

Abstract
Progression through the G1-phase of the cell cycle requires that cyclin D and CDK4 phosphorylate pRB and the other pocket proteins, p107 and p130. Cyclin E and CDK2 further phosphorylate pRB to complete its inactivation and allow the cell to enter S-phase. These phosphorylation events lead to the inactivation of the antiproliferative effect of the pocket proteins. The pocket proteins are the main targets of CDK4, and its unregulated activity can contribute to carcinogenesis. Mip/LIN9 is a recently described protein with growth suppressor, as well as growth promoting effects due to its ability to stabilize B-Myb and induce genes required for S phase and mitosis. The finding that a mutation that deletes the first 84 amino acids of Mip/LIN-9 corrects the defects of the CDK4 knockout mouse suggests that it should have a growth repressor effect that is blocked by CDK4. However, overexpression of cyclin D only partially blocks the inhibitory effect of Mip/LIN-9 on cell proliferation. Here, we performed experiments to further understand the antiproliferative effect of Mip/LIN-9 within the context of the pocket proteins. Our results suggest that there is a pocket protein-independent mechanism of the Mip/LIN-9 antiproliferative effect since it can be observed in cells with ablation of the three members of the family, and in NIH3T3 cells expressing the adenovirus E1A-12S protein. Altogether, the independence from the pocket proteins and the partial blockade of the antiproliferative effect produced by expression of cyclin D suggest that the role of Mip/LIN-9 downstream of CDK4 may be more closely related to the activation of B-Myb and the induction of S/M genes. Importantly, the regulatory effect of CDK4 is not due to direct phosphorylation of Mip/LIN-9 by this kinase or even CDK2, suggesting an indirect mechanism.
Keywords: Mip/LIN-9, retiboblastoma, pocket proteins, cell cycle, CDK4
INTRODUCTION
Quiescent cells are induced to divide following the addition of growth factors. In order for cell division to be autonomous of external stimuli it must overcome a regulatory mechanism termed the restriction point [1–3]. A key event at this point represents the inactivation of the retinoblastoma gene product, pRB, by phosphorylation thereby freeing the E2F transcription factor to activate the genes necessary for cell cycle progression [4–6]. This phosphorylation is accomplished by specific cyclin/CDK modules. CyclinD/CDK4 is the primary kinase to phosphorylate pRB in early G1 [7, 8]. Following expression of cyclin E, pRB is further phosphorylated by cyclinE/CDK2 leading to its complete inactivation [9–12]. The importance of this event is highlighted by activating mutations of the cyclin D/CDK4 machinery commonly found in cancer cells [13–15]. Furthermore, overexpressing pRB in tissue culture can lead to a G1 cell cycle arrest [16].
The CDK4 knockout mouse has provided insight to the importance of this G1 kinase [17, 18]. The phenotype of the CDK4 knockout mouse was characterized by small body mass, infertility in both sexes and diabetes. Each characteristic could be related to a defect in proliferation. During continuous culture in complete medium CDK4−/− MEFs are completely indistinguishable from wild-type MEFs. However, when they are serum starved to quiescence and then released, the CDK4−/− MEFs have a delay in cell cycle reentry demonstrating the role of CDK4 in initiating the cell cycle [17, 18].
Mip/LIN-9 is the mammalian member of a new family of proteins, which also includes C. elegans LIN-9, Drosophila mip130 and aly, and A. thaliana always early [19–22]. C. elegans LIN-9 was the first member discovered [19]. It participates in a pRB-like pathway that negatively regulates vulva differentiation and was shown to partially correct a cyclin D mutant in C. elegans [23, 24]. Mip/LIN-9 was first described by Gagrica et al. as a suppressor of oncogenesis in collaboration with pRB [25]. Sandoval et al. described that expression of Mip/LIN-9 inhibits cell proliferation and that this effect is partially overcome by cyclin D [26]. Furthermore, the CDK4−/− phenotype could be corrected by crossing with mice carrying an 84 amino acid N-terminal deletion of Mip/LIN-9, termed Δ84. Remarkably, MEFs generated from this cross had a restored cell cycle reentry following serum-starvation and a normalization of the expression of E2F-regulated genes. These findings demonstrate that the antiproliferative effect of Mip/LIN-9 is negatively regulated by Cyclin D/CDK4.
Interestingly, Mip/LIN-9 is also critical for the progression through S-phase and mitosis through an interaction with the transcription factor B-Myb [27]. In the absence of Mip/LIN-9, B-Myb is likely degraded leading to an impaired expression of S/M-phases genes such as cyclin A, cyclin B and CDK1. Additionally, co-expression of Mip/LIN-9 and B-Myb leads to activation of the same promoters in cells with unregulated CDK4 activity.
We recently have determined that Mip/LIN-9 specifically interacts with p107, p130 and E2F4 but not with pRB or activating E2Fs [28]. Moreover, Mip/LIN-9 has mutually exclusive interaction with p107,p130/E2F4 in G0/G1 and with B-Myb in G1/S phases of the cell cycle [28]. The dissociation of Mip/LIN-9 from p107,p130/E2F4 is likely driven by an increase in CDK4 activity, since it does not re-associate with the repressor complex until the activity of this CDK falls to baseline levels in the next cell cycle. These findings support a model in which CDK4 regulates the expression of S/M genes via Mip/LIN-9 [28].
Importantly, although the mutation of Mip/LIN-9 corrected the CDK4 null phenotype, overexpression of cyclin D resulted in only a partial block of the antiproliferative effect of Mip/LIN-9 in NIH/3T3 cells [26]. This finding supports the concept that Mip/LIN-9 may affect cell proliferation by different mechanisms. In this report, we tested whether Mip/LIN-9 was able to inhibit cell proliferation only within the context of the pocket proteins and the effect of CDK4 on Mip/LIN-9 phosphorylation. Our results demonstrate that overexpression of Mip/LIN-9 in cells that lack pRB, p107 and p130 still inhibits cell proliferation. Moreover, the effect of Mip/LIN-9 is not abrogated by E1A-12S, which encodes a protein capable of blocking the growth inhibitory effect of all pocket proteins, further supporting a pocket protein-independent component in the antiproliferative effect of Mip/LIN-9. Surprisingly, the 13S form of E1A was able to inhibit the growth inhibitory effect of Mip/LIN-9 indicating that the zinc finger region, present only in the 13S form, is responsible for overcoming the antiproliferative effect of Mip/LIN-9 [29]. Finally, our results also suggest that the regulatory effect of CDK4 is not exerted via direct phosphorylation of Mip/LIN-9. Altogether, these data supports the concept that the growth inhibitory effect of Mip/LIN-9 has a component that is independent of cyclin D/CDK4 and the pocket proteins, and that the role of this G1 kinase may be more closely related to the release of Mip/LIN-9 from the p107,p130/E2F4 repressor complex to allow the association with B-Myb and the induction of S/M genes.
MATERIAL AND METHODS
Cell Culture
All cell lines were cultured in DMEM supplemented with 10% FBS. pLXSN retrovirus was produced using the BOSC23 packaging cell line or PA317.
Colony formation
Cell lines were plated in 100mm plates and infected the next day with pLXSN, pLXSN-Mip/LIN-9L, or pLXSN-Mip/LIN-9S. Following two days of infection the medium was changed to selection medium containing 500µµg/mL G418. Selection continued for up to two weeks with the medium changed every 3 days. Following selection plates were fixed with methanol, stained with crystal violent, and destained with methanol.
BrdU incorporation assay
Transfections were carried out in NIH/3T3s stably expressing empty vector, E1A-12S or E1A-13s or 3T3s generated from Rb−/− MEFs or MEFs deficient in all three pocket proteins (TKO) [30]. Cells were seeded on coverslips and transfected with the indicated vector in triplicate. Twenty-four hours later the cells were pulsed with BrdU (10µM) for twenty-four hours. Coverslips were fixed with paraformaldehyde and stained with mouse anti-myc (UBI 05-724) or rat anti-BrdU (Abcam ab6326) counterstained with anti-mouse alexa 488 or anti-rat alexa 568 (molecular probes). At least one hundred myc positive cells were counted per coverslip and assessed for BrdU incorporation.
In Vitro kinase assay
293 lysate was used as a source of CDK4/cyclin D complexes. CDK4 was immunoprecipitated with anti-CDK4 (Santa Cruz, sc-260). Immunoprecipitations were incubated with GST-Mip/LIN-9, or GST-Rb (sc-4112). In vitro kinase assay was incubated for 30’ in in vitro kinase buffer (50mM Tris pH7.4, 12mM MgCl2, 200µM orthovanadate, 100mM NaCl, 25mM β-glycerol phsosphate, 2mM MnCl2) at room temperature in the presence of 2µCi [32]P γ-ATP (Amersham). Kinase reaction was quenched with 2x SDS-PAGE loading buffer, separated by SDS-PAGE, transferred to a PVDF membrane and exposed to autoradiography film. The membrane was then blotted with Mip/LIN-9 monoclonal Ab [26].
RESULTS
Previous reports indicated that Mip/LIN-9 has a negative effect on cell growth [25, 26]. To further assess the effect of Mip/LIN-9 on cell proliferation, we performed colony assays on a broad range of cell lines. A retroviral vector, pLXSN, containing either the full length Mip/LIN-9-L (MW approximately 62,000) or the splice variant Mip/LIN-9-S (MW approximately 54,000) or empty vector was used to infect a variety of cell lines. Following selection for ten to fourteen days, the colonies were stained with crystal violent. Fig. 1 shows that a large number of colonies were evident in the pLXSN infected plates (Fig. 1, pLXSN). However, significantly fewer colonies were found in the Mip/LIN-9 infected plates across all cell lines. The expression of the 54 kDa splice variant appears to be less inhibitory than the full length form (Fig. 1). These data support the concept that Mip/LIN-9 has a suppressive effect on cell proliferation. Importantly, the effect was observed in cells such as HeLa in which the function of the pocket proteins is inhibited by the E7 protein of human papilloma virus.
Figure 1. Mip/LIN-9 inhibits colony formation.
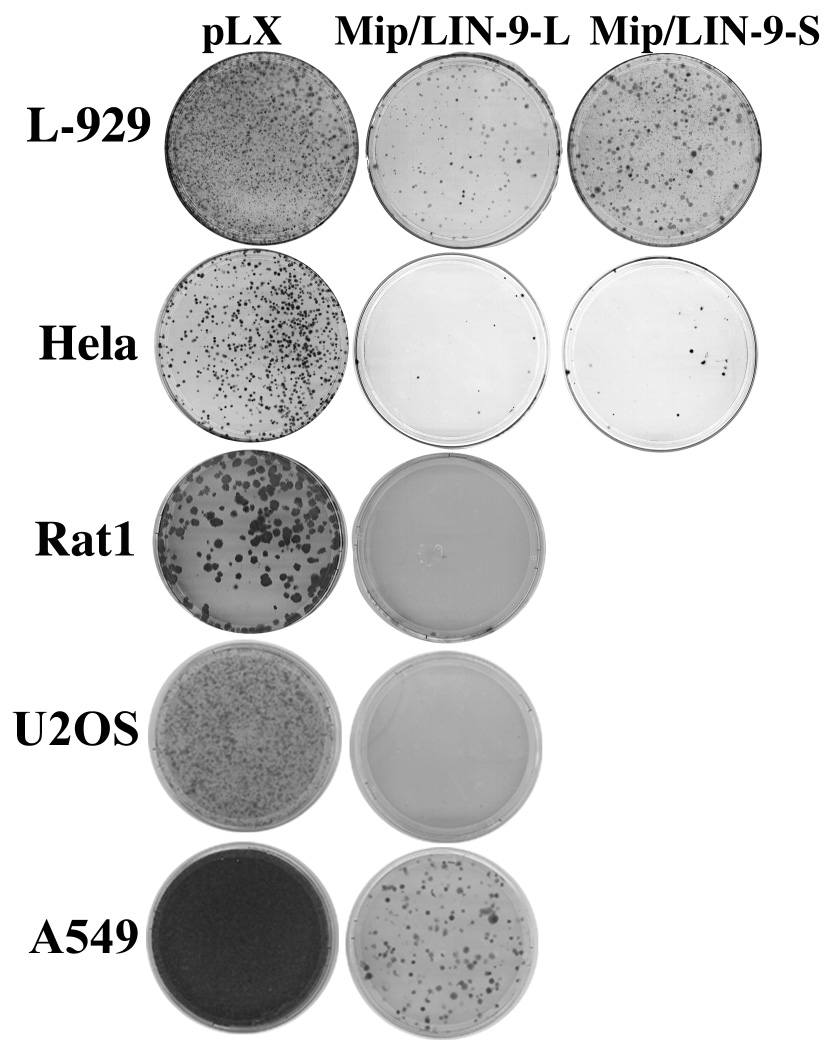
Cell lines were infected with control pLXSN retrovirus, pLXSN-Mip/LIN-9L or pLXSN-Mip/LIN-9S. Following two days of infection, cells were selected with G418 for up to two weeks. Plates were fixed and stained with crystal violet.
It was suggested by Gagrica et al that Mip/LIN-9 impairs oncogenic transformation through an interaction with pRB [25]. However, we have shown that Mip/LIN-9 interacts with p107 and p130 rather than with pRB [28]. Additionally, in NIH/3T3 cells Mip/LIN-9 can inhibit proliferation and this effect is partially counteracted by coexpressing cyclin D [26], which together with CDK4 are responsible for inactivation of the pocket proteins. To determine if the ability of Mip/LIN-9 to stop proliferation is dependent on pRB, p107 or p130, we used NIH/3T3 stably expressing the adenovirus 12S or 13S E1A oncoproteins. E1A encodes a protein that binds to pRB, as well as p107 and p130, and blocks its tumor suppressor effect [31]. Two splice variant proteins, 12S and 13S are encoded by the E1A gene. NIH/3T3 cells containing the different forms of E1A or only the neor-gene were plated on coverslips, transfected with myc-tagged Mip/LIN-9 or myc-caveolin 1 (myc-CAV1) as a negative control, and pulsed with 10µM BrdU. Myc positive transfectants were scored for BrdU positivity. Fig 2 shows that Mip/LIN-9 has the same antiproliferative effect on 3T3-12S as in control 3T3-neo cells and that this effect is partially blocked by cyclin D. This suggests that the anti-proliferative effect of Mip/LIN-9 is not dependent on pRB or the other pocket proteins and that there is a growth inhibitory component independent of cyclin D. Surprisingly, the inhibition of BrdU incorporation by the expression of Mip/LIN-9 was not observed in NIH/3T3 expressing the 13S form of E1A. This splice variant may have a stronger proliferative mechanism as it contains a zinc-finger domain not found in 12S that could contribute to transcriptional activation of proliferative genes [29].
Figure 2. Mip/LIN-9 inhibits cell proliferation independent of the pocket proteins.
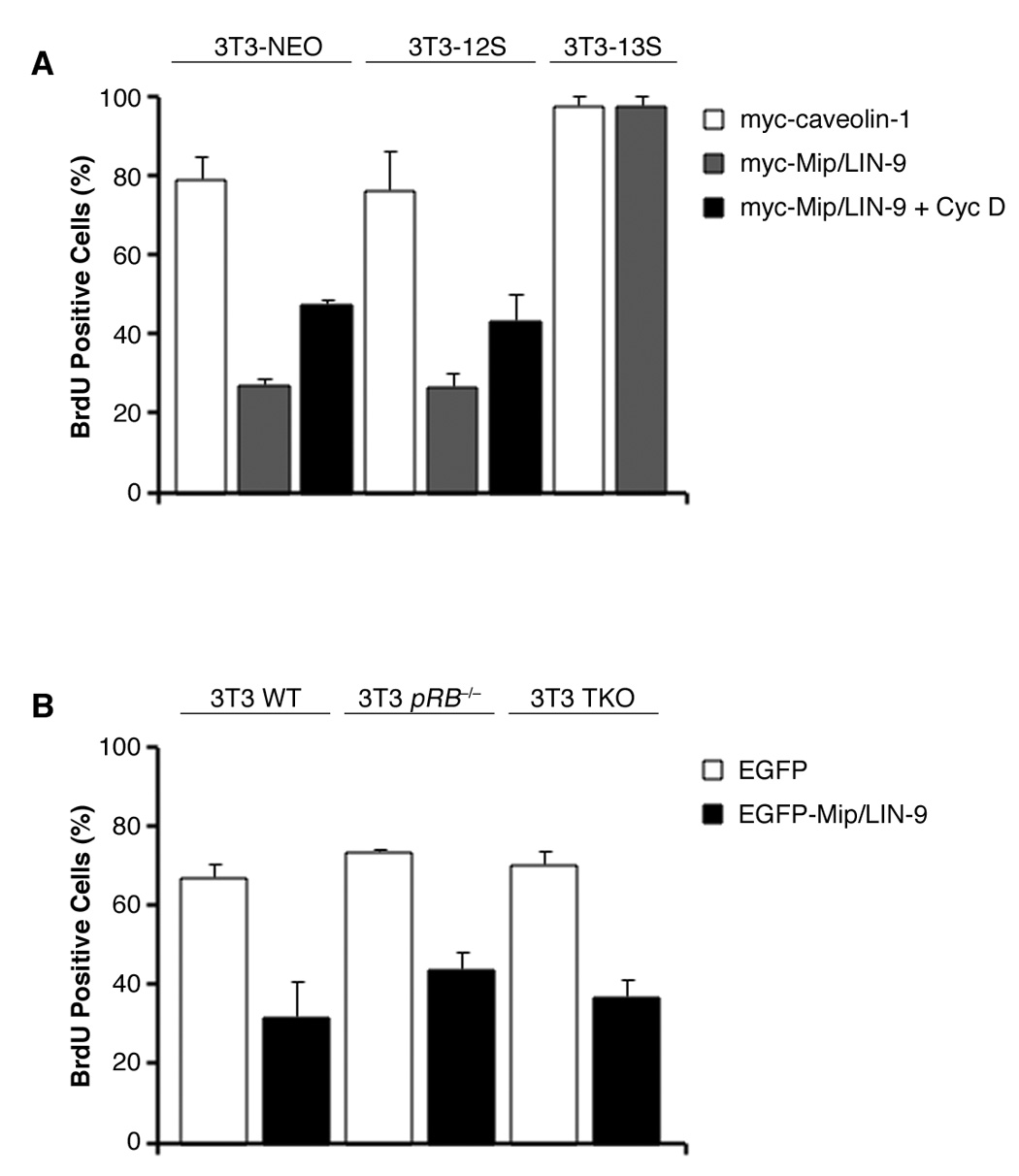
A) NIH/3T3 stably transfected with E1A-12S, -13S or empty vector were transfected with myc-Mip/LIN-9 or myc-CAV1 (control). Cells were pulsed with BrdU and immunostained with anti-myc and anti-BrdU antibodies. B) 3T3 cells deficient for Rb (Rb−/−) or all three pocket proteins (TKO) were transfected and immunostained as above.
To further confirm that this effect is independent of the pocket proteins, we expressed Mip/LIN-9 in 3T3 cells derived from embryos null for pRB or all three of the pocket proteins (TKO) [30, 32]. Fig. 3 shows that Mip/LIN-9 was able to arrest proliferation despite the absence of pRB, p107 and p130. These data demonstrate that part of the growth inhibitory effect of Mip/LIN-9 is pocket protein-independent.
Figure 3. Mip/LIN-9 is not a substrate of CDK4.

In vitro kinase assay. GST-Mip/LIN-9, or GST-Rb was incubated with active CDK4 or CDK2 complex in the presence of γ-ATP. Proteins were separated by SDS-PAGE and transferred to PVDF. The membrane was exposed to X-ray film and subsequently blotted with anti-Mip/LIN-9 mAb.
Since a mutation of Mip/LIN-9 can correct the CDK4 null phenotype, we next tested whether G1 CDKs were able to phosphorylated Mip/LIN-9. To achieve this goal, we performed immunoprecipitations with antibodies against G1 cyclins and CDKs followed by in vitro kinase assays using as a substrate GST-Mip/LIN-9 or GST-RB as a positive control. There was no detectable phosphorylation of Mip/LIN-9 despite the presence of active kinase (see pRB lanes). Therefore, we conclude, that while Mip/LIN-9 acts downstream of CDK4, it is not a direct substrate for CDK4 or even CDK2. The finding that Mip/LIN-9 interacts with p107 and p130 strongly suggests that CDK4 regulates Mip/LIN-9 via phosphorylation of these pocket proteins [28].
DISCUSSION
Several lines of evidence place Mip/LIN-9 downstream of CDK4. First, the CDK4−/− mouse has proliferative defects that are corrected by crossing with Δ84 Mip/LIN-9 animals, [17, 18, 26]. Second, overexpression of Mip/LIN-9 in NIH/3T3 causes a cell cycle arrest and this is partially overcome by coexpressing cyclin D [26]. In this paper, we explored the relationship between Mip/LIN-9 and the best-known substrates of CDK4, the pocket protein, as well as the relationship between CDK4 activity and the antiproliferative effect of Mip/LIN-9. We first demonstrated that Mip/LIN-9 inhibits cell growth across a broad range of mammalian cell types by showing its negative effect on colony formation assays. Importantly, the finding that Mip/LIN-9 also inhibited the growth of HPV-E7 positive HeLa cells suggested that at least part of the mechanism did not involve the pocket proteins.
Therefore we sought to determine if the anti-proliferative effect of Mip/LIN-9 was at least in part independent on pRB. Surprisingly, Mip/LIN-9 was able to inhibit proliferation of not only pRB−/− cells, but also cells that lack the entire family of pocket proteins. Whether the pocket proteins are inactivated by E1A-12S or are genetically ablated, overexpression of Mip/LIN-9 will nonetheless block cell proliferation. Therefore, at least part of the mechanism employed by Mip/LIN-9 to control cell proliferation goes beyond the CDK4 pathway, since inhibition of cell growth cannot be completely reverted by cyclin D.
Since we previously demonstrated that Mip/LIN-9 functions downstream of CDK4 within the context of p107,p130 [28], it was important to establish if it was directly phosphorylated by this G1 CDK. However, neither endogenous Mip/LIN-9 nor GST-Mip/LIN-9 was phosphorylated in vitro in the presence of active CDK4 or CDK2. Therefore, the mechanism by which Mip/LIN-9Δ84 corrects the CDK4 knockout is not by mutating a substrate site of CDK4. We are currently investigating whether the rescue of the CDK4 null phenotype involves an alteration in the association or recruitment of Mip/LIN-9 to the p107,p130/E2F4 repressor complex.
In summary, the antiproliferative effect of Mip/LIN-9 has a component that is regulated by cyclin D/CDK4 and a CDK4- and pocket protein-independent arm. It is likely that the main regulatory effect of CDK4 is more closely related to the release of Mip/LIN-9 from the p107,p130/E2F4 complex to allow it to interact and stabilize B-Myb, a critical step for the induction of S/M genes [27, 28, 33]
Acknowledgements
We thank Drs. M. Classon and T. Jacks for the pRB and TKO cells. We also appreciate Dr. J. Cook for the NIH/3T3 cells expressing different forms of the adenovirus E1A protein.
This work has been supported by The National Institutes of Health Grant GM54709 (ORC). MP was supported by an NRSA/NIH Institutional T32 training grant, "Training Program in Signal Transduction and Cellular Endocrinology", T32 DK07739 from the NIDDK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This paper is based on a presentation at a Focused Workshop of the 4th Myb Workshop entitled: “Mip/LIN-9 links negative and positive regulators of the cell cycle” sponsored by The Leukemia & Lymphoma Society held in Civitella Alfedena, May 20–24th, 2007
References
- 1.Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol. 1996;8:805–814. doi: 10.1016/s0955-0674(96)80081-0. [DOI] [PubMed] [Google Scholar]
- 2.Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle. 2002;1:103–110. [PubMed] [Google Scholar]
- 3.Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol. 1996;16:6917–6925. doi: 10.1128/mcb.16.12.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 5.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 6.Brown VD, Phillips RA, Gallie BL. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol Cell Biol. 1999;19:3246–3256. doi: 10.1128/mcb.19.5.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 8.Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol. 2000;12:676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 9.Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–12746. doi: 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- 10.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 12.Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF. Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol. 2001;21:4773–4784. doi: 10.1128/MCB.21.14.4773-4784.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho A, Dowdy SF. Regulation of G(1) cell-cycle progression by oncogenes and tumor suppressor genes. Curr Opin Genet Dev. 2002;12:47–52. doi: 10.1016/s0959-437x(01)00263-5. [DOI] [PubMed] [Google Scholar]
- 14.Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, Sherr CJ. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 15.Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 16.Knudsen ES, Buckmaster C, Chen TT, Feramisco JR, Wang JY. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 1998;12:2278–2292. doi: 10.1101/gad.12.15.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999;19:7011–7019. doi: 10.1128/mcb.19.10.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin J, Hunt SL, Dubus P, Sotillo R, Nehme-Pelluard F, Magnuson MA, Parlow AF, Malumbres M, Ortega S, Barbacid M. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene. 2003;22:5261–5269. doi: 10.1038/sj.onc.1206506. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson EL, Horvitz HR. The multivulva phenotype of certain Caenorhabditis elegans mutants results from defects in two functionally redundant pathways. Genetics. 1989;123:109–121. doi: 10.1093/genetics/123.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Aasland R, White-Cooper H, Dyson N, Brehm A. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell. 2004;119:181–193. doi: 10.1016/j.cell.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 21.White-Cooper H, Leroy D, MacQueen A, Fuller MT. Transcription of meiotic cell cycle and terminal differentiation genes depends on a conserved chromatin associated protein, whose nuclear localisation is regulated. Development. 2000;127:5463–5473. doi: 10.1242/dev.127.24.5463. [DOI] [PubMed] [Google Scholar]
- 22.Bhatt AM, Zhang Q, Harris SA, White-Cooper H, Dickinson H. Gene structure and molecular analysis of Arabidopsis thaliana ALWAYS EARLY homologs. Gene. 2004;336:219–229. doi: 10.1016/j.gene.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Horvitz HR. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell. 1998;95:981–991. doi: 10.1016/s0092-8674(00)81722-5. [DOI] [PubMed] [Google Scholar]
- 24.Boxem M, van den Heuvel S. C. elegans class B synthetic multivulva genes act in G(1) regulation. Curr Biol. 2002;12:906–911. doi: 10.1016/s0960-9822(02)00844-8. [DOI] [PubMed] [Google Scholar]
- 25.Gagrica S, Hauser S, Kolfschoten I, Osterloh L, Agami R, Gaubatz S. Inhibition of oncogenic transformation by mammalian Lin-9, a pRB-associated protein. Embo J. 2004;23:4627–4638. doi: 10.1038/sj.emboj.7600470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandoval R, Xue J, Tian X, Barrett K, Pilkinton M, Ucker DS, Raychaudhuri P, Kineman RD, Luque RM, Baida G, Zou X, Valli VE, Cook JL, Kiyokawa H, Colamonici OR. A mutant allele of BARA/LIN-9 rescues the cdk4−/− phenotype by releasing the repression on E2F-regulated genes. Exp Cell Res. 2006;312:2465–2475. doi: 10.1016/j.yexcr.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Pilkinton M, Sandoval R, Song J, Ness SA, Colamonici OR. Mip/LIN-9 regulates the expression of B-Myb and the induction of cyclin A, cyclin B, and CDK1. J Biol Chem. 2007;282:168–175. doi: 10.1074/jbc.M609924200. [DOI] [PubMed] [Google Scholar]
- 28.Pilkinton M, Sandoval R, Colamonici OR. Mammalian Mip/LIN-9 Interacts with either the p107,p130/E2F4 repressor complex or B-Myb in a Cell Cycle-Phase Dependent Context Distinct from the Drosophila dREAM complex. Oncogene. doi: 10.1038/sj.onc.1210562. in press. [DOI] [PubMed] [Google Scholar]
- 29.Yang Y, McKerlie C, Borenstein SH, Lu Z, Schito M, Chamberlain JW, Buchwald M. Transgenic expression in mouse lung reveals distinct biological roles for the adenovirus type 5 E1A 243- and 289-amino-acid proteins. J Virol. 2002;76:8910–8919. doi: 10.1128/JVI.76.17.8910-8919.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003;24:159–169. doi: 10.1093/carcin/24.2.159. [DOI] [PubMed] [Google Scholar]
- 32.Classon M, Salama S, Gorka C, Mulloy R, Braun P, Harlow E. Combinatorial roles for pRB, p107, and p130 in E2F-mediated cell cycle control. Proc Natl Acad Sci U S A. 2000;97:10820–10825. doi: 10.1073/pnas.190343497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osterloh L, von Eyss B, Schmit F, Rein L, Hubner D, Samans B, Hauser S, Gaubatz S. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. Embo J. 2007;26:144–157. doi: 10.1038/sj.emboj.7601478. [DOI] [PMC free article] [PubMed] [Google Scholar]