A). It is known that these five β-strands are structurally conserved among AAA+ ATPases (Bochtler et al. 2000). Although the tilts and lengths of the α helices in the α/β subdomain are quite different in the two cases, the overall structural folds are similar.
Figure 2.

The GAFTGA insertion forms an unusual structural motif. (A) Comparison of the central ATPase domain of NtrC1RC (left) and NSF D2 (right). The α/β and α-helical subdomains are colored gray and light blue, respectively, whereas the helix carrying the GAFTGA motif and the β-hairpin insertion in NtrC1 are colored blue and green, respectively. The putative Sensor I residue (N280) and the Sensor II residue (R357) of NtrC1 are shown using a ball-and-stick representation. Note that ADP is bound in NtrC1RC, but ATP is bound in NSF D2. Arrows indicate the insertions in NtrC1 relative to NSF D2. (B) A close view of the GAFTGA insertion. Two H bonds, typical of anti-parallel β-strands, are shown as broken lines. Sequence alignments among σ54 activators and other AAA+ ATPases are shown with the conserved sequences highlighted. The sequences that form the last turn of H8 in other AAA+ ATPases align well with those of σ54 activators. (C) Omit map of the GAFTGA region including H8 (residues 203-232) with density contoured at 3.5σ.
There are two notable insertions in NtrC1 that have no counterpart in NSF D2 (Fig. 2). These two ordered structures are in helix 8 (H8) and after helix 9 (H9). The β-hairpin loop after H9 contacts the insertion in H8. There are β-hairpin loops after H9 in two other AAA+ ATPases, HslU and RuvB (Bochtler et al. 2000; Han et al. 2001; Yamada et al. 2001). The insertion in H8 forms an unusual structural motif, an ordered loop that projects out of the middle of this helix (Fig. 2B). At the base, it is connected to H8 by two extended strands (containing two H bonds typical of anti-parallel β sheets). The segments of H8 before and after the insertion have a similar helical axis, but are separated by one helical turn as a result of the insertion. Comparison with structures of the corresponding regions of other AAA+ ATPases shows that the two helical segments of H8 in NtrC1 are normally a single helix (e.g., H8 from NSF D2 and H4 from the δ subunit of the DNA clamp loader). The insertion begins after the second turn of H8 (AEL) and is followed by residues (FELA) that complete the helix. The portion of the inserted sequence corresponding to the ordered loop has been referred to as the GAFTGA motif in NtrC1 and other σ54 activators (Fig. 2B).
Several interesting features stabilize the unusual GAFTGA-containing structural motif. First, the GAFTGA loop itself is composed of two back-to-back type-II β-turns. Second, there are two glycine residues (G210 and G225) in the extended strands that flank the GAFTGA loop and allow for the distortion of the main chain configuration of H8 required to accommodate the insertion. These are highly conserved among σ54 activators. Third, there are two conserved phenylalanine residues (F209 and F227) before and after the conserved glycine residues. These pack against L208 and other neighboring hydrophobic residues to support the structure of H8 (Fig. 2B,C). Two proposed roles for the novel GAFTGA structural motif—energy coupling and oligomerization—are discussed below.
Structure of the heptamer rings
The central ATPase domain of NtrC1 alone (NtrC1C), which is competent for assembly, ATP hydrolysis, and transcriptional activation (B.T. Nixon, in prep.), formed a ring-like heptamer (Fig. 1B). There were two such heptamers in the P1 unit cell, which were stacked on each other in a head-to-head fashion with an offset of ∼30 Å (data not shown). The diameter and height of the rings are ∼124 Å and ∼40 Å, respectively. The fold of the ATPase domain in the rings is identical to that in the dimer, except for a subdomain reorientation discussed in detail below. Calculation of rotation angles relating the seven protomers in the heptamer indicated that the heptamer rings are not perfectly symmetrical. Four of the protomers (chains D-G in Fig. 1B) are related to each other by rotations of 51°-55°, and the other three protomers (A-C) are related to each other by rotations of 53°-55°, but the rotation angles between these two groups is 47°. There are small gaps between the groups of three (A,-C) and four (D-G) protomers. Whether this is due to crystal packing is unclear. The rotation angles and spacings between protomers are quite similar in the two rings.
Interfaces involved in formation of dimers and heptamers
In general, AAA+ ATPases function as oligomeric rings (Vale 2000). Nature has used three ways to create such rings as follows: (1) regulated assembly of oligomers from monomers or dimers;(2) formation of stable rings that remain oligomerized;or (3) formation of a ring from one polypeptide chain containing six AAA+ ATPase domains (Vale 2000). Activators of σ54-RNA polymerase fall in the first category. The formation of oligomers by these activators is often regulated by phosphorylation of receiver domains in response to environmental signals. Receiver domains can control oligomerization either positively (in which case, phosphorylation is required for activity; Drummond et al. 1990; Weiss et al. 1992) or negatively (in which case, phosphorylation or deletion of the receiver domain is required to relieve inhibition; Huala et al. 1992; Gu et al. 1994). Using an in vivo transcription assay and an in vitro ATPase assay, we have shown that deletion of the receiver domain of A. aeolicus NtrC1 increases both transcriptional activation and ATPase activity several hundredfold (B.T. Nixon, in prep.). Thus, regulation of the activity of NtrC1 is negative and is analogous to that of S. meliloti DctD rather than S. typhimurium NtrC (Weiss et al. 1992; Lee et al. 1994; Meyer et al. 2001).
The extensive dimeric interface between receiver domains of NtrC1RC is very similar to one observed previously for the receiver domain of DctD (henceforth DctDR; Meyer et al. 2001). In both cases, the interface involves contacts between H4, β5, and H5 of the two monomers, in addition to contacts between the crossed extensions of H5. Nixon and colleagues refer to this extension as a linker, because not all σ54 activators with N-terminal receiver domains have such a structured extension between their receiver and output domains (e.g., S. typhimurium NtrC does not;Keener and Kustu 1988; Wootton and Drummond 1989), whereas some σ54 activators (and other proteins) with different sorts of regulatory domains apparently do [e.g., the Xylene (Xyl) R protein from Pseudomonas putida] (Garmendia and de Lorenzo 2000; Meyer et al. 2001; O'Neill et al. 2001). Many mutations screened for increasing transcriptional activation by intact DctD in the absence of its physiological phosphodonor resulted in weakening of the extensive dimer interface in DctDR, indicating that this interface is important for maintaining the off state of the ATPase domain (Meyer et al. 2001). Activated DctDR formed a very different dimer(s) that did not contain the crossed helices found in the off-state dimer (Park et al. 2002a,b).
As discussed above, contacts mediated by the crossed linkers of NtrC1RC appear to be major dimerization determinants. In support of this view, amino acid substitutions predicted to cause destabilization of contacts between them—on the basis of findings with DctDR—increased transcriptional activation by NtrC1RC in vivo (B.T. Nixon, in prep.). However, there are also regions of contact between central domains and the receiver domains (including linkers) of opposite monomers in the dimer and between the two central ATPase domains of NtrC1RC (Fig. 1A). Strand β2 and helix H8 contact the linker in the partner monomer, and both the insertion in H8 and the β-hairpin after H9 contact the C-terminal parts of H2 and H3 in this opposite receiver domain (Fig. 1A). Contacts between the ATPase domains are primarily in the α-helical subdomain. The dimerization interface spans ∼7100 Å2 (3550 Å2 per monomer).
The amount of buried surface area in the heptamer of NtrC1C is very similar to that in the dimer of NtrC1RC. One surface of the ATPase domain that participates in forming the heptamer overlaps with the surface that participates in forming the dimer (Fig. 3). In the heptamer, protomers pack front to back, so that there are then two surfaces in each ATPase domain participating in intermolecular interactions. This increases the intermolecular contact surface between ATPase domains, which would drive oligomerization once phosphorylation had altered the conformation of the receiver and linker domains. The novel structural motif inserted into H8, and H8 itself, also participate in oligomerization. In the NtrC1RC dimer, part of the β hairpin after H9 and the insertion in H8 contact the receiver domain, and, thus, are buried, whereas in the NtrC1C heptamer, these regions are involved in contacts with other protomers (Figs. 1, 3).
Figure 3.

Surface representations of both the inactive NtrC1RC dimer and the active NtrC1C heptamer. (A) Surface plot of NtrC1RC dimer. The monomers are colored gray and green. (B) The central domain of one monomer in the inactive dimer. Buried regions of contact with the other central domain in the dimer are shown in gray. (C) Same as B, but rotated 90°. The common region that is buried in both dimers and heptamers is colored red. (D) Surface plot of NtrC1C heptamer. Two protomers from the heptamer are colored gold and blue. (E) One protomer from the active heptamer. The contact regions with the preceding and following protomers in the heptamer are colored gray and blue, respectively. (F) Same as E, but rotated 90°.
Subdomain motions between dimer and heptamer and their effect on the nucleotide-binding pocket
Comparison of the central ATPase domain in the inactive dimer (NtrC1RC) to that in the assembled oligomer (NtrC1C) shows a hinge motion between the α/β subdomain and the α-helical subdomain, even though both forms have the same nucleotide (ADP) bound (Fig. 4A). In HslU, an analogous hinge motion between these subdomains was observed when the nucleotide bound to the hexamer was changed (Bochtler et al. 2000; Sousa et al. 2000; Wang et al. 2001a,b). The rotations between these subdomains in NtrC1 are ∼10° counterclockwise in going from dimer to heptamer (Fig. 4A), with the rotation axis at the midpoint between H7 and residue 308 and running parallel to H7. The α-helical subdomain, along with H6, opens relative to the α/β subdomain, leading to a flatter molecule that may have a preference for heptameric packing.
Figure 4.

Subdomain motion in the central ATPase domain between inactive NtrC1RC and active NtrC1C. (A) The hinge motion between the α/β subdomain and the α-helical subdomain. Residues 160-306 of both inactive and active NtrC1 are superimposed. The central domain of the inactive NtrC1RC (dimer) is colored blue, and that of the NtrC1C (heptamer) is colored green. The red dot indicates the rotation axis, which is perpendicular to the plane of the paper. (B) The nucleotide-binding sites for both NtrC1RC (blue) and NtrC1C (green) are shown. R293 (orange) from the adjacent protomer is located near the nucleotide-binding site in the active NtrC1C heptamer, whereas it is located on the opposite side in the inactive NtrC1RC dimer (see Fig. 1A). The single Mg2+ present in the dimer is indicated by a green dot.
A close view of the nucleotide-binding pocket shows that there are some rearrangements of ADP and the residues that contact it in going from dimer to heptamer (Fig. 4B). Because the domain rotation axis is located near the N terminus of H7, these do not involve the Walker A (GXXXGKE;also called the P-loop) or Walker B (DEXX) motifs, or the two phosphates of ADP. However, residues R357 and K360, which are located in the long helical region referred to by Neuwald et al. (1999) as Sensor II, and the ribose and adenine base of ADP, do show significant changes. The ADP in both NtrC1RC and NtrC1C has the anticonfiguration, as was the case for p97 (Zhang et al. 2000) and several other AAA+ proteins (Ogura and Wilkinson 2001).
Correlation of structure with genetic and biochemical studies of σ54 activators
Structures of NtrC1 allow us to map amino acid substitutions identified in genetic studies of other σ54 activators. Mutational changes that affect ATP binding and/or hydrolysis are shown in Figure 5A. Most of those that abolish ATP binding are located in the Walker A (P-loop) region or the sensor II region (e.g., G354 and R357;numbering in NtrC1 from A. aeolicus;Perez-Martin and de Lorenzo 1996; Gao et al. 1998; Rombel et al. 1999; Chaney et al. 2001; Lew and Gralla 2002; Zhang et al. 2002). In NtrC1, the side-chain of R357, which has been called the sensor II residue, contacts the β-phosphate of ADP, implying that this residue plays an important role in binding the phosphate region of the nucleotide. G354 is located in the loop preceding the sensor II helix. This glycine, which is highly conserved among σ54 activators, may orient the sensor II helix so that R357 can contact the β-phosphate of the nucleotide (Figs. 4B, 5A). Amino acid substitutions that prevent ATP hydrolysis without disrupting binding are located in the Walker B motif, at or around R293, which was predicted to be a catalytic arginine, or in residues of H8, just preceding the GAFTGA insertion (e.g., A206, which is an S in many other activators, and E207;Rombel et al. 1998, 1999; Chaney et al. 2001; Zhang et al. 2002). D238 in Walker B, which is thought to be required for divalent cation binding (Rombel et al. 1999), interacts with Mg2+, although Mg2+ was seen in only one monomer of the NtrC1RC dimer, and was not seen in the lower resolution structure of the NtrC1C heptamer. This Mg2+ also interacts with ADP (Fig. 4B). R293, which is highly conserved among σ54 activators, is located close to ADP bound by the neighboring protomer in the heptamer, and could probably contact the γ-phosphate of ATP if it were present (Fig. 5A). R293 is far from the nucleotide in the NtrC1RC dimer (Fig. 1A). It has been proposed that the catalytic arginine for the AAA+ superfamily is one close to R293 (R299; Neuwald et al. 1999).
Figure 5.

Correlation of structure with genetic and biochemical studies. (A) Mutations that affect the function of σ54 activators are mapped onto the structure of the central ATPase domain. Mutations that affect ATP binding, ATP hydrolysis, and transcriptional activation are colored red, green, and blue, respectively. Residues shown in ball-and-stick representation are as follows: E174 and R357 (red);E207, L208, F209, D237, D238, and R293 (different subunit;green);and A215, F216, T217, and A219 (blue). (B) The seven conserved regions (C1-C7) of the central ATPase domain of σ54 activators are shown. These regions cover residues 167-174 (C1), 191-198 (C2), 205-219 (C3), 232-242 (C4), 251-256 (C5), 273-310 (C6), and 352-361 (C7). The significance of each region is discussed in the text.
The GAFTGA portion of the insertion into H8 is highly conserved among σ54 activators, and has been shown to be involved in coupling the energy available from ATP hydrolysis to a conformational change of σ54-holoenzyme (Wang et al. 1997; Gonzalez et al. 1998; Rombel et al. 1998; Chaney et al. 2001; Lew and Gralla 2002; Zhang et al. 2002). Mutations that change the GAFTGA sequence often abolish transcriptional activation, but do not affect ATP binding or hydrolysis. Buck and colleagues recently showed that the GAFTGA loop contacts σ54 when the ATPase domain of phage shock protein F (PspF) is bound to ADP-AlFx (Chaney et al. 2001; Bordes et al. 2003), a nucleotide analog that mimics ATP in the transition state for hydrolysis. Notably, the GAFTGA loop is localized in the central pore region of the heptamer structure of NtrC1C (Fig. 1B). In this oligomer structure, the inserted loops can form an extended binding surface for σ54, and presumably play a role in mediating the energy coupling that allows NtrC1 to change the conformation of σ54-RNA polymerase at promoters. One of the mutational amino acid substitutions in the GAFTGA region of S. typhimurium NtrC (which corresponds to G218K in NtrC1) increases nonspecific DNA binding by ∼10-fold (North et al. 1996). This lesion provides evidence that the GAFTGA loop is near the DNA, perhaps near the distorted DNA in the -12 promoter region to which σ54 is initially bound (Guo et al. 1999, 2000; Chaney et al. 2001). As mentioned above, some lesions that decrease ATP hydrolysis affect residues in H8 just prior to the GAFTGA insertion. These residues may be involved in communication between active sites and sites of strong contact with the σ factor. They have also been implicated in a contact with σ54 that does not depend on the presence of nucleotide and occurs even in (inactive) dimers of DctD (Wang et al. 1997; Kelly and Hoover 2000), and they appear to play a role in oligomerization of S. typhimurium NtrC and the ATPase domain of PspF (Li et al. 1999; Chaney et al. 2001; Lew and Gralla 2002; Zhang et al. 2002).
In S. typhimurium, NtrC mutational amino acid substitutions that apparently favor oligomerization at low concentrations of the phosphorylated protein occur throughout the central ATPase domain (Li et al. 1999; Yan and Kustu 1999). In particular, such substitutions have been found in the GAFTGA loop (residues corresponding to A215C and G218C of A. aeolicus NtrC1) and in and around R293.
Discussion
It has been postulated that transcription by σ54-holoenzyme confers one notable advantage over transcription by σ70-holoenzyme, the capacity to vary transcriptional efficiency at a given promoter over a wide range (Rombel et al. 1998; Buck et al. 2000; Zhang et al. 2002). This wide range of control—not usually accessible to σ70-holoenzyme at a single promoter—is achieved by the use of AAA+ activators that couple energy to the process of open complex formation. Rather than melting the DNA themselves, activators remodel σ54-RNA polymerase bound to promoters in transcriptionally silent closed complexes so that it can form open complexes. The structures of A. aeolicus NtrC1C (active) and NtrC1RC (inactive) allow us to return to the questions posed about activators in the introduction. We begin with the related questions of how receiver domains of σ54 activators, which are a large subclass of their regulatory domains, control the activity of the adjacent ATPase domains and how the dimeric forms of the activators form active oligomers.
Oligomerization of NtrC1
Remarkably, receiver domains can control the output of their adjacent ATPase domains either positively or negatively (Drummond et al. 1990; Huala et al. 1992; Weiss et al. 1992; Gu et al. 1994). This is the case, even though activators such as S. meliloti DctD (negatively regulated) and S. typhimurium NtrC (positively regulated) have 38% identity and 53% sequence similarity over their entire lengths. In the case of A. aeolicus NtrC1, control is negative and appears to be very similar to that in DctD. Contacts between the receiver domain and linker of NtrC1RC and regions of the central ATPase domain of the opposite monomer in the dimer effectively bury one of the two surfaces required for the central domain to form heptamers (Figs. 1A, 3, 6). In addition, the central ATPase domains themselves are held in a front-to-front orientation in the dimer, whereas they must reorient in a front-to-back orientation to form the heptamer (Fig. 6). Hence, in their crossed state, the long helices at the ends of the receiver domains (linkers) prevent oligomerization. Deletion of the receiver domain and linker yields an active oligomeric ATPase, in which the active site for ATP hydrolysis has been completed and a surface has been created for interaction with σ54.
Figure 6.

Oligomerization of NtrC1. Surface representations of NtrC1 changing from a dimer to a heptamer. In brackets, the surface representations for the receiver and central domains are separated to illustrate how they adopt different conformations upon phosphorylation. The broken lines indicate the connections between the receiver and central domains. The brackets indicate a transient species. For the phosphorylated receiver domain, we used the structure of 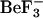 -
- (PDB code: 1L5Y), the activated form (see text).
(PDB code: 1L5Y), the activated form (see text).
When phosphorylation alters the conformation of the receiver domains of NtrC1, the dimerization interfaces between the receiver, and ATPase domains are presumably disrupted. Structural studies of DctDR indicate that the linker helices are no longer crossed in activated dimers, and the contact area between monomers is reduced. The ATPase domains of this protein, were they attached, would be free to repack in the front-to-back orientation required for oligomer formation (Fig. 6;Park et al. 2002a,b), although the relative postitions of the ATPase and receiver domains cannot be predicted. The presence of two contact surfaces between ATPase domains in the heptamer, one of which involves many residues that form the single contact surface in the dimer, would be sufficient to drive oligomerization (Figs. 3, 6). Use of the same contact surface in the on and off states provides a mechanism for minimizing both unwanted interactions and the free energy cost of switching between states.
In the presence of dimeric receiver domains, it is possible that the oligomerization could lead to a hexamer or octamer, rather than the heptamer found for the ATPase domain alone. A homoheptamer (ClpB), a heteropentamer (DNA clamp loader), and a mixture of homoheptamer and homohexamer (HslU) have been observed among AAA+ ATPases (Rohrwild et al. 1997; Kim et al. 2000; Miyata et al. 2000; Jeruzalmi et al. 2001). Our current interpretations do not depend on knowing the precise number of protomers in the active oligomer;further measurements are needed to determine this. Neither the inactive construct from NtrC1 (NtrC1RC) nor the active construct (NtrC1C) carries the C-terminal DNA-binding determinants of the protein. Where these are located and how they affect oligomerization and function of the intact protein, remain to be determined.
Novel structural motif and energy coupling
The GAFTGA motif in NtrC1 and other activators of σ54-RNA polymerase plays a direct role in contact with the polymerase and coupling of energy to a change in its conformation (Chaney et al. 2001; Zhang et al. 2002; Bordes et al. 2003). This sequence motif is part of an insertion with respect to other AAA+ ATPases (Fig. 2). The insertion forms an unusual structural motif consisting of two extended strands and an ordered loop (GAFTGA itself) projecting from the middle of H8 in the α/β subdomain of the ATPase. Although a somewhat similar insertion was observed in a helix of actin (PDB entry 1ijj), in that case, the inserted loop is shorter and is not connected to the helix with strands (Bubb et al. 2002). In addition, the helical segments before and after the loop have different helical axes. To our knowledge, the unusual structural motif containing the GAFTGA loop has not been seen before.
The ordered GAFTGA loop is found in the central pore of the NtrC1C heptamer. Loops from different subunits form a surface that could contact σ54 and mediate the energy coupling between NtrC1 and σ54-holoenzyme that allows the polymerase to initiate transcription. The only residue shown to be essential for contact between the polymerase and a short GAFTGA-containing peptide was T217 (Bordes et al. 2003), which is in the middle of the loop (Fig. 2).
There are a number of other AAA+ ATPases in which functionally important sequences are located in the region of H8 in NtrC1 and are found in the pore at the center of the oligomer. One of these is HslU, which contains a long insertion (I-domain) after H8 (Bochtler et al. 2000) that binds to partially unfolded proteins. HslU completes the unfolding process by means of a hinge motion between the α/β and helical subdomains that occurs as a result of ATP hydrolysis (Wang et al. 2001a,b). A second example is the DNA clamp loader γ complex, for which the structure of the AAA+ domain has been determined both with and without its target, the β protein (clamp;Jeruzalmi et al. 2001a,b). In the complex between the two proteins, H4 of the catalytic δ-subunit of the clamp loader (which corresponds to H8 of NtrC1) undergoes a drastic conformational change, as does the β subunit (Jeruzalmi et al. 2001a,b). Finally, a comparison of the structures of NSF D2 and p97 leads to the proposal that H8 in p97 relocates upon ATP hydrolysis, again through a domain motion involving the α/β and helical subdomains (Zhang et al. 2000). Recently, it was shown using cryo-electron microscopy that there are dramatic changes in the central pore of the p97 oligomer through the ATPase cycle (Rouiller et al. 2002). Taken together with the structure of NtrC1C, these other structures support the view that the inserted GAFTGA loop in H8 of NtrC1, which is implicated in binding σ54, will undergo changes in position and/or orientation as ATP is hydrolyzed. These changes are presumably driven by subdomain reorientations in the ATPase domain of NtrC1 and are coupled to a conformational change in σ54 RNA polymerase, but the detailed mechanism of energy coupling remains to be determined.
Conserved regions of σ54 activators
Morett and colleagues noted that activators of σ54-holoenzyme have seven regions of conserved sequence in their ATPase domains, which they designated C1 through C7 (Morett and Segovia 1993). Together with the analysis of Neuwald et al. (1999), the structure of the NtrC1C oligomer leads to a better understanding of the roles of these conserved regions (Fig. 5B). The C1 region is the Walker A motif, which, as expected, contacts the nucleotide. Region C2, which was previously of unknown function, clearly participates in contacts between the protomers of the NtrC1C heptamer, as does region C5. They contact one another, thereby stabilizing the oligomer. A number of mutational amino acid substitutions in and around these regions appear to increase oligomerization of phosphorylated S. typhimurium NtrC (Li et al. 1999). Region C3 carries the GAFTGA motif that is important for output coupling (vida infra), and region C4 is the Walker B motif, that contacts nucleotides. Region C6 carries the putative sensor I region and sensor I residue (N280), the catalytic arginine, (also called an arginine finger), and the hinge between the two subdomains. Although Neuwald et al. (1999) predicted that the catalytic arginine was conserved R299, there is genetic evidence that it may be conserved R293;a lesion in S. typhimurium NtrC that changed the corresponding residue to cysteine yielded a protein that bound nucleotide and oligomerized normally, but failed to hydrolyze ATP (Rombel et al. 1999). The structure supports this possibility, but is not definitive. In the oligomer, either R299 or R293 presumably completes the active site of an adjacent protomer (Figs. 1, 5A). Region C7 carries the sensor II residue (R357) and the sensor II helix, and is the only conserved region located in the helical subdomain of the ATPase. The position of this helix and its sensor residue allows the subdomain to respond to changes in nucleotide state.
Future directions
Unlike the case for many other AAA and AAA+ ATPases (Vale 2000), σ54 activators do not bind tightly to their target, the polymerase, in the presence of ATP (or ADP). Tight binding, which has been achieved for PspF and the ATPase domain of Klebsiella pneumoniae nitrogen fixation protein A (NifA;Chaney et al. 2001) depends on the presence of ADP-AlFx, an analog of ATP in the transition state for hydrolysis. In addition, it requires that activator and polymerase be mixed before this analog is added, implying that the proteins must first associate weakly. Under normal circumstances, contacts between activators and polymerase are apparently short-lived, allowing for very high levels of transcription from σ54-dependent promoters. Together with additional structures in the presence of various nucleotides, the detailed biochemical and genetic analyses that have been made of σ54 activators should facilitate progress in defining the mechanochemical action of these unusual transcriptional regulators and of AAA+ proteins generally.
Coordinates
Coordinates and reflection data for NtrC1RC and NtrC1C have been deposited at the Protein Data Bank under accession codes 1NY5 and 1NY6, respectively.
Materials and methods
Protein expression and purification
The A. aeolicus genomic DNA was a gift from R. Huber (Lehrstuhl für Mikrobiologie, Universitat Regensburg, Regensburg, Germany). Both NtrC1RC (A. aeolicus, GI #2983588, residues 1-387) and NtrC1C (residues 121-387) were cloned from the genomic DNA into pET21a vectors (Novagen). A selenomethionine derivative of the NtrC1RC protein was obtained by growing Escherichia coli B834 (DE3) carrying plasmid SJS1244 in M9 minimal medium supplemented with selenomethionine. NtrC1C protein was expressed using E. coli BL21 (DE3) with Rosetta.pLysS (to provide rare tRNAs). NtrC1RC and NtrC1C were purified in a similar manner. Cells were harvested and disrupted by sonication. Cell lysate in 50 mM Tris buffer (pH 8.2), 1 mM MgCl2, 1 mM ATP, and 1 mM tris-2-carboxy-ethylphosphine (TCEP) was heated at 75°C-80°C for 25 min, and the supernatant was dialyzed against 50 mM Tris buffer (pH 8.0) and 5 mM EDTA overnight. The protein was further purified on a Q-sepharose column, and then a gel-filtration column. The purified protein was dialyzed extensively against 5 mM ammonium bicarbonate (2-L chamber, three buffer changes for ∼24 h), and then lyophilized. Lyophilized NtrC1RC was redissolved in 50 mM Tris (pH 8.0), 20 mM ATP, 10 mM TCEP, 5 mM MgCl2, and 5% glycerol (vol/vol). NtrC1C was dissolved in the same buffer, except without glycerol. The pH of TCEP and ATP solutions was adjusted to 7.0 before mixing with the protein solution, and the final pH of the protein solution was 8.0. Protein solutions were filtered with a 0.2-μm filter before use in crystallization trials. Gel filtration chromatography (Superdex200, 10/30) and dynamic light scattering confirmed the dimeric state of NtrCRC and the higher oligomeric state (M.W. ∼220 kD) of NtrC1C. The oligomeric state of NtrC1C is dependent on salt concentration with oligomer stabilized at low salt concentration (data not shown).
Crystallization and structure determination
Crystals of NtrC1RC were grown at room temperature using the hanging drop vapor-diffusion method. A total of 6 μL of protein solution (10 mg/mL) was mixed with an equal volume of well solution containing 0.6-0.7 M NaH2PO4/KH2PO4, 100 mM citric acid (pH 6.5), 10 mM imidazole, and 3% (v/v) methanol. Crystals appeared after 1 d and grew to ∼0.2 × 0.2 × 1.0 mm (hexagonal rod shape) after 1 wk. Crystals were transferred to well solution plus 10% glycerol for 1 min, and then transferred to well solution plus 20% glycerol for 5 min, after which they were flash frozen and stored in liquid nitrogen until being used for data collection. These crystals had the space group P3221, with unit cell dimensions a = b = 94.8Å and c = 195.0Å with two molecules in the asymmetric unit. SAD (single-wavelength anomalous diffraction) data were collected at the beam line 5.0.2 of the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory. The data were processed and scaled with DENZO and SCALEPACK from the HKL program suite (Otwinowski and Minor 1997). Heavy atom sites (Se) were found with SOLVE at 3 Å (Terwilliger 2000). A total of 11 of 12 SeMet sites were found, and 10 sites were related by twofold rotations that are not related to crystallographic symmetry. Phasing was calculated with SHARP (de La Fortelle and Bricogne 1997) up to 3 Å resolution, and was improved and extended by RESOLVE (Terwilliger 2000) using twofold noncrystallographic symmetry (NCS) averaging and solvent flattening. Model building was done using O (Jones et al. 1991). Using partial models, two separate NCS operators for both regulatory and central ATPase domains were calculated, and two-domain NCS-averaging was performed using DM (Wang 1985). This improved the map quality significantly and allowed us to complete the model building. The GAFTGA loop that is inserted into H8 was not initially visible, but the connecting strands were clear in the very early stage of the experimental map before NCS averaging. The loop was built after completion of the rest of the chain. The structure was refined with CNS (Brunger et al. 1998). Anisotropic B factors and bulk solvent corrections, as well as the cross-validation methods, were applied throughout the refinement. NCS restraints were applied only in the initial refinement stage and were released after the R-working and R-free factors dropped to 33%/35%. Water and solvent picking was performed after the Rworking and Rfree dropped to 25%/28.5% using CNS. PROCHECK was used to monitor geometric parameters (Laskowski et al. 1993).
Crystals of NtrC1C were grown at room temperature using the hanging drop vapor-diffusion method. A total of 2 μL of protein solution (20 mg/mL) was mixed with an equal volume of well solution containing (pH 6.5), 50 mM diammonium tartrate, and 8% (vol/vol) PEG 3350. Crystals grew to ∼0.2 × 0.2 × 0.2 mm (diamond shape) after 1 d. Crystals were transferred to well solution plus 10% glycerol for 2 min, and then transferred to well solution plus 25% glycerol for 5 min, after which they were flash frozen and stored in liquid nitrogen until being used for data collection. These crystals had the space group P1, with unit cell dimensions a = 106.8 Å, b = 108.3 Å, c = 110.0 Å, α = 70.3°, β = 85.9°, and γ = 73.3°, with 14 molecules in the unit cell. Data were collected at the beam line 8.3.1 of the Advanced Light Source (ALS). The data were processed and scaled with MOSFLM and SCALA using the Elves interface (J.M. Holton, in prep.). Seven twofold NCS peaks were found (at about 5σ) using the self-rotation function of Molrep (Vagin and Isupov 2001). Locked cross-rotation was performed using the self-rotation solutions by Molrep at several different resolution ranges. Three sets of solutions (10 solutions each) were found at several different resolution ranges and sphere radius (25 Å-40 Å). Solutions that were not located to form rings were discarded, and the correct solutions were confirmed by the decrease of Rfree in the initial refinement with CNS. Eleven solutions were found in this way, and the remaining three molecules were placed manually in the 2Fo-Fc electron density map. After all 14 solutions had been found, two-domain rigid body refinement (α/β subdomain and α-helical subdomain) improved Rworking/Rfree factor significantly. Refinement was done with CNS.
Acknowledgments
We thank Donghae Shin for excellent advice in crystallography, Ho S. Cho for help with sample preparation, David King for help in characterizing NtrC1 by mass spectrometry, the beam line staff at the ALS for help with data collection, and Seth Rubin for critical reading of the manuscript. This work was supported by National Institute of Health grants GM62163 to D.W. and GM38361 to S.K., and National Science Foundation grant MCB9727745 to B.T.N.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1125603.
References
- Bochtler M., Hartmann, C., Song, H.K., Bourenkov, G.P., Bartunik, H.D., and Huber, R. 2000. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature 403: 800-805. [DOI] [PubMed] [Google Scholar]
- Bordes P., Wigneshweraraj, S.R., Schumacher, J., Zhang, X., Chaney, M., and Buck, M. 2003. The ATP hydrolyzing transcription activator phage shock protein F of Escherichia coli: Identifying a surface that binds σ54. Proc. Natl. Acad. Sci. 100: 2278-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905-921. [DOI] [PubMed] [Google Scholar]
- Bubb M.R., Govindasamy, L., Yarmola, E.G., Vorobiev, S.M., Almo, S.C., Somasundaram, T., Chapman, M.S., Agbandje-McKenna, M., and McKenna, R. 2002. Polylysine induces an antiparallel actin dimer that nucleates filament assembly: Crystal structure at 3.5-A resolution. J. Biol. Chem. 277: 20999-21006. [DOI] [PubMed] [Google Scholar]
- Buck M., Gallegos, M.T., Studholme, D.J., Guo, Y., and Gralla, J.D. 2000. The bacterial enhancer-dependent σ54 (σN) transcription factor. J. Bacteriol. 182: 4129-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney M., Grande, R., Wigneshweraraj, S.R., Cannon, W., Casaz, P., Gallegos, M.T., Schumacher, J., Jones, S., Elderkin, S., Dago, A.E., et al. 2001. Binding of transcriptional activators to σ54 in the presence of the transition state analog ADP-aluminum fluoride: Insights into activator mechanochemical action. Genes & Dev. 15: 2282-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Fortelle E. and Bricogne, G. 1997. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Academic Press, Inc., San Diego, CA. [DOI] [PubMed]
- Deckert G., Warren, P.V., Gaasterland, T., Young, W.G., Lenox, A.L., Graham, D.E., Overbeek, R., Snead, M.A., Keller, M., Aujay, M., et al. 1998. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature 392: 353-358. [DOI] [PubMed] [Google Scholar]
- Drummond M.H., Contreras, A., and Mitchenall, L.A. 1990. The function of isolated domains and chimaeric proteins constructed from the transcriptional activators NifA and NtrC of Klebsiella pneumoniae. Mol. Microbiol. 4: 29-37. [DOI] [PubMed] [Google Scholar]
- Gao Y., Wang, Y.K., and Hoover, T.R. 1998. Mutational analysis of the phosphate-binding loop of Rhizobium meliloti DctD, a σ54-dependent activator. J. Bacteriol. 180: 2792-2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmendia J. and de Lorenzo, V. 2000. The role of the interdomain B linker in the activation of the XylR protein of Pseudomonas putida. Mol. Microbiol. 38: 401-410. [DOI] [PubMed] [Google Scholar]
- Gonzalez V., Olvera, L., Sobernon, X., and Morett, E. 1998. In vivo studies on the positive control function of NifA: A conserved hydrophobic amino acid patch at the central domain involved in transcriptional activation. Mol. Microbiol. 28: 55-67. [DOI] [PubMed] [Google Scholar]
- Gu B., Lee, J.H., Hoover, T.R., Scholl, D., and Nixon, B.T. 1994. Rhizobium meliloti DctD, a σ54-dependent transcriptional activator, may be negatively controlled by a subdomain in the C-terminal end of its two-component receiver module. Mol. Microbiol. 13: 51-66. [DOI] [PubMed] [Google Scholar]
- Guo Y., Wang, L., and Gralla, J.D. 1999. A fork junction DNA-protein switch that controls promoter melting by the bacterial enhancer-dependent sigma factor. EMBO J. 18: 3736-3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Lew, C.M., and Gralla, J.D. 2000. Promoter opening by σ54 and σ70 RNA polymerases: σ factor-directed alterations in the mechanism and tightness of control. Genes & Dev. 14: 2242-2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.W., Iwasaki, H., Miyata, T., Mayanagi, K., Yamada, K., Morikawa, K., and Shinagawa, H. 2001. A unique β-hairpin protruding from AAA+ ATPase domain of RuvB motor protein is involved in the interaction with RuvA DNA recognition protein for branch migration of Holliday junctions. J. Biol. Chem. 276: 35024-35028. [DOI] [PubMed] [Google Scholar]
- Huala E., Stigter, J., and Ausubel, F.M. 1992. The central domain of Rhizobium leguminosarum DctD functions independently to activate transcription. J. Bacteriol. 174: 1428-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeruzalmi D., O'Donnell, M., and Kuriyan, J. 2001. Crystal structure of the processivity clamp loader gamma (γ) complex of E. coli DNA polymerase III. Cell 106: 429-441. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47: 110-119. [DOI] [PubMed] [Google Scholar]
- Keener J. and Kustu, S. 1988. Protein kinase and phosphoprotein phosphatase activities of nitrogen regulatory proteins NTRB and NTRC of enteric bacteria: Roles of the conserved amino-terminal domain of NTRC. Proc. Natl. Acad. Sci. 85: 4976-4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M.T. and Hoover, T.R. 2000. The amino terminus of Salmonella enterica serovar typhimurium σ54 is required for interactions with an enhancer-binding protein and binding to fork junction DNA. J. Bacteriol. 182: 513-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern D., Volkman, B.F., Luginbuhl, P., Nohaile, M.J., Kustu, S., and Wemmer, D.E. 1999. Structure of a transiently phosphorylated switch in bacterial signal transduction. Nature 402: 894-898. [DOI] [PubMed] [Google Scholar]
- Kim K.I., Cheong, G.W., Park, S.C., Ha, J.S., Woo, K.M., Choi, S.J., and Chung, C.H. 2000. Heptameric ring structure of the heat-shock protein ClpB, a protein-activated ATPase in Escherichia coli. J. Mol. Biol. 303: 655-666. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283-291. [Google Scholar]
- Lee J.H., Scholl, D., Nixon, B.T., and Hoover, T.R. 1994. Constitutive ATP hydrolysis and transcription activation by a stable, truncated form of Rhizobium meliloti DCTD, a σ54-dependent transcriptional activator. J. Biol. Chem. 269: 20401-20409. [PubMed] [Google Scholar]
- Lenzen C.U., Steinmann, D., Whiteheart, S.W., and Weis, W.I. 1998. Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell 94: 525-536. [DOI] [PubMed] [Google Scholar]
- Lew C.M. and Gralla, J.D. 2002. New roles for conserved regions within a σ54-dependent enhancer-binding protein. J. Biol. Chem. 277: 41517-41524. [DOI] [PubMed] [Google Scholar]
- Li J., Passaglia, L., Rombel, I., Yan, D., and Kustu, S. 1999. Mutations affecting motifs of unknown function in the central domain of nitrogen regulatory protein C. J. Bacteriol. 181: 5443-5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M.G., Park, S., Zeringue, L., Staley, M., McKinstry, M., Kaufman, R.I., Zhang, H., Yan, D., Yennawar, N., Yennawar, H., et al. 2001. A dimeric two-component receiver domain inhibits the σ54-dependent ATPase in DctD. FASEB J. 15: 1326-1328. [DOI] [PubMed] [Google Scholar]
- Miyata T., Yamada, K., Iwasaki, H., Shinagawa, H., Morikawa, K., and Mayanagi, K. 2000. Two different oligomeric states of the RuvB branch migration motor protein as revealed by electron microscopy. J. Struct. Biol. 131: 83-89. [DOI] [PubMed] [Google Scholar]
- Morett E. and Segovia, L. 1993. The σ54 bacterial enhancer-binding protein family: Mechanism of action and phylogenetic relationship of their functional domains. J. Bacteriol. 175: 6067-6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald A.F., Aravind, L., Spouge, J.L., and Koonin, E.V. 1999. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9: 27-43. [PubMed] [Google Scholar]
- Nixon B.T., Ronson, C.W., and Ausubel, F.M. 1986. Two-component regulatory systems responsive to environmental stimuli share strongly conserved domains with the nitrogen assimilation regulatory genes ntrB and ntrC. Proc. Natl. Acad. Sci. 83: 7850-7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North A.K., Weiss, D.S., Suzuki, H., Flashner, Y., and Kustu, S. 1996. Repressor forms of the enhancer-binding protein NtrC: Some fail in coupling ATP hydrolysis to open complex formation by σ54-holoenzyme. J. Mol. Biol. 260: 317-331. [DOI] [PubMed] [Google Scholar]
- Ogura T. and Wilkinson, A.J. 2001. AAA+ superfamily ATPases: Common structure—diverse function. Genes Cells 6: 575-597. [DOI] [PubMed] [Google Scholar]
- O'Neill E., Wikstrom, P., and Shingler, V. 2001. An active role for a structured B-linker in effector control of the σ54-dependent regulator DmpR. EMBO J. 20: 819-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307-326. [DOI] [PubMed] [Google Scholar]
-
Park S., Meyer, M., Jones, A.D., Yennawar, H.P., Yennawar, N.H., and Nixon, B.T. 2002a. Two-component signaling in the AAA+ ATPase DctD: Binding Mg2+ and
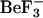 selects between alternative dimeric states of the receiver domain. FASEB J. 16: 1964-1966. [DOI] [PubMed] [Google Scholar]
selects between alternative dimeric states of the receiver domain. FASEB J. 16: 1964-1966. [DOI] [PubMed] [Google Scholar] - Park S., Zhang, H., Jones, A.D., and Nixon, B.T. 2002b. Biochemical evidence for multiple dimeric states of the Sinorhizobium meliloti DctD receiver domain. Biochemistry 41: 10934-10941. [DOI] [PubMed] [Google Scholar]
- Perez-Martin J. and de Lorenzo, V. 1996. ATP binding to the σ54-dependent activator XylR triggers a protein multimerization cycle catalyzed by UAS DNA. Cell 86: 331-339. [DOI] [PubMed] [Google Scholar]
- Rohrwild M., Pfeifer, G., Santarius, U., Muller, S.A., Huang, H.C., Engel, A., Baumeister, W., and Goldberg, A.L. 1997. The ATP-dependent HslVU protease from Escherichia coli is a four-ring structure resembling the proteasome. Nat. Struct. Biol. 4: 133-139. [DOI] [PubMed] [Google Scholar]
- Rombel I., North, A., Hwang, I., Wyman, C., and Kustu, S. 1998. The bacterial enhancer-binding protein NtrC as a molecular machine. Cold Spring Harb. Symp. Quant. Biol. 63: 157-166. [DOI] [PubMed] [Google Scholar]
- Rombel I., Peters-Wendisch, P., Mesecar, A., Thorgeirsson, T., Shin, Y.K., and Kustu, S. 1999. MgATP binding and hydrolysis determinants of NtrC, a bacterial enhancer-binding protein. J. Bacteriol. 181: 4628-4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouiller I., DeLaBarre, B., May, A.P., Weis, W.I., Brunger, A.T., Milligan, R.A., and Wilson-Kubalek, E.M. 2002. Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat. Struct. Biol. 9: 950-957. [DOI] [PubMed] [Google Scholar]
- Sousa M.C., Trame, C.B., Tsuruta, H., Wilbanks, S.M., Reddy, V.S., and McKay, D.B. 2000. Crystal and solution structures of an HslUV protease-chaperone complex. Cell 103: 633-643. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 55: 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.A. and Isupov, M.N. 2001. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr. D Biol. Crystallogr. 57: 1451-1456. [DOI] [PubMed] [Google Scholar]
- Vale R.D. 2000. AAA proteins. Lords of the ring. J. Cell Biol. 150: F13-F19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.C. 1985. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 115: 90-112. [DOI] [PubMed] [Google Scholar]
- Wang Y.K., Lee, J.H., Brewer, J.M., and Hoover, T.R. 1997. A conserved region in the σ54-dependent activator DctD is involved in both binding to RNA polymerase and coupling ATP hydrolysis to activation. Mol. Microbiol. 26: 373-386. [DOI] [PubMed] [Google Scholar]
- Wang J., Song, J.J., Franklin, M.C., Kamtekar, S., Im, Y.J., Rho, S.H., Seong, I.S., Lee, C.S., Chung, C.H., and Eom, S.H. 2001a. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 9: 177-184. [DOI] [PubMed] [Google Scholar]
- Wang J., Song, J.J., Seong, I.S., Franklin, M.C., Kamtekar, S., Eom, S.H., and Chung, C.H. 2001b. Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure 9: 1107-1116. [DOI] [PubMed] [Google Scholar]
- Weiss D.S., Klose, K.E., Hoover, T.R., North, A.K., Porter, S.C., Wedel, A.B., and Kustu, S. 1992. Prokaryotic transcriptional enhancers. In Transcriptional regulation. (eds. S. McKnight and K. Yamamoto), pp. 667-694.Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Wootton J.C. and Drummond, M.H. 1989. The Q-linker: A class of interdomain sequences found in bacterial multidomain regulatory proteins. Protein Eng. 2: 535-543. [DOI] [PubMed] [Google Scholar]
- Yamada K., Kunishima, N., Mayanagi, K., Ohnishi, T., Nishino, T., Iwasaki, H., Shinagawa, H., and Morikawa, K. 2001. Crystal structure of the Holliday junction migration motor protein RuvB from Thermus thermophilus HB8. Proc. Natl. Acad. Sci. 98: 1442-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D. and Kustu, S. 1999. “Switch I” mutant forms of the bacterial enhancer-binding protein NtrC that perturb the response to DNA. Proc. Natl. Acad. Sci. 96: 13142-13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.C., Hanson, P.I., Jahn, R., and Brunger, A.T. 1998. Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat. Struct. Biol. 5: 803-811. [DOI] [PubMed] [Google Scholar]
- Zhang X., Shaw, A., Bates, P.A., Newman, R.H., Gowen, B., Orlova, E., Gorman, M.A., Kondo, H., Dokurno, P., Lally, J., et al. 2000. Structure of the AAA ATPase p97. Mol. Cell 6: 1473-1484. [DOI] [PubMed] [Google Scholar]
- Zhang X., Chaney, M., Wigneshweraraj, S.R., Schumacher, J., Bordes, P., Cannon, W., and Buck, M. 2002. Mechanochemical ATPases and transcriptional activation. Mol. Microbiol. 45: 895-903. [DOI] [PubMed] [Google Scholar]