

Abstract
Substitutions on the 2-position of the imidizole ring of histamine have proven useful in a number of biochemical settings. Current art for the synthesis of these constructs relies upon a cumbersome and low-yielding condensation reaction. Here-in we report a new procedure for the synthesis of a series of substituted 2-phenylhistamines utilizing a microwave-promoted Suzuki coupling.
Keywords: heteroaromatic Suzuki coupling, microwave promoted Suzuki coupling, 2-phenylhistamine
The utility of histamine analogues in various biochemical settings is firmly established. Derivatives of histamine based upon alterations to the 2-aminoethyl side chain have been employed as substrates for histamine N-methyltransferase and as antagonists of the H1- receptor.1,2 Methyl substitution at the 4 position of histamine generates a potent agonist of the histamine H4-receptor.3 Substitutions at the 2 position of histamine have been widely explored both synthetically and biochemically, primarily as pharmacological tools for the histamine receptors. Of particular interest has been the use of 2-phenylhistamines as potent agonists of the H1-receptor.4,5 Recent efforts by Leurs and coworkers have identified 2-(3-trifluoromethyl)phenylhistamine (3a) as the sole agonist of a mutant form of the histamine H1-receptor.6 This combination of a synthetic ligand and mutant receptor provides researchers with a tool to study the relationship between function and phenotype of a cellular target free of the selectivity issues which plague studies utilizing the native ligand and unaltered protein.
Modified native ligands such as the aforementioned histamine analogues have proven to be among the most frequently explored pharmacological tools. A key to this strategy is the ability to quickly explore numerous alterations and structural sub-types to ascertain the effectiveness of these constructs in a variety of biochemical settings. It is imperative that the strategies employed to construct these compounds are appropriately convergent to maximize synthetic efficiency. The full understanding of the biochemical utility of substituted 2-phenylhistamine analogues is likely unknown given the limited number of analogues explored to date. The synthesis of substituted 2-phenylhistamines has relied upon a condensation between amidines/imidates and 2-oxo-4-phthalimido-1-butyl acetate to afford phthalimide protected histamine analogues.5,7 Thus, a separate syntheses of substituted amidines or imidates are required for individual substituted 2-phenylhistamine analogues. Recognizing the biochemical utility of these constructs and the need for efficient methods to generate larger numbers of related analogues, we sought out alternate synthetic methods.
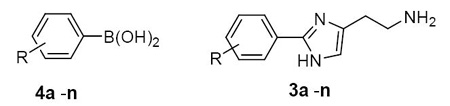
The utility of palladium-catalyzed cross coupling reactions has evolved to become a major foundation by which new carbon-carbon bonds are formed.8 The inclusion of microwave heating has aided the speed and efficiency of these transformations, particularly in the case of aryl-aryl Suzuki couplings.9 It was our goal to develop a synthetic method for the formation of substituted 2-phenylhistamines utilizing a microwave-promoted Suzuki coupling between substituted phenyl boronic acids and a common 2-halohistamine derivative. Suzuki couplings between halo-heteroaromatic constructs and aryl-boronic acids are frequently reported and successful examples include couplings between aryl-boronic acids and benzimidazoles,10 imidazoles,11 pyrimidines,12 and oxazoles.13 A method for the construction of substituted 2-phenylhistamines using this approach would allow for the rapid advancement of numerous analogues via a final stage coupling from a common precursor.
Our method relies upon the facile synthesis of a 2-halo-histamine derivative for the implementation of the final Suzuki reaction (Scheme 1). To achieve this a protocol from Cohen and coworkers14 was utilized whereby Boc protected (α-nitrogen) histamine was iodinated with 2.2 equivalents of N-iodosuccinimide to achieve Nα-Boc-2,4-diiodohistamine (1) in quantitative yield. Monoiodinations utilizing stoichiometric quantities of N-iodosuccinimide resulted in complex mixtures of the mono and diiodinated products. Treatment of 1 in refluxing 1N HCl for 72 hours followed by replacement of the Boc protecting group effectively yielded the necessary Nα-Boc-2-iodohistamine (2) in modest yields over two steps. This key intermediate was then subjected to Suzuki type conditions {PdCl2(PPh3)2 [5 mol%] in DME for 30 minutes followed by 3 equivalents of boronic acids 4a-n in aqueous sodium bicarbonate}.15 The mixture was heated via microwave irradiation to 110 °C for 2h at which time a 4 N solution of aqueous HCl was added to the reaction mixture and microwave irradiation was continued for an additional 0.5 h. Higher temperatures with shorter reaction times resulted in decomposition of the iodohistamine and an inability to recover the coupled biaryl product. The resulting products (3a-n) were purified immediately (failure to immediately purify the final products was consistently detrimental to the overall yield). Table 1 describes the scope and generality of the procedure utilizing variously substituted phenyl boronic acids to yield numerous substituted 2-phenylhistamine analogues.16 Yields were consistently greater than 60% for the two step, one pot synthetic procedure. Various catalysts were explored, as well as surveys of the inorganic base, solvent combinations and temperature ranges. While the results from these appraisals were not universal, the end choice of 5 mol% PdCl2(PPh3)2 and aqueous sodium bicarbonate consistently gave the best results. Further, we considered the need for protection of the τ-nitrogen and found that such attempts were detrimental to the Suzuki coupling. Protection of the α-nitrogen was essential for the reaction to progress. Use of alternate protecting groups for the α-nitrogen was also noted to confer unfavorable results on the procedure.
Scheme 1.
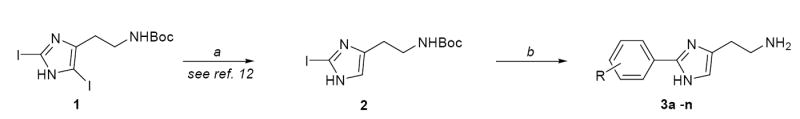
Reagents and conditions: (a) 1N HCl, reflux 72 h; then (Boc)2O, 4M NaOH, dioxane/H2O (50% over two steps); (b) PdCl2(PPh3)2 5 mol%, DME, 30 min.; then 4a-n (3 equiv.), aq. Na2CO3, microwave irradiation (110 °C, 2h.); then 4 N HCl microwave irradiation (110 °C, 30 min.).
Table 1.
 | |||
|---|---|---|---|
| Reactant | Product | R | Yield (%)* |
| 4a | 3a | m-CF3 | 62% |
| 4b | 3b | H | 66% |
| 4c | 3c | o-OCH3 | 69% |
| 4d | 3d | p-OCH3 | 67% |
| 4e | 3e | p-CH3 | 61% |
| 4f | 3f | p-tbutyl | 64% |
| 4g | 3g | o-F | 60% |
| 4h | 3h | p-F | 66% |
| 4i | 3i | 3,5-CF3 | 42% |
| 4j | 3j | o-OCH3, 5-F | 66% |
| 4k | 3k | 3,5-F | 51% |
| 4l | 3l | m-NO2 | 58% |
| 4m | 3m | m-NO2, p-CH3 | 59% |
| 4n | 3n | o-Cl | 59% |
Yields calculated from intermediate 2 following the one-pot Suzuki coupling and Boc deprotection
In summary, we have developed a concise, efficient and general method for the synthesis of substituted 2-phenylhistamine analogues from a common precursor.
Acknowledgments
We thank J. Lloyd for HRMS data. The authors would like to thank the Intramural Research Programs of the NHGRI and the NIDDK, NIH for supporting this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dent C, Nilam F, Smith IR. Biochem Pharm. 1982;31:2297–2300. doi: 10.1016/0006-2952(82)90118-6. [DOI] [PubMed] [Google Scholar]
- 2.Govoni M, Bakker RA, van de Wetering I, Smit MJ, Menge WMBP, Timmerman H, Elz S, Schunack W, Leurs R. J Med Chem. 2003;46:5812–5824. doi: 10.1021/jm030936t. [DOI] [PubMed] [Google Scholar]
- 3.Lim HD, van Rijn RM, Ling P, Bakker RA, Thurmond RL, Leurs R. J Pharmacol Exp Ther. 2005;314:1310–1321. doi: 10.1124/jpet.105.087965. [DOI] [PubMed] [Google Scholar]
- 4.Zingel V, Elz S, Schunack W. Eur J Med Chem. 1990;25:673–680. [Google Scholar]
- 5.Leschke C, Elz S, Garbarg M, Schunack W. J Med Chem. 1995;38:1287–1294. doi: 10.1021/jm00008a007. [DOI] [PubMed] [Google Scholar]
- 6.Bruysters M, Jongejan A, Akdemir A, Bakker RA, Leurs R. J Biol Chem. 2005;280:34741–34746. doi: 10.1074/jbc.M504165200. [DOI] [PubMed] [Google Scholar]
- 7.Elz S, Kramer K, Pertz HH, Detert H, ter Laak AM, Kühne R, Schunack W. J Med Chem. 2000;43:1071–1084. doi: 10.1021/jm991056a. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki A. Chem Commun. 2005:4759–4763. doi: 10.1039/b507375h. [DOI] [PubMed] [Google Scholar]
- 9.Kappe CO. Angew Chem Int Ed. 2004;43:6250–6284. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- 10.Ezquerra J, Lamas C, Pastor A, García-Navío JL, Vaquero JJ. Tetrahedron. 1997;37:12755–12764. [Google Scholar]
- 11.Langhammer I, Erker T. Heterocycles. 2005;65:2721–2728. [Google Scholar]
- 12.Ceide SC, Montalban AG. Tetrahedron Lett. 2006;47:4415–4418. [Google Scholar]
- 13.Flegeau EF, Popkin ME, Greaney MF. Org Lett. 2006;8:2495–2498. doi: 10.1021/ol060591j. [DOI] [PubMed] [Google Scholar]
- 14.Jain R, Avramovitch B, Cohen LA. Tetrahedron. 1998;54:3235–3242. [Google Scholar]
- 15.General Experimental. All microwave reactions were performed in a Biotage 0.5-2.0 mL vessel with a crimped top. 1,2-Dimethoxyethane (DME) was used as purchased. The 4M hydrochloric acid in dimethoxyethane (HCl in DME) solution was used as purchased. The 2M aqueous sodium carbonate solution was freshly prepared for each iteration. All organic reagents were used as purchased. The catalyst, transdichlorobis(triphenylphosphine) palladium(II) [Pd(PPh3)2Cl2] was purchased from Strem Chemicals and stored in a desiccator. The products were purified via flash chromatography, using 32 – 63 μm silica gel. Methylene chloride for chromatography (CH2Cl2) was used as purchased. Saturated ammonia/methanol (CH3OH/(NH3)) was prepared by sparging methanol with ammonia gas for approx. 1 h. A Biotage Initiator 1.2 microwave was utilized for the Suzuki coupling reactions. HRMS were obtained by The Proteomics and Mass Spectrometry Facility at NIDDK/NIH/DHHS on a Waters LCT Premier time-of-flight (TOF) mass spectrometer, using the internal reference standard method. General Procedure for Suzuki Coupling: To a solution of N-tertbutylcarbonyl-2-iodohistamine (1eq) in dimethoxyethane (0.04M) was added PdCl2(PPh3)2 (5 mol%). The solution was sparged with nitrogen stirred at room temperature for 0.5 h, at which time the boronic acid (3 eq) and aq. sodium carbonate (2M) were added. The reaction solution was sparged again with nitrogen and then placed in the microwave and heated for 2 h at 110 °C. When TLC and LC-MS showed full consumption of starting materials, HCl/dioxane (4M) was added and the reaction tube was again placed in the microwave and heated for 0.5 h. The crude product was directly purified by column chromatography (without additional work-up; 0-20% CH3OH(NH3)/CH2Cl2) to isolate the histamines as free bases. All products showed greater than 95% purity by LCMS analysis, however, it should be noted that each product eluted in the void volume of a 100% buffered aqueous gradient.
- 16.2-(3-trifluoromethylphenyl)-histamine (3a): Using the general procedure and starting with 0.10 g (0.297 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.047 g (62%) of 3a was obtained; 1H NMR (CD3OD, 300 MHz) δ 8.29 (s, 1H) 8.21-8.17 (m, 1H), 7.78-7.72 (m, 2H), 7.10 (s, 1H), 3.09 (t, J = 6.9 Hz, 2H), 2.92 (t, J = 6.9 Hz, 2H); 13C NMR (CD3OD, 150 MHz) δ 144.8, 131.1, 130.8, 129.4, 128.1, 125.0, 124.6, 124.5, 123.2, 121.4 40.7, 28.9; HRMS calcd for C12H13N3F3 (M + H) 256.0983, found 256.1060. 2-(phenyl)-histamine (3b): Using the general procedure and starting with 0.030 g (0.089 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0011 g (66%) of 3b was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.86-7.81 (m, 2H), 7.47-7.32 (m, 3H), 6.94 (s, 1H), 2.98 (t, J = 6.9 Hz, 2H), 2.80 (t, J = 6.9 Hz, 2H); HRMS calcd for C11H14N3 (M + H) 188.1109, found 188.1185. 2-(2-methoxyphenyl)-histamine (3c): Using the general procedure and starting with 0.031 g (0.093 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.014 g (69%) of 3c was obtained; 1H NMR (CD3OD, 300 MHz) δ 8.00 (dd, J = 1.7, 7.7 Hz, 1H), 7.34 (ddd, J = 1.7, 7.3, 8.3 Hz, 1H), 7.23 (d, J = 8.4 Hz, 1H), 7.03 (ddd, J = 1.0, 7.4, 7.7 Hz, 1H), 7.07 (s, 1H), 4.08 (s, 3H), 3.15 (t, J = 6.9 Hz, 2H), 2.95 (t, J = 6.9 Hz, 2H); HRMS calcd for C12H16N3O (M + H) 218.1215, found 218.1296. 2-(4-methoxyphenyl)-histamine (3d): Using the general procedure and starting with 0.027 g (0.082 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.012 g (67%) of 3d was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.88-7.85 (m, 2H), 7.11-7.07 (m, 3H), 6.97 (s, 1H), 3.93 (s, 3H), 3.05 (t, J = 6.9 Hz, 2H), 2.88 (t, J = 6.9 Hz, 2H); HRMS calcd for C12H16N3O (M + H) 218.1215, found 218.1292. 2-(4-methylphenyl)-histamine (3e): Using the general procedure and starting with 0.030 g (0.090 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.011 g (61%) of 3e was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.82 (d, J = 8.1 Hz, 2H), 7.35 (d, J = 7.8 Hz, 2H), 7.00 (s, 1H), 3.06 (t, J = 6.9 Hz, 2H), 2.95 (t, J = 6.9 Hz, 2H), 2.47 (s, 3H); HRMS calcd for C12H16N3 (M + H) 202.1266, found 202.1342. 2-(4-tert-butylphenyl)-histamine (3f): Using the general procedure and starting with 0.028 g (0.083 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.013 g (64%) of 3f was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.76 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.7 Hz, 2H), 6.91 (s, 1H), 2.99 (t, J = 6.9 Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H), 1.42 (s, 9H); HRMS calcd for C15H22N3 (M + H) 244.1735, found 244.1823. 2-(2-fluorophenyl)-histamine (3g): Using the general procedure and starting with 0.030 g (0.089 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.011 g (60%) of 3g was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.53 (ddd, J = 1.5, 7.5, 9.3 Hz, 1H), 7.55-7.47 (m, 1H), 7.40-7.30 (m, 2H), 7.09 (s, 1H), 3.07 (t, J = 6.9 Hz, 2H), 2.92 (t, J = 6.9 Hz, 2H); HRMS calcd for C11H13N3F (M + H) 206.1015, found 206.1098. 2-(4-fluorophenyl)-histamine (3h): Using the general procedure and starting with 0.029 g (0.088 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.012 g (66%) of 3h was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.98-7.93 (m, 2H), 7.31-7.26 (m, 2H), 7.04 (s, 1H), 3.11 (t, J = 6.9 Hz, 2H), 2.92 (t, J = 6.9 Hz, 2H); HRMS calcd for C11H13N3F (M + H) 206.1015, found 206.1101. 2-(3,5-bis-trifluoromethylphenyl)-histamine (3i): Using the general procedure and starting with 0.030 g (0.088 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.012 g (42%) of 3i was obtained; 1H NMR (CD3OD, 300 MHz) δ 8.58 (s, 1H), 8.05 (app. s, 2H), 7.18 (s, 1H), 3.15 (t, J = 6.9 Hz, 2H), 2.97 (t, J = 6.9 Hz, 2H); HRMS calcd for C13H12N3F6 (M + H) 324.0857, found 324.0932. 2-(2-methoxy-5-fluorophenyl)-histamine (3j): Using the general procedure and starting with 0.028 g (0.084 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.013 g (66%) of 3j was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.75 (dd, J = 2.7, 9.6 Hz, 1H), 7.14-7.03 (m, 2H), 6.98 (s, 1H), 3.98 (s, 3H), 3.03 (t, J = 6.9 Hz, 2H), 2.84 (t, J = 6.9 Hz, 2H); HRMS calcd for C12H15N3OF (M + H) 236.1121, found 236.1207. 2-(3, 5-difluorophenyl)-histamine (3k): Using the general procedure and starting with 0.030 g (0.088 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.010 g (51%) of 3k was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.49-7.44 (m, 2H) 7.04 (s, 1H), 7.0-6.92 (m, 1H), 3.10 (t, J = 6.9 Hz, 2H), 2.88 (t, J = 6.9 Hz, 2H); HRMS calcd for C11H12N3F2 (M + H) 224.0921, found 224.1007. 2-(3-nitrophenyl)-histamine (3l): Using the general procedure and starting with 0.030 g (0.089 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.012 g (58%) of 3l was obtained; 1H NMR (CD3OD, 300 MHz) δ 8.77-8.76 (m, 1H), 8.32-8.22 (m, 2H), 7.84-7.78 (m, 1H), 7.10 (s, 1H), 3.17 (t, J = 6.9 Hz, 2H), 2.93 (t, J = 6.9 Hz, 2H); HRMS calcd for C11H13N4O2 (M + H) 233.0906, found 233.1038. 2-(3-nitro-4-methylphenyl)-histamine (3m): Using the general procedure and starting with 0.028 g (0.083 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.012 g (59%) of 3m was obtained; 1H NMR (CD3OD, 300 MHz) δ 8.57 (d, J = 1.8, 1H), 8.15-8.09 (m, 1H), 7.67-7.62 (m, 1H), 7.13 (s, 1H), 3.15 (t, J = 6.9 Hz, 2H), 2.97 (t, J = 6.9 Hz, 2H), 2.69 (s, 3H); HRMS calcd for C12H15N4O2 (M + H) 247.1117, found 247.1201. 2-(2-chlorophenyl)-histamine (3n): Using the general procedure and starting with 0.031 g (0.092 mmol) of N-tertbutylcarbonyl-2-iodohistamine, 0.012 g (59%) of 3n was obtained; 1H NMR (CD3OD, 300 MHz) δ 7.71-7.68 (m, 1H), 7.53-7.50 (m, 1H), 7.42-7.38 (m, 2H), 6.99 (s, 1H), 2.98 (t, J = 6.9 Hz, 2H), 2.81 (t, J = 6.9 Hz, 2H); HRMS calcd for C11H13ClN3 (M + H) 222.0720, found 222.0797.