

Abstract
Transmission bottlenecks occur in pathogen populations when only a few individual pathogens are transmitted from one infected host to another in the initiation of a new infection. Transmission bottlenecks can dramatically affect the evolution of virulence in rapidly evolving pathogens such as RNA viruses. Characterizing pathogen diversity with the quasispecies concept, we use analytical and simulation methods to demonstrate that severe bottlenecks are likely to drive down the virulence of a pathogen because of stochastic loss of the most virulent pathotypes, through a process analogous to Muller’s ratchet. We investigate in this process the roles of host population size, duration of within-host viral replication, and transmission bottleneck size. We argue that the patterns of accumulation of deleterious mutation may explain differing levels of virulence in vertically and horizontally transmitted diseases.
Keywords: disease evolution, quasispecies, RNA virus, Muller’s ratchet, mutation–selection balance
Disease biologists have long been concerned with the manner in which pathogen virulence evolves through the coevolutionary processes dictated by continued host–pathogen interaction. It has traditionally been argued that, over time, the interaction between a given pathogen and its host should become less severe; that is, virulence should decline to a point of benign coexistence. Much recent theoretical and empirical work, however, has challenged this conventional wisdom. It appears that the evolution of virulence is a complex interaction between within-host pathogen replication and among-host pathogen transmission (1–13).
A particularly intriguing hypothesis, popularized by Ewald (4, 14), concerns the relative effects of vertical and horizontal transmission on the evolution of virulence. Under vertical transmission, pathogens are transmitted across generations, and each host infects only its own progeny. Under horizontal transmission, pathogens are transmitted to all susceptible hosts in the population regardless of descent. Ewald suggests that virulence should be low in vertically transmitted diseases, because the pathogen’s reproduction is limited by the host’s reproductive success. The more progeny available for infection, the greater the success of the pathogen. High virulence reduces the health of the parent (thereby reducing offspring production) and consequently reduces the available pool of new infections. Virulence can be higher in horizontally transmitted pathogens, which do not rely exclusively on the host’s descendants for their future survival. Some empirical evidence supports this basic hypothesis (5, 15).
Two distinctions should be made between vertical and horizontal transmission. The first pertains to the coupling of host and pathogen fitness described above: vertically transmitted parasites, unlike horizontally transmitted strains, are strictly dependent on the survival and successful reproduction of their hosts, and hence have an interest in keeping virulence low. The second regards the relative degree of parasite–parasite competition in vertically and horizontally transmitted parasite populations. Vertically transmitted parasites never move among host lineages, and competition therefore only occurs at the intrahost level. Horizontally transmitted parasites, by contrast, can conceivably spread to every member of the host population in a short period of time, and competition can occur at the interhost level. Under horizontal transfer, more virulent strains are able to export their extra productivity to other host lineages (16). As we argue below, this means that natural selection will more effectively maintain high virulence in horizontally transmitted pathogens.
An independent line of investigation can also shed some light on the role of vertical transmission on the evolution of virulence. In a large number of laboratory studies using viral pathogens in cell culture, plaque-to-plaque serial transfer reduces viral replication rate and virulence (reviewed in ref. 17). However, the mechanism proposed for the loss in virulence in these serial transfers is distinct from that proposed by Ewald.
Any clonally reproducing lineage, such as that produced by a viral or bacterial pathogen, will tend to accumulate deleterious mutations over time. This will be especially true in RNA viruses, which experience a million fold higher mutation rate than their DNA counterparts (17). Most of these mutations will be harmful, and if the most fit (i.e., the most quickly reproducing) class of pathogens is lost from the population by chance, then the mean fitness of the population of pathogens will decrease relative to the original population. This continual decrease in mean fitness of the clonal lineage by stochastic loss of the least mutated class is the process described by Muller’s ratchet (18). In general, researchers conducting serial-transfer experiments invoke Muller’s ratchet as the basic mechanism driving the loss of pathogen virulence in the laboratory system (19–23); it has also been suggested that this mechanism may effect virulence evolution in vivo (20, 21). The loss of virulence, in this case, is not mediated through any effects on the host (as suggested by Ewald) but rather through the direct genetic consequences of the transmission dynamics.
Muller’s ratchet requires significant stochastic losses of a population’s genetic variability. In pathogen populations, these stochastic losses usually are associated with transmission bottlenecks in which only a few pathogen particles are transmitted from one host to another to initiate infection. In this process, only a limited subset of the pathogen diversity present in the source host is transmitted to the new host.
Transmission is thought to act as a tight bottleneck for a number of diseases; as a general phenomenon this may be quite common. In HIV, new infections typically contain only a small fraction of the viral sequences found in the infectious source (24). In respiratory droplet-transmitted diseases, droplets typically contain only one or two viral particles (25, 26), with a single particle being sufficient to initiate a new infection. In other diseases, hosts must be exposed to a large number of pathogen particles before an infection is initiated, simply because of the very low probability that any single pathogen particle will successfully invade. Indeed, infection after large doses of inoculum are often initiated by a single pathogen particle (27). Transmission bottlenecks may be especially severe in cases of vertical transmission where pathogens must cross a variety of barriers intended to protect the developing embryo.
A MODEL OF QUASISPECIES EVOLUTION
The Virus.
RNA viruses feature extremely rapid mutation rates, on the order of 10−3–10−5 per base per replication. As a consequence, even a single-founder population of virions within a single host is likely to represent a large number of genotypes after a few generations of viral replication. This rapid production of intrahost diversity may have a significant impact on selection and evolutionary dynamics for these viruses. Several authors (20–23, 28, 29) have suggested that these features of RNA virus biology can be incorporated into evolutionary models using the quasispecies concept (30). The virus is represented not as a few distinct genotypes but rather as a distribution of genotypes produced by a joint action of mutation and selection. In practice, this distribution will span a multidimensional genotype space; in the analysis, genotype space is often condensed to a single one-dimensional axis representing replication rate. The advantages and hazards inherent in this approach are discussed in detail in ref. 31. A schematic diagram of the interacting mutation and growth processes is presented in Fig. 1.
Figure 1.

The forces driving quasispecies evolution. Any given viral pathotype N is more likely to undergo deleterious mutation to pathotype N − 1 than it is to undergo advantageous mutation to pathotype N + 1. However, pathotypes with fewer deleterious mutations tend to reproduce more quickly. This results in a mutation–selection balance that determines the quasispecies distribution.
Mathematically, a quasispecies distribution can be described by a vector representing the number of individuals of each pathotype. The dynamics of viral growth are then described by a transition matrix representing the probability of mutating from one pathotype to another and a growth vector representing the reproductive rate of each strain. In this paper, we examine a model in which there are 10 viral pathotypes, indexed 1 through 10. Higher numbered pathotypes have higher replication rates and therefore higher virulence. Type 10 is the master sequence, the pathotype with the highest replication rate. To further simplify the model, we assume that all mutations are stepwise. For example, a pathotype of type 3 can only mutate to type 2 or 4, not to type 7. This gives us a tri-diagonal transition matrix:
 |
1 |
Here, d2 is the probability of a beneficial mutation (which increases viral reproduction rate), d1 is the probability of a deleterious mutation (which decreases viral reproduction rate), and b = 1 − d1 − d2 is probability that no mutation occurs.
Letting s be the selective consequence of each mutation, the vector representing the growth rates of each pathotype, a, is the column vector
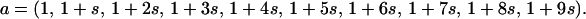 |
2 |
Let the matrix A be the product of a and the row vector (1, 1, … , 1), so that A is a 10 × 10 matrix with each column equal to a. In each generation of viral reproduction, each virion undergoes growth followed by mutation. Mathematically, a single generation is expressed by the equation x′ = Wx, where x is the initial viral pathotype distribution, x′ is the final pathotype distribution, and W ≡ Q ○ A is the Schur (or pairwise) matrix product of Q and A. The vector describing the pathogen distribution after t periods of viral replication and mutation is then x′ = Wtx.
In the analysis that follows, we use example values of d1 = 0.1, d2 = 0.01, and s = 0.1 for a mutation rate of 0.11 per virion per replication with a 10:1 ratio of deleterious to beneficial mutations. These values were chosen to be conservative estimates. At 10:1, these parameters generously favor beneficial mutations. A reduced rate of beneficial mutation will serve to accelerate the phenomena studied in this paper, as it will accelerate the rate of mutational decline in fitness while reducing the opportunity for the beneficial mutations that oppose the ratchet. Our total mutation rate is at the low end of estimated rates (10−1 to 101 per virion per replication [17]) and hence is also conservative with regard to the rate of mutationally induced virulence decline. Finally, the linear form of the growth vector a is not essential; the results of simulations using multiplicative and subadditive growth vectors (data not shown) do not differ qualitatively from those presented below. The mutation process is also simplified relative to the actual mutation matrix for any given species. However, relaxing the assumption of stepwise mutation is unlikely to change qualitatively the conclusions of our models, provided that deleterious mutations are generally more likely than advantageous ones. Asymmetry in the mutation matrix is the most essential feature of the quasispecies dynamics.
The Transmission Process.
In this paper, we examine both a horizontal transmission model (Fig. 2) and a vertical transmission model (Fig. 3). In each model, the transmission cycle begins with a population of n newly infected hosts. Within each host, the virus population mutates and replicates for t periods, as specified by Eqs. 1 and 2. After this viral growth phase is a viral transmission phase; this may be horizontal or vertical, depending on the transmission model (Figs. 2 and 3). The transmission bottleneck occurs here, with b virus particles initiating each new infection. The transmission phase gives rise to a population of n newly infected hosts; the n previously infected hosts are assumed to be removed from the population, either by death or immunity. (For simplicity, we refer to n as the host population size, although in fact n is the size of the newly infected population.) At this point, the cycle begins anew. In both models, viral titer has no effect on host survival. As a consequence, there is no selection on the virus for reduced virulence at the interhost level, and we can examine the consequences of transmission bottlenecks in the absence of Ewald’s mechanism of selection for reduced virulence.
Figure 2.

The horizontal transmission model. Starting with a population of n newly infected hosts, the transmission cycle proceeds as follows. (1) Within each host, the virus undergoes t periods of intrahost process of replication and mutation. (2) Hosts carrying mature virus populations release viral particles into the environment. In this model, all hosts contribute viral particles, in proportion to their viral titer, to a common pool (e.g., a host carrying twice as many virions as a second host contributes twice as many viral particles to the common pool). (3) Susceptible hosts are infected by b viral particles drawn at random from this common pool, thereby generating a new set of newly infected hosts.
Figure 3.

The vertical transmission model. (i) Within each of n newly infected hosts, the virus population undergoes t periods of mutation and growth, giving rise to a population of hosts carrying mature virus populations. (ii) A total of n newly infected offspring are produced by sampling with replacement from the set of “parent” hosts. Each offspring carries b virion particles drawn at random from the pathotype frequency distribution in its parent.
These two models represent the extremes of the continuum between purely vertical and purely horizontal transmission. Each permits certain heuristically useful simplifications. In the horizontal case, the allocation of parasites into specific hosts is, in a sense, irrelevant. There are no density-dependent effects on the pathogen (such as carrying capacity, immune response, or host-level selection), and the pathotypes from all hosts are perfectly mixed before infection. Consequently, the entire population of all virions in all hosts can be treated as a single unstructured population. In the vertical model, by contrast, each host lineage contains a parasite subpopulation that is effectively evolving in complete isolation from the parasite subpopulations in the other host lineages. This model allows us to infer the properties of the evolutionary dynamics of the entire pathogen population from those of a single subpopulation.
ANALYSIS
What Happens When Inoculum Size Is Large?
Let us first consider the vertical transmission case. Here, pathogens cannot move among host lineages, and thus we can examine what happens in any particular lineage. Matrix theory tells us that with successive viral generations, the mutation–selection process described above will converge to a steady-state distribution of pathotypes, regardless of the initial pathotype distribution. This steady-state distribution—at the deterministic mutation–selection balance—will be the eigenvector associated with the leading eigenvalue of the transmission matrix W. Fig. 4 shows this distribution for the parameter values introduced above.
Figure 4.

The pathotype distribution at the deterministic mutation-selection balance, given by the leading eigenvector of the matrix W.
As inoculum size becomes large, the distribution of pathotypes in each inoculum approaches the actual distribution found in the individual transmitting the infection. Pathotype frequency changes caused by sampling effects become insignificant by the law of large numbers. Therefore, as inoculum size grows large, the pathotype distribution within a single vertical lineage of k host generations (each allowing t rounds of viral replication) approaches the pathotype distribution in a single individual after kt viral replications. We already know that this latter distribution converges to the distribution at the deterministic mutation–selection balance. A parallel argument can be used to show that horizontal transmission results in convergence to the same distribution when inoculum size is large.
What Happens When Inoculum Size Is Small?
When inoculum size is small, random sampling causes the pathotype frequency distribution in the newly infected host to deviate from that in the transmitting host. The evolutionary dynamics of horizontally and vertically transmitted pathogens then differ in two significant ways.
As mentioned above, in the vertical model, the pathogens in each individual host compose an evolutionarily separate population from those in every other host. Members of pathogen lineages from different hosts at time t1 will never share a common host at time t2 > t1. Hence, the larger population of all pathogens in all hosts is separated into n entirely isolated subpopulations. The vertical model features n separate pathogen populations each passing through a bottleneck of size b at each transmission event. In the horizontal model, pathogens from separate hosts at time t1 may share a common host at time t2 > t1. Indeed, the packaging into distinct hosts is irrelevant to the dynamics, and thus the horizontal model features a single population passing through a bottleneck of size nb.
Second, vertically and horizontally transmitted populations differ in the potential for differences in growth rate to counterbalance the incessant downward bias to mutation. Consider the evolutionary fate of a novel mutation relative to that of its progenitor. Immediately after the mutation occurs, differences in growth rate distinguish the mutant from the progenitor in both models. In the horizontal model, the two types will continue competing to infect the same set of hosts until one is lost to extinction. In the vertical model, by contrast, the two types can compete only as long as they reside in the same individual. Once they are passed into separate individuals, they reside in effectively isolated populations, and selection on growth rate no longer serves to distinguish among them. As a consequence, mutations are exposed to a longer period of selection in the horizontal model. Phrased another way, selection has more time to oppose downward mutation in horizontally transmitted pathogens.
For both of these reasons, we would expect mutation to more severely reduce virulence in vertically transmitted pathogens than in their horizontally transmitted counterparts. In the interest of brevity, we look ahead to Fig. 6 Lower Right. This figure depicts a typical pair of vertical (shaded bars) and horizontal (open bars) steady-state pathotype distributions, for a population of n = 25 host individuals, with the virus replicating for t = 10 viral generations in between each transmission event. In the horizontal transmission model, virulence is maintained at a level just below that of the quasispecies equilibrium. As expected, virulence in the vertical model drops to a much lower level. The time course leading to these distributions can be summarized by a plot of the mean pathotype as a function of time (Fig. 5).
Figure 6.

The effect of host population size on the pathotype distributions. Long-term distributions of pathotype frequencies, generated by computer simulation, are shown for the vertical transmission model (shaded bars) and horizontal transmission model (open bars). Parameter values are t = 10 and b = 1. (Upper Left) n = 1; (Upper Right) n = 5; (Lower Left) n = 10; (Lower Right) n = 25. The distributions shown are averaged over 1,800 transmission events, after a 200-generation settling period that allows the system to approach equilibrium.
Figure 5.

The change in mean pathotype over time in transmission events, for the horizontal (solid line) and the vertical (dashed line) transmission models with n = 25, t = 10, and b = 1. For comparison, the mean pathotype at the quasispecies equilibrium is 8.47.
The Role of Host Population Size.
In the vertical model, each host lineage is composed of an effectively distinct subpopulation and hence the pathogen dynamics within any particular lineage are unaffected by the number of subpopulations. Therefore, we expect that the host population size will not affect the balance between downward-biased mutation and differential growth in the vertical model; the dynamics will always be those of a subpopulation passed through a bottleneck of size b.
By contrast, virus particles can move among distinct host lineages in the horizontal transfer model. As mentioned above, the consequences are twofold. The effective bottleneck size between two successive host cohorts is now nb, and therefore increases with n. Moreover, in order to found a new infection, a given virion competes with virions from all of the hosts. As the number of hosts n increases, so does the amount of competition. The more virulent pathotypes not only enjoy a replicative advantage over less virulent types within the same host, but also directly outcompete less virulent types in other hosts. Fig. 6 shows the effect of host population size on horizontally and vertically transmitted pathogen populations. As expected, the pathogen distribution in the vertical model is unaffected by the host population size, whereas the steady-state distribution in the horizontal model shifts toward higher virulence with increasing host population size.
The Role of Viral Replication Time.
In both the horizontal and the vertical models, the distribution of viral pathotypes will depend crucially on the number of periods t of viral replication between each transmission event. The pathotype distribution will approach the quasispecies distribution as t increases regardless of the initial pathotype distribution at the time of infection. However, the sampling process may disrupt this convergence by the stochastic loss of more fit pathotypes. The viral pathotype distribution will go through a cycle in which it approaches the quasispecies equilibrium through within-host replication and then drifts away from this distribution through interhost transfer and the associated bottleneck. Therefore, the frequency of transmission events and the duration of the intervening periods of viral reproduction will determine the degree to which the actual pathotype distribution is able to approach the quasispecies equilibrium.
In the horizontal transmission model, the number of viral generations between transmission events simply determines the rate of bottleneck-induced drift; as t decreases, the rate of drift increases, and consequently mean pathotype will decline. In the vertical model, a similar effect operates. In addition, t also determines the degree of virus–virus competition. Recall that in this model two pathotypes compete only while the residing in the same host. For illustrative purposes, consider the competition experienced by a new mutant when the bottleneck size is 1. At transmission, the mutant will inevitably be separated from its progenitor, and selection on differential growth rate will cease. The expected time from the genesis of a new mutant to this separation is simply t/2, and hence the strength of selection to oppose downward mutation increases with t. This is illustrated in Fig. 7.
Figure 7.

The effect of the number of within-host viral replication events between each transmission event, on the pathotype distributions for n = 25 and b = 1 in the vertical (shaded bars) and horizontal (open bars) models. (Upper Left) t = 1; (Upper Right) t = 10; (Lower Left) t = 25; (Lower Right) t = 50. The pathotype distributions were generated by computer simulation; each histogram represents the pathotype distribution averaged over 45,000 viral replication periods, after an initial settling-time of 5,000 viral replication periods.
The Role of Transmission Bottleneck Size.
Novella et al. (22) suggest that inoculum size may affect not only the risk of invasion into a particular host but also the rate of genetic drift in the pathogen and thus longer term evolution of pathogen virulence. We can examine this possibility formally for both vertically transmitted and horizontally transmitted pathogens.
In the horizontal model, the effective transmission bottleneck size on the entire viral population is nb. Therefore, when n is small, the inoculum size b may play an important role, with virulence declining as inoculum size decreases. When n is large, the ratchet will operate very slowly even for b = 1, and inoculum size will have little effect. In the vertical model, by contrast, the bottleneck size affects the expected length of time that two pathotypes share a common host. Because bottleneck size consequently affects the degree of competition as well as the rate of the ratchet in this model, we expect a stronger reduction in virulence with declining bottleneck size for vertically transmitted pathogens. This is illustrated in Fig. 8.
Figure 8.

The effect of transmission bottleneck size on the pathotype distributions for n = 25 and t = 10 in the vertical (shaded bars) and horizontal (open bars) models. (Upper Left) b = 1; (Upper Right) b = 2; (Bottom Left) b = 4; (Bottom Right) b = 8. The simulation procedure is as described in Fig. 6.
The deterministic mutation–selection balance distribution (Fig. 4) represents, in a sense, the maximum ability of selection to oppose downward-biased mutation. We can see how the actual strength of selection is affected by innoculum size by considering the expected change in virulence over the course of a single transmission and replication cycle, starting at this distribution. In the horizontal model, the strength of selection is entirely unaffected: the expected virulence is unchanged from that at the quasispecies equilibrium, regardless of the number of generations of viral replication or the bottleneck size. In the vertical model, by contrast, the strength of selection is reduced by the partitioning of virions into evolutionarily separate host lineages. As Fig. 9 shows, tighter bottlenecks more powerfully obstruct selection and hence allow greater declines in mean virulence. As the number of viral replications increases, mean fitness takes an initial dip. This is because it takes numerous generations of replication and downward mutation to cause maximum damage to the viral pathotype distribution within a particular host. The maximum decline in pathotype per viral generation occurs in the first viral replication after the bottleneck, and successively eases from that point on. In the absence of another bottleneck, the viral pathotype distribution can actually begin to improve, as the viral titer within a single host becomes very large and the distribution within the single host begins to approach the deterministic mutation–selection balance distribution. We see this in the eventual return of expected fitness toward the deterministic mutation–selection balance level, as the number of viral replications becomes very large.
Figure 9.

The expected virulence after a single round of host-to-host transfer, starting at the deterministic mutation–selection balance distribution, for the vertical transmission model. Curves correspond to bottleneck sizes of 1, 2, 3, and 4. Horizontal line gives the mean virulence at the deterministic mutation-selection balance.
The results illustrated in Figs. 8 and 9 may shed light on the intriguing results presented in refs. 20, 22, and 23. In a series of plaque-to-plaque transfer experiments using vesicular stomatitis virus, these authors observed that for most strains, successive transmission bottlenecks of size 1 led to dramatic reductions in mean fitness, whereas transmission bottlenecks of size 5 led to little, if any, reduction in fitness. These results are in marked contrast to conventional expectations regarding the population sizes at which ratchet effects disappear (18).
Why does the ratchet effect disappear at such low bottleneck sizes? We can offer at least three points of qualitative explanation in addition to our numerical results. First, our selective coefficient s is large. Second, population size grows large between bottlenecks, and moreover, there will be almost no drift effects between bottlenecks because the populations are undergoing exponential growth. Third, in our model, the ratchet is actually somewhat reversible, because of beneficial mutations.
CONCLUSIONS
In this study, we have shown that transmission bottlenecks can significantly impact the fitness and consequently the virulence of rapidly mutating pathogens. While some decrease in fitness occurs even under horizontal transmission, vertically transmitted pathogens suffer the most dramatic declines in fitness. Because fitness and virulence are often tightly associated in viruses, this provides an alternative explanation for the oft-cited observation that vertically transmitted pathogens evolve lower virulence than do their horizontally transmitted relatives, an explanation that relies not on selection for improving host survival but rather on the mechanics of the process of genetic transmission for the pathogen.
Our models demonstrate that the cause of this virulence decline is twofold. First, as bottleneck sizes become smaller, a process akin to Muller’s ratchet can cause the stochastic loss of more virulent pathotypes. Because vertically transmitted pathogens are effectively partitioned into many evolutionary distinct populations, they will face tighter effective bottlenecks than their horizontally transmitted counterparts and thus will suffer a faster rate of virulence decline. Second, in vertically transmitted pathogen populations, transmission bottlenecks serve to separate competing pathotypes and reduce the net power of natural selection to favor rapid viral reproduction, further reducing the virulence of these populations. Together, these effects may play an important role in determining pathogen virulence in natural systems.
Acknowledgments
We thank the following of individuals for their helpful discussion and comments: R. Antia, E. Domingo, E. Halloran, B. Levin, M. Lipsitch, J. Paulsson, J. Pritchard, D. Rosen, and two anonymous referees. C.B. is partially supported by National Institutes of Health/National Institute of Allergy and Infectious Diseases Grant T32-AI0742. L.R. is supported by National Science Foundation Grant DEB 9629748.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Antia R, Levin B R, May R M. Am Nat. 1994;144:457–472. [Google Scholar]
- 2.Bonhoeffer S, Nowak M A. Proc R Soc London Ser B. 1994;258:133–140. [Google Scholar]
- 3.Bull J J, Molineaux I J, Rice W R. Evolution. 1991;45:875–882. doi: 10.1111/j.1558-5646.1991.tb04356.x. [DOI] [PubMed] [Google Scholar]
- 4.Ewald P W. Ann N Y Acad Sci. 1987;503:295–306. doi: 10.1111/j.1749-6632.1987.tb40616.x. [DOI] [PubMed] [Google Scholar]
- 5.Herre E A. Science. 1993;259:1442–1445. doi: 10.1126/science.259.5100.1442. [DOI] [PubMed] [Google Scholar]
- 6.Lenski R E, May R M. J Theoret Biol. 1994;169:253–265. doi: 10.1006/jtbi.1994.1146. [DOI] [PubMed] [Google Scholar]
- 7.Levin B R, Svanborg Eden C. Parasitology. 1990;100:S103–S115. doi: 10.1017/s0031182000073054. [DOI] [PubMed] [Google Scholar]
- 8.Lipsitch M, Herre E A, Nowak M A. Evolution. 1995;49:743–748. doi: 10.1111/j.1558-5646.1995.tb02310.x. [DOI] [PubMed] [Google Scholar]
- 9.Lipsitch M, Nowak M A, Ebert D, May R M. Proc R Soc London Ser B. 1995;260:321–327. doi: 10.1098/rspb.1995.0099. [DOI] [PubMed] [Google Scholar]
- 10.Lipsitch M, Siller S, Nowak M A. Evolution. 1996;50:1729–1741. doi: 10.1111/j.1558-5646.1996.tb03560.x. [DOI] [PubMed] [Google Scholar]
- 11.May R M, Nowak M A. Proc R Soc London Ser B. 1995;261:209–215. [Google Scholar]
- 12.Nowak M A, May R M. Proc R Soc London Ser B. 1994;255:81–89. [Google Scholar]
- 13.Read A F. Nature (London) 1993;362:500–501. doi: 10.1038/362500a0. [DOI] [PubMed] [Google Scholar]
- 14.Fine P E F. Ann N Y Acad Sci. 1975;26:173–194. doi: 10.1111/j.1749-6632.1975.tb35099.x. [DOI] [PubMed] [Google Scholar]
- 15.Clayton D H, Tompkins D M. Proc R Soc London Ser B. 1994;256:211–217. doi: 10.1098/rspb.1994.0072. [DOI] [PubMed] [Google Scholar]
- 16.Wilson D S, Pollack G B, Dugatkin L A. Evol Ecol. 1992;6:331–341. [Google Scholar]
- 17.Domingo E, Holland J J. Annu Rev Microbiol. 1997;51:151–178. doi: 10.1146/annurev.micro.51.1.151. [DOI] [PubMed] [Google Scholar]
- 18.Haigh J. Theoret Pop Biol. 1978;14:251–267. doi: 10.1016/0040-5809(78)90027-8. [DOI] [PubMed] [Google Scholar]
- 19.Chao L. Nature (London) 1990;348:454–455. doi: 10.1038/348454a0. [DOI] [PubMed] [Google Scholar]
- 20.Duarte E, Clarke D, Moya A, Domingo E, Holland J. Proc Nat Acad Sci USA. 1992;89:6015–6019. doi: 10.1073/pnas.89.13.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duarte E, Clarke D K, Moya A, Elana S F, Domingo E, Holland J. J Virol. 1993;67:3620–3623. doi: 10.1128/jvi.67.6.3620-3623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novella I S, Elena S F, Moya A, Domingo E, Holland J J. J Virol. 1995;69:2869–2872. doi: 10.1128/jvi.69.5.2869-2872.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novella I S, Elena S F, Moya A, Domingo E, Holland J J. Mol Gen Genet. 1996;252:733–738. doi: 10.1007/BF02173980. [DOI] [PubMed] [Google Scholar]
- 24.Nowak M A, Anderson R M, McLean A R, Wolfs T F, Goudsmit J, May R M. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 25.Artenstein M S, Miller W S. Bacteriol Rev. 1966;30:571–572. doi: 10.1128/br.30.3.571-572.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerone P J, Couch R B, Ketter G V, Douglas R G, Derrenbacher E B, Knight V. Bacteriol Rev. 1966;30:576–584. doi: 10.1128/br.30.3.576-588.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubin L G. Rev Infect Dis. 1987;9:488–492. doi: 10.1093/clinids/9.3.488. [DOI] [PubMed] [Google Scholar]
- 28.Domingo E, Martinez-Salas E, Sobrino F, de la Torre J C, Portela A, Ortin J, Lopez-Galindez C, Perez-Brena P, Villanueva N, Najera R, et al. Gene. 1985;40:1–8. doi: 10.1016/0378-1119(85)90017-4. [DOI] [PubMed] [Google Scholar]
- 29.Domingo E, Escarmis C, Sevilla N, Moya A, Elena S F, Quer J, Novella I S, Holland J J. FASEB J. 1996;10:859–864. doi: 10.1096/fasebj.10.8.8666162. [DOI] [PubMed] [Google Scholar]
- 30.Eigen M, Schuster P. Naturwissenschaften. 1977;64:541–565. doi: 10.1007/BF00450633. [DOI] [PubMed] [Google Scholar]
- 31.Tsimring L S, Levine H, Kessler D A. Phys Rev Lett. 1996;76:4400–4443. doi: 10.1103/PhysRevLett.76.4440. [DOI] [PubMed] [Google Scholar]