

Abstract
Although the number of pathologies known to arise from the inappropriate folding of proteins continues to grow, mechanisms underlying the recognition and ultimate disposition of misfolded polypeptides remain obscure. For example, how and where such substrates are identified and processed is unknown. We report here the identification of a specific subcellular structure in which, under basal conditions, the 20S proteasome, the PA700 and PA28 (700- and 180-kD proteasome activator complexes, respectively), ubiquitin, Hsp70 and Hsp90 (70- and 90-kD heat shock protein, respectively) concentrate in HEK 293 and HeLa cells. The structure is perinuclear, surrounded by endoplasmic reticulum, adjacent to the Golgi, and colocalizes with γ-tubulin, an established centrosomal marker. Density gradient fractions containing purified centrosomes are enriched in proteasomal components and cell stress chaperones. The centrosome-associated structure enlarges in response to inhibition of proteasome activity and the level of misfolded proteins. For example, folding mutants of CFTR form large inclusions which arise from the centrosome upon inhibition of proteasome activity. At high levels of misfolded protein, the structure not only expands but also extensively recruits the cytosolic pools of ubiquitin, Hsp70, PA700, PA28, and the 20S proteasome. Thus, the centrosome may act as a scaffold, which concentrates and recruits the systems which act as censors and modulators of the balance between folding, aggregation, and degradation.
Keywords: proteasome, protein misfolding, inclusions, CFTR, centrosome
Many pathologies result from the inability of a mutant protein to properly fold (Thomas et al., 1995). It is therefore important to understand the mechanisms by which misfolded proteins are recognized, processed, and ultimately degraded by the cell. The 26S proteasome catalyzes the intracellular degradation of damaged or misfolded proteins, the rapid degradation of short-lived regulatory proteins such as those involved in cell cycle control and signaling, as well as the generation of antigenic peptides for presentation to MHC-I (Coux et al., 1996). Degradation of most substrates by the proteasome requires the covalent conjugation of ubiquitin, an event catalyzed by the sequential action of three enzymes (Ciechanover, 1994; Hochstrasser, 1996). The ubiquitination machinery acts progressively, resulting in a polyubiquitin chain that serves as a signal for degradation of the modified protein by the 26S proteasome.
The 26S proteasome is composed of two major subcomplexes. The 20S proteasome, a 700-kD cylinder, constitutes the catalytic core of the protease (Coux et al., 1996; Baumeister et al., 1998). Structural (Lowe et al., 1995; Groll et al., 1997) and biochemical evidence (Wenzel and Baumeister, 1995) indicate the catalytic sites are located within a hollow cavity of the cylinder. Access to these sites is achieved through narrow openings at the ends of the cylinder which may be gated by regulatory proteins. This topology limits substrates to short peptides and completely unfolded proteins.
The regulatory complex of the 26S proteasome is a 20-subunit, 700-kD activator called PA7001 or 19S cap (Coux et al., 1996), which associates with either (or both) end(s) of the 20S proteasome to form the 26S complex (Yoshimura et al., 1993; Coux et al., 1996; Adams et al., 1997). Such binding probably activates proteasome function by opening access to the central cavity, thereby increasing access of substrates to the catalytic sites. PA700 has multiple ATPase activities (Coux et al., 1996), binds to and cleaves polyubiquitin (Lam et al., 1997), and functions to enhance degradation of proteins, perhaps by unfolding and/or translocating its substrates to the central cavity (Ma et al., 1994; Adams et al., 1997).
A second regulator of the 20S proteasome is PA28 or 11S, a 180-kD heterohexameric ring-shaped complex that binds to the proteasome in an orientation similar to that of PA700 (Dubiel et al., 1992; Ma et al., 1993; Gray et al., 1994). PA28 does not require ubiquitinated substrates for activity and functions to increase proteasomal processing of short peptides rather than larger proteins in vitro. Synthesis of PA28 is stimulated by γ-interferon, implicating its probable functional importance in antigen production and presentation (Realini et al., 1994; Ahn et al., 1995; Dick et al., 1996). New data suggest the proteasome may exist in vitro in a complex simultaneously with PA28 and PA700, although the physiological relevance of such an observation is unclear (Hendil et al., 1998).
A role has been established for the proteasome in the degradation of incompletely folded or misfolded proteins (Kopito, 1997). For example, both cystic fibrosis (CF)- causing mutant forms (Riordan et al., 1989) and up to 70% of the wild-type cystic fibrosis transmembrane conductance regulator (CFTR) (Ward and Kopito, 1994; Jensen et al., 1995; Ward et al., 1995) have been shown to be degraded in an ATP-dependent, ubiquitin/proteasome-mediated manner (Jensen et al., 1995; Ward et al., 1995). CFTR is a member of the ATP-binding cassette (ABC) supergene family of membrane transport proteins (Higgins, 1992), and consists of five domains, including two nucleotide binding domains (NBD1 and NBD2), two transmembrane domains (TMD1 and TMD2), and a PKA-sensitive regulatory domain (R) (Riordan et al., 1989).
Notably, of the more than 800 CF-causing CFTR mutations identified to date, several initiate the CF pathology by affecting the ability of the nascent polypeptide to fold into a functional, stable native state (Qu et al., 1997). One such mutation, the deletion of phenylalanine 508 (ΔF508) is by far the most common, accounting for ∼70% of the disease causing alleles (Tsui, 1992). This mutation, located in NBD1, results in a folding defective protein which is incompletely glycosylated and fails to efficiently traffic to the apical membrane (Cheng et al., 1990), and is degraded by the proteasome (Jensen et al., 1995; Ward et al., 1995). Several other CF-causing folding mutations (Gregory et al., 1991; Sheppard et al., 1996) including P205S which is located in the third membrane spanning helix (Wigley et al., 1998) of TMD1, also result in inefficient processing and maturation and increased degradation.
Significantly, both the ΔF508 and P205S folding mutants are functional when they assume a native conformation, thus raising the possibility that overcoming the maturation deficiency by correcting the underlying folding defect or by circumventing a proteolytic recognition step may be of therapeutic benefit. Unfortunately, inhibition of the proteasome in HEK 293 cells expressing either wild-type or ΔF508 CFTR results in the accumulation of polyubiquitinated CFTR including insoluble, perhaps aggregated, species (Jensen et al., 1995; Ward et al., 1995).
Heat shock proteins, such as Hsp70 and Hsp90, have been shown to play a role in both preventing protein aggregation and promoting folding or degradation of a wide range of proteins. Recently, the ubiquitination of certain proteins has been shown to depend upon Hsp70, implicating it in the targeting of misfolded or mutant proteins for degradation by the proteasome (Bercovich et al., 1997). In this regard, Hsp70 has been shown to interact transiently with immature, incompletely folded CFTR (Yang et al., 1993). While this paper was under review, a report appeared that demonstrated that the Hsp90 inhibitor geldanamycin prevents the association of immature CFTR with the chaperone, blocks its maturation, and enhances its degradation by the proteasome (Loo et al., 1998).
In spite of the large body of evidence demonstrating the proteasome machinery's involvement in the degradation of a diverse range of substrates, it is unclear whether its functions are distributed homogeneously throughout the cell or if they are segregated. We present evidence herein of the centrosome as a unique location in which the proteasome machinery, Hsp70, and Hsp90 are concentrated in resting HEK 293 and HeLa cell lines. In addition, the centrosome, a structure thought previously to function primarily during cell division, responds to inhibition of the proteasome and increases in the level of misfolded proteins by expanding and recruiting cellular pools of the proteasome components and Hsp70.
Materials and Methods
Materials
Plasmids pCMVNot6.2 and pCMVNot6.2-ΔF containing expressible human CFTR cDNAs were the generous gift of Dr. Johanna Rommens (The Hospital For Sick Children, Toronto). Two anti–human (COOH terminus) CFTR antibodies were used: mouse monoclonal antibody 24-1 from Genzyme Diagnostics and rabbit polyclonal antibody R3194 (Zeng et al., 1997). Mouse monoclonal anti–rat Grp78 (BiP) was obtained from StressGen Biotechnologies Corp. Monoclonal anti-ubiquitin and anti–γ-tubulin antibodies and rhodamine-conjugated wheat germ agglutinin were purchased from Sigma Chemical Co. Mouse monoclonal anti-Hsp70 and anti-Hsp90 antibodies were from Affinity Bioreagents, Inc. Goat polyclonal anti-aldolase was from Biodesign International. Mouse monoclonal anti– lamin B1 antibody was from Zymed Laboratories, Inc. Mouse monoclonal anti–β-cop antibodies contained in media from secreting hybridoma cells were the generous gift of Dr. George Bloom (Department of Cell Biology, University of Texas Southwestern Medical Center). Fluorescein-conjugated concanavalin A was from Molecular Probes, Inc. Fluorescently labeled secondary antibodies were from Jackson ImmunoResearch Laboratories, Inc. Lactacystin was purchased from Calbiochem. All other materials were of the highest quality commercially available.
Generation and Characterization of Antisera
Polyclonal anti-PA28 (Ma et al., 1993), anti–PA28-α (Song et al., 1996), and anti-20S proteasome (Ma et al., 1994) were generated as described. Polyclonal antibodies were prepared in chickens against highly purified bovine PA700. Chicken IgY was purified from egg yolks of immunized birds. The antibodies specifically recognized multiple subunits of the PA700 complex, including p112, p97, p58, p56, and p45, upon Western blot analysis of crude cell lysates and could specifically immunoprecipitate PA700 from solution using an agarose bound anti-IgY antibody. Polyclonal antibodies were prepared in rabbits against an HPLC-purified peptide representing the 16 COOH-terminal amino acids of human p31 (Nin1p), a subunit of PA700 (Kominami et al., 1995).
Preparation of P205S Mutant Expression Construct
Oligonucleotide-directed mutagenesis as described (Andrews and Lesley, 1998) was used to generate the mutant CFTR from the parent expression vector pCMVNot6.2. In brief, mutants were selected based upon the incorporation of a second-site mutation in β-lactamase which alters its substrate specificity leading to resistance of transformed bacteria to cefotaxime and ceftriaxone in addition to ampicillin. The sequence of the mutagenic primer used to create P205S was 5′-CGTGTGGATCGCTTCTTTGCAAGTGGC-3′. Incorporation of the mutation was verified by DNA sequencing. Transfection-quality plasmid DNA was prepared using reagents supplied by Qiagen Inc.
Cell Culture and Transfection
Wild-type and mutant CFTR cDNAs were transfected into human embryonic kidney (HEK) 293 or HeLa cells (American Type Culture Collection) using the Fugene Mammalian Transfection Reagent (Boehringer Mannheim). Cell lines were maintained in DME supplemented with 10% FCS, 50 μg/ml streptomycin, and 50 units/ml penicillin.
Preparation of Centrosomes
Centrosomes were isolated from HEK 293 and HeLa cells by discontinuous gradient ultracentrifugation according to the method of Moudjou and Bornens (1998). In brief, cells in the exponential phase of growth were treated with 1 μg/ml cytochalasin D and 0.2 μM nocodazole for 1 h. Cells were collected by trypsinization and centrifugation and the resulting pellet was washed in TBS followed by 0.1× TBS/8% sucrose. Cells were resuspended in 2 ml of 0.1× TBS/8% sucrose followed by addition of 8 ml lysis buffer (1 mM Hepes, pH 7.2, 0.5% NP-40, 0.5 mM MgCl2, 0.1% β-mercaptoethanol, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, and 1 mM PMSF). The suspension was gently shaken and passed five times through a 10-ml narrow-mouth serological pipette to lyse the cells. The lysate was spun at 2,500 g for 10 min to remove swollen nuclei, chromatin aggregates, and unlysed cells. The resulting supernatant was filtered through a nylon membrane followed by addition of Hepes buffer and DNase 1 to a final concentration of 10 mM and 1 μg/ml, respectively, and incubated on ice for 30 min. The mixture was gently underlaid with 1 ml of 60% sucrose solution (10 mM Pipes pH 7.2, 0.1% Triton X-100, and 0.1% β-mercaptoethanol containing 60% [wt/wt] sucrose) and spun at 10,000 g for 30 min to sediment centrosomes onto the cushion. The upper 8 ml of the supernatant was removed and the remainder, including the cushion, containing the concentrated centrosomes was gently vortexed and loaded onto a discontinuous sucrose gradient consisting of 70, 50, and 40% solutions from the bottom, respectively, and spun at 120,000 g for 1 h. Fractions were collected and stored at –70°C before further analysis.
SDS-PAGE and Western Blotting
Density gradient fractions were diluted into 1 ml of 10 mM Pipes, pH 7.2, and centrosomes were sedimented at 14,000 rpm in a microfuge for 15 min at 4°C. Centrosome pellets were resuspended in Laemmli sample buffer containing 5% β-mercaptoethanol and electrophoresed on 10% SDS-PAGE gels. Proteins were transferred onto nitrocellulose membranes in the presence of Towbin transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol, pH 8.3). Membranes were blocked in TBS-T (Tris-buffered saline/0.1% Tween-20) containing 10% nonfat dry milk for 1 h and then incubated in fresh blocking buffer containing primary antibody at the desired concentration. Membranes were washed several times in TBS-T followed by incubation with the appropriate horseradish peroxidase–conjugated secondary antibody. Immunoreactive proteins were visualized with enhanced chemiluminescence.
Inhibition of 20S Proteasome
The proteolytic activity of the endogenous proteasomal pool was inhibited by the addition of the potent fungal product lactacystin. The inhibitor was added to 293 cells 48–72 h after transfection at a concentration of 10 μM. Treatment of cells was carried out for 2 to 12 h and was immediately followed by several washes with PBS before fixation for immunocytochemistry.
Immunocytochemistry
For immunofluorescent subcellular localization of CFTR (Lee et al., 1997), transiently transfected cells attached to glass coverslips were rinsed three times with PBS followed by fixation and permeabilization with 1 ml of ice-cold methanol for 10 min at –20°C. Upon removal of methanol, cells were again rinsed three times with PBS and incubated 10 min in 1 ml of PBS supplemented with 50 mM glycine (this and all subsequent manipulations were carried out at room temperature). This buffer was then removed and nonspecific sites were blocked with 0.1 ml of blocking medium (PBS supplemented with 5% goat serum, 1% BSA, and 0.1% gelatin) for 1 h in a humidified chamber. After blocking, the medium was replaced with 0.1 ml of blocking medium containing a 1/100 dilution of the respective primary antibody for 1 h. Cells were next washed three times with blocking medium and incubated for 1 h with blocking medium containing a 1/100 dilution of the appropriate fluorescently labeled secondary antibody. For double-labeling experiments, these primary and secondary incubations were repeated with antibodies against the second protein of interest. For lectin affinity staining involving either fluorescently tagged wheat germ agglutinin or concanavalin A, these reagents were added at either a 1/250 dilution with the secondary antibody, or at 1 μg/ml in PBS for 30 min before the initial fixation and permeabilization, respectively. Fluorescent images were obtained using a Bio-Rad MRC 1024 confocal microscope. Expression levels of cells expressing CFTR were divided into two categories, those which required a gain of 1,150 or higher to visualize (low expressers) and those which required a gain setting of 950 or less (high expressers). In each case the Iris was held constant at 3.5. CMV-driven CFTR expression levels can be categorized as either low or high. At the high expression level, some cells form CFTR inclusions without lactacystin treatment. This is observed for the folding mutants as well as for wild-type CFTR. This is likely due to an overload of the cellular folding machinery and may account, in part, for the low (∼30%) maturation efficiency observed for wild-type CFTR overexpressed in cultured cells (Ward and Kopito, 1994). We have therefore restricted our study to cells expressing low levels of CFTR.
Subcellular Morphometry
Due to periodic irregularities in the shape of the perinuclear structure, morphometric analysis describing their apparent diameter, as delineated by confocal imaging of immunostained HEK 293 cells, was limited to cells containing reasonably symmetric centrosomes. Measurements of diameter were performed using software supplied by the manufacturer. Three fields (×400) of each experiment were randomly selected and the diameters were measured from all the cells in each field. In the case of elliptically shaped structures, the geometric mean of the diameters were used. Data presented are reported as the mean ± SEM.
Analysis of Soluble and Insoluble Cellular Fractions
2.5 × 105 HeLa cells per 3-cm dish were either mock-transfected or transfected with P205S mutant CFTR expression plasmid. 48 h post-transfection, one of each was treated with 10 μM lactacystin for 12 h. Cells were washed with PBS, collected by trypsinization, and pelleted in a microfuge at 4°C. Each pellet was washed twice with PBS and resuspended in 100 μl PBS supplemented with Complete protease inhibitor cocktail (Boehringer Mannheim). After lysis by passage 10–20 times through a 27-gauge needle, lysates were spun at 14,000 g for 1 h at 4°C. The supernatants were collected and the pellets were washed in supplemented PBS followed by centrifugation at 14,000 g for 30 min. After aspiration of the wash solution, the pellets were resuspended in 100 μl supplemented PBS. Both pellets and supernatants were stored at –70°C until analysis. Equal volumes of each pellet and supernatant were analyzed by SDS PAGE through 4–20% gels followed by Western immunoblotting versus the indicated antibodies as described above.
Results
Concentration of Proteasome Machinery at the Centrosome
A central question with respect to proteasome-mediated degradation of misfolded integral membrane proteins is where in the cell does proteolysis occur. To address this question, we employed immunocytochemistry to delineate the subcellular distribution of several key components of the proteasome pathway including: the 20S proteasome, the PA700 and PA28 activator complexes, ubiquitin, and Hsp70.
In HEK 293 cells, antibodies directed against each of these proteasome components identified multiple distinct subcellular pools. The 20S proteasome (Fig. 1, A and D), the PA700 complex (Fig. 1, B and E), and ubiquitin (Fig. 1 G) are each clearly discernible in both a nuclear pool and a cytoplasmic pool. In contrast, PA28-associated immunofluorescence was identified mainly in the nucleus (Fig. 1, C and F), and Hsp70 was primarily found in a reticular pattern in the cytosol (Fig. 1 H). In addition to these general distributions, staining with each of these specific antibodies revealed the existence of a unique perinuclear site in which all of the studied components concentrate (Fig. 1, A–H). The structure is surrounded by but does not colocalize with staining against the ER lumenal chaperone BiP (Fig. 1 F), and is proximal to the Golgi apparatus as illustrated by staining with the lectin, WGA (Fig. 1, D and E). These same subcellular distributions were also observed in HeLa, COS, and CHO cells suggesting generality of the observation (data not shown).
Figure 1.
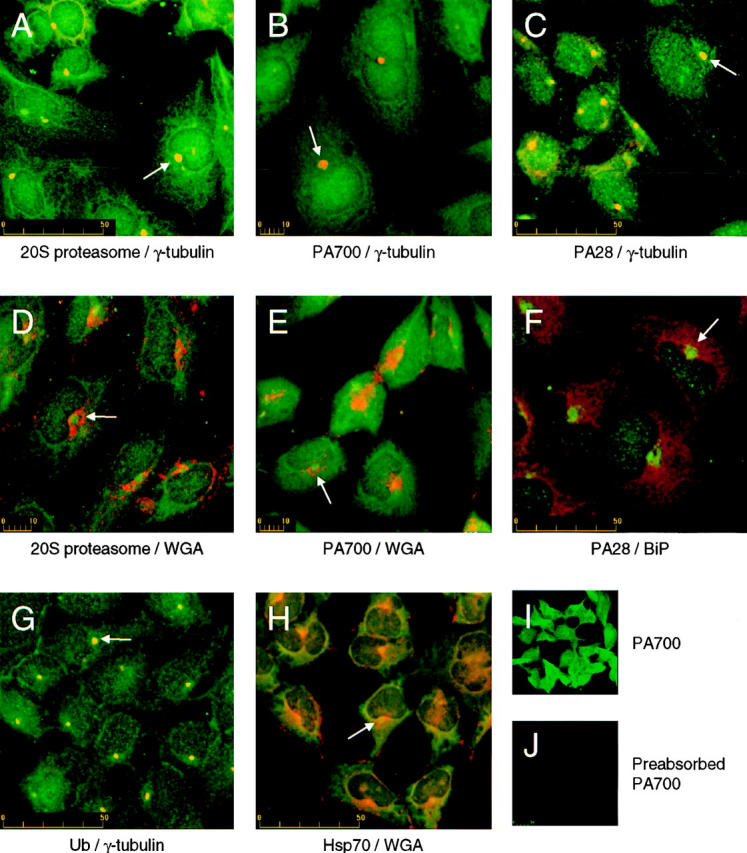
Localization of proteasome components at the centrosome. HEK 293 cells were immunostained with antibodies against (A and D) 20S proteasome and γ-tubulin (A) or fluorescently labeled WGA (D). (B and E) PA700 and γ-tubulin (B) or fluorescently labeled WGA (E). (C and F) PA28 and γ-tubulin (C) or BiP (F). (G) Ubiquitin (Ub) and γ-tubulin, and (H) Hsp70 and fluorescently labeled WGA. Confocal images were obtained as described under Materials and Methods. Specificity of antisera was investigated by staining HEK 293 cells with (I) polyclonal chicken anti-PA700, or (J) polyclonal chicken anti-PA700, preabsorbed at room temperature against purified bovine PA700. Confocal fluorescent images were obtained on the same day at identical magnification, iris settings, and gain. Note: For clarity of presentation in each panel, immunofluorescence of the proteasomal machinery, Hsp70, and Ub is consistently presented in green while the counter staining for γ-tubulin, WGA, and BiP is consistently presented in red. Arrows in panels A–H indicate locations of perinuclear proteasome-enriched centrosomes. Scale bars indicate size in μm.
Further investigation of this perinuclear structure using double-labeled immunofluorescence revealed exact colocalization of the 20S proteasome, PA700, PA28, ubiquitin, and Hsp70 with γ-tubulin, an established centrosomal marker (Mitchison and Kirschner, 1984; Oakley and Oakley, 1989). The majority of γ-tubulin staining was restricted to the centrosome, although an expected faint diffuse cytosolic staining was also observed corresponding to the soluble fraction of this protein. These data implicate the centrosome as a unique site for the colocalization and concentration of the proteasomal machinery and certain cell stress chaperones under basal conditions, suggesting a novel function for the centrosome.
The specificity of these proteasomal antibodies for immunocytochemistry is supported by several control experiments. First, using indirect immunofluorescence and confocal microscopy, staining with non- and preimmune rabbit sera or secondary antibody alone revealed virtually no detectable signal (data not shown). Second, preabsorption of chicken anti-PA700 antiserum against purified antigen completely eliminated immunocytochemical fluorescence (Fig. 1, I and J). Likewise, a similar preabsorption of rabbit anti-PA28 and Western blot analysis demonstrated the specificity of this antisera. And third, staining with anti-p31, a rabbit polyclonal antibody directed against a single subunit of PA700, yielded a staining pattern that is indistinguishable from that generated with the chicken anti-PA700 complex antibodies. Similarly, staining with polyclonal antisera raised against a peptide from the α subunit of PA28 (Song et al., 1996) was identical to that obtained with whole anti-PA28 (data not shown).
To confirm our immunocytochemical observations, centrosomes were purified from nocodazole/cytochalasin D–treated HEK 293 and HeLa cells by sucrose gradient ultracentrifugation and subjected to Western blotting (Fig. 2). Analysis of the final purification step revealed a peak of γ-tubulin immunoreactivity in fractions 3 through 6 corresponding to sucrose densities of between 50 and 60%. In agreement with the immunocytochemical data, 20S proteasome, p31 (PA700), PA28, and Hsp70 were observed in fractions containing γ-tubulin, indicating their copurification with the centrosome in the absence of an intact cytoskeleton. In addition, Hsp90 also copurified with γ-tubulin. Interestingly, Hsp70, Hsp90, and to a lesser extent PA28, were also observed in lighter gradient fractions, where faint γ-tubulin immunoreactivity was detected. This observation may be explained either by a heterogeneous population of centrosomes or lower-affinity binding of the chaperones and PA28 to the centrosome relative to that of 20S and PA700. Sucrose fractions were devoid of other subcellular markers, including BiP (ER), aldolase (cytosol), Lamin B1 (nucleus), and β-cop (Golgi), establishing the purity of the centrosome preparations.
Figure 2.

Proteasomal components and heat shock proteins cosediment with γ-tubulin on a sucrose gradient. Centrosomes were prepared from 7 × 107 293 cells and fractions were collected from a discontinuous sucrose gradient as detailed in Materials and Methods. Equal amounts of individual fractions were subjected to Western blot analysis. Fractions 1–10 are the gradient fractions collected from the bottom up, i.e., 70–30% sucrose. W indicates 10 μg of whole cell lysate as a control for each blot. BiP, aldolase, lamin B1, and β-cop, the markers for ER lumen, cytosol, nucleus and Golgi, respectively, are detected only in whole cell lysates, but not in centrosome fractions, establishing the purity of the centrosome prep. Sizes of each protein are denoted in kD.
Expansion of the Centrosome in Response to Proteasome Inhibition
Treatment of cells with lactacystin, a potent and specific inhibitor of the proteasome (Fenteany et al., 1995) resulted in a significant increase in size of the centrosome. Representative images of cells treated and untreated with lactacystin, were stained for PA28 and the lectin Con A (Fig. 3, A and B). Similar results were obtained for cells stained for 20S proteasome, Ub, PA700, Hsp70, and γ-tubulin. These results were quantified by morphometric analysis of centrosome diameters (identified by fluorescent staining for γ-tubulin or perinuclear proteasomal components). Comparison of lactacystin-treated and untreated HEK 293 cells revealed a twofold increase in mean diameter in response to the proteasome inhibition (Fig. 3 C). These data suggest that the centrosome is a dynamic structure, capable of expansion in response to inhibition of the proteasome. This expansion may be due to a build up of misfolded proteins, which would otherwise be degraded by the proteasome concentrated at the centrosome. To further test this hypothesis, we expressed in HEK 293 cells wild-type CFTR and two variants known to misfold, namely ΔF508 and P205S, and examined their effect on the centrosome and the proteasome machinery.
Figure 3.

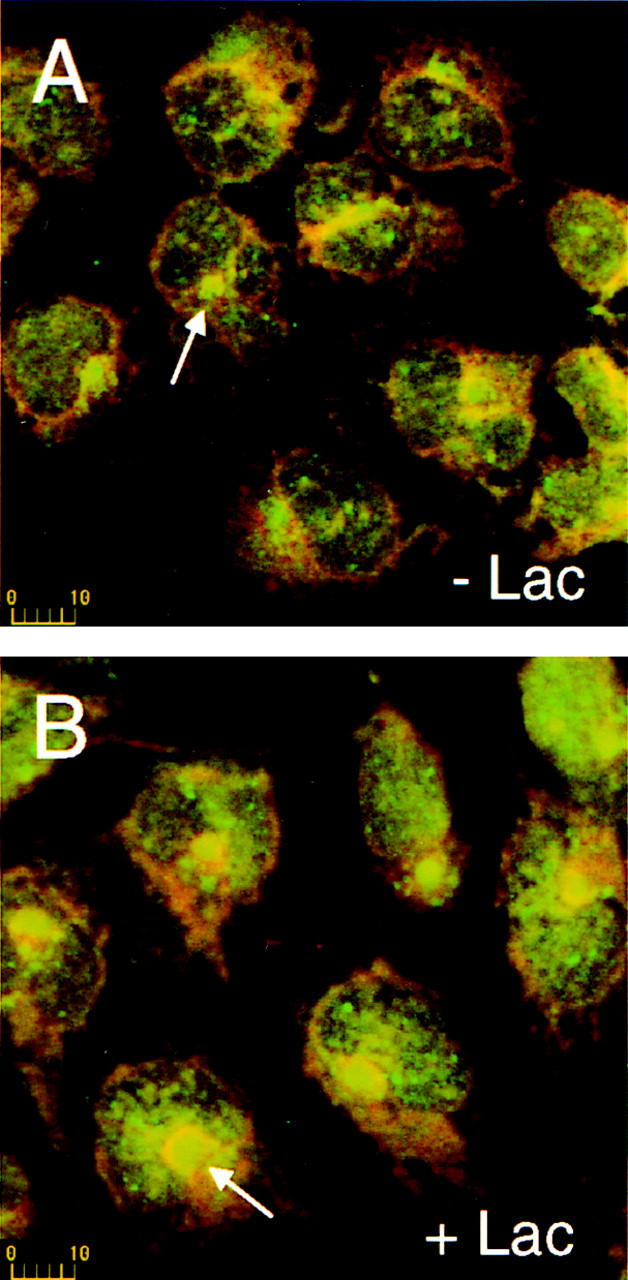
The centrosome expands in response to proteasome inhibition. (A and B) HEK 293 cells were stained with antibodies against PA28 (green) and fluorescently labeled Con A (red). Cells in B were treated with 10 μM lactacystin as described in Materials and Methods. Arrows in each panel indicate the location of representative centrosomes. Scale bars are in μm. (C) Morphometric measurements quantifying and comparing apparent centrosome diameter of lactacystin-treated (+) and control cells (−), as stained with antisera against the indicated proteins, is presented as the mean ± SEM of 9–16 determinations.
Expansion in Response to Expression of Mutant Protein
Consistent with previous reports (Cheng et al., 1990), immunolocalization of CFTR in transiently transfected HEK 293 cells clearly differentiates the wild-type CFTR subcellular locale from that of the folding mutants. Antiserum directed against the COOH terminus of CFTR recognized wild-type CFTR at the plasma membrane of transfected cells (Fig. 4 A) and at an early stage of maturation that colocalized with the ER-resident chaperone BiP (Fig. 4 B). The ER is the site of initial membrane translocation and integration of nascent polytopic membrane proteins. In sharp contrast, ΔF508 and P205S mutant CFTR are detected predominantly in the ER of transfected cells as illustrated by the ER pattern of CFTR staining (Fig. 4, C and E) and the complete colocalization with BiP staining (Fig. 4, D and F).
Figure 4.

Intracellular localization of CFTR by immunofluorescence. Transiently transfected HEK 293 cells expressing the indicated CFTR constructs were stained with rabbit polyclonal anti-CFTR followed by rhodamine-labeled goat anti–rabbit IgG, and mouse monoclonal anti-BiP followed by fluorescein-labeled donkey anti–mouse IgG. A and B, C and D, and E and F are pairs from the same fields. Scale bars indicate size in μm.
Cells expressing low levels of the wild-type CFTR do not significantly perturb either the PA28 distribution or size of the centrosome, although in highly expressing cells a fraction of CFTR colocalizes within this structure as seen in Fig. 5 A. Similar results were obtained with other proteasome components studied (data not shown). In striking contrast, expression of P205S (Fig. 5 B) or ΔF508 (data not shown) expands the centrosome in a manner similar to that observed when cells are treated with lactacystin alone (Fig. 3). This observation provides further support for the accumulation of misfolded proteins at the site of the centrosome even when the proteasome is functional.
Figure 5.

Centrosomes expand in response to misfolded protein. Transiently transfected HEK 293 cells expressing either wild-type or mutant P205S CFTR (as indicated) were stained with antibodies against CFTR (red) and either PA28 (A) or 20S proteasome (B) (each in green). Arrows indicate the location of centrosomes in cells expressing CFTR. Arrowheads indicate centrosomes in nonexpressing cells from the same fields for relative comparison. Scale bars are in μm.
Recruitment of Proteasomal Components to a Centrosome-Associated Inclusion
In cells overexpressing mutant CFTR (P205S) and treated with lactacystin, we observed the formation of large, perinuclear aggregates of misfolded CFTR which appear to arise from the centrosome as indicated by colocalization with γ-tubulin (Fig. 6). Identical results were observed with ΔF508 expressing cells (data not shown). In addition, a remarkable and comprehensive recruitment of the cytosolic pools of the 20S proteasome (Fig. 7 A), PA700 (B), ubiquitin (D), and Hsp70 (E) to the site of CFTR aggregate formation was observed. This recruitment is coincident with a depletion of the cytosolic pools implicating this as the source. PA28 is also recruited to the inclusion in response to the build up of misfolded CFTR (Fig. 7 C). Morphometric analysis of the CFTR aggregate formed in lactacystin treated cells is presented in Fig. 7 F. A composite of data from Fig. 3 E describing centrosome expansion due to proteasome inhibition alone relative to nontreated cells is included for comparison. The diameter of the aggregate formed in lactacystin-treated cells expands to four to six times the size of the centrosome from which it is apparently derived.
Figure 6.

Intracellular inclusions of misfolded proteins arise from the centrosome. Cells expressing moderate levels of CFTR-ΔF508 were treated with 10 μM lactacystin for 12 h and stained with (A and C) rabbit polyclonal anti-CFTR followed by fluorescein-labeled goat anti–rabbit IgG and (B and D) mouse monoclonal anti–γ-tubulin followed by rhodamine-labeled donkey anti–mouse IgG. A and B, C and D are pairs from the same fields. Arrows indicate γ-tubulin–visualized centrosomes centered at the sites of mutant CFTR aggregate formation. The scale bar indicates size in μm.
Figure 7.
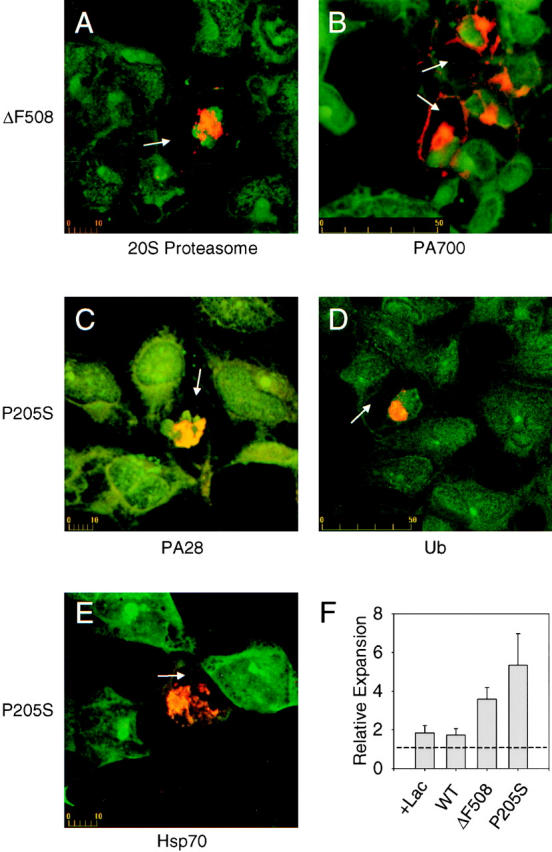
Proteasome machinery is recruited to CFTR aggregates. HEK 293 cells transiently transfected with the indicated CFTR expression construct were treated with 10 μM lactacystin as described in Materials and Methods. Cells were then stained with (A) mouse monoclonal anti-CFTR (red), and rabbit polyclonal anti-20S proteasome (green), (B) rabbit polyclonal anti-CFTR (red), and chicken polyclonal anti-PA700 (green), (C) mouse monoclonal anti-CFTR (red), and rabbit polyclonal anti-PA28 (green), (D) mouse monoclonal anti-CFTR (red), and rabbit polyclonal anti-ubiquitin (green), or (E) rabbit polyclonal anti-CFTR (red), and mouse monoclonal anti-Hsp70 (green). Arrows in each panel indicate cells whose cytosol has been cleared of detectable proteasomal components due to recruitment to centrosome-associated inclusions. A morphometric analysis of the relative expansion of the centrosome and CFTR aggregates is presented in panel (F). The expansion due to inhibition of the proteasome with lactacystin alone and lactacystin with low expression of wild-type, ΔF508, and P205S CFTR was determined from at least eight measurements. The dashed line represents the relative size of an untreated centrosome. Errors are ± SD. Scale bars indicate size in μm.
Next, to further investigate the observed redistribution of the proteasomal machinery and Hsp70 in response to the formation of lactacystin-induced intracellular inclusions of misfolded CFTR, we separated cell lysates into soluble and insoluble cellular fractions. In mock-transfected cells, γ-tubulin was observed primarily in the soluble fraction (Fig. 8). However, in cells expressing P205S mutant CFTR and treated with lactacystin, γ-tubulin was observed distributed between the soluble and insoluble fractions. PA28, PA700, and Hsp70 redistributed in a manner parallel to γ-tubulin (Fig. 8). An intermediate degree of redistribution was observed for mock-transfected cells treated with lactacystin and untreated mutant transfected cells (data not shown).
Figure 8.

Coredistribution of the centrosome, Hsp70, and the proteasome in response to inclusion formation. 2.5 × 105 HeLa cells expressing P205S mutant CFTR and treated with 10 μM lactacystin for 12 h and untreated mock-transfected control cells were lysed and separated into supernatant (S) and pellet (P) fractions as described in Materials and Methods. Fractions were separated by SDS-PAGE and analyzed by Western blotting versus each of the indicated antibodies.
Discussion
The work reported here describes the immunocytochemical (Fig. 1) and biochemical (Fig. 2) identification of the centrosome as a unique subcellular location in which, under basal conditions, components of the proteasome proteolytic pathway and certain relevant heat shock chaperones concentrate. This localization was observed under normal growth conditions in HEK 293, HeLa, COS, and CHO cells. Moreover, the association of the proteasome with the centrosome does not require an intact F-actin nor microtubular network as indicated by their presence in the purified centrosomal fractions obtained from nocodazole/ cytochalasin D–treated cells (Fig. 2), indicating that their localization at this site is not simply a result of clustering at the minus-ends of microtubules. This does not, however, exclude the possibility that the cytoskeleton may be required for trafficking to and from this location.
When the cellular level of misfolded protein is high, either due to the overexpression of a misfolded mutant protein (such as ΔF508 or P205S CFTR) or the inhibition of the proteasome, the cell responds by expanding the diameter of the centrosome up to twofold (Figs. 3 and 5). Assuming a spherical, three-dimensional shape, this would translate to more than a fourfold increase in its volume. Under high loads of misfolded substrate and/or insufficient proteasome activity, the centrosome, Hsp70, and the proteolytic machinery undergo correspondent redistribution to a sedimentable fraction (Fig. 8). The presence of the perinuclear proteasome concentration in control cells argues that the simplest explanation for this expansion involves the targeting of misfolded proteins to this centralized locale for rapid and efficient degradation. Consistent with this hypothesis, the centrosomal-associated proteasomal machinery is active (Fabunmi, R.P., W.C. Wigley, P.J. Thomas, and G.N. DeMartino, unpublished observations). When degradation is insufficient the misfolded proteins accumulate at and proximal to this site eventually forming a large inclusion. In light of this finding and the proximity of the centrosome to the Golgi and lysosomes, care should be taken when interpreting subcellular localization of overexpressed proteins.
It is particularly interesting that under the highest loads of misfolded protein, when the proteasome is inhibited and a mutant protein is overexpressed, the cell responds by extensively recruiting additional proteasomal machinery from the cytosolic pools to the centrosome-associated inclusion (Fig. 7). The function of the centrosome in nucleating and organizing microtubules (Mitchison and Kirschner, 1984) suggests the involvement of microtubule-based motors in the recruitment process. Consistent with this notion is the observation that the nuclear pools of proteasomal component remain unperturbed (Fig. 7). Further experiments will be required to determine if the recruitment is an active process or simply due to diffusion although the latter possibility is unlikely because molecules the size of the 26S proteasome are too large to simply diffuse through the cytosol (Janson et al., 1996).
Similar questions exist as to the targeting of the substrates to this site. While this manuscript was under review, an interesting and complementary study reported the formation of CFTR aggregates at the centrosomes (Johnston et al., 1998). Formation of the aggregates required an intact microtubular system implicating a motor-based translocation of substrate to the centrosome, analogous to the potential mechanism of proteasome recruitment suggested here. Interestingly, fibrillar extensions, as observed by CFTR and proteasomal staining (Figs. 6 A and 7), radiate from the centrosomally localized aggregates. These fibrils are sensitive to treatment with nocodazole suggesting their microtubular origin. However, the centrosomal localization of the aggregates and associated proteasomal components persist in lactacystin treated cells after nocodazole treatment. The mechanisms employed for assembly and, potentially, disassembly of these structures deserves further study. Regardless of the means of assembly, it is clear that this structure concentrates and recruits proteins that would be expected to perform a censor function by monitoring and perhaps controlling the balance between folding, degradation, and aggregation of nascent membrane proteins in the cell.
Understanding the composition and assembly of the proteasome-enriched centrosomes should provide new insight into the mechanisms of quality control employed by eukaryotic cells. For example, it is unclear if PA700, free or in complex with the 20S proteasome, participates directly in the recognition of the misfolded CFTR, altered localization, or the formation of the inclusions. However, it is possible that free PA700 uses its poly-Ub binding domains and other cues to identify substrates not only for the proteasome, but also for transport to the centrosome. Subsequently or coincidentally, ATP-dependent association with the 20S proteasome, PA700-mediated Ub isopeptidase activity, and other likely activities such as active unfolding would then serve to reexamine the substrate and perhaps, as an alternative to degradation, allow a second attempt at folding before degradation. Such iterative steps are known to occur in the Hsp60 class of chaperones (Bukau and Horwich, 1998) and the Clp family of proteases (Kessel et al., 1996).
The accumulation of overexpressed mutant CFTR at the centrosome upon inhibition of proteolysis suggests that it may serve as a terminal point in the pathway of misfolded polypeptides suggesting that they either assemble or nucleate at this location. It is interesting to note that similar inclusions (Russell, 1890; Valetti et al., 1991) have been previously observed in heat-stressed (Vidair et al., 1996), protease-inhibited cells (Wójcik et al., 1996; Johnston et al., 1998), and a growing family of pathologies related to protein misfolding such as Alzheimer's, Huntington's, amyotrophic lateral sclerosis, and type-1 spinocerebellar ataxia (SCA1) (Bruijn et al., 1998; Cummings et al., 1998; Sisodia, 1998). The detailed structural and functional relationships among these inclusions are unknown and warrants further investigation.
The centrosomal localization of the proteolytic machinery under basal conditions described in this study may play an important role in the degradation of proteins involved in progression through the cell cycle (King et al., 1996). In addition, this position also places the proteasome and chaperones proximal to the cellular organelles directly involved in the production, maturation, and trafficking of membrane proteins, a strategic locale for machinery involved in the recognition and processing of mutant proteins. Interestingly, recent studies in yeast, which lack the resolution of the current work, place the 26S proteasome at either the nuclear envelope-endoplasmic reticulum (Enenkel et al., 1998) or, in fission yeast, at the nuclear periphery at rest and in a nuclear-associated spot during meiosis (Wilkinson et al., 1998). Although the ER is generally considered the quality control organelle, based on BiP, WGA, and Con A staining, the current higher resolution studies indicate that in mammalian cells the proteasome concentrates in a compartment probably post-ER, lacking the complex glycoproteins of the Golgi. Dissecting the biochemical function of the centrosome should reveal if it is the site of proteasome-mediated degradation of misfolded and mutant membrane proteins. We are actively addressing these issues.
Acknowledgments
We thank the members of the Thomas, DeMartino, and Muallem Laboratories for advice and helpful discussions; George Bloom and Elena Kaznacheyeva for excellent technical advice; and Helen Yin and Bruce Horazdovsky for helpful comments.
This work was supported by research grants from the American Heart Association (9740033N) and National Institute of Arthritis, Diabetes, and Digestive and Kidney Diseases (NIDDK; DK49835) to P.J. Thomas, NIDDK (DK46181) to G.N. DeMartino, National Institute of Dental Research (NIDR; DE12309), and NIDDK (DK38938) to S. Muallem. P.J. Thomas is an Established Investigator of the American Heart Association.
Abbreviations used in this paper
- 20S
20S proteasome
- cAMP
adenosine 3′,5′-cyclic monophosphate
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- Hsp70/Hsp90
heat shock protein (70 and 90 kD, respectively)
- NBD
nucleotide binding domain
- PA700/ PA28
proteasome activator complex (700 and 180 kD, respectively)
- PKA
cAMP-dependent protein kinase
- SCA1
type-1 spinocerebellar ataxia
- TMD
transmembrane domain
- Ub
ubiquitin
- TBS-T
tris-buffered saline/0.1% Tween-20
References
- Adams GM, Falke S, Goldberg AL, Slaughter CA, DeMartino GN, Gogol EP. Structural and functional effects of PA700 and modulator protein on proteasomes. J Mol Biol. 1997;273:646–657. doi: 10.1006/jmbi.1997.1334. [DOI] [PubMed] [Google Scholar]
- Ahn J, Tanahashi N, Akiyama K, Hisamatsu H, Noda C, Tanaka K, Chung CH, Shibmara N, Willy PJ, Mott JD, et al. Primary structures of two homologous subunits of PA28, a γ-interferon-inducible protein activator of the 20S proteasome. FEBS Lett. 1995;366:37–42. doi: 10.1016/0014-5793(95)00492-r. [DOI] [PubMed] [Google Scholar]
- Andrews CA, Lesley SA. Selection strategy for site-directed mutagenesis based on altered β-lactamase specificity. Biotechniques. 1998;24:972–980. doi: 10.2144/98246st03. [DOI] [PubMed] [Google Scholar]
- Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz A, Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitrorequires the molecular chaperone Hsc70. J Biol Chem. 1997;272:9002–9010. doi: 10.1074/jbc.272.14.9002. [DOI] [PubMed] [Google Scholar]
- Bruijn S, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1853. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nature Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Dick T, Ruppert T, Groettrup M, Kloetzel P-M, Kuehn L, Koszinowski UH, Stevanovic S, Schild H, Rammensee H-G. Coordinated dual cleavage induced by the proteasome regulator PA28 lead to dominant MHC ligands. Cell. 1996;86:253–262. doi: 10.1016/s0092-8674(00)80097-5. [DOI] [PubMed] [Google Scholar]
- Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Purification of an 11S regulator of the multicatalytic protease. J Biol Chem. 1992;267:22369–22377. [PubMed] [Google Scholar]
- Enenkel C, Lehmann A, Kloetzel P-M. Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelope-ER network in yeast. EMBO (Eur Mol Biol Organ) J. 1998;17:6144–6154. doi: 10.1093/emboj/17.21.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Gray CW, Slaughter CA, DeMartino GN. PA28 activator protein forms regulatory caps on proteasome stacked rings. J Mol Biol. 1994;236:7–15. doi: 10.1006/jmbi.1994.1113. [DOI] [PubMed] [Google Scholar]
- Gregory RJ, Rich DP, Cheng SH, Souza DW, Paul S, Manavalan P, Anderson MP, Welsh MJ, Smith AE. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991;11:3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Hendil KB, Khan S, Tanaka K. Simultaneous binding of PA28 and PA700 activators to 20S proteasomes. Biochem J. 1998;332:749–754. doi: 10.1042/bj3320749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Janson LW, Ragsdale K, Luby-Phelps K. Mechanism and size cutoff for steric exclusion from actin-rich cytoplasmic domains. Biophysical J. 1996;71:1228–1234. doi: 10.1016/S0006-3495(96)79367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel M, Wu W, Gottesman S, Kocsis E, Steven AC, Maurizi MR. Six-fold rotational symmetry of ClpQ, the E. colihomolog of the 20S proteasome, and its ATP-dependent activator, ClpY. FEBS Lett. 1996;398:274–278. doi: 10.1016/s0014-5793(96)01261-6. [DOI] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters J-M, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1658. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Kominami K-I, DeMartino GN, Moomaw CR, Slaughter CA, Shimbara N, Fujimuro M, Yokosawa H, Hisamatsu H, Tanahashi N, Shimizu Y, et al. Nin1p, a regulatory subunit of the 26S proteasome, is necessary for activation of Cdc28p kinase of Saccharomyces cerevisiae. . EMBO (Eur Mol Biol Organ) J. 1995;14:3105–3115. doi: 10.1002/j.1460-2075.1995.tb07313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. ER quality control: the cytoplasmic connection. Cell. 1997;88:427–430. doi: 10.1016/s0092-8674(00)81881-4. [DOI] [PubMed] [Google Scholar]
- Lam YA, Xu W, DeMartino GN, Cohen RE. Editing of ubiquitin conjugates by an isopeptidase in the 26S proteasome. Nature. 1997;385:737–740. doi: 10.1038/385737a0. [DOI] [PubMed] [Google Scholar]
- Lee MG, Xu X, Zeng W, Diaz J, Wojcikiewicz RJH, Kuo TH, Wuytack F, Racymaekers L, Muallem S. Polarized expression of Ca2+channels in pancreatic and salivary gland cells. J Biol Chem. 1997;272:15765–15770. doi: 10.1074/jbc.272.25.15765. [DOI] [PubMed] [Google Scholar]
- Loo MA, Jensen TJ, Cui L, Hou Y-X, Chang X-B, Riordan JR. Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO (Eur Mol Biol Organ) J. 1998;17:6879–6887. doi: 10.1093/emboj/17.23.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268:533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- Ma C-P, Vu JH, Proske RJ, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a high molecular weight, ATP-dependent activator (PA700) of the 20 S proteasome. J Biol Chem. 1994;269:3539–3547. [PubMed] [Google Scholar]
- Ma C-P, Willy PJ, Slaughter CA, DeMartino GN. PA28, an activator of the 20S proteasome, is inactivated by proteolytic modification of its carboxyl terminus. J Biol Chem. 1993;268:22514–22519. [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Microtubule assembly nucleated by isolated centrosomes. Nature. 1984;312:232–237. doi: 10.1038/312232a0. [DOI] [PubMed] [Google Scholar]
- Moudjou, M., and M. Bornens. 1998. Method of centrosome isolation from cultured animal cells. In Cell Biology: A Laboratory Handbook. Academic Press, Inc., New York. 111–119.
- Oakley CE, Oakley BR. Identification of gamma tubulin, a new member of the tubulin superfamily encoded by mipA gene of Aspergillus nidulans. . Nature. 1989;338:662–664. doi: 10.1038/338662a0. [DOI] [PubMed] [Google Scholar]
- Qu BH, Strickland E, Thomas PJ. Cystic fibrosis: a disease of altered protein folding. J Bioenerg Biomem. 1997;29:483–490. doi: 10.1023/a:1022439108101. [DOI] [PubMed] [Google Scholar]
- Realini C, Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Molecular cloning and expression of a γ-interferon-inducible activator of the multicatalytic protease. J Biol Chem. 1994;269:20727–20732. [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Russell W. Address on a characteristic organism of cancer. Brit Med J. 1890;2:1356–1360. doi: 10.1136/bmj.2.1563.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Travis SM, Ishihara H, Welch MJ. Contribution of proline residues in the membrane-spanning domains of cystic fibrosis transmembrane conductance regulator to chloride channel function. J Biol Chem. 1996;271:14995–15001. doi: 10.1074/jbc.271.25.14995. [DOI] [PubMed] [Google Scholar]
- Sisodia SS. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? . Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- Song X, Mott JD, von Kampen J, Pramanik B, Tanaka K, Slaughter CA, DeMartino GN. A model for the quaternary structure of the proteasome activator PA28. J Biol Chem. 1996;271:26410–26417. doi: 10.1074/jbc.271.42.26410. [DOI] [PubMed] [Google Scholar]
- Thomas PJ, Qu B-H, Pedersen PL. Defective protein folding as a basis of human disease. Trends Biochem Sci. 1995;20:456–459. doi: 10.1016/s0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- Tsui I-C. The spectrum of cystic fibrosis mutations. Trends Genet. 1992;8:392–398. doi: 10.1016/0168-9525(92)90301-j. [DOI] [PubMed] [Google Scholar]
- Valetti C, Grossi CE, Milstein C, Sitia R. Russell bodies: a general response of secretory cells to synthesis of a mutant immunoglobulin which can neither exit from, nor be degraded in, the endoplasmic reticulum. J Cell Biol. 1991;115:983–994. doi: 10.1083/jcb.115.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidair CA, Huang RN, Doxsey SJ. Heat shock causes protein aggregation and reduced protein solubility at the centrosome and other cytoplasmic locations. Int J Hyperther. 1996;12:681–695. doi: 10.3109/02656739609027676. [DOI] [PubMed] [Google Scholar]
- Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degragation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Wenzel T, Baumeister W. Conformational constraints in protein degradation by the 20S proteasome. Nat Struct Biol. 1995;2:199–204. doi: 10.1038/nsb0395-199. [DOI] [PubMed] [Google Scholar]
- Wigley WC, Vijayakumar S, Jones JD, Slaughter C, Thomas PJ. The transmembrane domain of CFTR: design, characterization and secondary structure of synthetic peptides m1-m6. Biochemistry. 1998;37:844–853. doi: 10.1021/bi972293n. [DOI] [PubMed] [Google Scholar]
- Wilkinson CRM, Wallace M, Morphew M, Perry P, Allshire R, Javerzat J-P, McIntosh JR, Gordon C. Localization of 26S proteasome during mitosis and meiosis in fission yeast. EMBO (Eur Mol Biol Organ) J. 1998;17:6465–6476. doi: 10.1093/emboj/17.22.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójcik C, Schroeter D, Wilk S, Lamprecht J, Paweleta N. Ubiquitin-mediated proteolysis centers in HeLa cells: indication from studies of an inhibitor of the chymotrypsin-like activity of the proteasome. Eur J Cell Biol. 1996;71:311–318. [PubMed] [Google Scholar]
- Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kameyama K, Takagi T, Ikai A, Tokunaga F, Koide T, Tanahashi N, Tamura T, Cejka Z, Baumeister W, Tanaka K, Ichihara A. Molecular characterization of the “26S” proteasome complex from rat liver. J Struct Biol. 1993;111:200–211. doi: 10.1006/jsbi.1993.1050. [DOI] [PubMed] [Google Scholar]
- Zeng W, Lee MG, Yan M, Diaz J, Benjamin I, Marino CR, Kopito R, Freedman S, Cotton C, Muallem S, Thomas P. Immuno and functional characterization of CFTR in submandibular and pancreatic acinar and duct cells. Am J Physiol. 1997;273:C442–C455. doi: 10.1152/ajpcell.1997.273.2.C442. [DOI] [PubMed] [Google Scholar]