

Abstract
The cause of Huntington's disease (HD) is a pathological expansion of the polyglutamine domain within the NH2-terminal region of huntingtin. Neuronal intranuclear inclusions and cytoplasmic aggregates composed of the mutant huntingtin within certain neuronal populations are a characteristic hallmark of HD. Because in vitro expanded polyglutamine repeats are glutaminyl-donor substrates of tissue transglutaminase (tTG), it has been hypothesized that tTG may contribute to the formation of these aggregates in HD. Therefore, it is of fundamental importance to establish whether tTG plays a significant role in the formation of mutant huntingtin aggregates in the cell. Human neuroblastoma SH-SY5Y cells were stably transfected with truncated NH2-terminal huntingtin constructs containing 18 (wild type) or 82 (mutant) glutamines. In the cells expressing the mutant truncated huntingtin construct, numerous SDS-resistant aggregates were present in the cytoplasm and nucleus. Even though numerous aggregates were present in the mutant huntingtin-expressing cells, tTG did not coprecipitate with mutant truncated huntingtin. Further, tTG was totally excluded from the aggregates, and significantly increasing tTG expression had no effect on the number of aggregates or their intracellular localization (cytoplasm or nucleus). When a YFP-tagged mutant truncated huntingtin construct was transiently transfected into cells that express no detectable tTG due to stable transfection with a tTG antisense construct, there was extensive aggregate formation. These findings clearly demonstrate that tTG is not required for aggregate formation, and does not facilitate the process of aggregate formation. Therefore, in HD, as well as in other polyglutamine diseases, tTG is unlikely to play a role in the formation of aggregates.
Keywords: Huntington's disease, inclusions, polyglutamine, polar zipper, isopeptide bond
Introduction
Huntington's disease (HD) is an autosomal-dominant neurodegenerative disorder caused by pathological expansion of polyglutamine repeats in the NH2-terminal region of a 350-kD protein of unknown function called huntingtin (The Huntington Disease Collaborative Research Group 1993). Although mutant huntingtin is a fundamental cause of HD, the specific molecular mechanisms responsible for the selective neuronal degeneration remain to be elucidated. A pathological hallmark of HD brain is the presence of cytoplasmic and nuclear aggregates in specific neuronal populations that contain the NH2-terminal region of mutant huntingtin (DiFiglia et al. 1997). Further, protein aggregates have been reported in the brains of transgenic HD mouse models, as well as in transfected cell models of HD (Davies et al. 1997; Saudou et al. 1998; Persichetti et al. 1999; Yamamoto et al. 2000). Although it is clear that cytoplasmic and nuclear inclusions occur in HD, and in other polyglutamine diseases, the role of the inclusions in the pathogenesis of the disease remains inconclusive. Indeed, there have been reports indicating that aggregates may be beneficial (Klement et al. 1998; Saudou et al. 1998; Gutekunst et al. 1999), while others studies have concluded that the aggregates are likely to be toxic (Hackam et al. 1998; Hackam et al. 1999; Li 2000).
Two mechanisms have been proposed to explain how expanded polyglutamine domains form insoluble aggregates. It has been hypothesized that the expanded polyglutamine repeats may interact with each other through a polar zipper and thus contribute to aggregate formation (Perutz et al. 1994). Further, it has been hypothesized that tissue transglutaminase (tTG), perhaps in conjunction with the polar zipper mechanism, may be a contributing factor in the formation of these aggregates (Cooper et al. 1997, Cooper et al. 1999; Kahlem et al. 1996, Kahlem et al. 1998). Because it has been proposed that tTG may be a potential therapeutic target in the treatment of polyglutamine diseases (Igarashi et al. 1998), it is of critical importance to determine the contribution of tTG to the formation of inclusions.
The transglutaminases are a family of calcium-dependent enzymes that catalyze the formation of ε-(γ-glutamyl)lysine isopeptide bonds between substrate proteins, rendering the resulting cross-linked protein complexes insoluble (Folk 1983; Lorand and Conrad 1984; Greenberg et al. 1991). Transglutaminases also catalyze the incorporation of polyamines into substrate proteins (Lorand and Conrad 1984; Greenberg et al. 1991). Because the polypeptide-bound glutamine is the primary determining factor for a transglutaminase-catalyzed reaction, it has been hypothesized that increasing the number of glutamines in a protein beyond a certain threshold may result in the protein becoming a transglutaminase substrate (Green 1993). tTG is found within neurons (Miller and Anderton 1986; Appelt et al. 1996; Lesort et al. 1999) and is increased in specific areas affected in HD brain (Karpuj et al. 1999; Lesort et al. 1999). Further, the increase in tTG expression in HD brain occurred within neurons (Lesort et al. 1999). Previously, it had been shown that tTG levels in SH-SY5Y cells are significantly increased by treatment with retinoic acid, and further tTG can be activated by increasing intracellular calcium levels (Zhang et al. 1998). Even though it has been demonstrated that polyglutamine repeat domains (Kahlem et al. 1996; Cooper et al. 1997) and mutant huntingtin (Kahlem et al. 1998) are substrates for tTG in vitro, it has not yet been shown that huntingtin interacts with or is modified by tTG in situ. To determine the potential role of tTG in aggregate formation, SH-SY5Y cell lines stably expressing mutant or wild-type truncated NH2-terminal huntingtin constructs were established. Using these cells, we demonstrate that tTG and the mutant truncated huntingtin do not interact, and further huntingtin is not modified by tTG in situ. Moreover, immunocytochemical analysis revealed that tTG was totally excluded from the aggregates that form in the cells expressing the mutant huntingtin construct. Finally, transient transfection of the YFP-tagged mutant huntingtin construct into cells that do not express detectable levels of tTG due to stable transfection with an antisense tTG construct, resulted in significant aggregate formation. In addition, the formation of mutant huntingtin aggregates was equivalent in tTG antisense cells and in cells stably transfected with vector only. These data clearly demonstrate that tTG is unlikely to be a contributing factor to the formation of aggregates in HD brain.
Materials and Methods
Construction of Expression Plasmids
Expression constructs of truncated huntingtin with 63 amino acids, pcDNA3.1-N63-18/82Q-Myc/His, were created by subcloning a fragment of the huntingtin cDNA (bp 314–503) generated by PCR and included BamHI and XhoI restriction sites. The 189-bp product of interest was digested with BamHI and XhoI, and then subcloned into pcDNA3.1-/His A vector (Cooper et al. 1998). The BamHI–XhoI huntingtin cDNA fragments were also subcloned into the Amersham Pharmacia Biotech and XhoI sites of the pECFP-N1 vector (CLONTECH Laboratories, Inc.) (N-Q18) (pECFP-N1-18Q) or into the NheI and XhoI sites of the pEYFP-N1 vector (CLONTECH Laboratories, Inc.) (N-Q82) (pEYFP-N1-82Q). The BamHI and NheI restriction sites were filled in with T4 DNA polymerase before ligation. All DNA constructs were verified by automated sequencing.
Cell Culture and Generation of Stable Cell Lines
Human neuroblastoma SH-SY5Y cells were transfected by electroporation (Gene Pulser II; Bio-Rad Laboratories) according to the supplier's instructions. SH-SY5Y cells stably expressing pcDNA3.1 vector alone, N-Q82 (wild-type truncated huntingtin) or N-Q18 (mutant truncated huntingtin) were selected based on their resistance to G418, subcloned, and maintained on Corning dishes in RPMI 1640 medium supplemented with 20 mM glutamine, 10 U/ml penicillin, 100 μg/ml streptomycin, 5% fetal clone II serum, 10% horse serum, and 100 μg/ml G418 (GIBCO BRL). To differentiate the cells, the cells were grown in medium containing 1% fetal clone II serum, 5% horse serum, and 20 μM retinoic acid for 5 d. Previous studies have shown that treatment of SH-SY5Y cells with retinoic acid results in a significant increase in tTG expression (Zhang et al. 1998). Except where indicated, all studies were carried out on cells that were treated with retinoic acid and therefore express high levels of tTG (Zhang et al. 1998). All experiments were carried out on sub-confluent cultures.
Cell Viability Assay
To determine whether truncated mutant huntingtin decreases basal cell viability, LDH release was measured in the cell lines (Decker and Lohmann-Matthes 1988; Davis et al. 1997). There was no significant difference among groups, indicating no loss of cell viability due to the expression of the mutant huntingtin (data not shown). Further, there was no evidence of increased apoptosis in the mutant huntingtin-expressing cells as determined by the presence of condensed chromatin as detected by Hoechst staining (data not shown).
Antibodies
A rabbit polyclonal huntingtin antibody was prepared using a synthetic peptide composed of the first 17 amino acids of huntingtin protein and affinity purified using the antigen (a gift from Dr. P. Detloff, University of Alabama at Birmingham, Birmingham, AL) (Lin et al. 2001). This huntingtin antibody was made and purified by Research Genetics. The antigen and reactivity of this NH2-terminal huntingtin antibody are identical to the NH2-terminal huntingtin antibody described by Sawa et al. 1999. Mouse monoclonal antibodies to C-myc (Zymed Laboratories), tTG (TG100 and CUB7402) (Neomarkers), Hsp70 (StressGen Biotechnologies), and ubiquitin (Zymed Laboratories) were also used in this study.
Immunoblotting
To evaluate the expression levels of tTG and the huntingtin proteins in naive and differentiated cells, extracts from cells were prepared and quantitatively immunoblotted. Cells were harvested in cold PBS, collected by centrifugation, resuspended in a homogenizing buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.1 mM phenylmethylsulfonyl fluoride, and a 10 μg/ml concentration each of aprotinin, leupeptin, and pepstatin), and sonicated on ice. Protein concentrations of the homogenates were determined using the BCA method (Pierce Chemical Co.) and diluted to a final concentration of 2 mg/ml with 2× reducing stop buffer (0.25 M Tris-HCl, pH 6.8, 5 mM EDTA, 5 mM EGTA, 25 mM dithiothreitol, 2% SDS, 10% glycerol, and bromophenol blue as the tracking dye). Samples (30 μg of protein) were resolved on 4–20% gradient SDS-polyacrylamide gels, and transferred to nitrocellulose. Blots were blocked in 5% nonfat dry milk in TBST (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.05% Tween 20) for 1 h at room temperature. The blots were then incubated with the anti–tTG monoclonal antibody TG100 (1:750; Neomarkers) or with a polyclonal huntingtin antibody (1:20,000) in the same buffer overnight at 4°C. The membranes were then washed three times with TBST and incubated with HRP-conjugated goat anti–mouse IgG (1:2,000) for tTG, or with HRP-conjugated goat anti–rabbit IgG (1:2,000) for the polyclonal huntingtin antibody for 2 h at room temperature. The membranes were rinsed three times for 30 min with TBST, followed by four quick rinses with distilled water, and developed with the enhanced chemiluminescence method (Amersham Pharmacia Biotech).
Coimmunoprecipitation
Cell lysates were prepared in homogenizing buffer and samples containing 200 μg of protein were precleared for 1 h at 4°C with protein A-Sepharose (Amersham Pharmacia Biotech) before immunoprecipitation of huntingtin. Precleared samples were immunoprecipitated overnight at 4°C with the polyclonal NH2-terminal huntingtin antibody. To control for nonspecific binding, some samples were immunoprecipitated with nonimmune rabbit IgG. Protein A–Sepharose was added, and the incubation continued for 3 h at 4°C. The precipitates were washed three times with 1.0% NP-40, 50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, and a 10 μg/ml concentration each of aprotinin, leupeptin, and pepstatin, 50 μl of 1× reducing stop buffer was added to each sample and the samples were placed in a boiling water bath for 15 min before SDS-PAGE and immunoblotting. Blots were probed with the monoclonal tTG antibody TG100, and then stripped by incubation in 100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 7.6, at 50°C for 30 min with agitation, rinsed thoroughly with TBST, blocked with 5% milk/TBST overnight, and reprobed with the monoclonal C-myc antibody.
Immunocytochemistry
Cells were seeded on poly-d-lysine–coated cover slips in 24-well plates. 24 h later, cells were fixed in 90% methanol, 50 mM EGTA, pH 6.0, for 5 min at −20°C (Melan and Sluder 1992), incubated for 10 min with 0.2% Triton X-100 in PBS, and rinsed three times with PBS, before incubation with 5% bovine serum albumin in PBS for 90 min to reduce the background. Cells were then incubated at room temperature for 90 min with the polyclonal NH2-terminal huntingtin antibody (1:20,000) in 5% BSA/PBS. Cells were then rinsed three times with PBS, incubated for 60 min at room temperature with FITC-conjugated anti–rabbit IgG (1:200) in 5% BSA/PBS. For colocalization studies, cells were treated with 20 μM retinoic acid for 5 d before fixation and permeabilization. Cells were then incubated at room temperature for 90 min with the polyclonal NH2-terminal huntingtin antibody (1:20,000) and the monoclonal tTG antibody CUB7402 (1:20), or the monoclonal Hsp70 antibody (1:200), or the monoclonal ubiquitin antibody (1:200). Cells were then rinsed three times with PBS and incubated for 60 min at room temperature with FITC-conjugated anti–rabbit IgG (1:200) and Texas red–conjugated anti–mouse IgG (1:100). Cells were then rinsed with PBS and incubated with 5 μg/ml Hoescht for 30 min at room temperature. Coverslips were then washed extensively in PBS and mounted. Cells were viewed using confocal microscopy and images were acquired by sequential scanning at the appropriate wavelengths. The digitally stored images were combined and displayed with the accompanying software and Adobe Photoshop 4.0.
Expression of C/YFP-tagged Huntingtin Constructs in Antisense tTG Cells
To essentially abolish tTG expression, SH-SY5Y cells were stably transfected with an antisense tTG construct as described previously (Tucholski et al. 2001). There is no detectable tTG expression or transglutaminase activity in these antisense tTG cells (Tucholski et al. 2001).
The pcDNA and antisense tTG cells were plated onto poly-d-lysine–coated coverslips in six-well plates 24 h before transfection with pECFP-N1-18Q or pEYFP-N1-82Q using FUGENE™ 6, according to the manufacturer's protocol. As a control, pcDNA and antisense tTG cells were transfected with pECFP-N1 or pEYFP-N1. At 24 and 48 h after transfection, the cells were washed with PBS and subsequently fixed for 30 min at room temperature in 70 mM Pipes, pH 6.9, 1 mM MgCl2, 1 mM EGTA, 2% paraformaldehyde, 0.2% glutaraldehyde, 30% (vol/vol) glycerol. Cells were washed with PBS and incubated with 5 μg/ml Hoeschst for 30 min at room temperature, rinsed three times with 0.1× PBS, and extensively with water before mounting the coverslips. After mounting the coverslips, the cells were viewed using confocal microscopy. All images were captured with the accompanying software and displayed using Adobe Photoshop 4.0.
In Situ TG Activity Assay and Detection of tTG-modified Proteins
Cells were labeled with 2 mM 5-(biotinamido)pentylamine (Pierce Chemical Co.), a biotinylated polyamine, for 45 min. To increase intracellular levels of calcium and therefore activate tTG, 2 nM maitotoxin was added to the cells for 20 min. Transglutaminase activity was quantified by measuring the transglutaminase-dependent incorporation of 5-(biotinamido)pentylamine into proteins by a microplate assay as described by Zhang et al. 1998.
To determine whether huntingtin or huntingtin-associated proteins were modified by tTG, cells were labeled with 2 mM 5-(biotinamido)pentylamine and treated with maitotoxin as described above. Clarified cell lysates were prepared and samples containing 200 μg of protein were immunoprecipitated with the polyclonal NH2-terminal huntingtin antibody. Protein A-Sepharose was added, and the incubation continued for 3 h at 4°C. After the precipitates were washed three times, 40 μl of 1× reducing stop buffer was added to each sample and the samples boiled before SDS-PAGE and immunoblotting. Blots were probed with the monoclonal myc antibody (1:2,000), developed with ECL, and then stripped and reprobed with HRP-conjugated neutravidin (1:2,000).
Results
To demonstrate the specificity of the polyclonal NH2-terminal huntingtin antibody, cell extracts from the vector control cells were immunoblotted. The immunoblot in Fig. 1 A demonstrates that the antibody recognizes endogenous huntingtin. However, the levels of endogenous huntingtin in these cells is low, and this immunoblot had to be exposed a relatively long time to bring up the signal. In Fig. 1 B immunocytochemical analyses with the polyclonal NH2-terminal huntingtin antibody in vector-transfected and N-Q18 cells are shown. These data demonstrate that immunoreactivity is extremely low in the vector cells; however, in the N-Q18 cells, expression is significantly elevated.
Figure 1.

(A) Representative immunoblot of endogenous huntingtin in vector-transfected SH-SY5Y cells probed with the polyclonal NH2-terminal huntingtin antibody. (B) Vector-transfected and N-Q18 cells stained with the polyclonal NH2-terminal huntingtin antibody. In the vector-transfected cells, immunoreactivity is extremely low in comparison with the strong signal in the N-Q18 cells.
To determine the putative role of tTG in aggregate formation, SH-SY5Y cells stably transfected with the truncated NH2-terminal huntingtin constructs N-Q18 (wild-type truncated huntingtin) or N-Q82 (mutant truncated huntingtin) were established. Immunoblot analysis revealed that in the N-Q18 cells there was a huntingtin immunoreactive band, as expected, at ∼14 kD (Fig. 2). In the N-Q82 cells, huntingtin immunoreactive bands were observed at ∼23 and 55 kD. The 55-kD band is likely be a complex containing the truncated mutant huntingtin. As expected, retinoic acid treatment increased the expression of tTG without effecting the expression of the transfected protein (Fig. 2). Further, treatment with retinoic acid had no effect on cell viability in either the N-Q18 or the N-Q82 cells (data not shown).
Figure 2.

Representative immunoblots of the expression of truncated NH2-terminal huntingtin (top) and tTG (bottom) in SH-SY5Y cells stably expressing truncated NH2-terminal huntingtin constructs, N-Q18 or N-Q82. Cells were incubated in the absence (−) or presence (+) of 20 μM retinoic acid (RA) for 5 d before immunoblotting for huntingtin or tTG. N-Q18 cells displayed a major huntingtin immunoreactive band at ∼14 kD and in N-Q82 cells, two truncated NH2-terminal huntingtin immunoreactive bands were detected at ∼23 and 55 kD. The 55-kD band is likely to be a complex containing the truncated NH2-terminal huntingtin. Treatment of the cells with RA resulted in a significant increase in tTG levels, but did not alter the expression or levels of the truncated NH2-terminal huntingtin protein.
To evaluate the intracellular distribution of N-Q18 and N-Q82, immunocytochemical analyses were carried out using the polyclonal NH2-terminal huntingtin antibody. In N-Q18 cells huntingtin immunoreactivity was diffuse through the cytoplasm in all the cells (Fig. 3 a). In the N-Q82 cells, diffuse huntingtin immunoreactivity was also observed; however, in ∼13% of the cells cytoplasmic and/or nuclear aggregates were observed (Fig. 3, b–d). There was also evidence of diffuse huntingtin staining in the nucleus of N-Q82 cells (Fig. 3 d). Identical results were obtained with the monoclonal antibody to c-myc (data not shown).
Figure 3.

Huntingtin immunostaining in cells stably transfected with N-Q18 or N-Q82. Huntingtin localization was detected with an NH2-terminal polyclonal huntingtin antibody. In the N-Q18 cells, huntingtin immunostaining was diffuse throughout the cytoplasm (a). In contrast, in the N-Q82 cells, huntingtin formed large aggregates in the cytoplasm (b and c) and also in the nucleus (b and d). Note also the presence of diffuse huntingtin immunoreactivity in the nucleus of N-Q82 cells (d).
To further evaluate the inclusions formed by N-Q82, cells were costained for huntingtin and ubiquitin or Hsp70, as inclusions in models of HD and other polyglutamine diseases are usually ubiquitinated and often reactive with antibodies to heat shock proteins (Davies et al. 1997; Saudou et al. 1998; Wyttenbach et al. 2000). The majority of the aggregates were immunopositive for both ubiquitin and hsp70 (Fig. 4). Interestingly, both the huntingtin and ubiquitin antibodies stained the peripheral areas of the inclusions, while the Hsp70 antibody stained the entire inclusion (Fig. 4). The reason for the peripheral staining pattern of the aggregates with the NH2-terminal huntingtin and ubiquitin antibodies is unknown, but may be due to the organization of the inclusion that results in “masking” of the NH2-terminal portion of the huntingtin at the center of the inclusion. In addition, when N-Q82 cells were costained for huntingtin and C-myc, there was complete overlap of the immunostaining, clearly demonstrating that the truncated huntingtin is a primary constituent of the aggregates (Fig. 5).
Figure 4.

Aggregates of N-Q82 are ubiquitinated and colocalize with Hsp70. Cells were double labeled with the polyclonal huntingtin antibody (green) and with the monoclonal ubiquitin or Hsp70 antibody (red), and nuclei were counterstained with Hoechst (blue). Aggregates of N-Q82 were ubiquitinated and ubiquitin immunoreactivity colocalized in the peripheral areas of the inclusion (a–c). Hsp70 immunoreactivity colocalized with the aggregates and stained the entire inclusion (d–f).
Figure 5.

Colocalization of huntingtin and C-myc immunoreactivity in N-Q82 cells. N-Q82 cells were double labeled with the polyclonal huntingtin antibody (green) and with the monoclonal C-myc antibody (red). Nuclei were counterstained with Hoechst (blue). Huntingtin and C-myc immunoreactivity showed complete overlap, especially at the levels of the aggregates.
To determine whether tTG and highly truncated huntingtin interact, coimmunoprecipitation assays were carried out. N-Q18 huntingtin coprecipitated with tTG; however, this interaction was not increased when tTG was activated (Fig. 6 A). tTG is associating with the N-Q18, as when endogenous full-length huntingtin was immunoprecipitated from the parental SH-SY5Y cells line, tTG did not coimmunoprecipitate with the huntingtin (data not shown). In contrast, N-Q82 huntingtin only showed a very weak interaction with tTG (Fig. 6 A). SDS-resistant material that contained the N-Q82 huntingtin was detected at the top of the gel, indicating that insoluble aggregates had formed. When the N-Q82 or N-Q18 cells were immunoprecipitated with nonimmune rabbit IgG, no huntingtin precipitated (Fig. 6 B). To determine whether tTG colocalized with the highly truncated huntingtin, costaining was carried out with the huntingtin and tTG antibodies. In the N-Q18 cells, both huntingtin and tTG immunoreactivity were diffuse through the cytoplasm (Fig. 7, a–c). In the N-Q82 cells, there was also no specific colocalization of tTG and huntingtin and, further, tTG immunoreactivity was completely excluded from the aggregates (Fig. 7, d–i). In addition, the number of N-Q82 cells with aggregates was similar in control conditions and after increased tTG expression due to retinoic acid treatment (Fig. 8). To determine whether tTG modifies highly truncated huntingtin or associated proteins, cells were prelabeled with 5-(biotinamido)pentylamine, incubated in the absence or presence of maitotoxin, and immunoprecipitated with the polyclonal huntingtin antibody, probed with a C-myc antibody, and then stripped and reprobed with neutravidin-HRP. The results of these experiments demonstrated that neither truncated huntingtin nor any huntingtin-associated proteins were modified by tTG (data not shown).
Figure 6.

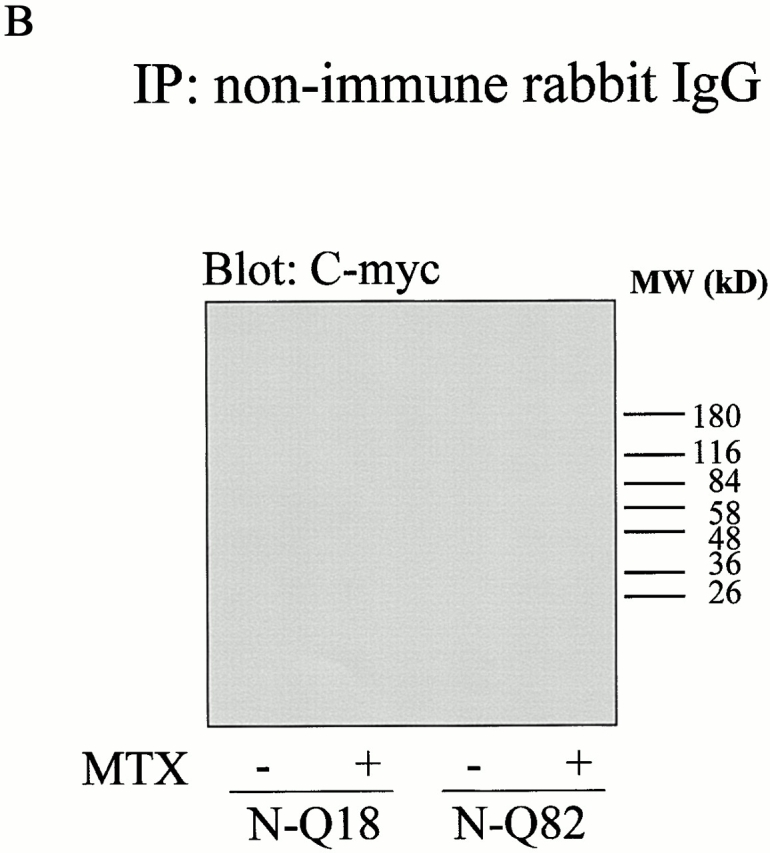
(A) In situ association between truncated NH2-terminal huntingtin and tTG. Cells stably expressing N-Q18 or N-Q82 were treated with 20 μM retinoic acid for 5 d and incubated in the absence (−) or presence (+) of MTX. Huntingtin was immunoprecipitated with the polyclonal huntingtin antibody and immunoblotted with a monoclonal tTG antibody (left). Immunoblots were stripped and reprobed with a monoclonal C-myc antibody (right). These results revealed that N-Q18 and tTG interact; however, there is only a very weak association between N-Q82 and tTG. Further, activation of tTG did not have any effect on the association of tTG and N-Q18. The bands at the top of the gel in the C-myc blot of the N-Q82 lane confirm the presence of SDS-insoluble huntingtin aggregates. The immunoreactive bands in the C-myc blot of N-Q18 cells at ∼55 kD is IgG, as the goat anti–mouse secondary antibody weakly recognizes the rabbit IgG used in the immunoprecipitation. (B) Cells stably expressing N-Q18 or N-Q82 were treated with 20 μM retinoic acid for 5 d, incubated in the absence (−) or presence (+) of MTX, immunoprecipitated with nonimmune rabbit IgG, and probed with the C-myc antibody. These data demonstrate that the precipitation of N-Q18 and N-Q82 is selective, as there was no C-myc reactivity present when the nonimmune IgG was used.
Figure 7.

Truncated NH2-terminal huntingtin and tTG do not colocalize. Cells were double labeled with the polyclonal huntingtin antibody (green) and with the monoclonal tTG antibody CUB7402 (red); nuclei were counterstained with Hoechst (blue). In cells stably expressing N-Q18, huntingtin (a) and tTG immunoreactivities (b) were diffuse throughout the cytoplasm and did not show specific colocalization (c). In contrast, cells stably expressing N-Q82 showed large huntingtin immunoreactive aggregates in the cytoplasm (d and g) and in the nucleus (g). In these cells, tTG immunoreactivity was also diffuse throughout the cytoplasm and, remarkably, appeared completely excluded from the huntingtin aggregates (e, f, h, and i). These results demonstrate that the huntingtin aggregates and tTG do not colocalize.
Figure 8.

The frequency of N-Q82 aggregates is not effected by tTG overexpression. Cells expressing N-Q82 were incubated in the absence or presence of 20 μM retinoic acid (RA) for 2 or 5 d. tTG expression levels were increased in a time-dependent manner (A). Treatment of the cells with RA for time periods >5 d did not increase tTG expression further (data not shown) (Zhang et al. 1998). The frequency of cells with aggregates was quantified in the different conditions. Approximately 13% of the cells formed aggregates in all cells regardless of the tTG expression level. There were no significant differences in the frequency of aggregates in either the cytoplasm or nucleus of cells expressing the different levels of tTG (B). Data represent three independent experiments; 600–1,400 cells were counted for each experiment. The data are expressed as mean% ± SEM.
To confirm that tTG does not play a role in the formation of mutant huntingtin inclusions, pEYFP-N1-82Q or pCYFP-N1-18Q were transiently transfected into cells stably transfected with vector (pcDNA3.1) only or into cells stably transfected with an antisense tTG construct. In the antisense tTG cells, tTG is undetectable and there is no measurable transglutaminase activity (Tucholski et al. 2001). As expected, transient transfection of pCYFP-N1-18Q into either the pcDNA or antisense tTG cells did not result in any aggregate formation (data not shown). In contrast, transient transfection of pEYFP-N1-82Q resulted in abundant aggregate formation in both the pcDNA and antisense cells (Fig. 9). These studies clearly demonstrate that it is unlikely that tTG plays a role in the formation of inclusions in HD brain.
Figure 9.

N-Q82 forms aggregates in the absence of tTG. pEYFP-N1-82Q was transiently transfected into cells stably transfected with vector only (pcDNA3.1) (a and b) or into cells stably transfected with an antisense tTG construct (c and d). Transient transfection of pEYFP-N1-82Q resulted in equivalent aggregate formation in both the pcDNA (a and b) and antisense tTG cells (c and d).
Discussion
The hypothesis that tTG contributes to the etiology of HD was first proposed by Green 1993 before the identification of huntingtin aggregates in HD brain. With the discovery of intranuclear and cytoplasmic inclusions of mutant huntingtin in HD brain (DiFiglia et al. 1997) and in transgenic mouse models for HD (Davies et al. 1997), it was further hypothesized that tTG may facilitate the formation of these aggregates by selectively cross linking mutant huntingtin (Cooper et al. 1999). Indeed, it has even been suggested that tTG may be a potential therapeutic target in the treatment of polyglutamine diseases (Igarashi et al. 1998). Additionally, tTG levels and transglutaminase activity are significantly increased in HD brain (Karpuj et al. 1999; Lesort et al. 1999) in a grade- and region-dependent manner, and there is a significant increase in the tTG immunoreactivity in specific neuronal populations in HD brain (Lesort et al. 1999). Although these findings are intriguing, there has been no direct demonstration that tTG is involved in the formation of aggregates in HD or other polyglutamine diseases. Therefore, the goals of this study were to determine whether or not tTG and huntingtin interact in situ, and whether tTG facilitates the formation of mutant huntingtin aggregates.
It has been well documented that in vitro polyglutamine constructs and mutant huntingtin are substrates for tTG (Kahlem et al. 1996, Kahlem et al. 1998; Cooper et al. 1997). However, the ability of tTG to modify mutant huntingtin in situ has not been demonstrated, and the putative contribution of tTG in the formation of polyglutamine aggregates is still controversial. It was reported that transglutaminase inhibitors suppressed aggregate formation and apoptosis in cells expressing truncated DRPLA protein with an expanded polyglutamine domain (Igarashi et al. 1998). These findings need to be interpreted with some caution as both transglutaminase inhibitors used in this study can also inhibit other enzymes (e.g., caspases), and in some cases the transglutaminase inhibitors reduced apoptotic cell death, but were ineffective in blocking the formation of aggregates (Lorand 1998). In another study, tTG overexpression was reported to increase the aggregate formation of synthetic fusion proteins containing 36 or 43 glutamines. However, the percent increase in aggregate formation induced by tTG overexpression was only ∼10–15% (de Cristofaro et al. 1999). These authors also presented data that cystamine, a transglutaminase inhibitor, at relatively high concentrations (0.25 and 0.50 mM) reduced aggregate formation (de Cristofaro et al. 1999). In another study, the same inhibitor (0.20 mM) had no effect in mutant huntingtin aggregate formation, and, when the inhibitor was used at higher concentrations (0.5 mM), it was cytotoxic (Kim et al. 1999). These findings indicate the need for further investigations before a reasonable case can be made for the involvement of tTG in the etiology of HD.
In the present study, we used human neuroblastoma SH-SY5Y cells stably transfected with wild-type and mutant truncated huntingtin to investigate the role of tTG in huntingtin modification and aggregate formation. SH-SY5Y cells are an excellent model system for these studies because they have neuronal-like features (Preis et al. 1988; Pahlman et al. 1990; Jalava et al. 1992), and tTG expression is robustly upregulated in response to retinoic acid (Zhang et al. 1998). Therefore, alterations in huntingtin can be evaluated in conditions when tTG levels are low, and also when the levels and activity of tTG are elevated. In these cells, N-Q82 did not increase cell death under basal conditions. However, it should be noted that overexpression of N-Q82 does sensitize the cells to apoptotic stressors (Chun, W., M. Lesort, and G.V.W. Johnson, unpublished observations, manuscript in preparation). This is in agreement with a previous study reporting that transient transfection of the N-Q82 construct into neuroblastoma N2a cells resulted in aggregate formation and increased sensitivity to staurosporine-induced apoptosis; however, no changes in cell viability under basal conditions were reported (Cooper et al. 1998). Similar findings were reported when mutant truncated huntingtin constructs were transiently transfected into 293T cells; there was significant aggregate formation and no changes in viability under basal conditions, while the presence of the truncated huntingtin constructs sensitized cells to the apoptotic stimuli (Hackam et al. 1998).
In the SH-SY5Y cells stably overexpressing N-Q82, tTG associated with the wild-type truncated huntingtin; however, this association was not affected by the activity state of tTG. In addition, there was no selective association between tTG and the mutant truncated huntingtin. In all cases, no in situ modification of huntingtin tTG was observed. This demonstrates that although huntingtin is an in vitro substrate of tTG (Kahlem et al. 1998), it is unlikely to be modified by tTG in vivo. Furthermore, we found that tTG was totally excluded from the insoluble huntingtin aggregates in the N-Q82 cells, although other proteins such as Hsp70 showed a strong colocalization with huntingtin at the level of the aggregates. In addition, the number and size of the aggregates in the N-Q82 cells expressing either low or high levels (due to retinoic acid treatment) of tTG were not significantly different. These findings demonstrate that it is unlikely that tTG plays a role in aggregate formation in HD brain. In addition, when pEYFP-N1-82Q was transiently transfected into cells that had been stably transfected with antisense tTG, and therefore express undetectable levels of the protein, insoluble aggregates were still formed. Further, tTG is virtually undetectable in mouse (C57BL) brain by immunoblot analysis (Lesort, M., unpublished observations), although inclusions are usually found in the brains of mouse models of HD (Davies et al. 1997; Schilling et al. 1999; Wheeler et al. 2000). Indeed, given the fact that the cross-linking and polyamination reactions catalyzed by tTG are competing reactions (Lorand and Conrad 1984; Greenberg et al. 1991), and the fact that the levels of polyamines in the brain are in the millimolar range (Morrison et al. 1995), it seems unlikely that tTG catalyzes cross-linking reactions within the cells. Therefore, another mechanism, such as the formation of β-sheets via the glutamine repeats acting as polar zippers (Perutz et al. 1994; Stott et al. 1995), is more likely to be responsible for the formation of huntingtin aggregates in HD brain. In vitro, polyglutamine constructs form β-sheets that are held together by hydrogen bonds (Perutz et al. 1994; Stott et al. 1995; Scherzinger et al. 1997). Congo red selectively stains β-sheet structure, and aggregates from HD brain (Huang et al. 1998), as well as aggregates formed from mutant huntingtin in vitro (Scherzinger et al. 1997), stain with Congo red. These data suggest that hydrogen bonds between the side chain amides of glutamine and the amides of the polypeptide backbone may be the essential process in huntingtin aggregate formation in HD (Perutz 1994; Perutz et al. 1994).
It should be noted that the appearance of the aggregates in the cells stably expressing N-Q82 compared with those in which the N-Q82 construct was transiently transfected was significantly different. In the transient transfection model, the protein is expressed rapidly and at very high levels, which likely results in the more amorphous appearance of the aggregates, as has been shown by others (Hackam et al. 1998). In the stable cells, expression of N-Q82 resulted in the formation of very well-defined aggregates, which is likely due to the fact that expression is more controlled in the stably transfected cells, and hence the proteins can organize into more well-defined structures (Lunkes and Mandel 1998).
In conclusion, the results of the present study demonstrate that tTG associates with wild-type, but not mutant, truncated huntingtin; however, tTG does not modify the huntingtin, either wild-type or mutant. Further, tTG is totally excluded from the inclusions in the mutant truncated huntingtin-expressing cells, and mutant huntingtin aggregates form in the absence of tTG. These findings clearly demonstrate that tTG is not essential for the formation of huntingtin aggregates in HD brain.
Acknowledgments
The authors thank Dr. Peter Detloff for the generous gift of the polyclonal huntingtin antibody.
This work was supported by National Institutes of Health grant AG12396 (G.V.W. Johnson) and a fellowship from the Hereditary Disease Foundation (M. Lesort).
Footnotes
Drs. Chun and Lesort contributed equally to this work and should be considered co-first authors.
Abbreviations used in this paper: HD, Huntington's disease; tTG, tissue transglutaminase.
References
- Appelt D.M., Kopen G.C., Boyne L.J., Balin B.J. Localization of transglutaminase in hippocampal neuronsimplications for Alzheimer's disease. J. Histochem. Cytochem. 1996;44:1421–1427. doi: 10.1177/44.12.8985134. [DOI] [PubMed] [Google Scholar]
- Cooper A.J., Sheu K.F., Burke J.R., Onodera O., Strittmatter W.J., Roses A.D., Blass J.P. Polyglutamine domains are substrates of tissue transglutaminasedoes transglutaminase play a role in expanded CAG/poly-Q neurodegenerative diseases? J. Neurochem. 1997;69:431–434. doi: 10.1046/j.1471-4159.1997.69010431.x. [DOI] [PubMed] [Google Scholar]
- Cooper A.J., Sheu K.F., Burke J.R., Strittmatter W.J., Gentile V., Peluso G., Blass J.P. Pathogenesis of inclusion bodies in (CAG)n/Qn-expansion diseases with special reference to the role of tissue transglutaminase and to selective vulnerability. J. Neurochem. 1999;72:889–899. doi: 10.1046/j.1471-4159.1999.0720889.x. [DOI] [PubMed] [Google Scholar]
- Cooper J.K., Schilling G., Peters M.F., Herring W.J., Sharp A.H., Kaminsky Z., Masone J., Khan F.A., Delanoy M., Borchelt D.R. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Davis P.K., Dudek S.M., Johnson G.V. Select alterations in protein kinases and phosphatases during apoptosis of differentiated PC12 cells. J. Neurochem. 1997;68:2338–2347. doi: 10.1046/j.1471-4159.1997.68062338.x. [DOI] [PubMed] [Google Scholar]
- de Cristofaro T., Affaitati A., Cariello L., Avvedimento E.V., Varrone S. The length of polyglutamine tract, its level of expression, the rate of degradation, and the transglutaminase activity influence the formation of intracellular aggregates. Biochem. Biophys. Res. Commun. 1999;260:150–158. doi: 10.1006/bbrc.1999.0851. [DOI] [PubMed] [Google Scholar]
- Decker T., Lohmann-Matthes M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods. 1988;115:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- DiFiglia M., Sapp E., Chase K.O., Davies S.W., Bates G.P., Vonsattel J.P., Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Folk J.E. Mechanism and basis for specificity of transglutaminase-catalyzed epsilon-(gamma-glutamyl) lysine bond formation. Adv. Enzymol. Relat. Areas Mol. Biol. 1983;54:1–56. doi: 10.1002/9780470122990.ch1. [DOI] [PubMed] [Google Scholar]
- Green H. Human genetic diseases due to codon reiterationrelationship to an evolutionary mechanism. Cell. 1993;74:955–956. doi: 10.1016/0092-8674(93)90718-6. [DOI] [PubMed] [Google Scholar]
- Greenberg C.S., Birckbichler P.J., Rice R.H. Transglutaminasesmultifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- Gutekunst C.A., Li S.H., Yi H., Mulroy J.S., Kuemmerle S., Jones R., Rye D., Ferrante R.J., Hersch S.M., Li X.J. Nuclear and neuropil aggregates in Huntington's diseaserelationship to neuropathology. J. Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackam A.S., Singaraja R., Wellington C.L., Metzler M., McCutcheon K., Zhang T., Kalchman M., Hayden M.R. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J. Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackam A.S., Singaraja R., Zhang T., Gan L., Hayden M.R. In vitro evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington's disease. Hum. Mol. Genet. 1999;8:25–33. doi: 10.1093/hmg/8.1.25. [DOI] [PubMed] [Google Scholar]
- Huang C.C., Faber P.W., Persichetti F., Mittal V., Vonsattel J.P., MacDonald M.E., Gusella J.F. Amyloid formation by mutant huntingtinthreshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell Mol. Genet. 1998;24:217–233. doi: 10.1023/b:scam.0000007124.19463.e5. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Igarashi S., Koide R., Shimohata T., Yamada M., Hayashi Y., Takano H., Date H., Oyake M., Sato T., Sato A. Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat. Genet. 1998;18:111–117. doi: 10.1038/ng0298-111. [DOI] [PubMed] [Google Scholar]
- Jalava A., Heikkila J., Lintunen M., Akerman K., Pahlman S. Staurosporine induces a neuronal phenotype in SH-SY5Y human neuroblastoma cells that resembles that induced by the phorbol ester 12-O-tetradecanoyl phorbol-13 acetate (TPA) FEBS Lett. 1992;300:114–118. doi: 10.1016/0014-5793(92)80176-h. [DOI] [PubMed] [Google Scholar]
- Kahlem P., Green H., Djian P. Transglutaminase action imitates Huntington's diseaseselective polymerization of Huntingtin containing expanded polyglutamine. Mol. Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- Kahlem P., Terre C., Green H., Djian P. Peptides containing glutamine repeats as substrates for transglutaminase-catalyzed cross-linkingrelevance to diseases of the nervous system. Proc. Natl. Acad. Sci. USA. 1996;93:14580–14585. doi: 10.1073/pnas.93.25.14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuj M.V., Garren H., Slunt H., Price D.L., Gusella J., Becher M.W., Steinman L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington's disease brain nuclei. Proc. Natl. Acad. Sci. USA. 1999;96:7388–7393. doi: 10.1073/pnas.96.13.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Lee H.-S., LaForet G., McIntyre C., Eileen J.M., Chang P., Kim T.W., Williams M., Reddy P.H., Tagle D. Mutant huntingtin expression in clonal striatal cellsdissociation on inclusion formation and neuronal survival by caspase inhibition. J. Neurosci. 1999;19:964–973. doi: 10.1523/JNEUROSCI.19-03-00964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klement I.A., Skinner P.J., Kaytor M.D., Yi H., Hersch S.M., Clark H.B., Zoghbi H.Y., Orr H.T. Ataxin-1 nuclear localization and aggregationrole in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Lesort M., Chun W., Johnson G.V., Ferrante R.J. Tissue transglutaminase is increased in Huntington's disease brain. J. Neurochem. 1999;73:2018–2027. [PubMed] [Google Scholar]
- Li X.J. The early cellular pathology of Huntington's disease. Mol. Neurobiol. 2000;20:111–124. doi: 10.1007/BF02742437. [DOI] [PubMed] [Google Scholar]
- Lin C.-H., Tallaksen-Greene S., Chein W.-M., Cearley J., Jackson W., Crouse A., Ren S., Li X.-J., Albin R., Detloff P. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Lorand L. DRPLA aggregation and transglutaminase, revisited. Nat. Genet. 1998;20:231. doi: 10.1038/3033. [DOI] [PubMed] [Google Scholar]
- Lorand L., Conrad S.M. Transglutaminases. Mol. Cell. Biochem. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- Lunkes A., Mandel J. A cellular model that recapitulates major pathogenic steps of Huntington's disease. Hum. Mol. Genet. 1998;7:1335–1361. doi: 10.1093/hmg/7.9.1355. [DOI] [PubMed] [Google Scholar]
- Melan M.A., Sluder G. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells. Implications for immunofluorescence microscopy. J. Cell Sci. 1992;101:731–743. doi: 10.1242/jcs.101.4.731. [DOI] [PubMed] [Google Scholar]
- Miller C.C., Anderton B.H. Transglutaminase and the neuronal cytoskeleton in Alzheimer's disease. J. Neurochem. 1986;46:1912–1922. doi: 10.1111/j.1471-4159.1986.tb08513.x. [DOI] [PubMed] [Google Scholar]
- Morrison L.D., Becker L., Ang L.C., Kish S.J. Polyamines in human brainregional distribution and influence of aging. J. Neurochem. 1995;65:636–642. doi: 10.1046/j.1471-4159.1995.65020636.x. [DOI] [PubMed] [Google Scholar]
- Pahlman S., Mamaeva S., Meyerson G., Mattsson M.E., Bjelfman C., Ortoft E., Hammerling U. Human neuroblastoma cells in culturea model for neuronal cell differentiation and function. Acta Physiol. Scand. Suppl. 1990;592:25–37. [PubMed] [Google Scholar]
- Persichetti F., Trettel F., Huang C.C., Fraefel C., Timmers H.T., Gusella J.F., MacDonald M.E. Mutant huntingtin forms in vivo complexes with distinct context-dependent conformations of the polyglutamine segment. Neurobiol. Dis. 1999;6:364–375. doi: 10.1006/nbdi.1999.0260. [DOI] [PubMed] [Google Scholar]
- Perutz M. Polar zipperstheir role in human disease. Prot. Sci. 1994;3:1629–1637. doi: 10.1002/pro.5560031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz M.F., Johnson T., Suzuki M., Finch J.T. Glutamine repeats as polar zipperstheir possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preis P.N., Saya H., Nadasdi L., Hochhaus G., Levin V., Sadee W. Neuronal cell differentiation of human neuroblastoma cells by retinoic acid plus herbimycin A. Cancer Res. 1988;48:6530–6534. [PubMed] [Google Scholar]
- Saudou F., Finkbeiner S., Devys D., Greenberg M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Sawa A., Wiegand G., Cooper J., Margolis R., Sharp A., Lawler J., Jr., Greenamyre J., Snyder S., Ross C. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat. Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G.P., Davies S.W., Lehrach H., Wanker E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Stott K., Blackburn J.M., Butler P.J., Perutz M. Incorporation of glutamine repeats makes protein oligomerizeimplications for neurodegenerative diseases. Proc. Natl. Acad. Sci. USA. 1995;92:6509–6513. doi: 10.1073/pnas.92.14.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucholski J., Lesort M., Johnson G.V.W. Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience. 2001;102:481–491. doi: 10.1016/s0306-4522(00)00482-6. [DOI] [PubMed] [Google Scholar]
- Wheeler V.C., White J.K., Gutekunst C.A., Vrbanac V., Weaver M., Li X.J., Li S.H., Yi H., Vonsattel J.P., Gusella J.F. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A., Carmichael J., Swartz J., Furlong R.A., Narain Y., Rankin J., Rubinsztein D.C. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc. Natl. Acad. Sci. USA. 2000;97:2898–2903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Zhang J., Lesort M., Guttmann R.P., Johnson G.V. Modulation of the in situ activity of tissue transglutaminase by calcium and GTP. J. Biol. Chem. 1998;273:2288–2295. doi: 10.1074/jbc.273.4.2288. [DOI] [PubMed] [Google Scholar]