

Abstract
The transition of laminin from a monomeric to a polymerized state is thought to be a crucial step in the development of basement membranes and in the case of skeletal muscle, mutations in laminin can result in severe muscular dystrophies with basement membrane defects. We have evaluated laminin polymer and receptor interactions to determine the requirements for laminin assembly on a cell surface and investigated what cellular responses might be mediated by this transition. We found that on muscle cell surfaces, laminins preferentially polymerize while bound to receptors that included dystroglycan and α7β1 integrin. These receptor interactions are mediated through laminin COOH-terminal domains that are spatially and functionally distinct from NH2-terminal polymer binding sites. This receptor-facilitated self-assembly drives rearrangement of laminin into a cell-associated polygonal network, a process that also requires actin reorganization and tyrosine phosphorylation. As a result, dystroglycan and integrin redistribute into a reciprocal network as do cortical cytoskeleton components vinculin and dystrophin. Cytoskeletal and receptor reorganization is dependent on laminin polymerization and fails in response to receptor occupancy alone (nonpolymerizing laminin). Preferential polymerization of laminin on cell surfaces, and the resulting induction of cortical architecture, is a cooperative process requiring laminin– receptor ligation, receptor-facilitated self-assembly, actin reorganization, and signaling events.
Keywords: laminin, matrix assembly, muscular dystrophy, dystroglycan, integrin
Basement membranes are a subset of extracellular matrices made unique by their distinct composition and close association with selected cell surfaces. Laminin and type IV collagen polymers are thought to be key structural elements of the basement membrane and are usually accompanied by entactin/nidogen, perlecan, and other glycoproteins (65). The laminins are a family of large multifunctional molecules thought to be essential for the development and stability of these specialized matrices. All laminins are heterotrimers composed of individual α, β, and γ subunits. Laminins assemble into trimers by forming a long coiled-coil interaction in the COOH-terminal half of the protein, designated as the long arm, whereas the NH2-terminal regions remain independent and make up the three short arms. There are currently five laminin alpha chains, three beta chains, and three gamma chains that were found in at least 11 different heterotrimer combinations in vivo (37). Depending on the tissue or organ, different laminin subunits appear to be required at crucial stages of embryogenesis, as well as for normal function in mature tissues such as muscle and skin (36, 46, 50).
It is becoming clear that different laminins have both overlapping and unique functions. To understand the mechanisms underlying the requirement for so many laminins, it will be important to determine how these different laminins locally alter the function of basement membranes and exert effects on adjacent cell layers. For instance, it was shown that various laminin isoforms differ in their ability to form polymers. Laminin isoforms 1–4 are capable of forming three-dimensional polymers through interactions among their three NH2-terminal short arms, whereas laminins 5–7, with one or more truncated short arms, are not (7, 48). An independent laminin polymer in conjunction with the collagen IV network provides structure and support for adjacent cells (7, 64). Furthermore, laminins provide these cells with signals and cues through multiple cell surface receptor interactions, influencing processes such as cell survival, proliferation, and migration. However, the relationship and possible interplay between the structural role of laminin (i.e., its architectural state) and its many cell interactive functions has remained unexplored.
In vitro laminin polymerization was shown to be a reversible process in which each of the three short arms interacts to form the basic polymer bond (7). This nucleation/propagation assembly process occurs in solution and has a critical concentration of 70–140 nm (66). Although laminins and collagens both form polymers in solution, laminin polymers are generated through weaker interactions, forming a more plastic polymer better suited to dynamic processes such as tissue remodeling and mechanochemical signaling. Studies using laminin in solution have been useful for elucidating the domain requirements and kinetics of laminin polymerization, but alone cannot describe how the process occurs in a living organism. For instance, it was observed that basement membranes do not form in just any empty space between cells, but are found closely adjacent to particular cell types or even particular regions of a cell type, such as the basolateral surface of epithelial cells. In fact, basement membrane constituents may be synthesized by one cell type but end up in the basement membrane of another, more distal cell type (26). An earlier study found that as laminin attached to synthetic phospholipid membranes, it polymerized at concentrations well below the solution critical concentration (32). These results hinted at a mechanism by which laminin– receptor interactions would generate very high local surface concentrations of laminins, thereby driving it above its critical concentration on the plasma membrane surface. Supporting a role for receptor interactions in basement membrane assembly, several mice with mutations in cell surface receptors were found to have various basement membrane defects (17, 19, 29, 47, 58). Although these basement membrane defects were not characterized at the level of laminin polymer formation, and none of these receptors (α3 and β1 integrin subunits, and dystroglycan) exclusively binds laminin, together these studies strongly suggested that cells might use receptors to facilitate and target laminin polymerization.
Recently, attention has focused on the role of laminin and its interactions in skeletal muscle. Many severe congenital muscular dystrophies are caused by mutations in the LAMA2 gene and are marked by basement membrane defects in both muscle and the nervous system (46). In the muscle sarcolemmal basement membrane, laminin has been proposed to provide a linkage between the extracellular matrix and the dystroglycan–glycoprotein complex. In fact, mutations in virtually any component of the dystroglycan–glycoprotein complex can result in some form of muscular dystrophy, from Duchenne's (dystrophin) to muscular dystrophies of the Limb-Girdle type (sarcoglycans; ref. 14). Another binding partner for laminin in skeletal muscle is the α7β1 integrin, particularly at the myotendinous junction. Mutations in the α7 integrin subunit can also cause skeletal muscle pathologies, although these appear to be less severe and are classified as myopathies, not dystrophies (28, 35). Interestingly, several LAMA2 mutations result in the expression of partially functional laminin molecules that maintain their basement membrane localization (1, 42, 49, 55, 59). One such mutation occurs in the dy2J mouse, where the laminin-2 expressed by these mice has been found to be defective in its ability to polymerize, even though it remains localized to the basement membrane (Colognato, H., and P.D. Yurchenco, manuscript in preparation).
While these disorders have established an important role for basement membranes, the underlying mechanisms behind the requirement for laminins have remained unclear. In the case of the dy2J mouse, laminin remains linked to the cell through receptor interactions and to the basement membrane through its interaction with entactin/ nidogen. Therefore, it seems likely that laminin is required not only to provide an anchor to neighboring tissues, but to somehow alter these tissues, and we should consider that polymerization may play a role in this process. To address these issues it will be important to understand how the many and diverse functions of laminin might cooperate to promote matrix assembly and to provide mechanical and chemical signals to adjacent cells. An emerging paradigm is that the architecture itself transmits information to cells, through mechanisms such as matrix rigidity, spatial arrangement of cell receptors, and tension exerted between the matrix and receptor (8, 18, 20, 33, 51). The structural properties and multiple functions of the laminin molecule would make it well suited to modulate cell interactions through these mechanisms.
In this study, we have evaluated the effects that laminin, as a monomeric or polymeric ligand, has on surface receptors and cortical cytoskeletal components using a muscle cell culture system (60). We present evidence that laminin polymerization occurs preferentially on cell surfaces through specific receptor interactions, providing a targeting mechanism for the assembly of basement membranes. Furthermore, we show that laminin polymerization, above and beyond receptor occupancy, is required to induce reorganization of laminin, its receptors, and cytoskeletal components. This polymer-mediated rearrangement may represent a general mechanism used wherever basement membranes are assembling or remodeling. In fact, a specific disruption in this architecture may be the molecular basis for many congenital muscular dystrophies.
Materials and Methods
Cell Culture
C2C12 myoblasts (American Type Culture Collection) were maintained in DME (Gibco Laboratories) containing 10% FBS in a 37°C incubator with 5% CO2. Subconfluent myoblasts were trypsinized, transferred to 35 × 10 mm tissue culture dishes, and switched to fusion medium upon confluency (DME, 5% horse serum). Myotubes were analyzed 3–5 d after fusion. Cytochalasin D (Sigma Chemical Co.) was used at 0.4 μM and genistein (Calbiochem-Novabiochem Corp.) was used at 25 μM; both inhibitors were added to cultures 2 h before the addition of ligand.
Immunocytochemistry
Fused myotubes were incubated at 37°C with laminins or other proteins suspended in DME containing 0.5% BSA. Unattached protein was removed by washing four times with PBS containing 1 mM CaCl2. Cells were fixed with 3% paraformaldehyde for 10 min at room temperature and, when using antibodies against intracellular epitopes, permeabilized with PBS containing 0.5% Triton X-100 at 0°C. Cells were blocked for 30 min in PBS containing 0.5% BSA and 5% normal goat serum and incubated with primary antibodies diluted in wash buffer (PBS, 0.5% BSA, 0.5% normal goat serum) for 1 h at room temperature. After several washes, cells were incubated with FITC- or rhodamine-conjugated secondary antibodies for 1 h at room temperature. Cells were washed, coverslipped in 1,4 diazabicyclo[2.2.2]octane (Sigma Chemical Co.), and imaged using an Olympus IX-70 inverted fluorescent microscope and a cooled CCD camera (Princeton Instruments Micromax).
Antibodies
Rabbit polyclonal antibodies against mouse laminin-1, human laminin-2, laminin-1 proteolytic fragments E4, E8, and E3, and recombinant laminin α2-G domain were generated as described previously (13). Anti-E4, anti-E8, anti-E3, and anti-α2(G) were found to be highly specific to these regions of laminins after affinity purification and extensive cross-absorption. CY8 mouse monoclonal IgG ascites fluid raised against the α7 integrin subunit was provided by Dr. Randy Kramer (University of California, San Francisco, CA) and was diluted 1:500 for both blocking experiments and indirect immunofluorescence. IIH6 mouse monoclonal IgM raised against rabbit dystroglycan, and used at a 1:2 dilution of hybridoma medium, was provided by Drs. Hiroki Yamada and Kevin Campbell (Howard Hughes Medical Institute, University of Iowa, Iowa City, IA). The following antibodies were obtained commercially. A mouse mAb specific for the COOH-terminal region of β-dystroglycan was used at 1:25 (Novocastra Laboratories). A hamster monoclonal IgM specific for the β1 integrin subunit was used at 10–20 μg/ml for blocking experiments, and at 5 μg/ml for indirect immunofluorescence (PharMingen). Mouse monoclonal IgM specific for the central domain of dystrophin was used at 1:50 dilution (Upstate Biotechnology). Mouse monoclonal IgG specific for sarcomeric α-actinin was used at 1:800 dilution (Sigma Chemical Co.). Rabbit polyclonal IgG specific for human fibronectin was used at 1:400 dilution (Sigma Chemical Co.). Mouse monoclonal IgG specific for vinculin was used at 1:100 dilution (Sigma Chemical Co.). Rhodamine- and fluorescein-conjugated secondary antibodies specific for mouse IgG, mouse IgM, hamster IgM, and rabbit IgG were used at the recommended dilutions (Jackson Immunochemicals and Sigma Chemical Co.).
Proteins
Mouse laminin-1 was extracted from lathyritic EHS tumor and purified as described (63). A preparation of laminin-2 and -4 (both containing the α2 chain subunit) was prepared from human placenta as described (7), using a modification of a procedure developed previously (4). Defined fragments of laminin were prepared after digestion of laminin-1 with elastase or cathepsin G (63). Elastase fragments include: E1′, a partial complex of all three NH2-terminal short arms associated with a nidogen fragment; E4, β-chain short arm domains V and VI; E8, distal long arm coiled-coil region and proximal G domain (G repeats 1–3); and E3, distal G domain (G repeats 4 and 5). The cathepsin G fragment C8-9 is similar to the elastase fragment E8 but is larger, including most of the coiled coil. Recombinant laminin α-chain proteins α1(VI-IVb), α2(VI-IVb), and α2(G) were generated and purified as described previously (12, 13, 45). Human fibronectin was purchased from Sigma Chemical Co.
Protein Analysis
100 mM serine protease inhibitor p-aminoethylbenzenesulfonyl fluoride (AEBSF,1 HCl) was incubated overnight on ice with laminin and used to inactivate laminin self-assembly (see Fig. 4). AEBSF-treated laminin was dialyzed to remove free AEBSF and taken through two rounds of polymerization conditions (37°C, 3 h) to remove any potentially active laminins. [14C]AEBSF (American Radiolabeled Chemicals Inc.) had a specific activity of 55 mCi/mmol. For binding studies, [14C]AEBSF was incubated overnight on ice with laminin and dialysis was performed to remove unbound material. Laminin was digested with elastase (Boehringer Mannheim) using an enzyme/substrate ratio of 1:100 for 24 h at 25°C. Bound [14C]AEBSF was determined using PhosphoImaging (Bio-Rad GS-250; Bio-Rad Laboratories) on fragments separated by SDS-PAGE. Assays to evaluate laminin polymer formation and laminin cell adhesion were performed as previously described (7, 12).
Figure 4.

Generation of nonpolymerizing laminin using AEBSF. (A) Binding of [14C]AEBSF to laminin proteolytic fragments. Laminin incubated with [14C]AEBSF was treated with elastase to generate standard laminin elastase fragments (Fig. 2 A, fragment map of laminin). Total laminin digest was separated using SDS-PAGE and carbon-14 incorporation was determined by phosphoanalysis, shown here relative to fragment migration positions. Most [14C]AEBSF bound to E1′, a fragment shown to contain sites essential for polymerization. (B) AEBSF-treated laminin (open circles) and untreated laminin (closed circles) were evaluated for their ability to form a polymer in solution. Laminin was incubated for 3 h at 37°C and the polymer fraction was separated by centrifugation (see Materials and Methods). Untreated laminin showed concentration-dependent polymerization, whereas AEBSF-treated laminin remained in solution. (C) AEBSF-treated laminin (open circles) and untreated laminin (closed circles) were compared for their ability to support adhesion of C2C12 myoblasts. Both treated and untreated laminin supported adhesion and showed a similar dependence on substrate concentration. In addition, laminin fragments (E3, E8, and C8 and 9) treated with AEBSF bound to fused myotubes (not shown).
Results
Laminin Binds to the Myotube Surface and Forms a Network
To understand how laminin interacts with muscle cells, we evaluated laminins on C2C12 cells, a mouse myogenic cell line (60). When deprived of mitogens, these cells fuse and form long, multinucleated myotubes that spontaneously beat in culture (Fig. 1 f, phase micrograph). Fortunately, these cells appear to produce only low levels of endogenous laminin, so added laminin was monitored easily. We incubated fused myotubes with laminin-1 (α1β1γ1) and found that laminin bound to the dorsal myotube surface (Fig. 1). Initially (15 min), laminin binding was diffuse (Fig. 1, a and a′), but later (1 h) appeared aggregated into a reticular pattern on the surface (Fig. 1, b and b′). By 4 h, laminin organization on the cell surface appeared predominantly in a repeating, polygonal network with dimensions generally between 1 and 3 μm (see higher magnification in c′ for greater detail). In addition, these longer incubations revealed a transition from widespread coverage of laminin networks to more focal regions of laminin (d shows another myotube at 4 h). This transition was accompanied by the appearance of compact networks of laminin surrounded by zones with little or no laminin. These regions maintained the repeating substructure seen in more widespread laminin networks, but the entire architecture was somewhat more densely packed. Constant replenishment of the incubation medium with fresh laminin did not alter this progression from ubiquitous to more focal laminin coverage, suggesting that a change was occurring in the accessibility, composition, or activation state of laminin receptors. In addition, myotubes incubated in the presence of laminin for up to 24 h remained healthy and maintained focal laminin networks.
Figure 1.

Laminin binds to the myotube surface and forms a network. Laminin attached to the surface of C2C12 myotubes was visualized using polyclonal antibodies against specific laminin subunits followed by rhodamine-conjugated secondary antibodies. After incubation with 1 μg/ml laminin-1 for 15 min, laminin was seen on the dorsal myotube surface in a diffuse punctate pattern (a and a′). In 1 h, laminin was organized in a more aggregated, reticular pattern (b and b′). Myotubes incubated with 10 μg/ml laminin-1 (c, c′, and d) for 4 h showed an extensive repeating polygonal network of laminin on their surface (see c′ for a higher magnification view of this network, arrow depicts polygonal unit). At 4 h, many myotubes also contain regions cleared of laminin, with surface networks appearing in more compact clusters (d). Control myotubes incubated with BSA show little laminin staining (e). Fibronectin (10 μg/ml for 4 h) forms fibrillar structures primarily in regions containing myoblasts, not myotubes (f, phase micrograph, f′, antifibronectin). Bar, 5 μm.
Laminin Uses Its COOH-terminal Long Arm to Bind to the Myotube Surface
First, we sought to determine which regions of the large multisubunit laminin molecule interacted directly with the myotube cell surface (Fig. 2). A battery of laminin proteolytic fragments and recombinant laminin domains was evaluated to determine which domains bound directly to myotube cell surface receptors. Here we considered the binding properties of both α1- and α2-chain–containing laminins, shown together in a composite model (Fig. 2 A). This model depicts the boundaries of laminin fragments and some of the sites known to interact with receptors and extracellular matrix molecules.
Figure 2.
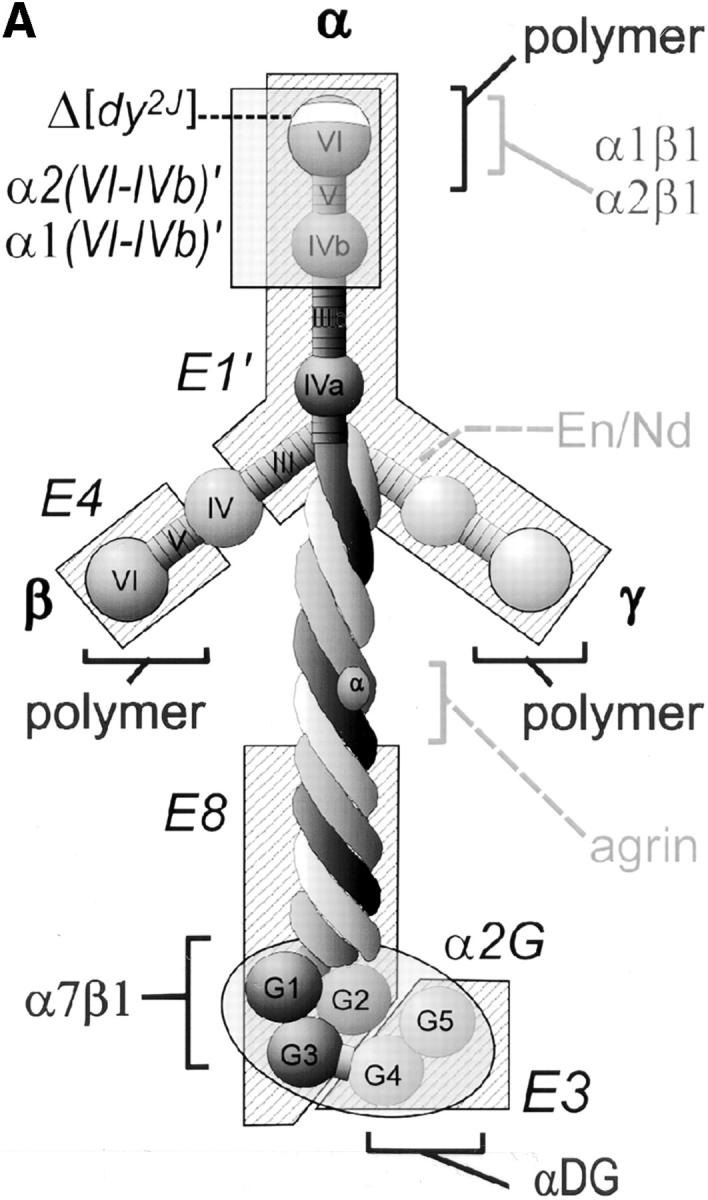

Laminin uses its COOH-terminal long arm to bind to the myotube surface. (A) Structure/function map of laminin proteolytic fragments and recombinant proteins, shown as a composite of features common to laminin-1 (α1β1γ1) and laminin-2 (α2β1γ1). Proteolytic fragments include: E1′, E4, E8, and E3. Recombinant proteins include: α1(VI-IVb), α2(VI-IVb), and α2(G). Receptor binding sites are: α1β1, α2β1, α7β1 integrin, and α-dystroglycan (αDG). Binding sites for extracellular matrix molecules: laminin polymer– forming regions (domains V and VI of α-, β-, and γ-short arms), entactin/nidogen (En/Nd), and agrin. Mutations used include the following: 57–amino acid region deleted in α2 chain of dystrophic dy2J mouse (Δdy2J). (B) Direct binding of laminin COOH-terminal long arm proteins to the myotube surface. Laminin-1 and laminin-2 bound myotubes and formed a reticular pattern after 1 h (insets). Recombinant laminin α-subunit proteins from the NH2-terminal short arm region (α1- and α2[VI-IVb]′) showed no detectable binding. COOH-terminal proteins, including proteolytic fragments E8 and E3, and recombinant α2-G domain protein showed widespread attachment to the myotube surface, but remained in a diffuse, punctate distribution. Insets show regions at two times the magnification.
We found that laminin proteins composed of distal COOH-terminal domains, but not those from NH2-terminal domains, were detected on the cell surface using indirect immunofluorescence (Fig. 2 B). The NH2-terminal short arm region from both the α1 and α2 laminin subunits (α1[VI-IVb] and α2[VI-IVb]) have been shown previously to possess active α1β1 and α2β1 integrin binding sites using PC12, HT1080, and primary rat Schwann cells (12). In contrast, we did not detect any significant level of binding between C2C12 myotubes and the α1 and α2 laminin short arms, indicating that fused muscle cells do not directly interact with these NH2-terminal domains. Also, we did not detect binding of short arm proteolytic fragments E1′ and E4 (data not shown). Proteolytic fragments E8 and E3, derived from the COOH-terminal long arm of mouse laminin-1 (34), bound to the cell surface, remained diffusely distributed, and had a punctate staining appearance. A recombinant laminin protein consisting of the entire α2 subunit G-domain (45) also bound to the surface and had a staining pattern similar to that of E3 and E8. Fragment E8 has been shown to mediate laminin binding to the α7β1 integrin (57), whereas fragment E3 has been shown to mediate binding to α-dystroglycan (22). Although both fragments bound to the cell surface, neither formed the large aggregates and networks seen with intact laminin-1 or laminin-2. One explanation for this difference could be that integrin and dystroglycan binding need to occur simultaneously, acting as coreceptors to stimulate surface aggregation. To test this possibility, we incubated cells with a mixture of E3 and E8. However, the staining pattern remained similar to that seen with individual fragments (not shown), indicating that occupancy of both receptor types was not sufficient to produce larger surface aggregates.
Next, we tested the ability of surface binding proteins or antibodies to block or alter the organized pattern of intact laminin on myotube surfaces (Fig. 3). First, cells were incubated for 1 h with COOH-terminal long arm fragments E3 or E8, or a combination of E3 and E8, or with blocking antibodies to each of the two principal receptors for laminin in mature muscle fibers, integrin and dystroglycan. Treatment with these reagents was followed by addition of intact laminin for 1 h. The cells were probed with antibodies specific for the NH2-terminal region of laminin, thereby only detecting intact laminin bound to the surface (i.e., not fragments E3 or E8). Surprisingly, function-blocking antibodies against the β1 integrin subunit or the α7 integrin subunit (not shown) had almost no effect on the ability of laminin to bind to and aggregate on myotube surfaces. These same antibodies completely blocked fragments E8 and C8-9 (larger than E8, fragment C8-9 also contains the agrin binding site) from binding to the myotube surface, demonstrating that these antibodies prevent α7β1 integrin binding interactions (not shown). In contrast, treatment with antibody that interferes with laminin– dystroglycan interactions (mAb IIH6) had a clear effect on laminin surface aggregation and network formation. Furthermore, fragment E3, which contains the dystroglycan binding site, significantly reduced laminin binding and aggregation on the myotube surface. Fragment E3 consistently had a greater blocking effect than antidystroglycan. One explanation may be that the E3 fragment contains additional, less prominent receptor binding sites. It is also possible that a partial or weak blocking effect exerted by this monoclonal IgM may be responsible for the difference. Nevertheless, a more prominent role in both direct binding and subsequent aggregation can be attributed to the E3-mediated interaction(s), compared with those mediated by E8. Finally, incubations combining fragment E3 with fragment E8 now completely blocked laminin from binding to the myotube surface. The extent of inhibition seen with single or multiple reagents persisted during longer laminin incubations: after treatment with laminin for 4 h, laminin network and cluster formation was significantly blocked by E3, very little by E8, and completely by a combination of the two (not shown).
Figure 3.

Blocking receptor interactions disrupts laminin surface networks. Laminin COOH-terminal proteins and/or receptor-blocking antibodies were used to compete for laminin binding sites. Myotubes were preincubated with the following proteins for 1 h: BSA (control), fragment E3, fragment E8, anti–β1 integrin blocking antibody, antidystroglycan (anti-DG), or a combination of fragments E3 and E8. Laminin incubated thereafter (5 μg/ml for 1 h) was visualized using an antibody specific for the NH2-terminal region of the laminin β1 subunit (anti-E4, which does not recognize COOH-terminal fragments E3 and E8). Blockade using fragment E3 alone resulted in a large decrease of laminin binding and network formation on the cell surface, whereas blockade of the long arm integrin binding sites using E8 or integrin blocking antibodies had minimal effect. Treatment with antibodies against α-dystroglycan disrupted laminin surface networks, to a lesser degree than blockade with fragment E3. Blockade of both E3- and E8-mediated cell recognition domains completely inhibited laminin binding to the surface. Bar, 10 μm.
Laminin Polymerization Is Necessary for Surface Networks to Form
To determine whether laminin–laminin polymer bonds were required to mediate laminin network formation on a cell surface, we used several methods to selectively analyze polymer formation. A nonpolymerizing laminin was generated by treating laminin with AEBSF, a serine protease inhibitor that we fortuitously found has the ability to bind covalently to laminin (Fig. 4). Binding specificity was determined by incubating laminin with [14C]AEBSF, followed by the standard elastase proteolysis conditions used to generate laminin fragments (34). Fig. 4 A shows the relative amount of isotope bound to various laminin elastase proteolytic fragments. We found that nearly all of the label was associated with E1′, a fragment consisting mostly of the α– and γ–NH2-terminal short arms and containing sites essential for laminin polymer formation (48, 63). In contrast, the COOH-terminal long arm fragments E3 and E8 bound little, if any, [14C]AEBSF. Although treatment with AEBSF abolished laminin's ability to form a polymer in solution (Fig. 4 B), its ability to interact with surface receptors (including α1β1, α2β1, α3β1, α6β1, and α7β1 integrins) in the cell types we have tested so far (HT1080, C2C12, 1072Sk fibroblasts, and primary Schwann cells) remained unchanged. Adhesion of C2C12 myoblasts to both untreated and AEBSF-treated laminin-1 is shown in Fig. 4 C. In addition, we found that the dystroglycan-binding fragment E3 retained its ability to interact with the muscle cell surface after treatment with AEBSF (not shown) and that the affinity of laminin for dystroglycan remained unchanged after AEBSF treatment (Combs, A., and J. Ervasti, personal communication). Essentially, this reagent produced a nonpolymerizing but otherwise functional laminin molecule, and allowed us to test directly the role of laminin polymer formation independent of its cell adhesive functions.
We found that nonpolymerizing AEBSF–laminin was unable to form aggregates and networks on the cell surface (Fig. 5, AEBSF-Lm). Conditions used to disrupt laminin polymer bonds selectively in solution (48) also were found to disrupt laminin cell surface organization, but had no effect on the binding of surface receptors (Fig. 5). Specifically, molar excess of either laminin short arm proteolytic fragments E1′or E4 prevented formation of laminin surface aggregates that appear after 1 h. Inhibition of polymerization also prevented development of laminin networks and clusters seen after longer incubations. After 4 h, polymerization-competent laminin appeared in both widespread networks and more compact clusters. However, AEBSF-treated laminin, or laminin blocked with fragment E1′ did not form these structures.
Figure 5.

Laminin polymer bonds are required for network formation on the cell surface. Laminins were treated with reagents that block laminin polymerization, but not cell adhesion activities. Myotubes were incubated with these polymer-deficient laminins for 1 h at 1 μg/ml (top) or for 4 h at 10 μg/ml (bottom) then evaluated for the presence of laminin aggregates. In the presence of short arm fragments E1′ and E4, laminin was unable to organize into reticular clusters in 1 h (untreated laminin shown at left). Similarly, AEBSF-treated laminin (Fig. 4) did not reorganize on the cell surface into aggregates. Formation of ordered laminin networks (at 4 h) was also blocked by polymer-disrupting reagents E1′ and AEBSF. Bar, 5 μm.
Laminin Polymerization Is Required for Induction of a Cortical Network of Receptors and Cytoskeletal Components
The organization of receptors and cortical cytoskeleton in the presence or absence of laminin was evaluated. Treatment of myotubes for 4 h with polymer-competent laminin resulted in the rearrangement of α7β1 integrin, dystroglycan, dystrophin, and cortical vinculin, but not α-actinin (Fig. 6). Laminin-treated myotubes were double-labeled to visualize the laminin network, sarcolemmal proteins (α7 and β1 integrin subunits, dystroglycan, vinculin, dystrophin, and α-actinin) and their overlapping distributions (Fig. 6, merge shown on left). Integrin (α7 and β1 subunits), dystroglycan, vinculin, and dystrophin formed repeating polygonal structures similar in appearance and location to those formed by laminin. Beneath the laminin network, both receptor types also codistributed (Fig. 6, dystroglycan and integrin double labeling). Myotubes incubated with BSA and those incubated with nonpolymerizing laminin are shown in the bottom two rows of Fig. 6. The organization of β1 integrin, dystroglycan, vinculin, and dystrophin was very different in these myotubes, appearing diffusely distributed with regions of small clusters. Therefore, laminin that could engage receptors, but could not form surface polymers, was insufficient to induce receptor and cytoskeleton rearrangements.
Figure 6.
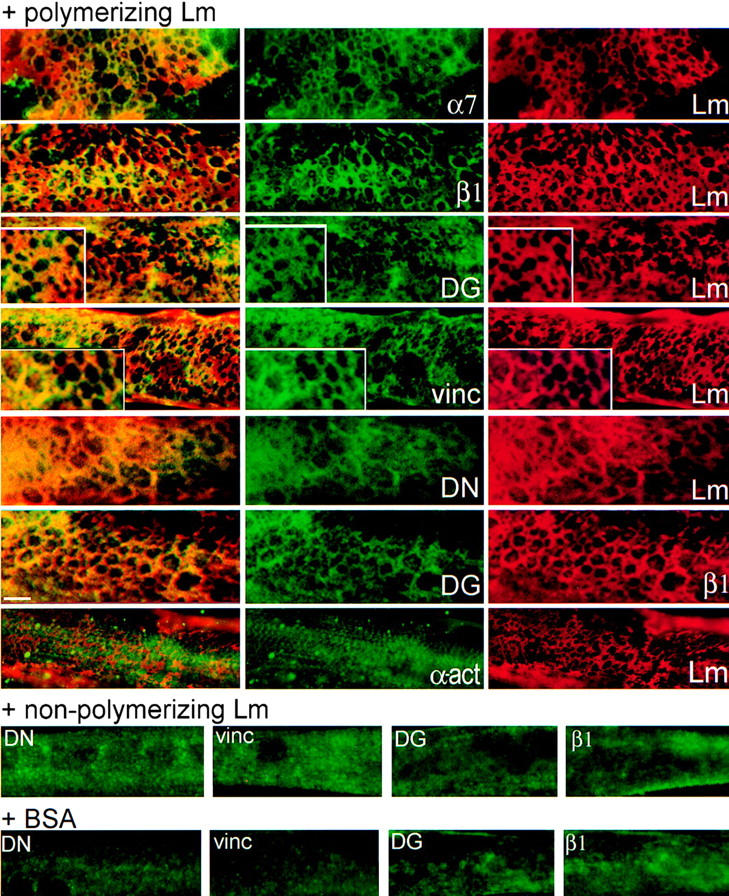
Laminin polymerization induces a selective reorganization of cell surface receptors and components of the cortical cytoskeleton. Myotubes were incubated with 10 μg/ml laminin-1 for 4 h, followed by double indirect immunofluorescence to visualize sets of bound laminin and various sarcolemmal proteins: Lm, laminin; α7, α7 integrin subunit; β1, β1 integrin subunit; DG, dystroglycan; vinc, vinculin; DN, dystrophin; α-act, α-actinin. Merged images of paired sets are shown to the far left in yellow. Sarcolemmal proteins in myotubes treated with BSA or with nonpolymerizing (AEBSF-treated) laminin are shown in the bottom two rows of images. In the presence of laminin that can polymerize, receptors and cytoskeletal proteins reorganize into a similar polygonal pattern, colocalizing with each other and with laminin. Bar, 5 μm; insets are twice the magnification.
Actin Reorganization and Tyrosine Phosphorylation Are Required for the Formation of Laminin Cell Surface Networks
The architecture of the laminin polymer, with its ∼35-nm strut meshwork, can be appreciated only at the level of electron microscopy. While triggered by polymerization, the organized semi-regular appearance of the laminin surface network suggested that creation of this architecture might be an active process perhaps requiring remodeling of the cortical actin network, or even signaling events. We investigated the role of actin by comparing the organization of laminin on myotubes incubated in the presence or absence of cytochalasin, an agent that prevents assembly and disassembly of filamentous actin (Fig. 7). In contrast to control myotubes, cytochalasin-treated myotubes did not show prominent rearrangement of surface laminin into complex networks, even after 4 h. On cytochalasin-treated myotubes, laminin coverage remained widespread and had a near continuous, almost mattelike quality. Inspection at higher magnification (Fig. 7, insets) revealed a delicate substructure to regions of this laminin covering, one that was very different from the larger, more sharply delineated polygonal units seen in control cultures. In addition, the clearance of laminin from large regions of the myotube surface was inhibited.
Figure 7.

Inhibition of actin reorganization or tyrosine phosphorylation prevents formation of laminin surface networks. Myotubes were preincubated with either cytochalasin, an agent that interferes with assembly and disassembly of actin filaments, or genistein, an agent that blocks tyrosine phosphorylation. Treated and untreated myotubes were incubated with 5 μg/ml laminin and evaluated for the presence of laminin surface networks after 4 h. Cytochalasin and genistein both prevented characteristic laminin surface networks from forming and disrupted laminin reorganization and clearance. Bar, 5 μm; insets are twice the magnification.
Next, we evaluated whether energy-dependent signaling cascades might be required to promote laminin network formation on the cell surface. We found that genistein, a general inhibitor of protein tyrosine kinases, had a large effect on laminin's ability to form a surface network (Fig. 7). On genistein-treated myotubes, laminin coverage appeared ubiquitous and without much complexity, similar to that seen on cytochalasin-treated cells. In contrast, several other inhibitors of signaling molecules had virtually no effect on laminin network formation (not shown). These were wortmannin, an irreversible inhibitor of phophatidylinositol 3-kinase, and ML-7 hydrochloride, a selective inhibitor of myosin light chain kinase that is often used to interfere with actin–myosin contractility. The observed requirement for actin remodeling suggested a role for the Rho family of small GTPases (27). To address this possibility, we used C3 transferase, a selective inhibitor of Rho, but this treatment was found to disrupt myotube adhesion and structure grossly. Therefore, it could not be used to evaluate the role of Rho-mediated signaling in laminin surface network formation.
Laminin Expressed by dy2J/dy2J Mice Is Defective in Forming Surface Networks
Mice homozygous for the dy2J LAMA2 allele were found to have a point mutation within the 5′ coding region that causes an abnormal splicing event (55, 59). This genetic defect causes a severe and progressive muscular dystrophy thought to be analogous to the many human congenital muscular dystrophies caused by LAMA2 mutations. Because of irregular splicing, dy2J/dy2J mice express a mutated laminin α2 chain lacking 57 amino acids within domain VI, the most NH2-terminal short arm domain (see laminin map, Fig. 2 A). Nonetheless, this aberrant laminin subunit forms a heterotrimer with the β and γ chains, is secreted, and is localized to the sarcolemmal basement membrane.
Subsequently, it has been found that the α2-chain–containing laminins (a combination of laminins-2 and -4) from the skeletal muscle of dy2J/dy2J mice is defective in its ability to form a polymer in solution (Colognato, H., and P.D. Yurchenco, manuscript in preparation). This polymerization defect most likely accounts for the dystrophy seen in these mice since receptor recognition sites in the laminin α2 domain VI do not seem to be used in mature skeletal muscle. In vivo, this mutant laminin appears to be held within the basement membrane solely through linkage to the collagen IV network, since we find that digestion of muscle tissue with collagenase liberates dy2J-laminin into solution. Normally, muscle laminin requires an additional treatment with EDTA to disrupt the calcium-dependent laminin polymer.
In Addition to Providing Insight into the Mechanism of LAMA2 Muscular Dystrophies, dy2J-Laminin Provides an Opportunity to Study a Naturally Occurring, Polymer-defective Laminin
We purified α2-laminin extracted from wild-type and dy2J/ dy2J muscle and evaluated the ability of these laminins to attach to muscle cell receptors and organize into a surface network (Fig. 8 A). Myotubes incubated for 4 h with wild-type mouse α2-laminin showed extensive formation of laminin surface networks. The inset at higher magnification (3×) clearly shows the characteristic laminin network architecture seen with laminin-1. On average, the spacing of repeating polygonal units was often more compact when compared with that of laminin-1, but the unit size range was similar for both. In contrast, dy2J–α2-laminin bound to the myotube surface but did not form these organized structures, remaining in a diffusely distributed punctate pattern. This pattern was similar to that seen with nonpolymerizing laminin-1 (Fig. 5).
Figure 8.

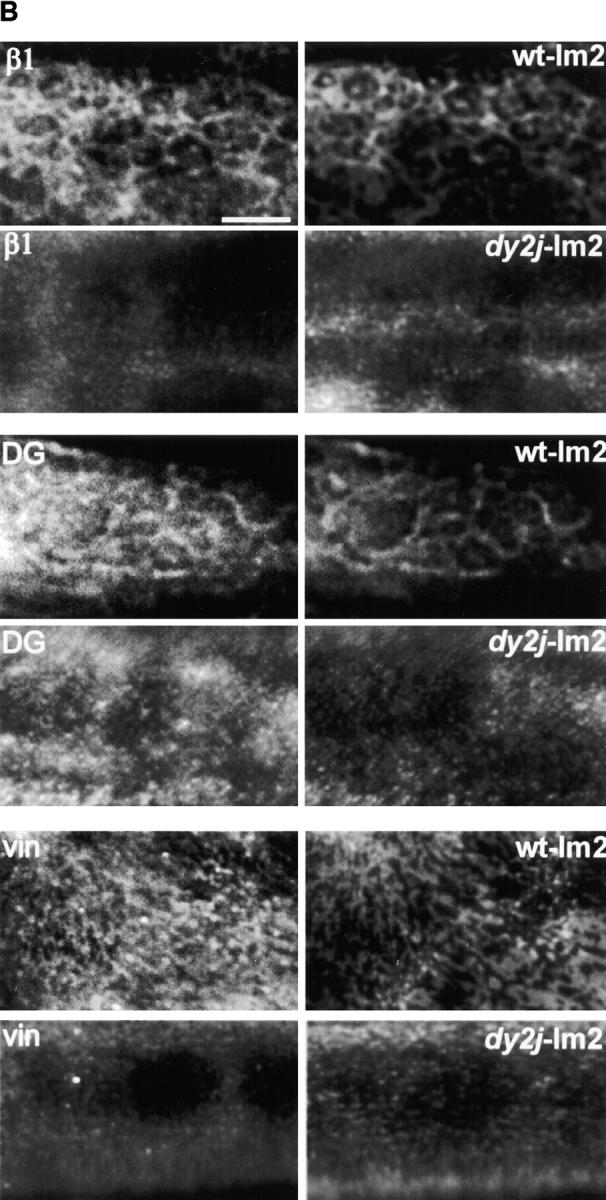
Polymer-defective laminin from dy2J/dy2J mice does not form surface networks and does not induce sarcolemmal reorganization. (A) The top frame shows an SDS-PAGE of laminin from skeletal muscle of +/+ and dy2J/dy2J mice. Proteins extracted from mouse skeletal muscle were separated by SDS-PAGE under reducing conditions and visualized with Coomassie blue. The arrow depicts dy2J laminin α2 subunit, roughly 25 kD smaller than native α2. Bottom frame: dy2J–α2-laminin binds to the myotube surface but fails to form networks. On myotubes incubated for 4 h, α2-laminin from wild-type mice formed networks (+/+), whereas dy2J–α2-laminin remained in a punctate, diffusely distributed pattern (dy2J/dy2J). Bar, 5 μm; insets, three times the magnification. (B) dy2J–α2-laminin does not induce reorganization of sarcolemmal proteins into a cortical network. Myotubes were incubated for 4 h with laminin from either wild-type (wt-lm2) or dy2J/dy2J mice (dy2j-lm2) and costained to visualize laminin α2 subunit and b1 integrin subunit (β1), dystroglycan (DG), or vinculin (vin). α2-Laminin induces a reorganization of integrin, dystroglycan, and vinculin, whereas dy2J–α2-laminin does not. Bar, 10 μm.
Polymer-defective dy2J–α2-Laminin Fails to Induce Reorganization of Receptors and Cortical Cytoskeletal Proteins
Myotubes were incubated with either wild-type or dy2J– α2-laminin and visualized by indirect immunofluorescence. Double staining revealed the location and relationship of laminin and the following sarcolemmal proteins: β1 integrin subunit, dystroglycan, and vinculin (Fig. 8 B). Induction of corresponding integrin, dystroglycan, and vinculin networks was observed in the presence of wild-type α2-laminin, but dy2J–α2-laminin failed to induce this reorganization. As in the presence of nonpolymerizing laminin-1, laminin receptors remained in a diffusely distributed, punctate pattern, as did vinculin. In addition, the intensity of vinculin staining at the cortical region appeared to be greater in myotubes incubated with polymerizing laminin-1 or -2, suggesting that vinculin was being recruited into the cortex and reorganized.
Discussion
The extracellular matrix molecule laminin has been described previously as having two major, though independent, functions: (1) a ligand for cell surface receptors, mediating such processes as neurite outgrowth and prevention of apoptosis, and (2) a structural molecule that forms a polymer and is required for basement membrane architecture, providing mechanical support to adjacent cells. In this study, we have presented evidence that these two types of functions are in fact integrated, acting synergistically to reorganize the cell surface and adjacent cortical cytoskeleton (Fig. 9). It seems likely that this laminin-induced process represents a specific mechanism for the transmission of complex cellular signals. For these cells, this mechanism appears to be laminin-specific, not induced by the addition of fibronectin, collagen IV, or even by the addition of divalent antibodies to artificially cluster nonpolymerizing laminin (not shown). We have shown that laminin attaches to the cell surface while bound to dystroglycan, integrin α7β1, and possibly other receptors for the laminin long arm. The concentration of receptor-engaged laminin is thereby selectively increased, now exceeding the critical concentration for laminin self-assembly and driving polymerization preferentially on cell surfaces (receptor-facilitated self-assembly). Furthermore, through cooperative polymer and receptor interactions, laminin organizes into complex polygonal arrays on the cell surface. This appears to be an active process that requires remodeling of actin filaments and tyrosine kinase signaling. Laminin polymerization induces reorganization of laminin receptors, both integrin and dystroglycan, and elements of the cortical cytoskeleton, vinculin and dystrophin. Moreover, we find that a mutated laminin causing muscular dystrophy and dysmyelination in the dy2J/dy2J mouse is defective in its ability to form a surface network and to induce these specific changes in the sarcolemmal architecture.
Figure 9.

A model for receptor-facilitated laminin polymerization on the sarcolemmal plasma membrane. Receptor-facilitated self-assembly of laminin occurs through synergistic laminin–laminin polymer interactions and laminin–receptor interactions. On the muscle sarcolemmal membrane, the three NH2-terminal short arms of laminin mediate polymer interactions, whereas the COOH-terminal long arm of laminin mediates receptor binding. This basement membrane assembly process in turn mediates an organizational signaling, rearranging the cortical cellular architecture and assembling matrix, receptor, and cytoskeletal elements into a polygonal meshwork. We find that formation of this organized network is an actively driven cooperative process that requires laminin receptor–facilitated self-assembly, actin reorganization, and tyrosine phosphorylation.
Laminin at the Muscle Sarcolemma: Receptor Ligation Is Not Sufficient for Induction
In the past few years, numerous mutations in the LAMA2 gene have been identified as the cause of congenital muscular dystrophy (CMD, ref. 53). Furthermore, the hallmark of this emerging class of disorders is the accompaniment of peripheral and central nervous system abnormalities, indicating a requirement for this laminin beyond the skeletal musculature. Although the isolation of these laminin mutations offers a significant breakthrough in the understanding of these congenital disease processes, insight into the molecular mechanism has been elusive. Many identified mutations result in absence of the laminin α2 protein altogether, but may be accompanied to varying degrees by upregulation of additional laminin chains such as α4 and α5, whose degree of overlapping function remains unclear (43). Mutations resulting in expressed protein with specific functional deletions, a smaller group, may lead to a more selective analysis of the contributions of specific laminin functions. Since laminin isoforms have many overlapping functions, structure/function correlation may permit identification of laminin candidates that could be upregulated or expressed to correct α2-deficient CMDs, similar to the rescue of dystrophin-negative dystrophic mdx mice using the dystrophin homologue, utrophin (44, 56).
The dystrophic dy2J mouse has an abnormally spliced laminin α2 transcript resulting from a point mutation in a splice donor site (55, 59). The resulting α2 protein is expressed, incorporated into a laminin heterotrimer, and localized to the sarcolemmal basement membrane. This α2-chain short arm defect in domain VI is analogous to several human CMDs that express partially functional α2 proteins with defects in the NH2-terminal short arm (1, 11, 42). In this study, we find that fused muscle cells bind to laminin through receptor-binding sites in the COOH-terminal long arm, ∼110 nm away from the short arm region that is defective in polymer formation (Colognato, H., and P.D. Yurchenco, manuscript in preparation). This laminin remains within the basement membrane through interactions with cell surface receptors and through linkage to the type IV collagen network, the latter mediated by entactin/ nidogen. In this context, it seemed unlikely that the prime role of laminin polymerization is simply to dock laminin into the basement membrane and provide a mechanical tether. Therefore, we sought to understand how laminin polymerization might alter the muscle sarcolemma in such a way that could promote muscle function and, ultimately, survival. The novel laminin function that we report here seems a likely candidate: the induction of dramatic changes in organization of matrix, receptors, and cortical cytoskeletal components in response to laminin polymerization. This induction may underlie a key requirement for laminin polymerization in maintenance of proper cellular architecture and function.
A Role for Ligand Architecture above and beyond Receptor Occupancy
Signaling pathways mediated by some growth factors not only have been found to be dependent on ligand occupancy of receptors, but also on dimerization of these receptors (3, 9, 62). In a related concept, receptor aggregation, mediated by fibronectin fibrils, is thought to recruit a large collection of cytoskeletal components and signaling molecules to these sites (38, 61). For this process, it has been shown that ligand occupancy alone (provided by RGD peptide) activates only a small subset of the responses mediated by a fibrillar fibronectin network. Similar or related mechanisms might play a role in laminin signaling, although laminin differs significantly from fibronectin in its distribution, receptor partners, and mode of supramolecular assembly. For instance, fibronectin forms a fibrillar rather than a meshlike polymer and its assembly process does not occur in the absence of receptor interactions. This difference turned out to be helpful to our study, as we were able to evaluate laminin's polymerization process independent of receptor occupancy and assess the extent to which laminin-induced changes were polymer-induced.
Traditionally, basement membranes have been thought of as supportive substrates for adjacent cells that enable cell adhesion or migration. The data presented here suggest a more dynamic, instructive role for the supramolecular organization of laminin in the basement membrane. Evidence has been building in recent years for the idea that cellular architecture has the ability to transmit information. At the forefront of this paradigm has been the concept of tensegrity, a model for mechanotransduction in which cells respond to mechanical stresses and changes in the physical environment through a matrix receptor–cytoskeleton nuclear continuum (30). In support of this model, spatial arrangement of matrix molecules has been shown to mediate processes such as proliferation, apoptosis, and arrangement of cytoskeletal architecture (6, 31, 33, 40). In this study we have shown that a change in laminin architecture is able to instruct muscle cells, leading to changes in receptor and cytoskeletal architecture. Interestingly, preliminary data suggest that laminin polymerization may activate tyrosine phosphorylation above and beyond that seen with nonpolymerizing laminin (Hanus, C., and P.D. Yurchenco, unpublished observations).
Studies in which certain laminin domains have been masked or proteolytically altered also support the notion that laminin architecture has the ability to influence cellular responses (5, 25). In these studies, the modified laminin domains were completely distinct from those regions interacting with the cell receptor(s) in question. In a study done by Giannelli and colleagues, it was found that proteolytic cleavage in the short arm of the γ2 subunit transformed cell interactions mediated by the G-domain of laminin-5, converting a static adhesion activity to an invasive, migratory activity used by cancer cells. The mechanism for this functional switching remains unclear, but information transmitted through the architectural arrangement of these ligands may very well contribute.
Cooperativity between Laminin Polymerization and Laminin–Receptor Interactions
The relationship between laminin–receptor interactions and basement membrane formation has been examined in recent years. Targeted gene disruptions of the β1 integrin subunit and dystroglycan, both lethal to embryonic development, have indicated a requirement for receptors in the proper assembly and subsequent function of embryonic basement membranes (19, 52, 58). Furthermore, mutations in the α3, α6, and β4 integrin subunits cause defects in the epidermal basement membrane of the skin, often leading to epidermolysis bullosa (17, 24, 41). Teratomas formed by β1-null embryonic stem cells injected into normal mice formed aberrant basement membranes that appeared to develop and slough off of adjacent cells, often in multiple and abnormally thickened layers (2, 47). Dystroglycan-null embryos die after implantation after failing to develop a functional extraembryonic Reichert's membrane, allowing maternal blood into the yolk sac cavity. Examination of this specialized basement membrane revealed only occasional patchy regions of laminin and collagen IV staining instead of the continuous staining observed normally. Furthermore, recent work has shown that dystroglycan −/− embryonic stem cells grown in culture to form embryoid bodies also have defects in basement membrane assembly (29).
How does the removal of dystroglycan, or other laminin receptors, lead to such profoundly defective basement membranes? Other molecular bonds, such as laminin–integrin or laminin–nidogen-collagen IV, could presumably be sufficient to retain laminin within the basement membrane of dystroglycan-null cells. This suggests that the laminin– dystroglycan interaction may provide an inductive role to cells in addition to providing a simple mechanical linkage. The ability of dystroglycan binding fragment E3 to disrupt markedly not only laminin surface binding but also its surface assembly is consistent with this hypothesis. The large effect seen by inhibiting dystroglycan, versus integrin, interaction sites may be due to a larger reduction in laminin accumulation on the surface, or a greater mobility of dystroglycan through the lipid bilayer compared with integrin, but it could also indicate an inductive role specific to the laminin–dystroglycan interaction.
The laminin–dystroglycan relationship also has been examined in the context of neuromuscular junction formation. It has been found that AChR receptor clustering is mediated in part by neural agrin through a mechanism dependent on the tyrosine kinase receptor MuSK (15, 21). Agrin, which has laminin-G domainlike repeats, has been shown to bind to both laminin and dystroglycan (16, 23). It has been suggested that laminin and dystroglycan may each serve as reservoirs for agrin at the neuromuscular endplate, perhaps facilitating agrin's ability to induce AChR clustering. Moreover, recent work has illustrated a MuSK-independent AChR clustering activity that is activated by the addition of exogenous laminin (54). In fact, the laminin–dystroglycan interaction specifically was required for this laminin-induced AChR clustering to occur (39). However, an earlier study by Cohen and colleagues found that by adding laminin to Xenopus muscle cultures, small dystroglycan clusters are converted to larger more focal clusters but that these laminin-induced clusters were not accompanied by accumulation of AChR (10). Using the C2C12 muscle culture system, we do observe an increase in AChR clusters upon addition of laminin, but only a subset of these clusters colocalizes with laminin (micrographs not shown). It should be interesting to learn whether the receptor and cytoskeletal rearrangements induced by laminin polymerization play a role in signaling this MuSK-independent pathway for AChR clustering.
In summary, we present evidence that laminin polymerization induces a cortical cellular architecture comprised of matrix, receptor, and cytoskeletal elements. This process requires a unique synergism between laminin architecture–forming and receptor–ligating functions, mediated by distal NH2-terminal and COOH-terminal epitopes, respectively. Furthermore, we demonstrate that a mutant laminin responsible for CMD and nervous system defects is defective in its ability to form surface networks and subsequently induce receptor–cytoskeleton rearrangement. This organizational signaling induced by laminin introduces a novel mechanism by which the supramolecular configuration of basement membranes instructs adjacent cells and tissues.
Acknowledgments
We thank Ms. Ariana Combs and Dr. James Ervasti for evaluating the dystroglycan-binding activity of AEBSF-treated laminin. We thank Dr. Hiroki Yamada and Dr. Kevin Campbell (IIH6) and Dr. Randy Kramer (CY8) for their gifts of antibodies.
Abbreviations used in this paper
- AEBSF
p-aminoethylbenzenesulfonyl fluoride
- CMD
congenital muscular dystrophy
Footnotes
This project was supported by National Institutes of Health grants R01-DK36425 (to P.D. Yurchenco) and R01-AR38454 (to D.A. Winkelmann).
References
- 1.Allamand V, Sunada Y, Salih MA, Straub V, Ozo CO, Al-Turaiki MH, Akbar M, Kolo T, Colognato H, Zhang X, Sorokin LM, Yurchenco PD, et al. Mild congenital muscular dystrophy in two patients with an internally deleted laminin alpha2-chain. Hum Mol Genet. 1997;6:747–752. doi: 10.1093/hmg/6.5.747. [DOI] [PubMed] [Google Scholar]
- 2.Bloch W, Forsberg E, Lentini S, Brakebusch C, Martin K, Krell HW, Weidle UH, Addicks K, Faessler R. β1 integrin is essential for teratoma growth and angiogenesis. J Cell Biol. 1997;139:265–278. doi: 10.1083/jcb.139.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boni-Schnetzler M, Pilch PF. Mechanism of epidermal growth factor receptor autophosphorylation and high-affinity binding. Proc Natl Acad Sci USA. 1987;84:7832–7836. doi: 10.1073/pnas.84.22.7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown JC, Wiedemann H, Timpl R. Protein binding and cell adhesion properties of two laminin isoforms (AmB1eB2e, AmB1sB2e) from human placenta. J Cell Sci. 1994;107:329–338. doi: 10.1242/jcs.107.1.329. [DOI] [PubMed] [Google Scholar]
- 5.Calof AL, Campanero MR, O'Rear JJ, Yurchenco PD, Lander AD. Domain-specific activation of neuronal migration and neurite outgrowth-promoting activities of laminin. Neuron. 1994;13:117–130. doi: 10.1016/0896-6273(94)90463-4. [DOI] [PubMed] [Google Scholar]
- 6.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 7.Cheng YS, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem. 1997;272:31525–31532. doi: 10.1074/jbc.272.50.31525. [DOI] [PubMed] [Google Scholar]
- 8.Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 9.Cochet C, Kashles O, Chambaz EM, Borrello I, King CR, Schlessinger J. Demonstration of epidermal growth factor-induced receptor dimerization in living cells using a chemical covalent cross-linking agent. J Biol Chem. 1988;263:3290–3295. [PubMed] [Google Scholar]
- 10.Cohen MW, Jacobson C, Yurchenco PD, Morris GE, Carbonetto S. Laminin-induced clustering of dystroglycan on embryonic muscle cells: comparison with agrin-induced clustering. J Cell Biol. 1997;136:1047–1058. doi: 10.1083/jcb.136.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohn RD, Herrmann R, Sorokin L, Wewer UM, Voit T. Laminin alpha2 chain-deficient congenital muscular dystrophy: variable epitope expression in severe and mild cases. Neurology. 1998;51:94–100. doi: 10.1212/wnl.51.1.94. [DOI] [PubMed] [Google Scholar]
- 12.Colognato-Pyke H, O'Rear JJ, Yamada Y, Carbonetto S, Cheng YS, Yurchenco PD. Mapping of network-forming, heparin-binding, and alpha 1 beta 1 integrin-recognition sites within the alpha-chain short arm of laminin-1. J Biol Chem. 1995;270:9398–9406. doi: 10.1074/jbc.270.16.9398. [DOI] [PubMed] [Google Scholar]
- 13.Colognato H, MacCarrick M, O'Rear JJ, Yurchenco PD. The laminin alpha2-chain short arm mediates cell adhesion through both the alpha1beta1 and alpha2beta1 integrins. J Biol Chem. 1997;272:29330–29336. doi: 10.1074/jbc.272.46.29330. [DOI] [PubMed] [Google Scholar]
- 14.Culligan KG, Mackey AJ, Finn DM, Maguire PB, Ohlendieck K. Role of dystrophin isoforms and associated proteins in muscular dystrophy (review) Int J Mol Med. 1998;2:639–648. doi: 10.3892/ijmm.2.6.639. [DOI] [PubMed] [Google Scholar]
- 15.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 16.Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve–muscle basal lamina via laminin. J Cell Biol. 1997;137:671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiPersio CM, Hodivala-Dilke KM, Jaenisch R, Kreidberg JA, Hynes RO. α3β1 integrin is required for normal development of the epidermal basement membrane. J Cell Biol. 1997;137:729–742. doi: 10.1083/jcb.137.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ezzell RM, Goldmann WH, Wang N, Parasharama N, Ingber DE. Vinculin promotes cell spreading by mechanically coupling integrins to the cytoskeleton. Exp Cell Res. 1997;231:14–26. doi: 10.1006/excr.1996.3451. [DOI] [PubMed] [Google Scholar]
- 19.Fassler R, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- 20.Felsenfeld DP, Choquet D, Sheetz MP. Ligand binding regulates the directed movement of beta1 integrins on fibroblasts. Nature. 1996;383:438–440. doi: 10.1038/383438a0. [DOI] [PubMed] [Google Scholar]
- 21.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 22.Gee SH, Blacher RW, Douville PJ, Provost PR, Yurchenco PD, Carbonetto S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin binding domain of laminin. J Biol Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- 23.Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan–alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 24.Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M. Absence of integrin alpha 6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- 25.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 26.Graham PL, Johnson JJ, Wang S, Sibley MH, Gupta MC, Kramer JM. Type IV collagen is detectable in most, but not all, basement membranes of Caenorhabditis elegansand assembles on tissues that do not express it. J Cell Biol. 1997;137:1171–1183. doi: 10.1083/jcb.137.5.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi YK, Chou FL, Engvall E, Ogawa M, Matsuda C, Hirabayashi S, Yokochi K, Ziober BL, Kramer RH, Kaufman SJ, et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet. 1998;19:94–97. doi: 10.1038/ng0598-94. [DOI] [PubMed] [Google Scholar]
- 29.Henry MD, Campbell KP. A role for dystroglycan in basement membrane assembly. Cell. 1998;95:859–870. doi: 10.1016/s0092-8674(00)81708-0. [DOI] [PubMed] [Google Scholar]
- 30.Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997;59:575–599. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- 31.Ingber DE, Prusty D, Sun Z, Betensky H, Wang N. Cell shape, cytoskeletal mechanics, and cell cycle control in angiogenesis. J Biomech. 1995;28:1471–1484. doi: 10.1016/0021-9290(95)00095-x. [DOI] [PubMed] [Google Scholar]
- 32.Kalb E, Engel J. Binding and calcium-induced aggregation of laminin onto lipid bilayers. J Biol Chem. 1991;266:19047–19052. [PubMed] [Google Scholar]
- 33.Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci USA. 1997;94:849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mann K, Deutzmann R, Timpl R. Characterization of proteolytic fragments of the laminin-nidogen complex and their activity in ligand-binding assays. Eur J Biochem. 1988;178:71–80. doi: 10.1111/j.1432-1033.1988.tb14430.x. [DOI] [PubMed] [Google Scholar]
- 35.Mayer U, Saher G, Fassler R, Bornemann A, Echtermeyer F, von der Mark H, Miosge N, Poschl E, von der Mark K. Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat Genet. 1997;17:318–323. doi: 10.1038/ng1197-318. [DOI] [PubMed] [Google Scholar]
- 36.Miner JH, Patton BL, Lentz SI, Gilbert DJ, Snider WD, Jenkins NA, Copeland NG, Sanes JR. The laminin alpha chains: expression, developmental transitions, and chromosomal locations of α1–5, identification of heterotrimeric laminins 8–11, and cloning of a novel α3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyamoto S, Akiyama SK, Yamada KM. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science. 1995;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- 39.Montanaro F, Gee SH, Jacobson C, Lindenbaum MH, Froehner SC, Carbonetto S. Laminin and alpha-dystroglycan mediate acetylcholine receptor aggregation via a MuSK-independent pathway. J Neurosci. 1998;18:1250–1260. doi: 10.1523/JNEUROSCI.18-04-01250.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mooney D, Hansen L, Vacanti J, Langer R, Farmer S, Ingber D. Switching from differentiation to growth in hepatocytes: control by extracellular matrix. J Cell Physiol. 1992;151:497–505. doi: 10.1002/jcp.1041510308. [DOI] [PubMed] [Google Scholar]
- 41.Niessen CM, van der Raaij-Helmer MH, Hulsman EH, van der Neut R, Jonkman MF, Sonnenberg A. Deficiency of the integrin beta 4 subunit in junctional epidermolysis bullosa with pyloric atresia: consequences for hemidesmosome formation and adhesion properties [published erratum appears in J. Cell Sci.1996. 109(8):preceding table of contents] J Cell Sci. 1996;109:1695–1706. doi: 10.1242/jcs.109.7.1695. [DOI] [PubMed] [Google Scholar]
- 42.Nissinen M, Helbling-Leclerc A, Zhang X, Evangelista T, Topaloglu H, Cruaud C, Weissenbach J, Fardeau M, Tome FM, Schwartz K, Tryggvason K, Guicheney P. Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am J Hum Genet. 1996;58:1177–1184. [PMC free article] [PubMed] [Google Scholar]
- 43.Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle-specific expression of a utrophin transgene rescues utrophin–dystrophin deficient mice. Nat Genet. 1998;19:79–82. doi: 10.1038/ng0598-79. [DOI] [PubMed] [Google Scholar]
- 45.Rambukkana A, Salzer JL, Yurchenco PD, Tuomanen EI. Neural targeting of Mycobacterium lepraemediated by the G domain of the laminin-alpha2 chain. Cell. 1997;88:811–821. doi: 10.1016/s0092-8674(00)81927-3. [DOI] [PubMed] [Google Scholar]
- 46.Ryan MC, Christiano AM, Engvall E, Wewer UM, Miner JH, Sanes JR, Burgeson RE. The functions of laminins: lessons from in vivo studies. Matrix Biol. 1996;15:369–381. doi: 10.1016/s0945-053x(96)90157-2. [DOI] [PubMed] [Google Scholar]
- 47.Sasaki T, Forsberg E, Bloch W, Addicks K, Fassler R, Timpl R. Deficiency of beta 1 integrins in teratoma interferes with basement membrane assembly and laminin-1 expression. Exp Cell Res. 1998;238:70–81. doi: 10.1006/excr.1997.3837. [DOI] [PubMed] [Google Scholar]
- 48.Schittny JC, Yurchenco PD. Terminal short arm domains of basement membrane laminin are critical for its self-assembly. J Cell Biol. 1990;110:825–832. doi: 10.1083/jcb.110.3.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sewry CA, Naom I, D'Alessandro M, Sorokin L, Bruno S, Wilson LA, Dubowitz V, Muntoni F. Variable clinical phenotype in merosin-deficient congenital muscular dystrophy associated with differential immunolabeling of two fragments of the laminin alpha 2 chain. Neuromuscul Disord. 1997;7:169–175. doi: 10.1016/s0960-8966(97)00425-2. [DOI] [PubMed] [Google Scholar]
- 50.Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–160. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stamenovic D, Fredberg JJ, Wang N, Butler JP, Ingber DE. A microstructural approach to cytoskeletal mechanics based on tensegrity. J Theor Biol. 1996;181:125–136. doi: 10.1006/jtbi.1996.0120. [DOI] [PubMed] [Google Scholar]
- 52.Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- 53.Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- 54.Sugiyama JE, Glass DJ, Yancopoulos GD, Hall ZW. Laminin-induced acetylcholine receptor clustering: an alternative pathway. J Cell Biol. 1997;139:181–191. doi: 10.1083/jcb.139.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sunada Y, Bernier SM, Utani A, Yamada Y, Campbell KP. Identification of a novel mutant transcript of laminin alpha 2 chain gene responsible for muscular dystrophy and dysmyelination in dy2Jmice. Hum Mol Genet. 1995;4:1055–1061. doi: 10.1093/hmg/4.6.1055. [DOI] [PubMed] [Google Scholar]
- 56.Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdxmice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 57.von der Mark H, Durr J, Sonnenberg A, von der Mark K, Deutzmann R, Goodman SL. Skeletal myoblasts utilize a novel beta 1-series integrin and not alpha 6 beta 1 for binding to the E8 and T8 fragments of laminin. J Biol Chem. 1991;266:23593–23601. [PubMed] [Google Scholar]
- 58.Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- 59.Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet. 1994;8:297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- 60.Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270:725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- 61.Yamada KM, Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr Opin Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- 62.Yarden Y, Schlessinger J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry. 1987;26:1443–1451. doi: 10.1021/bi00379a035. [DOI] [PubMed] [Google Scholar]
- 63.Yurchenco PD, Cheng YS. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J Biol Chem. 1993;268:17286–17299. [PubMed] [Google Scholar]
- 64.Yurchenco, P.D., and J.J. O'Rear. 1994. Basement membrane assembly. In Methods in Enzymology. Vol. 245. E. Ruoslahti and E. Engvall, editors. Academic Press, New York. 489–518. [DOI] [PubMed]
- 65.Yurchenco PD, Cheng YS, Colognato H. Laminin forms an independent network in basement membranes [published erratum appears in J. Cell Biol.1992. 118:493] J Cell Biol. 1992;117:1119–1133. doi: 10.1083/jcb.117.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yurchenco PD, Tsilibary EC, Charonis AS, Furthmayr H. Laminin polymerization in vitro. Evidence for a two-step assembly with domain specificity. J Biol Chem. 1985;260:7636–7644. [PubMed] [Google Scholar]