

Abstract
Recent findings suggest that methamphetamine (METH) functions acutely to inhibit N-methyl-d-aspartate (NMDA) receptor function. Protracted withdrawal from METH exposure may increase the sensitivity of NMDA receptors to agonist exposure, promoting neuronal excitability. However, the relevance of METH effects on NMDA receptor activity with regard to neuronal viability has not been studied. The present studies examined the effects of protracted METH exposure (6 or 7 days; 1.0-100 μM) and withdrawal (1 or 7 days) on NMDA receptor-dependent neurotoxicity, determined with use of the non-vital fluorescent marker propidium iodide, in organotypic slice cultures of male and female rats. Prolonged exposure to METH (100 μM) produced only modest toxicity in the granule cell layer of the dentate gyrus. Withdrawal from METH exposure (1 or 7 days) did not produce overt neuronal injury in any region of slice cultures. Exposure to NMDA (5 μM) produced marked neurotoxicity in the CA1 pyramidal cell layer. Neither co-exposure to METH nor 1 day of METH withdrawal in combination with NMDA exposure altered NMDA-induced neurotoxicity. In contrast, protracted withdrawal from METH exposure (7 days) was associated with a marked (~400%) increase in NMDA-induced neurotoxicity in CA1 region pyramidal cells. This potentiation of neurotoxicity was prevented by co-exposure to the selective NMDA receptor antagonist 5-2-amino-5-phoshonovaleric acid (20 μM) and was markedly attenuated by co-exposure of slices to xestospongin C (1 μM), an antagonist of IP3 receptors. The results of the present studies suggest that long-term METH withdrawal functionally sensitizes the NMDA receptor to agonist exposure and requires the co-activation of NMDA and IP3 receptors.
Keywords: stimulants, drug abuse, amphetamine, glutamate, brain injury
1. Introduction
Methamphetamine (METH) use represents a significant public health concern in the United States. In 2005, 4.3% of Americans 12 years or older reported using METH for nonmedical purposes at least once their lifetime (Substance Abuse and Mental Health Services Administration, 2005). METH use has been associated with several significant health risks, including cardiac arrhythmia, stroke, increased blood pressure, hyperthermia, and CNS abnormalities. (Davidson et al., 2001, Ricaurte et al., 1982). CNS abnormalities associated with METH use, including volumetric deficits, have been reported in a variety of brains regions (Ernst et al., 2000; Thompson et al., 2004; Volkow et al., 2001) and are thought to reflect changes in the signaling and metabolism of multiple neurotransmitters, including dopamine, serotonin and glutamate (Davidson et al., 2001, Spina & Cohen, 1989, Rocher and Gardier, 2001; Yamamoto & Zhu, 1998; Zhang et al., 2006, reviewed by Cadet et al. 2007; Tat and Yamamoto 2007). In some individuals, these abnormalities correlate with cognitive deficits during both active METH use and early abstinence (Kalechstein et al, 2003, Simon et al, 2000).
It has become increasingly evident that N-Methyl-d-Aspartate-type glutamate receptors are targets of METH actions (Moriguchi et al, 2002; Smith et al., 2007; Yeh et al., 2002). Previous literature has suggested that METH may directly or indirectly influence the NMDA receptor in a biphasic manner (Moriguchi et al., 2002; Smith et al., 2007; Yeh et al., 2002). Short-term, repeated administration of METH to rodents has been shown to increase hippocampal and striatal glutamate content (Mark et al. 2007; Rocher and Gardier, 2001) while long-term METH administration was shown to deplete hippocampal glutamate content (Kaiya et al. 1982). Amphetamines have been shown to displace [3H]N-[1-(2-thienyl)cyclohexyl] piperidine (TCP) binding to the NMDA receptor ion channel, in addition to, decreasing NMDA-induced intracellular 45Ca accumulation in rat cortical neurons (Yeh et al., 2002). A recent report similarly demonstrated that short-term METH (≥1.0 μM) exposure antagonizes NMDA-induced neurotoxicity in organotypic hippocampal slice cultures, suggesting direct or indirect modulation of NMDA receptor activity (Smith et al., 2007). However, METH antagonism of the NMDA receptor does not appear to occur at the MK-801 sensitive channel site. Evidence that METH may act as a functional antagonist of the NMDA receptor, either by direct interactions or subsequent to METH-induced glutamate release (ie. desensitization, after Krupp et al. 1996), suggest that long-term METH exposure may result in adaptive changes of NMDA receptor abundance or function during withdrawal. In fact, Moriguchi et al. (2002) have reported that long-term METH exposure followed by long-term withdrawal may reduce Mg2+ blockade of the NMDA receptor resulting in increased EPSPs.
Although previous work suggests that METH either directly or indirectly influences function of some NMDA receptors, little is know of the consequences of this influence with regard to METH withdrawal. Therefore, the present studies examined the ability of long-term METH exposure or withdrawal to influence NMDA receptor mediated neurotoxicity in organotypic hippocampal slice cultures.
2. Results
2.1: Continuous Methamphetamine Exposure & NMDA Toxicity
Organotypic hippocampal slice cultures were initially exposed to METH (1.0-100 μM) for 7 days. During the final day of METH exposure, one-half of cultures were also exposed to NMDA (5 μM). Twenty-four hours later, no significant increases in propidium iodide (PI) uptake were observed in control (CTRL) or METH treated tissue in the CA1 and CA3 pyramidal cell layers. PI is a highly polar nucleic acid marker that only labels cells with marked membrane compromise (Zimmer et al. 2000). However, 7 days of exposure to the highest concentration of METH did produce a modest but significant increase in PI uptake in the granule cell layer of the DG (Table 1). Exposure to NMDA for 24 hr produced a marked increase in PI uptake (~200%) in the CA1 pyramidal cell region (F(7,136)=6.711, P<0.001; Fig. 1), an effect that was prevented by co-exposure to the selective NMDA receptor antagonist 5-2-amino-5-phoshonovaleric acid (20 μM). METH co-exposure did not affect NMDA induced toxicity.
Table 1.
PI uptake in DG granule cell layer following 7 days of METH (100 μM) exposure or following six days of METH exposure (100 μM), and 24 hours of withdrawal. * = P<0.05 vs. CTRL.
| CTRL | 1 μM METH | 10 μM METH | 100 μM METH | |
|---|---|---|---|---|
| No Withdrawal | 97.44 ± 2.82 | 95.46 ± 5.02 | 99.27 ± 4.44 | 120.38 ± 10.86* |
| 24hr Withdrawal | 103.39 ± 5.04 | 97.24 ± 3.89 | 98.16 ± 3.83 | 114.78 ± 5.82 |
Figure 1.
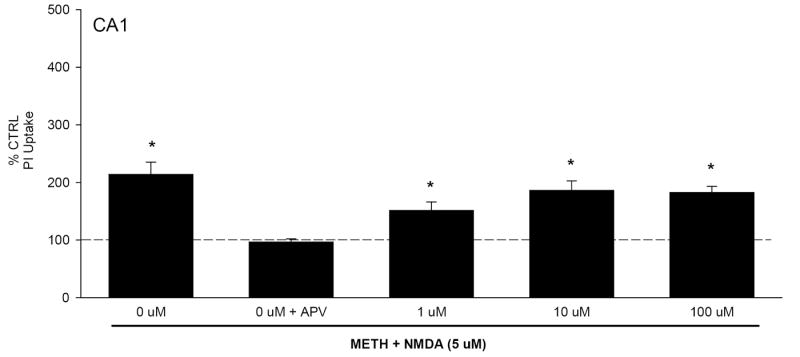
PI uptake in CA1 pyramidal cell layer following 7 days of METH (1-100 μM) exposure with the addition of NMDA (5 μM) on the seventh day. Treatment with NMDA, regardless of METH co-exposure, resulted in significant PI uptake. *=P<0.05 vs control.
2.2: Methamphetamine Withdrawal & NMDA Toxicity
Organotypic hippocampal slice cultures were exposed to 6 days of METH treatment, followed by 1 or 7 days of withdrawal. On the final day of withdrawal, cultures were then exposed to 5 μM NMDA. After 1 day of withdrawal, there were no significant increases in PI uptake in control and METH withdrawn tissue (Table 1). Tissue treated with NMDA displayed significant increases in PI uptake, in the CA1 pyramidal cell layers, regardless of pretreatment (F(7,204)=9.897, P<0.001; Fig. 2; F(7,204)=9.897, P<0.001). The DG and CA3 did not display significant increases in PI uptake.
Figure 2.

PI uptake in CA1 pyramidal cell layer following 6 days of METH exposure (100 μM), and 24 hours of withdrawal with the addition of NMDA (5 μM). Exposure to NMDA, regardless of pretreatment, resulted in significant PI uptake. *=P<0.05 vs control.
After 7 days of withdrawal from METH, there were no significant increases in PI uptake in NMDA naïve tissue. The CA1 pyramidal cell layer, displayed significant PI uptake in NMDA treated tissue, an effect that was significantly increased by prior METH exposure, at all concentrations (F(9,150)=25.652, P<0.001; 100 μM shown in Fig.3). PI uptake in METH withdrawn tissue (100 μM) treated with NMDA was approximately 80% more than control tissue treated with NMDA. All increases in PI uptake were blocked by exposure to 5-2-amino-5-phoshonovaleric acid, a selective NMDA receptor antagonist (20 μM; APV) (Fig. 3). The DG and CA3 regions did not display significant increases in PI uptake.
Figure 3.

PI uptake in CA1 pyramidal cell layer following 6 days of METH exposure (100 μM), followed by 7 days of withdrawal and subsequent exposure to NMDA (5 μM). METH withdrawal (1-100 μM) resulted in a potentiated NMDA-induced response when compared to CTRL tissue exposed to NMDA. Potentiated response to NMDA is displayed as increases beyond the solid black line. APV (20 μM) reduced all NMDA-induced PI uptake to CTRL levels. Xestospongin C (1 μM) significantly reduced the observed potentiated effect following METH withdrawal. * = P<0.001 vs. CTRL + NMDA, ** = p<0.001 vs. METH withdrawal + NMDA.
Exposure to the inositol triphosphate (IP3) receptor antagonist xestospongin C (1 μM) with NMDA did not alter PI uptake in control or METH withdrawn tissue. Additionally, xestospongin C treatment did not significantly influence PI uptake in CTRL tissue treated with NMDA, suggesting the lack of involvement of Ca2+ release from IP3-sensitive stores in the observed toxicity. However, xestospongin C co-exposure did significantly reduced METH-potentiated (100 μM) NMDA-induced neurotoxicity (F(7,139)=24.049, P<0.001; Fig. 3) in the CA1 pyramidal cell layer, by approximately 23%. Representative images of PI uptake are included in Fig. 4.
Figure 4.

Representative images of PI uptake in control cultures and those exposed to Xestospongin C (20 M), NMDA (5 μM) or a combination of both during protracted methamphetamine withdrawal.
3. Discussion
Previous literature has suggested that both short-term and long-term METH exposure may result in changes to glutamatergic signaling (Kaiya et al., 1983; Rocher & Gardier, 2001). Furthermore, some studies suggest that METH may act as an NMDA receptor antagonist (Smith et al., 2007; Yeh et al., 2002). Yeh et al. (2002) reported that METH reduced NMDA-induced intracellular Ca2+ accumulation as well as displaced [H3]TCP binding. Antagonism of NMDA-induced neurotoxicity by METH was reported in organotypic hippocampal slice cultures (Smith et al., 2007), further suggesting that METH may act as a NMDA receptor antagonist. Long-term NMDA receptor antagonism by METH may result in neuronal excitability during withdrawal. In fact, Moriguchi et al. (2002) reported increased amplitudes of NMDA mediated excitatory postsynaptic potentials (EPSPs) in neostriatal slices following withdrawal from METH. Evidence of neuronal excitability during protracted METH withdrawal suggests that compensatory responses to chronic METH administration may be similar to other NMDA antagonists. For example, compensatory responses to chronic ethanol exposure, such as increased cell surface expression of NR subunits, contribute to increased excitability and a loss of GABAergic tone during ethanol withdrawal (Alele & Devaud et al., 2005; Davidson et al., 1995; Maler et al., 2005; Prendergast et al., 2000). Such effects are likely to contribute to neurotoxicity, hippocampal deficits, cognitive problems, and possibly cue-induced craving during withdrawal (Bartels et al., 2007; Prendergast et al., 2004; Schneider et al., 2001).
The present studies examined the effects of long-term METH exposure, followed by withdrawal, on NMDA-induced neurotoxicity in organotypic hippocampal slice cultures. Long-term exposure (7 days) to METH did not produce overt neurotoxicity in pyramidal cells of the CA1 or CA3 regions, though a small but significant increase in toxicity was observed in granule cells of the DG with exposure to 100 μM METH. This finding is potentially of interest in that an isolated tissue model was employed that was devoid of METH effects on body temperature. However, the relevance of this high concentration of METH to the study of recreational METH use has not been fully addressed. Chung et al. (2004) did report that plasma levels of METH reached high μM to low mM in a sample of South Korean individuals suffering acute lethality associated with METH use (ie. from traumatic injury or cardiac arrhythmia). Further, Melega et al. (1995) demonstrated that brain concentrations of METH were approximately 10-fold greater than were plasma concentrations following a single METH injection in rodents, raising the possibility that METH may be sequestered in brain lipids.
Withdrawal (1 or 7 days) from METH did not result in any observable toxicity. However, long-term withdrawal (7 days) did result in a markedly potentiated neurotoxic response to NMDA exposure, a finding that is consistent with the electrophysiological findings of Moriguchi et al. (2002). Long-term withdrawal does appear to be an essential component of the observed neurotoxicity, since short-term withdrawal and chronic exposure without withdrawal did not result in a potentiated NMDA effect. It is unclear as to why evidence of sensitized NMDA receptor-mediated response was not evident within 24 hr of the start of METH withdrawal, though it is possible that METH may be sequestered in brain lipid (after Melega et al. 1995), requiring a protracted time period to achieve complete clearance of METH. Furthermore, CA1 pyramidal cells appear more susceptible to NMDA-induced neurotoxicity. This effect may reflect the higher receptor composition of the CA1 region, since NMDA receptors are extremely dense in the basal and apical dendritic fields of the CA1 (Martens et al, 1998; Mulholland et al, 2003; Prendergast et al, 2004; Shin & Lee, 1999). The observed potentiation was reduced by exposure to APV, an NMDA receptor antagonist. Furthermore, inhibition of IP3 mediated Ca2+ stores blocked the potentiated neurotoxicity to NMDA. These latter data, in particular, suggest that following NMDA receptor activation, Ca2+-induced Ca2+ release from endoplasmic reticulum (ER) is a key mediator of this METH withdrawal-potentiated NMDA response.
Changes in intracellular Ca2+ signaling reflecting activation of IP3 receptors may result from METH interactions with ER-bound sigma receptors (Hayashi et al., 2005). Related findings demonstrate that following long-term cocaine exposure, ER-bound sigma receptors promote dissociation of a cytoskeletal adaptor protein ankyrin from IP3 receptors (Su & Hayashi, 2001). Dissociation of ankyrin leads to increases of IP3 mediated intracellular Ca2+ accumulation. METH has been found to bind directly to sigma receptors (Su and Hayashi, 2003). Following METH self administration, sigma receptors were shown to be upregulated in the midbrain of rats (Stefanski et al., 2004). Furthermore, pharmacological antagonism of sigma receptors blocked the development of METH induced behavioral sensitization (Takahashi et al., 2000; Ujike et al., 1996). Thus, one possible interpretation of the present findings is that METH exposure produces a persisting increase in the sensitivity of ER Ca2+ stores to agonist-induced release into cytosol. Though the present studies did not examine the biochemical pathway(s) involved in the neuronal injury observed, METH administration has been shown to induce neuronal apoptosis, including that occurring after ER stress (Davidson et al., 2001; Jayanthi et al., 2004). For example, 30 min following METH administration (40mg/kg), calpain, a cytosolic cysteine protease associated with ER-dependent cell death, was activated and continued to remain activated for seven days (Jayanthi et al., 2004). It will be of interest in future studies to examine activation of calpain during protracted METH withdrawal.
While ER functioning does appear to be integral in the observed effects, other METH-induced signaling changes may also contribute to the initiation of neuronal excitability during withdrawal. Loss of GABAergic tone has been suggested to influence behavioral sensitization to METH (Zhang et al., 2006). Following long-term METH administration (25mg/kg/day) and withdrawal (28 days), GABAA α2 subunit protein levels were decreased in the nucleus acumbens. Glutamic acid decarboxylase (GAD) protein levels were also decreased in the nucleus acumbens (Zhang et al., 2006). Downregulation of GABA receptors, as well as, decreases in GABAergic signaling could result in increased excitability and possibly explain the observed effects. Further, METH may directly interact with specific NR subunits of NMDA receptors, producing compensatory upregulation(s) in their expression or function, though one recent study did demonstrate that METH does not appear to interact with an MK-801 channel binding site (Smith et al. 2007).
It is intriguing that protracted METH withdrawal produces a hypersensitivity of glutamatergic systems in the hippocampus, as this may be related to drug craving. During METH withdrawal, increases in craving have been reported by regular METH users (McGregor et al, 2005). Cue-induced craving, in particular, is a significant risk factor for relapse in abstinent METH users (Chang and Haning, 2006; Newton et al., 2006) and has been suggested to involve activation of the hippocampus in stimulant and depressant users (Kilts et al., 2001; Schneider et al., 2001). Though the specific means by which METH may influence NMDA receptor-mediated responses has not been fully elucidated, this receptor system represents a molecular target of METH that should be further studied as it may represent a novel therapeutic target to be exploited in the maintainence of abstinence from METH use.
4. Methods
4.1 Hippocampal Cell Culture
Eight day old, male and female, Sprague-Dawley rat pups (Harlan Laboratories; Indianapolis, ID, USA) were sacrificed and brains were aseptically removed. After the brains were extracted, they were transferred to dissecting medium (containing Minimum Essential Medium (MEM) plus glutamine, 25 mM HEPES, 200 mM glutamine, and 50 μM streptomycin/penicillin) (Gibco BRL, Gaithersburg, MD) and the bilateral hippocampi were dissected out. Hippocampi were then cleaned of extra tissue and placed in culture media (containing dissection medium with the addition of 36 mM glucose, 25% Hanks’ balanced salt solution (HBSS) (Gibco BRL, Gaithersburg, MD), and 25% heat-inactivated horse serum (HIHS) (Sigma, St. Louis, MO)). Afterward, unilateral hippocampi were then sectioned coronally at 200 μm using the McIllwain Tissue Chopper (Campden Instruments Ltd; Lafayette, ID, USA). Once sectioned intact hippocampi were plated onto Millicell-CM biopore membrane inserts, with one ml of culture medium added to the bottom of each well. Plates were then place in an incubator, at 37 °C and with a gas composition of 5% C02, 21% oxygen, and 74% nitrogen, for five days to allow hippocampi to affix to the teflon membrane.
4.2 Drug Exposure
Slice cultures were exposed to METH (1.0-100 μM; Sigma; St. Louis, MO, USA) for 6 or 7 days and uptake of propidium iodide (3.74 μM; Molecular Probes), a highly polar nucleic acid stain used to detect cell damage, was assessed at several time points after METH exposure (0, 1, or 7 days later). Concentrations of METH employed were chosen to reflect the range of concentrations observed in both plasma and brain of rats following a single i.v. injection of METH (Melega et al. 1995), as well as, plasma and hair samples of humans who had ingested METH for recreational purposes (Nakashima et al. 2003). Additional cultures were exposed to NMDA (5 μM; Sigma; St. Louis, MO, USA) at the end of the withdrawal period and were also labeled with PI. D,L,-2-amino-5-phosphovalerate, an NMDA antagonist, (APV) (20 μM; Sigma; St. Louis, MO, USA) was utilized to confirm the role for NMDA receptor involvement in NMDA effects. Xestospongin C (1 μM; EMD Biosciences; Darmstadt, Germany), an inositol triphospate (IP3) receptor antagonist, was also utilized on the final day of withdrawal to block IP3 mediated intracellular Ca2+ accumulation. One ml increments of drugs were applied to the bottom of culture plates.
4.3 Cytotoxicity assessment
PI only penetrates cell membranes of damaged or potentially dying cells, binding to DNA to produce a bright, intensified red fluorescence at 630 nm (Zimmer et al, 2000). Hippocampi were visualized with SPOT Advanced version 4.0.2 software for Windows (W. Nuhsbaum Inc.; McHenry, IL, USA) at a 5X objective with a Leica DMIRB microscope (W. Nuhsbaum Inc.; McHenry, IL, USA) that is fitted for fluorescent detection (Mercury-arc lamp) using blue-green light, and connected to a personal computer through a SPOT 7.2 color mosaic camera (W. Nuhsbaum Inc.; McHenry, IL, USA). The emission wavelength of PI is 620 nm in the visual range, and has a peak excitation wavelength of 536 nm. PI is excited using a band-pass filter exciting wavelengths between 510 and 560 nm. Intensity of the PI fluorescence was then analyzed by densitometry using Image J. Pictures were then quantified by detecting optical intensity of the CA1 pyramidal cell layer, CA3 pyramidal cell layer, and dentate gyrus granule cell layer of the hippocampus following background subtraction. To minimize variability in PI uptake between replications, each replication was converted to percent control before analysis.
4.4 Statistics
Two-way ANOVAs were initially employed analyzing the effects of exposure to NMDA and/or METH in each slice culture subregion separately, based on the a priori hypothesis that the CA1 region will selectively demonstrate NMDA-induced neurotoxicity. Previous studies have shown that the CA1 region of the hippocampus is more sensitive to excitotoxic stimuli (Avignone et al, 2005; Gee et al, 2006; Self et al, 2005). As no sex differences in NMDA or METH effects were observed in any subregion of slice cultures, data were collapsed across sex and one-way ANOVAs of the different areas were performed to compare treatments within each individual area of the hippocampus. Statistical outliers were discovered using the Grubb’s test for outliers (Grubbs, 1969). Post hoc comparisons were made using the Fisher LSD Method (Hayter, 1986). Significance was set at P<0.05. All analyses were conducted on data converted to percent control, to compensate for variability observed across weeks.
Acknowledgments
The authors acknowledge the support of DA 016176
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alele DE, Devaud LL. Differential adaptations in GABAergic and glutamatergic systems during ethanol withdrawal in male and female rats. Alcohol Clin Exp Res. 2005;29(6):1027–1034. doi: 10.1097/01.alc.0000167743.96121.40. [DOI] [PubMed] [Google Scholar]
- Avignone E, Frenguelli BG, Irving AJ. Differential responses to NMDA receptor activation in rat hippocampal interneurons and pyramidal cells may underlie enhanced pyramidal cell vulnerability. Eur J Neurosci. 2005;22(12):3077–3090. doi: 10.1111/j.1460-9568.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- Bartels C, Kunert HJ, Stawicki S, Kroner-Herwig B, Ehrenreich H, Krampe H. Recovery of hippocampus-related functions in chronic alcoholics during monitored long-term abstinence. Alcohol. 2007;42(2):92–102. doi: 10.1093/alcalc/agl104. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN, Jayanthi S, Lyles J. Neurotoxicity of substituted amphetamines: Molecular and cellular mechanisms. Neurotox Res. 2007;11(34):183–202. doi: 10.1007/BF03033567. [DOI] [PubMed] [Google Scholar]
- Chang L, Haning W. Insights from recent positron emission tomographic studies of drug abuse and dependence. Curr Opin Psychiatry. 2006;19(3):246–252. doi: 10.1097/01.yco.0000218594.46431.2f. [DOI] [PubMed] [Google Scholar]
- Chung H, Park M, Hahn E, Choi H, Choi H, Lim M. Recent trends of drug abuse and drug-associated deaths in Korea. Ann N Y Acad Sci. 2004;1025:458–464. doi: 10.1196/annals.1316.056. [DOI] [PubMed] [Google Scholar]
- Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: Necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Davidson M, Shanley B, Wilce P. Increased NMDA-induced excitability during ethanol withdrawal: a behavioural and histological study. Brain Res. 1995;674(1):91–96. doi: 10.1016/0006-8993(94)01440-s. [DOI] [PubMed] [Google Scholar]
- Devaud LL, Alele DE. Differential effects of chronic ethanol administration and withdrawal on gamma-aminobutyric acid type A and NMDA receptor subunit proteins in male and female rat brain. Alcohol Clin Exp Res. 2004;28(6):957–965. doi: 10.1097/01.alc.0000128225.83916.40. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chang L, Leonido-Yee M, Speck O. Evidence for long-term neurotoxicity associated with methamphetamine abuse: A 1H MRS Study. Neurology. 2000;54:1344–1349. doi: 10.1212/wnl.54.6.1344. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Raineteau O, Rietschin L, Kirbach SW, Gerber U. NMDA receptors and the differential ischemic vulnerability of hippocampal neurons. Eur J Neurosci. 2006;23(10):2595–2603. doi: 10.1111/j.1460-9568.2006.04786.x. [DOI] [PubMed] [Google Scholar]
- Grubbs F. Procedures for detecting outlying observations in samples. Technometrics. 1969;11(1):1–21. [Google Scholar]
- Hayashi T, Su TP. The potential role of sigma-1 receptors in lipid transport and lipid raft reconstitution in the brain: Implication for drug abuse. Life Sci. 2005;77:1612–1624. doi: 10.1016/j.lfs.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hayter AJ. The maximum familywise error rate of Fisher’s least significant difference test. J Am Stat Assoc. 1986;81:1000–1004. [Google Scholar]
- Jayanthi S, Deng X, Noailles P-IH, Ladenheim B, Cadet JL. Methamphetamine induces neuronal apoptosis via cross-talks between endoplasmic reticulum and mitochondria-dependent death cascades. FASEB J. 2004;18:238–251. doi: 10.1096/fj.03-0295com. [DOI] [PubMed] [Google Scholar]
- Kaiya H, Takeuchi K, Yoshida H, Kondo T, Sanpei F, Okada Y, Namba M. Effects of subchronic treatment of methamphetamine haloperidol on the rat brain levels of GABA, glutamate and aspartate. Folia Psychiatr Neurol Jpn. 1983;37(1):107–13. doi: 10.1111/j.1440-1819.1983.tb00309.x. [DOI] [PubMed] [Google Scholar]
- Kalechstein AD, Newton TF, Green M. Methamphetamine dependence is associated with neurocognitive impairment in the initial phases of abstinence. J Neuropsychiatry Clin Neurosci. 2003;15:215–220. doi: 10.1176/jnp.15.2.215. [DOI] [PubMed] [Google Scholar]
- Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Gaber TL, Muhammad F, Ely TD, Hoffman JM, Drexler KP. Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry. 2001;58(4):334–341. doi: 10.1001/archpsyc.58.4.334. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Heinemann SF, Westbrook GL. Calcium-dependent inactivation of recombinant N-methyl-D-aspartate receptors is NR2 subunit specific. Mol Pharmacol. 1996;50(6):1680–1688. [PubMed] [Google Scholar]
- Maler JM, Esselmann H, Wiltfang J, Kunz N, Lewczuk P, Reulbach U, Bleich S, Rǘther E, Kornhuber J. Memantine inhibits ethanol-induced NMDA receptor up-regulation in rat hippocampal neurons. Brain Res. 2005;1052(2):156–162. doi: 10.1016/j.brainres.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Mark KA, Quinton MS, Russek SJ, Yamamoto BK. Dynamic changes in vesicular glutamate transporter 1 function and expression related to methamphetamine-induced glutamate release. J Neurosci. 2007;27(25):6823–6831. doi: 10.1523/JNEUROSCI.0013-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens U, Capito B, Wree A. Septotemporal distribution of [3H]MK-801, [3H]AMPA, and [3H]Kainate Binding Sites in the Rat Hippocampus. Anat Embryol. 1998;198:195–204. doi: 10.1007/s004290050177. [DOI] [PubMed] [Google Scholar]
- McGregor C, Srisurapanont M, Jittiwutikarn J, Laobhripatr S, Wongtan T, White JM. The nature, time course, and severity of methamphetamine withdrawal. Addiction. 2005;100:1320–1329. doi: 10.1111/j.1360-0443.2005.01160.x. [DOI] [PubMed] [Google Scholar]
- Melega WP, Williams AE, Schmitz DA, DiStefano EW, Cho AK. Pharmacokinetic and pharmacodynamic analysis of the actions of D-amphetamine and D-methamphetamine on the dopamine terminal. J Pharmacol Exp Ther. 1995;274(1):90–96. [PubMed] [Google Scholar]
- Moriguchi S, Watanabe S, Kita H, Nakanishi H. Enhancement of N-methyl-D-aspartate receptor-mediated excitatory postsynaptic potentials in the neostriatum after methamphetamine sensitization. Exp Brain Res. 2002;144:238–246. doi: 10.1007/s00221-002-1039-3. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Prendergast MA. Transection of intrinsic polysynaptic pathways reduces N-methyl-D-aspartate neurotoxicity in hippocampal slice cultures. Neurosci Res. 2003;46:369–376. doi: 10.1016/s0168-0102(03)00102-0. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Kaddoumi A, Ishida Y, Itoh T, Taki K. Determination of methamphetamine and amphetamine in abusers’ plasma and hair samples with HPLC-FL. Biomed Chromatogr. 2003;17(7):471–476. doi: 10.1002/bmc.278. [DOI] [PubMed] [Google Scholar]
- Newton TF, Roache JD, De La Garza R, Fong T, Wallace CL, Li SH, Elkashef A, Chiang N, Kahn R. Bupropion reduces methamphetamine-induced subjective effects and cue-induced craving. Neuropsychopharmacology. 2006;31(7):1537–1544. doi: 10.1038/sj.npp.1300979. [DOI] [PubMed] [Google Scholar]
- Office of National Drug Control Policy Drug policy information clearinghouse fact sheet: Methamphetamine. Executive Office of the President. 2003:1–6. [Google Scholar]
- Prendergast MA, Harris BR, Blanchard JA, Mayer S, Gibson DA, Littleton JM. In vitro effects of ethanol withdrawal and spermidine on viability of hippocampus from male and female rat. Alcohol Clin Exp Res. 2000;24(12):1855–1861. [PubMed] [Google Scholar]
- Prendergast MA, Harris BR, Mulholland PJ, Blanchard JA, II, Gibson DA, Holley RC, Littleton JM. Hippocampal CA1 region neurodegeneration produced by ethanol withdrawal requires activation of intrinsic polysynaptic hippocampal pathways and function of N-methyl-D-aspartate receptors. Neuroscience. 2004;124:869–877. doi: 10.1016/j.neuroscience.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Rocher C, Gardier AM. Effects of repeated systemic administration of D-fenfluramine on serotonin and glutamate release in rat ventral hippocampus: comparison with methamphetamine using in vivo microdialysis. Naunyn-Schmiedeberg’s Arch Pharmacol. 2001;363(4):422–428. doi: 10.1007/s002100000381. [DOI] [PubMed] [Google Scholar]
- SAMHSA Office of Applied Studies The NSDUH report. Drug Abuse Warning Network. 2004:1–3. [Google Scholar]
- Schneider F, Habel U, Wagner M, Franke P, Salloum JB, Shah NJ, Toni I, Sulzbach C, Honig K, Maier W, Gaebel W, Zilles K. Subcortical correlates of craving in recently abstinent alcoholic patients. Am J Psychiatry. 2001;158(7):1075–1083. doi: 10.1176/appi.ajp.158.7.1075. [DOI] [PubMed] [Google Scholar]
- Self RL, Smith KJ, Mulholland PJ, Prendergast MA. Ethanol exposure and withdrawal sensitizes the rat hippocampal CA1 pyramidal cell region to beta-amyloid (25-35)-induced cytotoxicity: NMDA receptor involvement. Alcoholism Clin Exp Res. 2005;29(11):2063–2069. doi: 10.1097/01.alc.0000187591.82039.b2. [DOI] [PubMed] [Google Scholar]
- Shin C, Lee KH. Epilepsy and Cell Death. In: Koliatsos VE, Ratan RR, editors. Cell Death and Diseases of the Nervous System. Totowa, N.: Humana Press Inc.; 1999. pp. 361–377. [Google Scholar]
- Simon SL, Domier C, Carnell J, Brethen P, Rawson R, Ling W. Cognitive impairment in individuals currently using methamphetamine. J Addict Dis. 2000;9(3):222–231. doi: 10.1080/10550490050148053. [DOI] [PubMed] [Google Scholar]
- Smith KJ, Self RL, Butler TR, Mullins MM, Ghayoumi L, Holley RC, Littleton JM, Prendergast MA. Methamphetamine exposure antagonized N-methyl- d-aspartate receptor-mediated neurotoxicity in organotypic hippocampal slice cultures. Brain Res. 2007;1157:74–80. doi: 10.1016/j.brainres.2007.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina MB, Cohen G. Dopamine turnover and glutathione oxidation: implications for Parkinson’s disease. Proc Natl Acad Sci USA. 1989;86(4):1398–1400. doi: 10.1073/pnas.86.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanski R, Justinova Z, Hayashi T, Takebayashi M, Goldberg SR, Su TP. Sigma(1) receptor upregulation after chronic methamphetamine self-administration in rats: a study with yoked controls. Psychopharmacology. 2004;75(1):68–75. doi: 10.1007/s00213-004-1779-9. [DOI] [PubMed] [Google Scholar]
- Su TP, Hayashi T. Cocaine affects the dynamics of cytoskeletal proteins via sigma(1) receptors. Trends Pharmacol Sci. 2001;22(9):456–458. doi: 10.1016/s0165-6147(00)01740-5. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Miwa T, Horikomi K. Involvement of sigma 1 receptors in methamphetamine-induced behavioral sensitization in rats. Neurosci Lett. 2000;289(1):21–24. doi: 10.1016/s0304-3940(00)01258-1. [DOI] [PubMed] [Google Scholar]
- Tata DA, Yamamoto BK. Interactions between methamphetamine and environmental stress: Role of oxidative stress, glutamate, and mitochondrial dysfunction. Addiction. 2007;1:49–60. doi: 10.1111/j.1360-0443.2007.01770.x. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, Simon SL, Geaga JA, Hong MS, Sui Y, Lee JY, Toga AW, Ling W, London ED. Structural abnormalities in the brains of human subjects who use methamphetamine. J Neurosci. 2004;24(26):6028–6036. doi: 10.1523/JNEUROSCI.0713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujike H, Kuroda S, Otsuki S. Sigma receptor antagonists block the development of sensitization to cocaine. Eur J Neurosci. 1996;296(2):123–128. doi: 10.1016/0014-2999(95)00693-1. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci. 2001;21(23):9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Yeh GC, Chen JC, Tsai HC, Wu HH, Lin CY, Hsu PC, Peng YC. Amphetamine inhibits the N-Methyl-D-Asparate receptor-mediated responses by directly interacting the receptor/channel complex. J Pharmacol Exp Ther. 2002;300(3):1008–1016. doi: 10.1124/jpet.300.3.1008. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lee TH, Xiong X, Chen Q, Davidson C, Wetsel WC, Ellinwood EH. Methamphetamine induces long-term changes in GABAA receptor α2 subunit and GAD67 expression. Biochem Biophys Res Commun. 2006;351:300–305. doi: 10.1016/j.bbrc.2006.10.046. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Kristensen BW, Jakobsen B, Noraberg J. Excitatory amino acid neurotoxicity and modulation of glutamate receptor expression in organotypic brain slice cultures. Amino Acids. 2000;19:7–21. doi: 10.1007/s007260070029. [DOI] [PubMed] [Google Scholar]