

Abstract
p120ctn binds to the cytoplasmic domain of cadherins but its role is poorly understood. Colo 205 cells grow as dispersed cells despite their normal expression of E-cadherin and catenins. However, in these cells we can induce typical E-cadherin–dependent aggregation by treatment with staurosporine or trypsin. These treatments concomitantly induce an electrophoretic mobility shift of p120ctn to a faster position. To investigate whether p120ctn plays a role in this cadherin reactivation process, we transfected Colo 205 cells with a series of p120ctn deletion constructs. Notably, expression of NH2-terminally deleted p120ctn induced aggregation. Similar effects were observed when these constructs were introduced into HT-29 cells. When a mutant N-cadherin lacking the p120ctn-binding site was introduced into Colo 205 cells, this molecule also induced cell aggregation, indicating that cadherins can function normally if they do not bind to p120ctn. These findings suggest that in Colo 205 cells, a signaling mechanism exists to modify a biochemical state of p120ctn and the modified p120ctn blocks the cadherin system. The NH2 terminus–deleted p120ctn appears to compete with the endogenous p120ctn to abolish the adhesion-blocking action.
Keywords: E-cadherin, catenin, colon carcinoma, Colo 205, p120ctn
Cell–cell adhesion is a dynamic process, especially during morphogenetic cell rearrangement such as intercalation and delamination of cells, and in many pathological phenomena including tumor invasion. To clarify the mechanisms of how cell–cell adhesion is regulated is an important issue for understanding such biological phenomena. Cadherin-mediated adhesion is thought to be controlled by catenins which are associated with the cytoplasmic domain of cadherin. A region in the COOH half of the cadherin cytoplasmic domain directly binds β-catenin, which in turn associates with α-catenin (Barth et al., 1997). α-Catenin interacts with the actin-based cytoskeleton directly or indirectly, involving other cytoskeletal proteins such as α-actinin and vinculin (Knudsen et al., 1995; Rimm et al., 1995; Watabe-Uchida et al., 1998). These molecular interactions are essential for this system to exert its full adhesion activity (Hirano et al., 1992; Watabe et al., 1994; Ozawa and Kemler, 1998a; Watabe-Uchida et al., 1998), although homophilic interactions between the cadherin extracellular domains per se can be achieved without catenins (Brieher et al., 1996; Yap et al., 1997; Chitaev and Troyanovsky, 1998). It is thought that cadherin function may be regulated by biochemical modifications of the catenins. Indeed, phosphorylation of catenins, in particular β-catenin, has been implicated in the instability of cell–cell adhesion (e.g., Matsuyoshi et al., 1992; Hamaguchi et al., 1993; Serres et al., 1997; Takahashi et al., 1997; Balsamo et al., 1998).
One such catenin is p120ctn, a member of the Armadillo/ β-catenin gene family (Peifer et al., 1994). The p120ctn protein was originally identified as a target for p60v-src kinase (Reynolds et al., 1989, 1992) and later found to be a protein associated with the cytoplasmic domain of cadherins (Reynolds et al., 1994; Daniel and Reynolds, 1995; Shibamoto et al., 1995; Staddon et al., 1995). Other proteins related to p120ctn, constituting a subfamily of Armadillo/ β-catenin, have also been identified (Heid et al., 1994; Hatzfeld and Nachtsheim, 1996; Mertens et al., 1996; Paffenholz and Franke, 1997; Sirotkin et al., 1997). Recent studies show that p120ctn binds to the juxtamembrane portion of the cadherin cytoplasmic domain, which is different from the region to which β-catenin binds (Finnemann et al., 1997; Lampugnani et al., 1997; Yap et al., 1998). In contrast to the well-known function of β-catenin, that of p120ctn remains largely unknown. However, some biological effects of its ectopic expression have been reported, e.g., overexpression of p120ctn induces extensive dendrite-like processes in fibroblasts (Reynolds et al., 1996) and perturbs gastrulation in Xenopus laevis embryos (Geis et al., 1998; Paulson et al., 1999). In addition, a recent report shows that overexpression of δ-catenin in MDCK cells, a protein related to p120ctn, alters their morphology and motility (Lu et al., 1999).
In the present work, we studied a unique aggregation property of colon carcinoma Colo 205 cells (Semple et al., 1978). They grow as dispersed cells not forming compact aggregates, despite the expression of all general components of the E-cadherin–catenin complex. We found that typical E-cadherin–dependent aggregation could be induced by treatment with staurosporine, a kinase inhibitor, or with low concentrations of trypsin. Correlating with this adhesive change, the electrophoretic mobility of p120ctn was altered. Furthermore, when NH2 terminus–deleted p120ctn molecules were introduced into Colo 205 cells, these constructs induced an E-cadherin–dependent compact aggregate formation, similar to effects induced by staurosporine and trypsin. Together with other findings, our results suggest that p120ctn can function as an inhibitory regulator in the cadherin adhesion system.
Materials and Methods
Antibodies and Other Reagents
Mouse mAbs HECD-1 (Shimoyama et al., 1989) and SHE78-7 to human E-cadherin (Takara Shuzo Co., Ltd.), rat mAb NCD-2 to chicken N-cadherin (Nakagawa and Takeichi, 1998), mouse mAb to p120ctn (Transduction Laboratories), mouse mAb M2 to FLAG (F-3165; Sigma Chemical Co.), rabbit polyclonal antiserum to FLAG (SC-807; Santa Cruz Biotechnology, Inc.), mouse mAb to αE-catenin (Transduction Laboratories), rat mAb α18 to αE-catenin (Watabe et al., 1994), mouse mAb 5H10 to β-catenin (Johnson et al., 1993), and rabbit polyclonal antiserum to β-catenin (Shibamoto et al., 1995) were used. Anti-MUC1 antibody MY.1E12 (Yamamoto et al., 1996) was a gift from T. Irimura (University of Tokyo, Tokyo, Japan).
Antibodies used for detection of primary antibodies were as follows: goat Cy-3–labeled species-specific antibody to mouse IgG (AP-124C; Chemicon International, Inc.), donkey biotinylated species-specific antibody to rabbit IgG (RPN1004; Nycomed-Amersham), FITC-labeled streptavidin (RPN1233; Nycomed-Amersham), sheep HRP-linked species-specific antibody to mouse IgG (NA9310; Nycomed-Amersham) and rat IgG (NA932; Nycomed-Amersham), goat HRP–linked antibody to rabbit IgG (NA934; Nycomed-Amersham), and Sepharose 4B–linked goat antibody to mouse IgG (62-6541; Zymed Labs, Inc.).
The following protein kinase inhibitors were used: staurosporine (#19-123; Upstate Biotechnology), calphostin C (C-159; Research Biochemicals Inc.), tyrphostin (EI-215; Biomol), genestein (G-103; Research Biochemicals International), and herbimycin A (OP-12, Kyowa Medex Co., Ltd.). UCN-01 (Takahashi et al., 1990) was a kind gift of T. Tamaoki (Kyowa Medex Co., Ltd.). O-sialoglycoprotein endopeptidase was purchased from Cedarlane Labs., Ltd.
Cells and cDNA Transfection
Human colon carcinoma cell lines Colo 205 (Semple et al., 1978) and HT-29 (Fogh and Trempe, 1975), and MDCK cells (Gaush et al., 1966) were used. These cells were cultured in a 1:1 mixture of DME and Ham's F12 supplemented with 10% FCS (DH10). For transfer of Colo 205 cells, an aliquot of the suspended cell culture was moved to new dishes with fresh DH10 medium. When necessary, they were trypsinized as described below. For HT-29 and MDCK cells, they were rinsed with 1 mM EDTA in Ca2+- and Mg2+-free saline, then treated with 0.05% crude trypsin (#0152-13; Difco Laboratories) and 1 mM EDTA in the same solution for 5 min at 37°C, and finally suspended in DH10.
Colo 205 cells were transfected by electroporation or by use of adenoviral expression vectors. For electroporation, trypsinized cells (4 × 105) were suspended in 200 ml of Hepes-buffered (pH 7.4) saline with 1 mM Ca2+ and 1 mM Mg2+ (HBS). 20 μg of an expression vector was added to the suspension, which was electrified at 1,160 μF at 250 V. For adenovirus-mediated transfection, 106 cells were suspended in 500 μl of DH10 with 5 × 107 plaque-forming units of adenovirus, and incubated for 4 h. Cells were washed twice with DH10, and after 48 h samples were collected. MDCK cells were transfected by electroporation under the same conditions as for Colo 205 cells, except that 1,060 μF at 200 V was used. HT-29 cells were transfected with adenoviral vectors only.
Cell Aggregation and Trypsin Treatment
Colo 205 cells were washed twice with Ca2+- and Mg2+-free Hepes-buffered saline supplemented with 1 mM EDTA, and completely dissociated into single cells by pipetting. Then, 2 × 105 cells were resuspended in 1 ml of DH10 with or without 1 μg/ml of SHE78-7, a blocking antibody to human E-cadherin, and placed in a Nunc 6-well plate (#152795). Cells were incubated for 3 h at 37°C on a gyratory shaker (KS6320; Marysol) at 80 rpm.
For trypsin treatment to induce cell adhesion, cells were rinsed twice with DH (DH10 without FCS), and incubated in DH with crystalline trypsin (T-8253; Sigma Chemical Co.) of various concentrations at 37°C. For biochemical analysis, the incubation was generally stopped at 30 min, and the cells were rinsed twice with HBS containing 0.1% trypsin inhibitor (T-6522; Sigma Chemical Co.) and subjected to further analysis. 0.01% trypsin in the presence (TC) or absence (TE) of 1 mM Ca2+ treatments were performed as described previously (Takeichi, 1977).
cDNA Construction
Using mouse p120ctn cDNA (GenBank accession number Z17804) as a template, we generated FLAG-tagged p120ctn (FLf)1 and other constructs by PCR. The following primers were used: primer N, 5′-GAATTCATGGACGACTCAGAGGTG-3′; primer A, 5′-GAATTCATGTTAGCAAGCTTGGATAGTTTG-3′; primer CR, 5′-GAATTCGATATCCTA CTTGTCATCGTCGTCCTTGTAGTCAATCTTCTGCATCAAGG-GTGC-3′; primer ΔAR, 5′-CTGCAGGAAATCCACTGTATCATT-3′ Primer CR and ΔAR were antisense primers. The primer CR contained an EcoRV restriction site, and complementary sequences for stop codon and FLAG epitope DYKDDDDK (Hopp et al., 1988) at the 5′ portion, as underlined. Primers N and CR were used for amplifying FLf, and primers A and CR for ΔN346f, a mutant molecule in which the NH2-terminal 346 amino acids (aa) of p120ctn were deleted. For construction of other NH2-terminal deletion mutants ΔN101f, ΔN157f, ΔN244f, and ΔN323f, the fragment FLf was internally digested with BglII/EcoRV, NcoI/EcoRV, SmaI/ EcoRV, and Bsu36I/EcoRV, respectively. We assume that translation of the constructs will start at the next ATG downstream of the deletion. For FLΔRf and ΔN346ΔRf, in which aa 641–819 were deleted, oligonucleotides corresponding to aa 346–640 were synthesized by PCR by use of primer A and ΔAR, digested by PstI, and inserted into the internal PstI sites of FLf or ΔN346f. The obtained fragments were subcloned into the pCA-pA expression vector (Niwa et al., 1991) and pAdV-CA-pA adenoviral construction vector (Nakagawa and Takeichi, 1998).
For construction of cN/JM(−), encoding a chicken N-cadherin mutant in which the juxtamembrane portion of the cytoplasmic region is deleted, the catenin binding region with COOH-terminal 70 aa and the extracellular–transmembrane region were amplified by PCR, respectively, using pCMV-cN/FLAG-pA (Nakagawa and Takeichi, 1998) as a template. For amplifying the catenin binding region, primers 5′-CGCCGTACGACCATGAGATCTAATGAGGGACTTAAAGCAGCC-3′ and 5′-GCGGCCGCTTACTTGTCATCGTCGTCCTTGTAGTC-3′ were used. For the extracellular–transmembrane region, primers 5′-GCGCGTACGACCATGTGCCGGATAGCGGGAACGCCG-3′ and 5′-CGCGAGA TCTCTGACGCTCCTTATCCGGCG-3′ were used. Obtained products were linked through the BglII sites underlined above, and subcloned into pCA-pA or pAdV-CA-pA. All PCR-generated fragments of DNA obtained above were completely sequenced, and no sequence error was detected.
Construction of Recombinant Adenovirus
Recombinant adenovirus AdV-CA-lacZ expressing β-galactosidase with the CAG promoter was a gift from K. Moriyoshi (Kyoto University, Kyoto, Japan). Construction of recombinant viruses was performed according to methods described previously (Moriyoshi et al., 1996). In brief, HEK 293 cells (ATCC CRL 1573) cultured with DH10 in 6-well plates (Iwaki Co.) were cotransfected with viral genome fragments (0.2 μg) and linearized adenoviral shuttle vector plasmids (1 μg) by use of Lipofectamine™ (#18324-012; Life Technologies, Inc.). The next day, the cells were divided into collagen-coated 24-well plates (Iwaki Co.). 10 d later, wells became full of dead cells, caused by viral propagation, and the debris was screened for proper protein expression by immunostaining and immunoblotting with the anti-FLAG antibody M2. We obtained AdV-CA-FLf, AdV-CA-ΔN101f, AdV-CA-ΔN157f, AdV-CA-ΔN244f, AdV-CA-ΔN346f, which expressed different forms of p120ctn proteins under the control of the CAG promoter (Niwa et al., 1991), and AdV-cN/JM. The full-length N-cadherin expression vector AdV-Ncad was described previously (Nakagawa and Takeichi, 1998). The recombinant adenoviruses were amplified, and purified by CsCl2 step gradient centrifugation (Kanegae et al., 1994). FCS was added to the purified adenovirus solutions at a final concentration of 10%. Aliquots of the virus solution were stored at −80°C until used.
Cell Extraction and Immunoprecipitation
Colo 205 cells were removed from dishes by pipetting, rinsed twice in HBS, and lysed in TBS-Ca (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1 mM CaCl2) supplemented with 1% Triton X-100, 1% NP-40, 1 mM PMSF, 1 mM NaVO3, and 50 mM NaF (TBS-Triton) for 30 min at 4°C on a rocking platform. The soluble fraction was collected by centrifugation and preincubated with 50 μl of secondary antibody–conjugated beads and 5% BSA on ice for 20 min. After preincubation, incubations with primary and secondary antibodies were sequentially performed on ice for 1 h each, followed by washing three times with TBS-Triton. Finally, samples were boiled in 150 μl of SDS sample buffer with 7.5 μl of β-mercaptoethanol for 5 min.
Gel Electrophoresis, Immunoblotting, and Immunohistochemistry
Proteins were separated in SDS-PAGE and electrophoretically transferred to membranes. The transferred proteins were detected by the enhanced chemiluminescence system (RPN 2106; Nycomed-Amersham). Immunohistochemical detection of cadherin and catenins was performed as described in Watabe-Uchida et al. (1998).
Phosphatase Treatment
Immunoprecipitated materials were washed twice with phosphatase reaction buffer (50 mM Tris-HCl, pH 8.5, 50 mM MgCl2, 1 mM PMSF, 1 mM DTT, 0.1% Triton X-100, 0.1% NP-40). 27 μl of the reaction buffer and 3 μl of alkaline phosphatase (#567-744; Boehringer Mannheim GmbH) were added to the precipitate, and the mixture was then incubated at 30°C for 1 h. After incubation, 1 μl of 0.5 M EDTA and 30 μl of 2× SDS sample buffer were added, and the mixture was boiled for 5 min.
Phosphorous-32 Metabolic Labeling and Phosphoamino Acid Analysis
For 32P-metabolic labeling, cells were cultured in phosphate-free DME (#21097-035; Life Technologies Inc.) with 0.5 mCi of 32P (NEX-053, New England Nuclear Life Science Products, Inc.) for 24 h. Then, cells were harvested and trypsinized when necessary. From their detergent extracts prepared as above, p120ctn was immunoprecipitated and separated by SDS-PAGE. From the gels, the labeled p120ctn-protein band was excised after comparing with their autoradiograms, and the collected gel pieces were homogenized in 500 μl of freshly prepared 50 mM NH4HCO3. After addition of 25 μl β-mercaptoethanol and 5 μl 10% SDS, the samples were boiled for 5 min and agitated at room temperature for 2 h. Supernatants were collected after centrifugation at 15,000 rpm for 5 min, mixed with 20 μg RNase A and 250 μl ice-cold TCA (100% wt/wt), and incubated on ice for 1 h. After centrifugation, the pellets were air-dried and proteins were digested by incubating with 50 μl 6 N HCl at 110°C for 90 min. Digested products were air-dried again and resuspended in buffer (2.2% formic acid, 7.8% glacial acetic acid), pH 1.9, with unlabeled phosphoserine, phosphothreonine, and phosphotyrosine. Samples were spotted on TLC plate and separated by two-dimensional electrophoresis at 1.5 kV for 40 min with the pH 1.9 buffer, and at 1.0 kV for 30 min with buffer (5% glacial acetic acid, 0.5% pyridine), pH 3.5. Finally, labeled phosphoamino acids were visualized by the BAS-1000 image analyzing system (FUJIX Inc.).
Results
Induction of Compact Aggregate Formation in Colo 205 Cells
Colo 205 cells grow as dispersed cells occasionally forming loose, small clusters (Fig. 1 A) as seen in catenin-deficient cells (Hirano et al., 1992; Oyama et al., 1994; Shimoyama et al., 1992), and they only lightly attach to the culture dish. Despite this behavior, they express an apparently normal set of E-cadherin, αE-catenin, β-catenin, and p120ctn proteins (Fig. 1 E). Moreover, the expression levels of these proteins are similar to those in HT-29 cells, which can organize epithelial sheets (see Fig. 7 A). To determine whether the cadherin–catenin system in Colo 205 cells is entirely inactive, we rotated the cultures to facilitate cell aggregation. Under these culture conditions, Colo 205 cells clumped into larger aggregates (Fig. 1 C) and this clumping was inhibited by addition of the E-cadherin blocking antibody SHE78-7 (Fig. 1 D), indicating that E-cadherin is functional. However, their aggregates were still loose and never formed tightly associated compact aggregates as generally produced by the cadherin adhesion system (Takeichi, 1988). These observations suggest that Colo 205 cells expose functional E-cadherin molecules on their surface, but their cadherin system has some deficit in exerting its full activity to organize compact cell aggregates. Interestingly, when the cultures of Colo 205 cells were starved by not refreshing the culture medium for a few days, they occasionally formed compact aggregates (Fig. 1 B), implying that their cadherin system is reversibly impaired and can be reactivated under certain physiological conditions.
Figure 1.
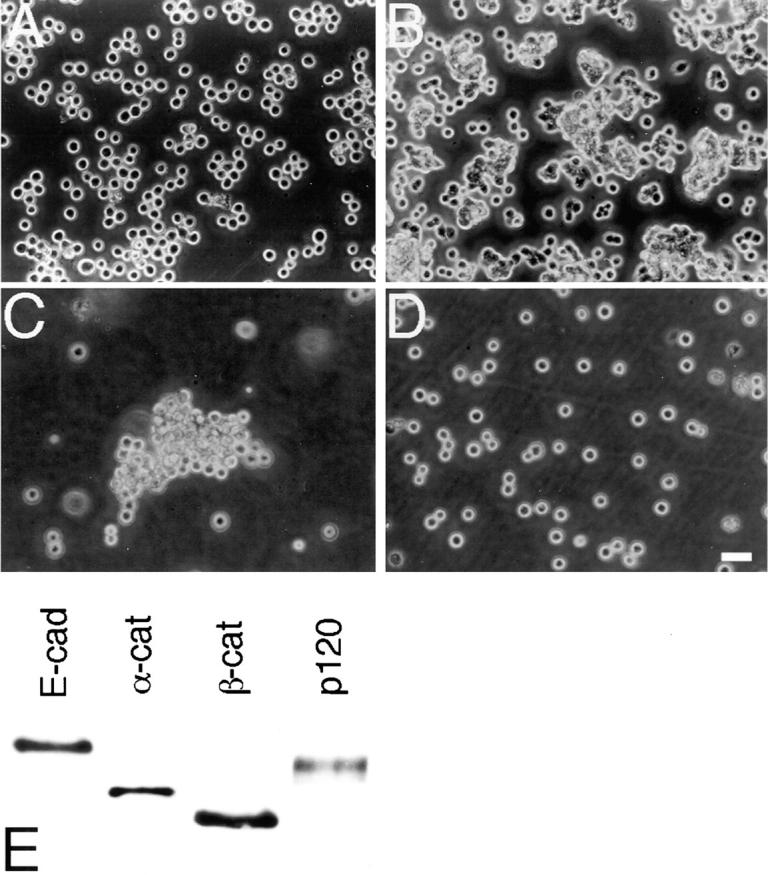
Aggregation profiles of Colo 205 cells. (A) Cells under normal culture conditions, showing their dispersed appearance. (B) Cells in a starved culture. The culture medium was not refreshed for 5 d. Compact aggregate formation was induced. (C and D) Aggregate formation in a rotation culture without (C) or with (D) the anti–E-cadherin antibody SHE78-7. E-Cadherin– dependent aggregation can take place, but cell association in the aggregates is loose. (E) Expression of E-cadherin and catenins detected by Western blotting. E-Cad, E-cadherin; α-cat, αE-catenin; β-cat, β-catenin; p120, p120ctn. Bar, 40 μm.
Figure 7.
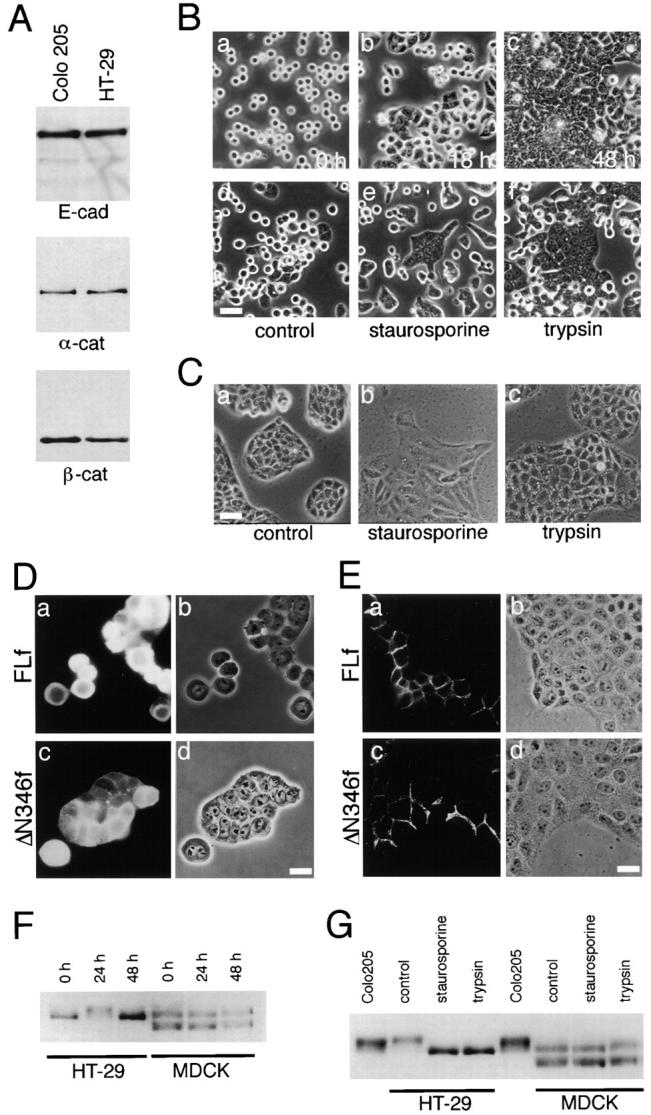
p120ctn in HT-29 and MDCK cells. (A) Immunoblot detection of E-cadherin (E-cad), α-catenin (α-cat), and β-catenin (β-cat) in HT-29 cells in comparison with Colo 205 cells. Samples with a similar cell number were loaded. (B) Morphological changes in HT-29 cells and effect of trypsin and staurosporine treatments. (a–c) Cells trypsinized as described in Materials and Methods were replated, and cultured for 0 (a), 18 (b), or 48 h (c). (d–e) Cultures preincubated for 18 h with nothing (d), 7 nM staurosporine (e), or 0.001% trypsin (f), followed by another incubation for 6 h (d and e), or 30 min (f). The culture medium was replaced with a serum-free DH when trypsin was added. (C) Effect of staurosporine and trypsin on MDCK colonies precultured for 18 h. a, Untreated; b, incubated with 7 nM staurosporine for 6 h; c, incubated with 0.001% trypsin for 30 min. (D and E) Effect of the ectopic expression of FLf and ΔN346f on HT-29 (D) or MDCK (E) cells. Cells were transfected with cDNA of each construct immediately after cell transfer and cultured for 24 h. The introduced molecules were detected with anti-FLAG antibody M2. Phase-contrast and immunofluorescence images are shown as a set for each result. In D, ΔN346f induced compaction in HT-29 aggregates, whereas FLf had no effect. In E, both FLf and ΔN346f were localized at cell–cell boundaries without affecting cell morphology. (F) Immunoblot analysis of p120ctn in HT-29 and MDCK cells. Cells were transferred as described in B, and cultured for 0, 24, or 48 h. Note the band shift of p120ctn at 24 h in the case of HT-29 cells. In MDCK cells, two p120ctn bands were always detected at the positions of 120 and 100 kD, as described by Ratcliffe et al. (1997), of which the lower band was likely a splicing variant. (G) Effect of staurosporine or trypsin treatment on the p120ctn band pattern. Cells precultured for 18 h were treated with these reagents as described in B and C. In HT-29, both treatments abolished the transient upward shift of the p120ctn band. In MDCK cells, staurosporine slightly induced a downward shift of the p120ctn band, although this may not be clearly seen in this particular blot. For comparison, Colo 205 samples were also loaded. Bars, 40 μm (B and C); 20 μm (D and E).
As an initial attempt to investigate how the cadherin system is impaired in Colo 205 cells, we examined the effect of various biochemical reagents, including kinase inhibitors and activators, on their aggregation. Among the reagents tested, staurosporine showed a marked effect. It induced compact cell clustering in Colo 205 cultures within a few hours after administration (Fig. 2, A–D), during which the initial sign of adhesion induction was observed within 2 h. This effect was saturated by 6 h. The compaction of cell clusters was E-cadherin–dependent, as it was inhibited by SHE78-7 (Fig. 2 E). Staurosporine also slightly promoted spreading of cells, but this was not inhibited by SHE78-7 (Fig. 2 E, arrows). Since staurosporine is known to inhibit protein kinase C (PKC), we tested other PKC inhibitors, but none were effective (Table I). Tyrosine kinase inhibitors were also negative, except herbimycin A showed a weak compaction-inducing activity, but only when cells were cultured overnight with this inhibitor (data not shown). PKC activators and phosphatase inhibitors, such as phorbol esters and okadaic acid, also had no effects (data not shown). Thus, among the reagents tested, staurosporine exhibited an exceptionally strong effect on Colo 205 aggregation. This effect of staurosporine was reversible as its removal caused redispersion of Colo 205 cells (data not shown).
Figure 2.
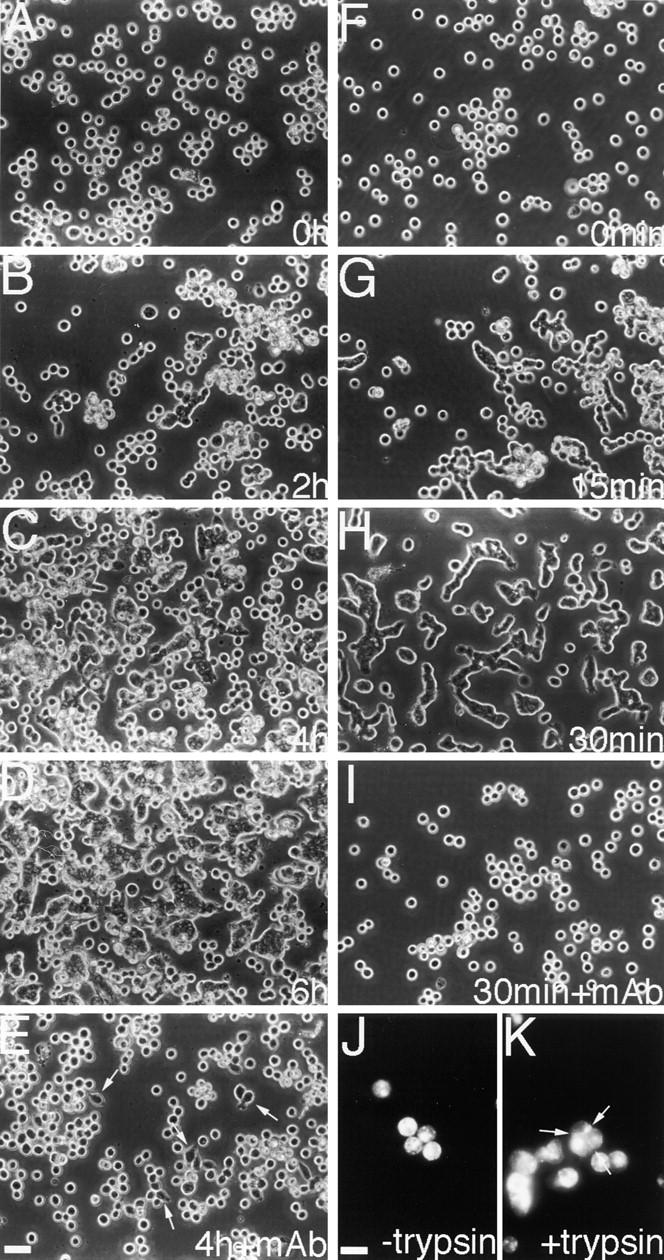
Induction of compact aggregate formation in Colo 205 cells by staurosporine or trypsin treatment. (A–E) Cells were incubated with 7 nM staurosporine for 0 h (A), 2 h (B), 4 h (C and E), or 6 h (D). (F–I) Cells were cultured with 0.001% trypsin for 0 min (F), 15 min (G), or 30 min (H and I). In E and I, SHE78-7 was added to the cultures. Both treatments induced cell–cell adhesion, unless the anti–E-cadherin antibody was present. Cell spreading was also induced by staurosporine in an SHE78-7–independent manner (arrows in E). (J and K) Immunofluorescence staining for MUC1 in cells treated with (K) or without (J) 0.001% trypsin. MUC1 can be detected in both samples, even at cell–cell boundaries (arrows in K). Bars, 40 μm (A–I); 20 μm (J and K).
Table I.
Effect of Protein Kinase Inhibitors on Aggregation of Colo 205 Cells
| Staurosporine | 35 nM | 15 nM | 7.0 nM | 3.5 nM | 0.7 nM | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| + | + | + | + | − | ||||||
| UCN-01 | 100 nM | 50 nM | 10 nM | 5 nM | 1 nM | |||||
| − | − | − | − | − | ||||||
| Calphostin C | 200 nM | 100 nM | 50 nM | 10 nM | 5 nM | |||||
| − | − | − | − | − | ||||||
| Herbimycin A | 2 μg/ml | 1 μg/ml | 0.5 μg/ml | 0.2 μg/ml | 0.1 μg/ml | |||||
| ≲+≳ | (+≳ | − | − | − | ||||||
| Tyrphostin | 150 μM | 75 μM | 30 μM | 15 μM | 7.5 μM | |||||
| − | − | − | − | − | ||||||
| Genestein | 78 μM | 39 μM | 26 μM | 13 μM | 6.5 μM | |||||
| − | − | − | − | − |
Cells were cultured in the presence of each inhibitor for 6 h, and their morphological changes were then observed. +, cell adhesion was induced; −, not induced. Herbimycin A was not effective at the end of the 6-h incubation, but it induced adhesion after 24 h. All others, except staurosporine, showed no effect even after the 24-h incubation. The specificity and concentrations of each inhibitor blocking 50% of enzymatic activity are as follows: staurosporine, 2.7 nM for PKC, 8.2 nM for PKA, and 6.4 nM for p60v-src; UCN-01, 4.1 nM for PKC, 42 nM for PKA, and 45 nM for p60v-src;calphostin C, 50 nM for PKC, >50 μM for PKA, and p60v-src; herbimycin A, 5 μg/ml for p60v-srcand p60v-abl; tyrphostin, 15 μM for EGF receptor; genestein, 22.6 μM for EGF receptor, and 25.9 μM for p60v-src.
Besides metabolic inhibitors, we found that trypsin is a strong inducer for compact Colo 205 aggregation. When low concentrations of trypsin (0.01–0.001%) were added to serum-free cultures, Colo 205 cells became tightly associated with each other, deforming their morphology (Fig. 2, F–H). This adhesion induction was quick, beginning within 15 min after the addition of trypsin, and the effect was almost saturated at 30 min. The trypsin-mediated induction of aggregation was completely blocked by SHE78-7 (Fig. 2 I), indicating that it was an E-cadherin– dependent process. Under these conditions, which included a physiological concentration of Ca2+, cadherins are not digested with trypsin (Takeichi, 1977). Trypsin concentration of 0.001% was sufficient for the above adhesion induction, whereas 0.0001% was not effective. Thus, we found two different classes of reagents to induce compact Colo 205 aggregation, trypsin as a quicker inducer and staurosporine as a slower inducer.
Localization of MUC1, E-Cadherin, and Catenins in Colo 205 Cells
Previous studies indicated that mucins, such as MUC1 (episialin; Wesseling et al., 1996; Kondo et al., 1998) and epiglycanin (Kemperman et al., 1994), reduce cell–cell adhesion presumably by steric hindrance. As Colo 205 cells express MUC1 (Baeckstrom et al., 1991), trypsin may remove such antiadhesive mucins. To check this possibility, we immunofluorescently stained for MUC1 before and after trypsin treatment, and found that this proteoglycan was equally present on the surface of both trypsinized and untrypsinized cells (Fig. 2, J and K), even being localized at intercellular contact sites in the adhesion-induced aggregates. Treatment of cells with O-sialoglycoprotein endopeptidase, which inactivates epiglycanin and enhances adhesion of mammary carcinoma cells (Kemperman et al., 1994), showed no effects on Colo 205 aggregation (data not shown). From these observations, we assumed that the effect of trypsin was not to remove steric hindrance molecules, but to digest some signaling proteins on the cell surface, such as receptors, affecting intracellular physiological states. This idea is supported by results of other experiments described below.
Immunostaining for E-cadherin in Colo 205 cells show a diffuse distribution of this molecule on their surface (Fig. 3, A and A′). When compaction was induced in their aggregates by staurosporine or trypsin treatment, E-cadherin became highly concentrated into cell–cell contact sites (Fig. 3, C, C′, E, and E′). Catenins displayed a similar distribution (see p120ctn in Fig. 3, B, B′, D, D′, F, and F′). These observations suggest that the E-cadherin–catenin complex can be redistributed to cell–cell contact sites under the above compaction-inducing conditions.
Figure 3.

Localization of E-cadherin and p120ctn in Colo 205 cells. (A and B) Control Colo 205 cells immunostained for E-cadherin (A) and p120ctn (B). (C–F) Cells treated with 7 nM staurosporine for 6 h (C and D) or with 0.001% trypsin for 30 min (E and F), and immunostained for E-cadherin (C and E) or p120ctn (D and F). (A′–F′) Phase-contrast images of A–F. Bar, 20 μm.
Changes in Electrophoretic Mobility of p120ctn
To study if any changes were induced in the cadherin– catenin system following the above treatments, we immunoprecipitated E-cadherin from Colo 205 cells untreated or treated with staurosporine or trypsin. The amount of catenins coprecipitating with E-cadherin was not changed after these treatments (Fig. 4 A). However, we noted a significant change in the pattern of the p120ctn band. In untreated Colo 205 cells, p120ctn was detected as a broad, diffuse band that appeared to comprise multiple components (Fig. 4 A, lanes p120 c), although the pattern varied slightly from experiment to experiment. After compaction-inducing treatments, the band pattern was altered. Its most intense portion was shifted to the position corresponding to the bottom (electrophoretic front) of the original band (Fig. 4 A, lanes p120, t and s). Similar electrophoretic patterns of these proteins were observed in Western blots of the whole cell lysates (Fig. 4 B). The changes in the p120ctn band pattern coincided with the onset of adhesion induction by each reagent, starting within 15 min with trypsin (Fig. 4 C) and within 2 h with staurosporine (Fig. 4 D). A certain level of p120ctn-band shift was already detected at 5 min of incubation with trypsin. When trypsin was removed from the culture, the original electrophoretic profile of p120ctn was eventually restored within overnight incubation (Fig. 4 C), together with a recovery of the dispersed cell morphology (data not shown). Removal of staurosporine gave similar results.
Figure 4.
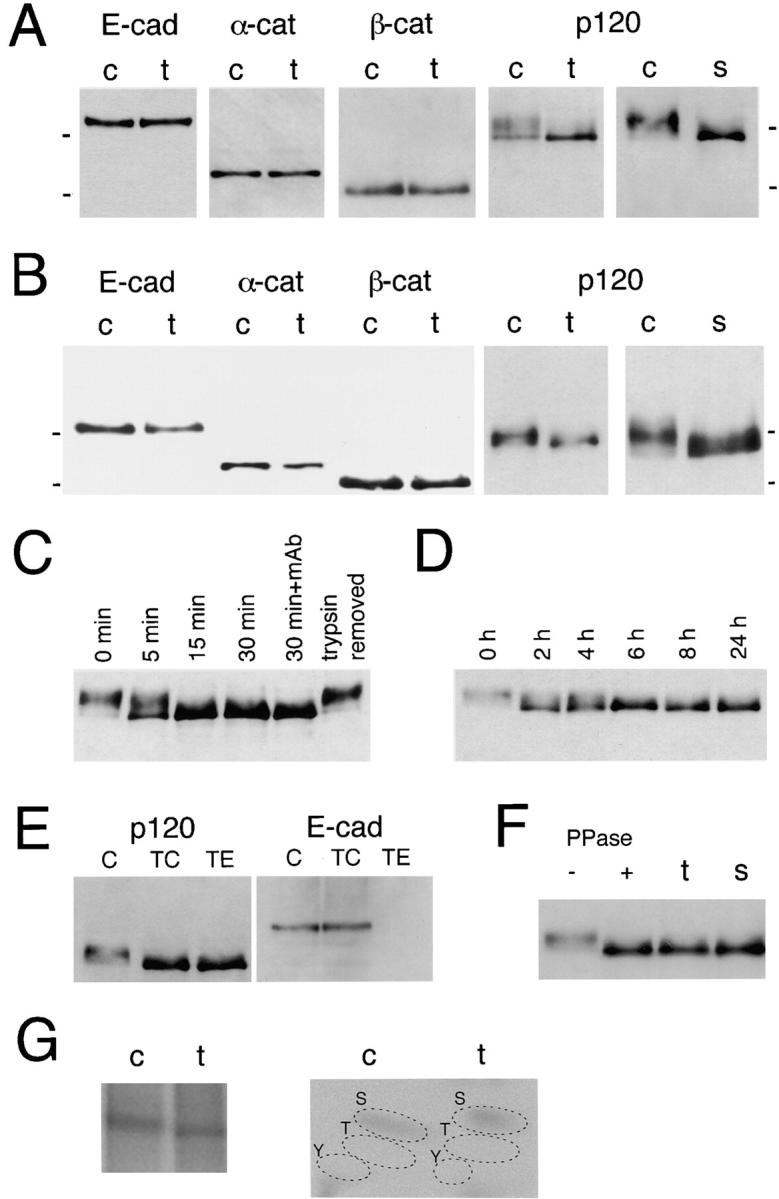
Analyses of E-cadherin and catenins before and after adhesion induction. (A) Immunoblot analysis of E-cadherin– catenin complexes. E-Cadherin was immunoprecipitated from Colo 205 cells treated with 0.001% trypsin for 30 min (t) or from untreated control cells (c), and copurified catenins were detected in each panel as indicated. Note the band shift in p120ctn after trypsin treatment but no changes in the other catenins. A similar result was obtained from cells treated with 7 nM staurosporine (s) for 6 h. (B) Immunoblot detection of E-cadherin and catenins from a whole lysate of cells treated as in A. E-Cad, E-cadherin; α-cat, α-catenin; β-cat, β-catenin; p120, p120ctn. (C and D) Time-dependent changes in the p120ctn band pattern after trypsin (C) or staurosporine (D) treatment. Lane 30 min + mAb, cells were incubated with trypsin in the presence of SHE78-7, an antibody to block E-cadherin, for 30 min; lane trypsin removed, trypsin was removed after 30 min, and then cells were further cultured overnight. Whole cell extracts were loaded. (E) p120ctn-band shift after treatment with 0.01% trypsin in the presence (TC) or absence (TE) of 1 mM Ca2+. The band pattern was similarly changed after both treatments. E-Cadherin was left intact in TC-treated cells, but digested in TE-treated cells. c, Untrypsinized control cells. (F) Effect of alkaline phosphatase treatment on the p120ctn band. p120ctn immunoprecipitated from nontreated Colo 205 cells was incubated with (+) or without (−) protein phosphatase (PPase) at 30°C for 1 h. For comparison, lysates of cells treated with trypsin (t) or staurosporine (s) were loaded. (G) 32P-metabolic labeling and phosphoamino acid (PAA) analysis. (Left) Autoradiogram of 32P-labeled p120ctn separated by SDS-PAGE. Cells were labeled with 32P for 24 h, and treated (t) or not treated (c) with 0.001% trypsin for 30 min. From their lysates, p120ctn was immunopurified, and separated by SDS-PAGE. (Right) PAA analysis. The p120ctn bands in the left panel were excised, and PAAs were separated by two-dimensional electrophoresis. S, phosphoserine; T, phosphothreonine; Y, phosphotyrosine. Molecular weight markers in A and B, 116 and 97.4 × 103.
Next, we asked whether the p120ctn change occurred before, or as a result of, adhesion induction. We treated Colo 205 cells with 0.01% trypsin in the presence (TC) or absence (TE) of 1 mM Ca2+. TC treatment leaves E-cadherin–mediated adhesion intact, whereas TE destroys E-cadherin–dependent adhesion (Takeichi, 1977). We found that both treatments resulted in a similar p120ctn-band shift (Fig. 4 E). This finding indicates that the p120ctn band change was not brought about as a result of adhesion induction but directly through trypsin treatment signals. We also found that the trypsin-induced p120ctn-band shift took place even when E-cadherin was blocked with antibodies (Fig. 4 C). This was also the case in the staurosporine treatment (data not shown). When other kinase inhibitors listed in Table I were tested, only herbimycin A induced a similar p120ctn-band shift at a 24-h incubation period (data not shown).
We sought to understand the molecular nature of the p120ctn-band shift. When p120ctn immunoprecipitates collected from untreated Colo 205 were incubated with alkaline phosphatase, the diffuse p120ctn band was transformed into a sharp band positioned at the front of the original (Fig. 4 F), which comigrated exactly with the p120ctn from staurosporine or trypsin treated cells. This suggests that the band shift observed may have been brought about by p120ctn dephosphorylation also. We then analyzed phosphorylated residues in p120ctn by 32P-metabolic labeling. The results showed that the major phosphorylated residues were serine (Fig. 4 G). However, their labeling intensity was however, not significantly reduced after trypsin treatment of cells. Antiphosphotyrosine antibodies detected only weak signals from the p120ctn bands in these cells (data not shown). It is possible that the p120ctn-band shift is caused by dephosphorylation of a subset of the phosphorylated residues. Alternatively, the quantitative differences in phosphorylation suggested by the phosphatase treatments may be underrepresented by amino acid analysis methodology.
Truncated p120 Induces Compaction in Cell Aggregates
The above findings suggest that the process of p120ctn-band shift could be associated with the induction of compact Colo 205 aggregation. To pursue this possibility, we attempted to modify the activity of p120ctn by expressing a series of its deletion constructs attached to a FLAG-tag at the COOH terminus in Colo 205 cells (Fig. 5 A). Transient expression of these molecules was achieved by electroporation of cDNAs or infection by adenoviral expression vectors. Generally, the adenoviral infection yielded much higher cDNA transfection efficiencies. Successfully transfected cells were detected by immunofluorescence staining with antibodies to the FLAG-tag. We first tested the full-length p120ctn as a control and found no particular effect on the aggregation of Colo 205 cells (Fig. 6 A). Then, we transfected them with ΔN346f, leaving the Armadillo repeat domain intact. Notably, cell groups expressing this construct were transformed into tightly associated aggregates in which the introduced molecules were sharply concentrated at cell–cell contact sites (Fig. 6 B). Then we tested other deletion constructs. Concerning shorter NH2-terminal deletions, ΔN323f displayed the same adhesion-inducing effect as ΔN346f (Fig. 6 C), and ΔN244f also showed a positive effect, but their aggregation appeared looser than that observed with the former (Fig. 6 D). On the other hand, ΔN157f and ΔN101f had no effect on cell adhesion (Fig. 6, E and F). These observations indicate that the critical sites are located between 245 and 323. We tested two other constructs, FLΔRf and ΔN346ΔRf, in which the Armadillo repeat domain was partially deleted, and found that they had no effect on cell aggregation (Fig. 6, G and H). The last two molecules did not bind to E-cadherin, as previously found (Daniel and Reynolds, 1995; data not shown), whereas all the others tested did. Consistently, FLΔRf and ΔN346ΔRf were distributed only in the cytoplasm (Fig. 6, G′ and H′) whereas the others were located along the cell membrane, as well as the cytoplasm (Fig. 6, A′–F′). Cell membrane distributions were categorized into two groups: the adhesion-inducing mutant molecules were concentrated into cell–cell contact sites (e.g., Fig. 6, B′and C′, arrows) whereas the noneffective proteins were located randomly along the cell membrane.
Figure 5.
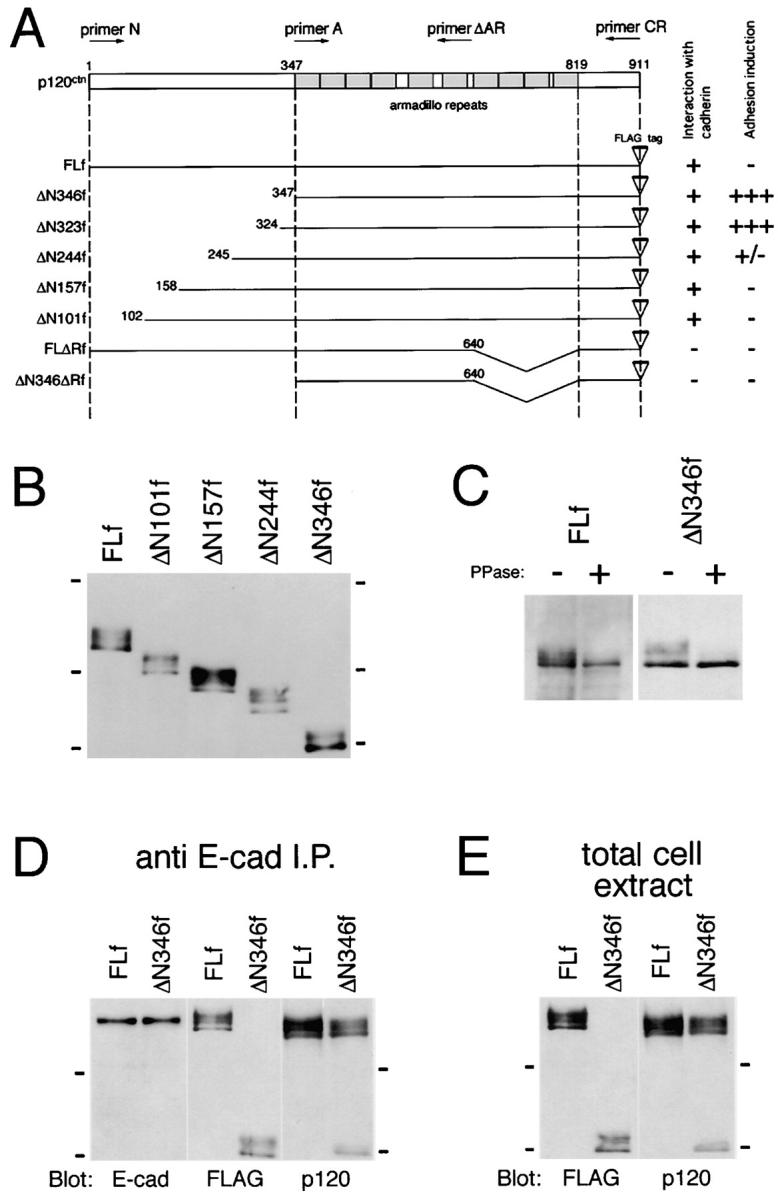
Deletion constructs of p120ctn and their expression. (A) A series of p120ctn mutant constructs used in this study. All constructs were attached to a FLAG-tag at the COOH terminus. (B) Immunoblot analysis of p120ctn mutant proteins expressed in Colo 205 cells. Cells were transfected with their cDNAs by use of adenoviral expression vectors. 2 d after viral infection the total cellular proteins were separated by SDS-PAGE and immunoblotted for detection of the FLAG-tag sequence. (C) Phosphatase treatment of ectopically expressed p120ctn molecules. FLf and ΔN346f expressed in Colo 205 cells were affinity-purified with anti-FLAG M2 mAb, and incubated with protein phosphatase as described in Fig. 4 F. (D and E) Binding of FLf and ΔN346f to E-cadherin. In D, E-cadherin was immunoprecipitated from Colo 205 cells transfected with FLf or ΔN346f, and copurified proteins were analyzed with antibodies to E-cadherin (E-cad), FLAG (FLAG), and p120ctn (p120). In E, the total extracts of the same Colo 205 transfectants as used in D were immunoblotted for FLAG and p120ctn. The anti-p120ctn mAb used recognizes the COOH-terminal region, thus detecting both FLf and ΔN346f, as well as the endogenous molecules, whereas the anti-FLAG antibody detected only the ectopic molecules. Molecular weight markers in B, 175, 83, and 62 × 103; in D and E, 83 and 62 × 103.
Figure 6.

Induction of compaction in Colo 205 aggregates by NH2 terminus–deleted p120ctn molecules. Colo 205 cells were transfected with cDNAs encoding FLf (A and A′), ΔN346f (B and B′), ΔN323f (C and C′), ΔN244f (D and D′), ΔN157f (E and E′), ΔN101f (F and F′), FLΔRf (G and G′), or ΔN346ΔRf (H and H′), and cultured for 72 h. Introduced molecules were detected with anti-FLAG antibody M2. (A–H) Phase-contrast images; (A′–H′) immunofluorescence staining for FLAG in the same fields as in A–H. ΔN346f and ΔN323f induced compaction in Colo 205 aggregates, and ΔN244f showed a partial effect. FLf, ΔN157f, ΔN101f, FLΔRf, and ΔN346ΔRf had no effect. Note that FLΔRf and ΔN346ΔRf are localized only in the cytoplasm, whereas the others also are distributed in the plasma membrane; the compaction-inducing constructs were concentrated at cell–cell contact sites. In most fields shown, not all cells express the ectopic molecules. In C and C′, arrows point to molecules concentrated at cell–cell adhesion sites, and arrowheads indicate cells not expressing the ectopic molecules. Bar, 20 μm.
In the preceding experiments, the p120ctn-band shift was correlated with adhesion induction. Therefore, we also analyzed the band profile of the NH2 terminus deletion constructs. In Western blotting of the lysates of cells transfected with these constructs, all NH2-terminally deleted molecules showed broad electrophoretic bands (Fig. 5 B), although the proportion of the bottom to the upper components in the band tended to increase in the adhesion-inducing construct ΔN346f. When FLf and ΔN346f were collected by immunoprecipitation and treated with phosphatase, only the bottom component remained undigested in both samples (Fig. 5 C), as found in endogenous p120ctn, indicating that the bottom bands of different p120ctn constructs were equivalent to each other in terms of phosphatase resistance.
We examined whether or not the binding of p120ctn to E-cadherin was altered by NH2-terminal deletions. We transfected Colo 205 cells with FLf or mutant ΔN346f, and immunoprecipitated E-cadherin from them (Fig. 5 D). Anti-FLAG antibody D8 detected only the ectopic proteins, and the antibodies recognizing the COOH-terminal region of p120ctn detected both the transfected and endogenous molecules (Fig. 5 D, middle and right panels). Comparison of the band profiles in these samples with those in Western blotting of whole cell lysates (Fig. 5 E) indicates that the proportion of the E-cadherin–bound p120ctn to its entire pool was not different between FLf and ΔN346f. The small difference seen in electrophoretic mobility between the endogenous and full-length ectopic molecules is probably due to their difference in species origin, the former from the mouse and the latter from the human. These results suggest that the binding affinity of p120ctn for E-cadherin was not altered by the NH2-terminal deletion.
p120ctn in Other Cell Lines
We tested two other epithelial lines, HT-29 and MDCK cells, to ask whether they also respond to the NH2 terminus–deleted p120ctn constructs, as well as the compaction-inducing reagents. Human colon carcinoma HT-29 cells, which express normal levels of E-cadherin and catenins (Fig. 7 A), organize into epithelial sheets in confluent cultures (Fig. 7 B, c; Fogh and Trempe, 1975; Chantret et al., 1988). When these cells are dispersed with trypsin in the absence of Ca2+ and seeded into new plates they require a lag period to reestablish epithelial sheets, e.g., at 18–24 h after cell transfer, the cultures still contain many round, dispersed cells (Fig. 7 B, b), although they eventually established epithelial sheets at 48 h. We analyzed p120ctn in these HT-29 cells, and found that its band pattern dynamically changed with the cycle of cell transfer. p120ctn derived from freshly trypsinized HT-29 cells showed a single band (Fig. 7 F). 24 h after the transfer p120ctn was shifted to an upper position (Fig. 7 F). At 48 h, when cells formed confluent epithelial sheets, the p120ctn band returned to the lowest position (Fig. 7 F). We tested if the p120ctn-band shift observed after overnight culture could be canceled by retreatment with trypsin (0.001%; 30 min) or staurosporine (7 nM; 6 h) by adding them to HT-29 cells precultured for 18 h. The results showed that both treatments abolished the mobility shift of the p120ctn band (Fig. 7 G). The range of the p120ctn-band shifting was similar to that found in Colo 205 cells. In correlation with this p120ctn band change, tight cell–cell association was induced in HT-29 cells preincubated for 18 h and then incubated with staurosporine (7 nM) for 6 h or with trypsin (0.001%) for 30 min (Fig. 7 B, e and f), as seen in the case of Colo 205 cells.
On the other hand, MDCK, which is widely used as a model of polarized epithelial cells, reorganized epithelial sheets within 18 h after transfer. Addition of staurosporine or trypsin to such MDCK cultures had no effects on cell– cell association (Fig. 7 C), although we observed subtle morphological alterations in the treated cultures. Analysis of p120ctn in MDCK cells detected two major bands as reported by Mo and Reynolds (1996), the upper band likely corresponds to the p120ctn band in human colon carcinoma lines (Fig. 7 F). The p120ctn pattern in MDCK was not changed by trypsin-mediated cell transfer. When MDCK cells were treated with staurosporine, the electrophoretic mobility of p120ctn was slightly enhanced (Fig. 7 G), as described by Ratcliffe et al. (1997). However, this position shift of the p120ctn band was subtle compared with that observed in the above carcinoma lines. Trypsin treatment of MDCK cells did not affect the mobility of p120ctn (Fig. 7 G).
Next, we transfected the above cell lines with the NH2 terminus–truncated ΔN346f, as well as with the control FLf immediately after cell transfer, and examined the effects after 24 h. ΔN346f, but not FLf, enhanced HT-29 cell–cell association (Fig. 7 D). ΔN346f-expressing cells formed compact aggregates, whereas FLf had no effects. Rather, we observed that high-level overexpressions of FLf tended to disperse epithelial sheets (data not shown). In MDCK cells, neither construct affected the cell–cell contact morphology, they simply accumulated at cell–cell contact sites (Fig. 7 E).
Induction of Colo 205 Aggregation by N-Cadherin Lacking the p120ctn-binding Site
These findings suggest that in particular cells such as Colo 205, p120ctn inhibits cadherin-mediated adhesion under certain physiological conditions. If this is the case, cadherin activity might be restored under circumstances where it cannot interact with p120ctn. To test this possibility, we transfected Colo 205 cells with two different constructs of N-cadherin cDNA, one encoding its entire portion and the other encoding a mutant molecule in which the juxtamembrane region of the cytoplasmic domain, which is known to contain the p120ctn-binding site (Yap et al., 1998), has been deleted (Fig. 8 A). Immunoprecipitation with anti–N-cadherin antibodies from the transfected cells confirmed that the mutant N-cadherin was unable to associate with p120ctn (Fig. 8 B), but coprecipitated normally with β-catenin. Morphological and immunostaining examinations of these transfectants showed that the expression of full-length N-cadherin had no effect on cell–cell adhesion, suggesting that cadherins were generally blocked in the Colo 205 environment, irrespective of their type. In contrast, the mutant N-cadherin induced a strong aggregation of Colo 205 cells (Fig. 8 C). These findings suggest that cadherins can function normally in these cells unless they interact with p120ctn, supporting the idea that p120ctn can act as an inhibitor of cadherin function.
Figure 8.
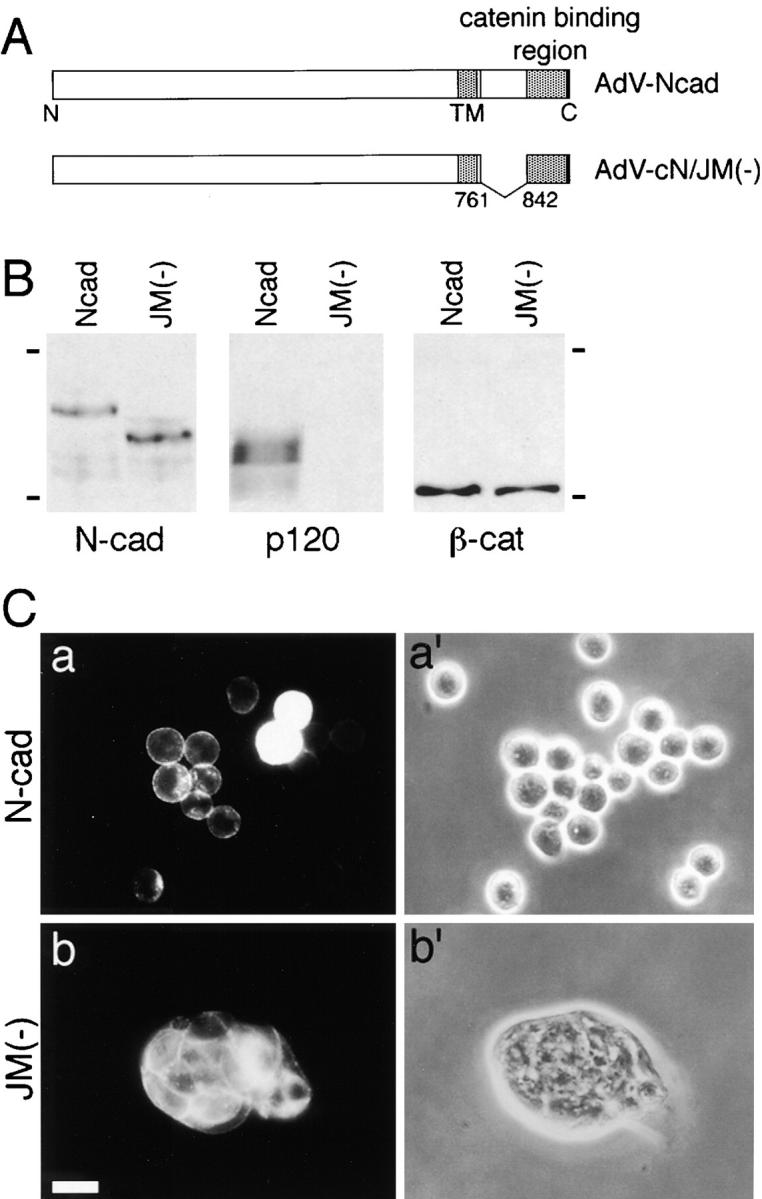
Induction of compact cell aggregation by expression of N-cadherin without the p120ctn-binding site. (A) Schematic drawing of full-length N-cadherin and a mutant N-cadherin in which the juxtamembrane portion of the cytoplasmic domain was deleted and used for construction of adenoviral expression vectors, AdV-Ncad and AdV-cN/JM(−), respectively. TM, transmembrane domain. (B) Immunoblot analysis of catenins coprecipitated with N-cadherin. Colo 205 cells were infected with AdV-Ncad or AdV-cN/JM(−) and cultured for 48 h. Expressed N-cadherin proteins (N-cad) were immunoprecipitated with NCD-2, and the immunoprecipitants were analyzed for p120ctn (p120) or β-catenin (β-cat). Molecular weight markers, 175 and 83 × 103. (C) Morphological changes in Colo 205 cell aggregates induced by the mutant N-cadherin. Cells infected with AdV-Ncad or AdV-cN/JM(−) were cultured for 24 h and stained for N-cadherin. a and b, Immunofluorescence staining for N-cadherin; a′ and b′, phase-contrast images in the same fields as in a and b. Bar, 20 μm.
Discussion
This work was initiated to understand why Colo 205 cells cannot undergo typical E-cadherin–dependent association despite their normal expression of E-cadherin and associated proteins. We found that the treatment of these cells with two independent reagents, trypsin and staurosporine, could reactivate the E-cadherin adhesion system. Although the initial mechanisms for their actions remain unknown, we assume that trypsin removes or truncates some cell surface proteins involved in intracellular signaling, leading to an activation or suppression of their downstream cascade and via this pathway, modulates the cadherin system. The response of Colo 205 cells to the trypsin treatment was rapid, suggesting that the proposed signaling system probably does not require transcription of new genes. As another possible mechanism of the trypsin action, we considered that this enzyme might have facilitated cell adhesion by digesting such surface components as mucins, known to physically prevent cell–cell contacts. However, this possibility is less likely, because we could induce compact cell aggregation by intracellular manipulation of Colo 205 cells without enzymatic treatment. Moreover, no correlation was found between the distribution of MUC1, a major Colo 205 mucin, and adhesion induction. Concerning staurosporine, its action could be connected with the proposed signaling cascade directly or indirectly. Of the many kinase inhibitors tested, only staurosporine effectively induced Colo 205 cell adhesion within a short incubation period. This suggests that staurosporine has a specific target in its action on Colo 205 adhesion, although we cannot specify it because this antibiotic can inhibit multiple classes of enzymes. It was reported that retinoic acid treatment results in a similar adhesion induction (Nakagawa et al., 1998), suggesting that this reagent is another effector to activate the cadherin system.
Since the initial or intermediate steps of the proposed signaling system could be complex, we focused our analysis on its putative terminal step, asking if the cadherin– catenin complex per se was modified during the adhesion induction, and we identified an alteration in the electrophoretic mobility of p120ctn. This change could be triggered in the absence of E-cadherin–mediated adhesion, implying that it resulted directly from the trypsin or staurosporine-triggered signaling cascade. A trypsin/staurosporine-sensitive mobility shift of p120ctn was also observed with another carcinoma line HT-29. Although transient in these cells, the shift nonetheless correlated perfectly with adhesion induction. In contrast, MDCK cells neither showed such modulation of p120ctn, nor responded to the adhesion-inducing reagents. Although staurosporine treatment slightly enhanced the electrophoretic mobility of p120ctn, even in MDCK cells (Ratcliffe et al., 1997), the magnitude of the band shifting in this case was much smaller as compared with that for Colo 205 and HT-29 cells.
These findings prompted us to further examine the role for p120ctn by structure–function analysis, and we found that NH2-terminally truncated p120ctn constructs by themselves could reactivate the E-cadherin system. This observation implies that p120ctn inhibits cadherin-mediated adhesion in Colo 205 cells. Thus, we postulate that the NH2-terminally deleted p120ctn lacks this activity, and competes with endogenous molecules, ultimately resulting in the reactivation of the cadherin system. This hypothesis is strongly supported by the finding that ectopic N-cadherin induced aggregation of Colo 205 cells only when it was unable to bind to p120ctn. This finding also suggests that different types of cadherin, in general, cannot function in the intracellular environment of Colo 205 if they bind to p120ctn.
The correlation between the band shifting of p120ctn and adhesion induction suggests that the putative inhibitory activity of this molecule might be elicited by certain biochemical modifications. In Colo 205 cells, the original p120ctn band was broad. Although multiple splicing products of p120ctn are expressed in many carcinoma lines (Mo and Reynolds, 1996), the broad band observed in Colo 205 cells appears to result from phosphorylation at various levels because phosphatase treatment of the Colo 205 p120ctn resulted in the generation of a sharp single band. This phosphatase-treated p120ctn was similar in electrophoretic mobility to that found in the adhesion-induced Colo 205 cells. These findings suggest the possibility that p120ctn acquires the inhibitory activity when hyperphosphorylated, and the staurosporine/trypsin-induced signals reduce the level of phosphorylation. We found that the major phosphorylated residue in p120ctn of Colo 205 cells was serine, consistent with a previous observation by Ratcliffe et al. (1997). Our results do not exclude the possibility that other residues are also phosphorylated at low levels, as it has been reported that tyrosine is phosphorylated in p120ctn of other cell lines (Kinch et al., 1995; Calautti et al., 1998; Hazan and Norton, 1998; Rosato et al., 1998). The net amount of serine phosphorylation in p120ctn did not appear reduced after its electrophoretic mobility shift, although phosphoamino acid analysis may not adequately detect small changes in phosphorylation status. If dephosphorylation is indeed involved in this process, one possibility is that only a specific subset of phosphorylation sites are required for the p120ctn change in motility. The NH2-terminally deleted p120ctn, which induced cell adhesion, still contained slower-migrating, phosphatase-sensitive components, although their proportion tended to be reduced. Possibly, the key phosphorylation sites directly affecting the electrophoretic mobility of p120ctn are located outside the NH2-terminal region. These observations also suggest that hyperphosphorylation may be insufficient to confer the adhesion inhibitory activity on p120ctn if its NH2 terminus is absent. A model for the signaling cascade to regulate p120ctn phosphorylation has been proposed by Ratcliffe et al. (1997).
How does p120ctn inhibit cadherin function? It is interesting to note that the juxtamembrane domain of the cadherin cytoplasmic region where p120ctn binds can inhibit cell adhesion when overexpressed (Kintner, 1992), and also is required for clustering of cadherin molecules (Yap et al., 1998) which is thought to be essential for cadherin-mediated cell adhesion. Another report shows that deletion of a similar region in E-cadherin resulted in activation of this adhesion molecule, enhancing its lateral interactions in the cell membrane of a leukemia line (Ozawa and Kemler, 1998b), suggesting that this domain can inhibit lateral clustering of cadherins. p120ctn is the only molecule known to bind to the juxtamembrane domain of cadherins and could be the major effector of these activities. For example, p120ctn may directly regulate lateral clustering of cadherins and this activity may be blocked by hyperphosphorylation or other mechanisms controlled by the NH2 terminus. In support of this hypothesis, our immunostaining observations suggest that clustering of E-cadherin into cell–cell contact sites is enhanced by the adhesion-induction treatments. Alternatively, p120ctn may have no function in the default state and might serve as an inhibitor of cadherin function only when posttranscriptionally modified. The results of our N-cadherin experiments, as well as the above leukemia study (Ozawa and Kemler, 1998b), indicate that cadherins can be active without p120ctn.
We have demonstrated a p120ctn-dependent inhibition of the cadherin system in carcinoma cells. It is known that cadherin-mediated adhesion is perturbed in tumors in a variety of ways, including mutation of cadherin or catenin genes, and these events have been implicated in cancer invasion and metastasis (reviewed by Shiozaki et al., 1996). Here, we have identified a novel physiological mechanism of inhibiting cadherin function. Many metastasizing tumors maintain strong cadherin and catenin expression. Such tumor cells must have a mechanism to destabilize cell–cell adhesion, at least transiently, for detaching from the original tumors. Biochemical modification of p120ctn could be used for such processes. Colo 205 cells may be an extreme case in which the p120ctn-dependent inhibitory mechanism is constitutively turned on, whereas in HT-29 it operates only transiently, as these cells can eventually form normal looking epithelial sheets. The mechanism for the transient modification of p120ctn in HT-29 remains unknown.
Finally, the question arises as to how such an activity of p120ctn is involved in normal cellular events. Cell–cell adhesion is dynamic and requires regulation during many types of morphogenetic processes. p120ctn could be implicated in such regulation as suggested by previous observations. The juxtamembrane domain of the cadherin cytoplasmic domain to which p120ctn binds was implicated in cell motility (Chen et al., 1997) and in axon outgrowth (Riehl et al., 1996). Overexpression of p120ctn in Xenopus embryos perturbs gastrulation (Geis et al., 1998; Paulsen et al., 1999). p120ctn, whose action can be modulated posttranscriptionally, could play a role in the control of such adhesion-related cellular behavior. As we suggested in light of the effect of trypsin, there might be a signaling cascade originating at the cell surface to regulate the p120ctn activity. Identification of components of such cascades is an important future issue for unraveling the morphogenetic regulatory mechanisms of cell–cell adhesion.
Acknowledgments
We thank T. Moriguchi, Y. Gotoh, and E. Nishida (Kyoto University) for advice and technical support on phosphoamino acid analysis. We also thank M.J. Wheelock (University of Toledo) for 5H10 antibody, T. Irimura (University of Tokyo) for anti-MUC1 antibody, K. Moriyoshi (Kyoto University) for recombinant adenoviruses, and T. Tamaoki (Kyowa Medex) for UCN-01.
This work was supported by the program Grants-in-Aid for Creative Fundamental Research, and for Specially Promoted Research of the Ministry of Education, Science, Sports, and Culture of Japan.
Abbreviations used in this paper
- aa
amino acid
- FLf
full-length FLAG-tagged p120ctn
Footnotes
Shinichi Nakagawa's current address is Department of Anatomy, Downing Street, University of Cambridge, Cambridge CB2 3DY, United Kingdom.
References
- Baeckstrom D, Hansson GC, Nilsson O, Johansson C, Gendler SJ, Lindholm L. Purification and characterization of a membrane-bound and a secreted mucin-type glycoprotein carrying the carcinoma-associated sialyl-Lea epitope on distinct core proteins. J Biol Chem. 1991;266:21537–21547. [PubMed] [Google Scholar]
- Balsamo J, Arregui C, Leung T, Lilien J. The nonreceptor protein tyrosine phosphatase PTP1B binds to the cytoplasmic domain of N-cadherin and regulates the cadherin–actin linkage. J Cell Biol. 1998;143:523–532. doi: 10.1083/jcb.143.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AIM, Näthke IS, Nelson WJ. Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997;9:683–690. doi: 10.1016/s0955-0674(97)80122-6. [DOI] [PubMed] [Google Scholar]
- Brieher WM, Yap AS, Gumbiner BM. Lateral dimerization is required for the homophilic binding activity of C-cadherin. J Cell Biol. 1996;135:487–496. doi: 10.1083/jcb.135.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calautti E, Cabodi S, Stein PL, Hatzfeld M, Kedersha N, Paolo G, Dotto Tyrosine phosphorylation and src family kinases control keratinocyte cell–cell adhesion. J Cell Biol. 1998;141:1449–1465. doi: 10.1083/jcb.141.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantret I, Barbat A, Dussaulx E, Brattain MG, Zweibaum A. Epithelial polarity, villin expression, and enterocytic differentiation of cultured human colon carcinoma cells: a survey of twenty cell lines. Cancer Res. 1988;48:1936–1942. [PubMed] [Google Scholar]
- Chen H, Paradies NE, Fedor-Chaiken M, Brackenbury R. E-cadherin mediates adhesion and suppresses cell motility via distinct mechanisms. J Cell Sci. 1997;110:345–356. doi: 10.1242/jcs.110.3.345. [DOI] [PubMed] [Google Scholar]
- Chitaev NA, Troyanovsky SM. Adhesive but not lateral E-cadherin complexes require calcium and catenins for their formation. J Cell Biol. 1998;142:837–846. doi: 10.1083/jcb.142.3.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JM, Reynolds AB. The tyrosine kinase substrate p120casbinds directly to E-cadherin but not to the adenomatous polyposis coli protein or α-catenin. Mol Cell Biol. 1995;15:4819–4824. doi: 10.1128/mcb.15.9.4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnemann S, Mitrik I, Hess M, Otto G, Wedlich D. Uncoupling of XB/U-cadherin–catenin complex formation from its function in cell–cell adhesion. J Biol Chem. 1997;272:11856–11862. doi: 10.1074/jbc.272.18.11856. [DOI] [PubMed] [Google Scholar]
- Fogh, J., and G. Trempe. 1975. New human tumor lines. In Human Tumor Cells In Vitro. J. Fogh, editor. Plenum Press, New York. 115–141.
- Gaush CR, Hard WL, Smith TF. Characterization of an established line of canine kidney cells (MDCK. ) Proc Soc Exp Biol Med. 1966;122:931–935. doi: 10.3181/00379727-122-31293. [DOI] [PubMed] [Google Scholar]
- Geis K, Aberle H, Kuhl M, Kemler R, Wedlich D. Expression of the Armadillo family member p120cas 1B in Xenopusembryos affects head differentiation but not axis formation. Dev Genes Evol. 1998;207:471–481. doi: 10.1007/s004270050138. [DOI] [PubMed] [Google Scholar]
- Hamaguchi M, Matsuyoshi N, Ohnishi Y, Gotoh B, Takeichi M, Nagai Y. p60v-srccauses tyrosine phosphorylation and inactivation of the N-cadherin–catenin cell adhesion system. EMBO (Eur Mol Biol Organ) J. 1993;12:307–314. doi: 10.1002/j.1460-2075.1993.tb05658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M, Nachtsheim C. Cloning and characterization of a new armadillo family member, p0071, associated with the junctional plaque: evidence for a subfamily of closely related proteins. J Cell Sci. 1996;109:2767–2778. doi: 10.1242/jcs.109.11.2767. [DOI] [PubMed] [Google Scholar]
- Hazan RB, Norton L. The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem. 1998;273:9078–9084. doi: 10.1074/jbc.273.15.9078. [DOI] [PubMed] [Google Scholar]
- Heid HW, Schmidt A, Zimbelmann R, Schafer S, Winter-Simanowski S, Stumpp S, Keith M, Figge U, Schnolzer M, Franke WW. Cell type-specific desmosomal plaque proteins of the plakoglobin family: plakophilin 1 (band 6 protein) Differentiation. 1994;58:113–131. doi: 10.1046/j.1432-0436.1995.5820113.x. [DOI] [PubMed] [Google Scholar]
- Hirano S, Kimoto N, Shimoyama Y, Hirohashi S, Takeichi M. Identification of a neural α-catenin as a key regulator of cadherin function and multicellular organization. Cell. 1992;70:293–301. doi: 10.1016/0092-8674(92)90103-j. [DOI] [PubMed] [Google Scholar]
- Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cerretti DP, Urdal DL, Conlon PJ. A short polypeptide marker sequence useful for recombinant protein identification and purification. Biotechnology. 1988;6:1204–1210. [Google Scholar]
- Johnson KR, Lewis JE, Li D, Wahl J, Soler AP, Knudsen KA, Wheelock MJ. P- and E-cadherin are in separate complexes in cells expressing both cadherins. Exp Cell Res. 1993;207:252–260. doi: 10.1006/excr.1993.1191. [DOI] [PubMed] [Google Scholar]
- Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- Kemperman H, Wijnands Y, Wesseling J, Niessen CM, Sonnenberg A, Roos E. The mucin epiglycanin on TA3/Ha carcinoma cells prevents α6β4-mediated adhesion to laminin and kalinin and E-cadherin–mediated cell–cell interaction. J Cell Biol. 1994;127:2071–2080. doi: 10.1083/jcb.127.6.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinch MS, Clark GJ, Der CJ, Burridge K. Tyrosine phosphorylation regulates the adhesions of ras-transformed breast epithelia. J Cell Biol. 1995;130:461–471. doi: 10.1083/jcb.130.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintner C. Regulation of embryonic cell adhesion by the cadherin cytoplasmic domain. Cell. 1992;69:225–236. doi: 10.1016/0092-8674(92)90404-z. [DOI] [PubMed] [Google Scholar]
- Knudsen KA, Soler AP, Johnson KR, Wheelock MJ. Interaction of α-actinin with the cadherin/catenin cell–cell adhesion complex via α-catenin. J Cell Biol. 1995;130:67–77. doi: 10.1083/jcb.130.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Kohno N, Yokoyama A, Hiwada K. Decreased MUC1 expression induces E-cadherin–mediated cell adhesion of breast cancer cell lines. Cancer Res. 1998;58:2014–2019. [PubMed] [Google Scholar]
- Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110:2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- Lu Q, Parades M, Medina M, Zhou J, Cavallo R, Peifer M, Orecchio L, Kosik KS. δ-Catenin, an adhesive junction-associated protein which promotes cell scattering. J Cell Biol. 1999;144:519–532. doi: 10.1083/jcb.144.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyoshi N, Hamaguchi M, Taniguchi S, Nagafuchi A, Tsukita S, Takeichi M. Cadherin-mediated cell–cell adhesion is perturbed by v-srctyrosine phosphorylation in metastatic fibroblasts. J Cell Biol. 1992;118:703–714. doi: 10.1083/jcb.118.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens C, Kuhn C, Franke WW. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomal plaque. J Cell Biol. 1996;135:1009–1025. doi: 10.1083/jcb.135.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo YY, Reynolds AB. Identification of murine p120 isoforms and heterogeneous expression of p120casisoforms in human tumor cell lines. Cancer Res. 1996;56:2633–2640. [PubMed] [Google Scholar]
- Moriyoshi K, Richards LJ, Akazawa C, O'Leary DD, Nakanishi S. Labeling neural cells using adenoviral gene transfer of membrane-targeted GFP. Neuron. 1996;16:255–260. doi: 10.1016/s0896-6273(00)80044-6. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M. Neural crest emigration from the neural tube depends on regulated cadherin expression. Development. 1998;125:2963–2971. doi: 10.1242/dev.125.15.2963. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Sogo S, Hioki K, Tokunaga R, Taketani S. Acquisition of cell adhesion and induction of focal adhesion kinase of human colon cancer Colo 201 cells by retinoic acid-induced differentiation. Differentiation. 1998;62:249–257. doi: 10.1046/j.1432-0436.1998.6250249.x. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Oyama T, Kanai Y, Ochiai A, Akimoto S, Oda T, Yanagihara K, Nagafuchi A, Tsukita S, Shibamoto S, Ito F, et al. A truncated β-catenin disrupts the interaction between E-cadherin and α-catenin: a cause of loss of intercellular adhesiveness in human cancer cell lines. Cancer Res. 1994;54:6282–6287. [PubMed] [Google Scholar]
- Ozawa M, Kemler R. Altered cell adhesion activity by pervanadate due to the dissociation of α-catenin from the E-cadherin–catenin complex. J Biol Chem. 1998a;273:6166–6170. doi: 10.1074/jbc.273.11.6166. [DOI] [PubMed] [Google Scholar]
- Ozawa M, Kemler R. The membrane–proximal region of the E-cadherin cytoplasmic domain prevents dimerization and negatively regulates adhesion activity. J Cell Biol. 1998b;142:1605–1613. doi: 10.1083/jcb.142.6.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paffenholz R, Franke WW. Identification and localization of a neurally expressed member of the plakoglobin/armadillo multigene family. Differentiation. 1997;61:293–304. doi: 10.1046/j.1432-0436.1997.6150293.x. [DOI] [PubMed] [Google Scholar]
- Paulson AF, Fang X, Ji H, Reynolds AB, McCrea P. Misexpression of the catenin p120ctn 1A perturbs Xenopusgastrulation. Develop Biol. 1999;207:350–363. doi: 10.1006/dbio.1998.9158. [DOI] [PubMed] [Google Scholar]
- Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. 1994;76:789–791. doi: 10.1016/0092-8674(94)90353-0. [DOI] [PubMed] [Google Scholar]
- Ratcliffe MJ, Rubin LL, Staddon JM. Dephosphorylation of the cadherin-associated p100/p120 proteins in response to activation of protein kinase C in epithelial cells. J Biol Chem. 1997;272:31894–31901. doi: 10.1074/jbc.272.50.31894. [DOI] [PubMed] [Google Scholar]
- Reynolds AB, Roesel DJ, Kanner SB, Parsons JT. Transformation specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol Cell Biol. 1989;9:629–638. doi: 10.1128/mcb.9.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AB, Herbert L, Cleveland JL, Berg ST, Gaut JR. p120, a novel substrate of protein tyrosine kinase receptors and of p60v-src, is related to cadherin-binding factors β-catenin, plakoglobin and armadillo. Oncogene. 1992;7:2439–2445. [PubMed] [Google Scholar]
- Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120casassociates with E-cadherin complexes. Mol Cell Biol. 1994;14:8333–8342. doi: 10.1128/mcb.14.12.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AB, Daniel JM, Mo YY, Wu J, Zhang Z. The novel catenin p120casbinds classical cadherins and induces an unusual morphological phenotype in NIH3T3 fibroblasts. Exp Cell Res. 1996;225:328–337. doi: 10.1006/excr.1996.0183. [DOI] [PubMed] [Google Scholar]
- Riehl R, Johnson K, Bradley R, Grunwald GB, Cornel E, Lilienbaum A, Holt CE. Cadherin function is required for axon outgrowth in retinal ganglion cells in vivo. Neuron. 1996;17:837–848. doi: 10.1016/s0896-6273(00)80216-0. [DOI] [PubMed] [Google Scholar]
- Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. α1(E)-Catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato R, Veltmaat JM, Groffen J, Heisterkamp N. Involvement of the tyrosine kinase fer in cell adhesion. Mol Cell Biol. 1998;18:5762–5770. doi: 10.1128/mcb.18.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple TU, Quinn LA, Woods LK, Moore GE. Tumor and lymphoid cell lines from a patient with carcinoma of the colon for a cytotoxicity model. Cancer Res. 1978;38:1345–1355. [PubMed] [Google Scholar]
- Serres M, Grangeasse C, Haftek M, Durocher Y, Duclos B, Schmitt D. Hyperphosphorylation of β-catenin on serine-threonine residues and loss of cell–cell contacts induced by calyculin A and okadaic acid in human epidermal cells. Exp Cell Res. 1997;231:163–172. doi: 10.1006/excr.1996.3443. [DOI] [PubMed] [Google Scholar]
- Shibamoto S, Hayakawa M, Takeuchi K, Hori T, Miyazawa K, Kitamura N, Johnson KR, Wheelock MJ, Matsuyoshi N, Takeichi M, et al. Association of p120, a tyrosine kinase substrate, with E-cadherin/catenin complexes. J Cell Biol. 1995;128:949–957. doi: 10.1083/jcb.128.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoyama Y, Hirohashi S, Hirano S, Noguchi M, Shimosato Y, Takeichi M, Abe O. Cadherin cell-adhesion molecules in human epithelial tissues and carcinomas. Cancer Res. 1989;49:2128–2133. [PubMed] [Google Scholar]
- Shimoyama Y, Nagafuchi A, Fujita S, Gotoh M, Takeichi M, Tsukita S, Hirohashi S. Cadherin dysfunction in a human cancer cell line: possible involvement of loss of α-catenin expression in reduced cell–cell adhesiveness. Cancer Res. 1992;52:5770–5774. [PubMed] [Google Scholar]
- Shiozaki H, Oka H, Inoue M, Tamura S, Monden M. E-cadherin mediated adhesion system in cancer cells. Cancer. 1996;77:1605–1613. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1605::AID-CNCR28>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Sirotkin H, O'Donnell H, DasGupta R, Halford S, St. Jore B, Puech A, Parimoo S, Morrow B, Skoultchi A, Weissman SM, et al. Identification of a new human catenin gene family member (ARVCF) from the region deleted in velo-cardio-facial syndrome. Genomics. 1997;41:75–83. doi: 10.1006/geno.1997.4627. [DOI] [PubMed] [Google Scholar]
- Staddon JM, Smales C, Schulze C, Esch FS, Rubin LL. p120, a p120-related protein (p100), and the cadherin/catenin complex. J Cell Biol. 1995;130:369–381. doi: 10.1083/jcb.130.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi I, Kobayashi E, Nakano H, Murakata C, Saitoh H, Suzuki K, Tamaoki T. Potent selective inhibition of 7-O-methyl UCN-01 against protein kinase C. J Pharmacol Exp Ther. 1990;255:1218–1221. [PubMed] [Google Scholar]
- Takahashi K, Suzuki K, Tsukatani Y. Induction of tyrosine phosphorylation and association of β-catenin with EGF receptor upon tryptic digestion of quiescent cells at confluence. Oncogene. 1997;15:71–78. doi: 10.1038/sj.onc.1201160. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Functional correlation between cell adhesive properties and some cell surface proteins. J Cell Biol. 1977;75:464–474. doi: 10.1083/jcb.75.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. The cadherins: cell–cell adhesion molecules controlling animal morphogenesis. Development. 1988;102:639–655. doi: 10.1242/dev.102.4.639. [DOI] [PubMed] [Google Scholar]
- Watabe M, Nagafuchi A, Tsukita S, Takeichi M. Induction of polarized cell–cell association and retardation of growth by activation of the E-cadherin–catenin adhesion system in a dispersed carcinoma line. J Cell Biol. 1994;127:247–256. doi: 10.1083/jcb.127.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Uchida N, Imamura Y, Nagafuchi A, Fujimoto K, Uemura T, Vermeulen S, van Roy F, Adamson ED, Takeichi M. α-Catenin–vinculin interaction functions to organize the apical junctional complex in epithelial cells. J Cell Biol. 1998;142:847–857. doi: 10.1083/jcb.142.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling J, van der Valk SW, Hilkens J. A mechanism for inhibition of E-cadherin–mediated cell–cell adhesion by the membrane-associated mucin episialin/MUC1. Mol Biol Cell. 1996;7:565–577. doi: 10.1091/mbc.7.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Bhavanandan VP, Nakamori S, Irimura T. A novel monoclonal antibody specific for sialylated MUC1 mucin. Jpn J Cancer Res. 1996;87:488–496. doi: 10.1111/j.1349-7006.1996.tb00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Pruschy M, Gumbiner BM. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol. 1997;7:308–315. doi: 10.1016/s0960-9822(06)00154-0. [DOI] [PubMed] [Google Scholar]
- Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn . J Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]