

Abstract
Endochondral ossification begins from the condensation and differentiation of mesenchymal cells into cartilage. The cartilage then goes through a program of cell proliferation, hypertrophic differentiation, calcification, apoptosis, and eventually is replaced by bone. Unlike most cartilage, articular cartilage is arrested before terminal hypertrophic differentiation. In this study, we showed that TGF-β/Smad3 signals inhibit terminal hypertrophic differentiation of chondrocyte and are essential for maintaining articular cartilage. Mutant mice homozygous for a targeted disruption of Smad3 exon 8 (Smad3 ex8/ex8) developed degenerative joint disease resembling human osteoarthritis, as characterized by progressive loss of articular cartilage, formation of large osteophytes, decreased production of proteoglycans, and abnormally increased number of type X collagen–expressing chondrocytes in synovial joints. Enhanced terminal differentiation of epiphyseal growth plate chondrocytes was also observed in mutant mice shortly after weaning. In an in vitro embryonic metatarsal rudiment culture system, we found that TGF-β1 significantly inhibits chondrocyte differentiation of wild-type metatarsal rudiments. However, this inhibition is diminished in metatarsal bones isolated from Smad3 ex8/ex8 mice. These data suggest that TGF-β/Smad3 signals are essential for repressing articular chondrocyte differentiation. Without these inhibition signals, chondrocytes break quiescent state and undergo abnormal terminal differentiation, ultimately leading to osteoarthritis.
Keywords: TGF-β/Smad3, mouse model, osteoarthritis, synovium, growth plate
Introduction
Osteoarthritis is the most common noninflammatory disease of synovial joints, which is distinct from rheumatoid arthritis, a systemic autoimmune disorder (Fujita 1997; Simon 1999; van den Berg 1999; Goldring 2000). Osteoarthritis is characterized by the progressive loss of articular cartilage, resulting in pain and loss of joint function. This condition affects a majority of people >75 yr of age, and the underlying mechanism is largely unknown.
Bone is formed through intramembranous and endochondral ossification (Erlebacher et al. 1995). Endochondral ossification initiates by the condensation and differentiation of mesenchymal cells into cartilage. The cartilage then proceeds through programmed cell proliferation, maturation, hypertrophic differentiation, calcification, apoptosis, and eventual replacement by bone tissues. Bone is continuously renewed by a process of bone remodeling that consists of cycles of bone formation and resorption. Unlike most cartilage, the articular cartilage is arrested before terminal hypertrophic differentiation and forms a smooth cartilage layer that covers the heads of bone and enables frictionless and pain-free movement of the joint.
Several members of the TGF-β super family play important roles in bone growth. The bone morphogenetic proteins (BMPs) induce early cartilage formation (Wozney 1989, Wozney 1992) and stimulate mesenchymal cells to differentiate into osteoblasts (Vukicevic et al. 1989; Yamaguchi et al. 1991; Wozney 1992). Mice harboring mutations in members of the TGF-β super family display multiple skeletal defects (Kingsley et al. 1992; Luo et al. 1995; Mikic et al. 1995; Storm and Kingsley 1996; Katagiri et al. 1998; Solloway et al. 1998; Ducy and Karsenty 2000). TGF-βs and their receptors are expressed during the development of the skeleton and play important roles in regulating chondrocyte proliferation and differentiation (Sandberg et al. 1988; Gatherer et al. 1990; Pelton et al. 1990; Millan et al. 1991; DiLeone et al. 1997; Fukumura et al. 1998; Horner et al. 1998; Kabasawa et al. 1998). It was shown that TGF-βs can stimulate cultured undifferentiated mesenchymal cells to differentiate into chondrocytes (Kulyk et al. 1989; Lafeber et al. 1993; Denker et al. 1995). Injection of TGF-βs in the periosteum of rat (Joyce et al. 1990) or mouse (Chimal-Monroy and Diaz de Leon 1997) femur induces chondrocyte differentiation and cartilage formation. On the other hand, TGF-βs can also inhibit hypertrophic differentiation of cultured chondrocyte and metatarsal bones (Ballock et al. 1993; Tschan et al. 1993; Dieudonne et al. 1994; Bohme et al. 1995). Notably, expression of dominant negative type II TGF-β receptors in cartilage and the synovium transgenic mice altered chondrocyte differentiation and resulted in phenotypes sharing some similarities with human osteoarthritis (Serra et al. 1997). Altogether, these studies indicate that TGF-β signals function as key regulators in bone formation, remodeling, and maintenance.
TGF-βs signals are transduced into nuclei by intracellular mediator SMADs (Heldin et al. 1997; Massague 1998; Datto and Wang 2000). At least eight different SMADs have been found in mammals so far. Based on their functions, these SMADs are categorized into three types: the receptor-activated SMADs, the common mediator SMAD (SMAD4), and the inhibitory SMADs (SMAD6 and 7). The receptor-activated SMADs include SMADs 1, 2, 3, 5, and 8. SMADs 2 and 3 respond to TGF-β and activins (Eppert et al. 1996; Graff et al. 1996; Macias-Silva et al. 1996; Zhang et al. 1996; Nakao et al. 1997), whereas SMAD1, 5, and 8 function in BMP signaling pathways (Lagna et al. 1996; Liu et al. 1996; Kretzschmar et al. 1997; Suzuki et al. 1997; Kawabata et al. 1998; Nakayama et al. 1998). Mutational analysis in mice using gene targeting has revealed multiple important functions for these genes in development (for review see Weinstein et al. 2000). Loss of Smad3 function results in diminished T cell response to TGF-β (Datto et al. 1999; Yang et al. 1999), accelerated wound healing (Ashcroft et al. 1999), and colon carcinoma (Zhu et al. 1998). Although 50–70% of Smad3 ex8/ex8 mice die 3 mo after birth due to infections adjacent to the mucosal surface, the remaining mutant mice overcome the infection and survive ≤10 mo (Yang et al. 1999). We now show that the Smad3 ex8/ex8 mice display skeletal abnormalities shortly after weaning, which become worse as the mice aged. One of the major characteristics of these mice is abnormal hypertrophic differentiation of articular chondrocytes, leading to the progressive loss of articular cartilage and formation of large osteophytes in synovial joints. Our further analysis indicates that Smad3-mediated signals are essential for cartilage maintenance rather than its formation. Loss of chondrocyte responsiveness to TGF-β signals during chondrogenesis in the Smad3 ex8/ex8 mice results in abnormal chondrocyte terminal differentiation, which causes progressive degenerative cartilage disease resembling osteoarthritis in human.
Materials and Methods
X-Ray Imaging and Skeleton Preparation
Smad3 ex8/ex8 mice (Yang et al. 1999) were killed by CO2. The skin and the viscera were removed. Photographs were taken in an x-ray machine (Faxitron X-ray, Co.). Preparation of whole mice skeletons was carried out using standard procedures (McLeod 1980). In brief, the mice without skin and viscera were fixed in 95% ethanol for 72 h and transferred into acetone for 48 h to remove fat and solidify the skeleton. After alcian blue and alizarin red S staining for 3 d, the samples were washed in distilled water and cleaned in 1% KOH and then processed through a series of 20, 50, and 80% glycerine/1% KOH solutions.
The Histologic and Immunohistologic Analysis of the Skeletal Tissues
The knee joints were fixed in 4% paraformaldehyde at 4°C overnight and decalcified in 0.5 M EDTA/PBS. Decalcified tissues were dehydrated and embedded in wax using standard procedures. The polyclonal antibody against type X collagen was provided by Bjorn Olsen (Harvard Medical School, Boston, MA). Zymed Laboratories Kit was used for immunohistologic staining.
To detect the expression of the proteoglycan in the skeletal tissues, safranine O staining was performed on slides. The slides were stained in hematoxylin for 1 min and destained in HCL/ethanol and NH4OH/ethanol solutions. The slides were then rinsed quickly in 1% acetic acid after staining in 0.2% Fast green for 2 min. After rinsing in distilled water, the slides were further stained in 0.1% safranine O for 5 min. Finally, the slides were dehydrated and mounted using standard procedures.
In Situ Hybridization
In situ hybridization was carried out using standard procedures. Probes used were Smad2 (Weinstein et al. 1998), Smad3 (Yang et al. 1999), FGF receptor (FGFR)3 (Deng et al. 1996), and others (see acknowledgments). Slides were dipped in emulsion (NTB-2; Eastman Kodak Co.) and exposed for 7–15 d before developing.
Embryonic Metatarsal Rudiment In Vitro Culture
Metatarsal rudiments were isolated from 17.5 pc pregnant Smad3+/ex8 female mice that were mated with Smad3 + /ex8 male mice. The three left–right paired central bones were used in experiments. Rudiments were cultured in 24-well plates with 350 μl BGJb medium (Cat. 12591–038; GIBCO BRL) supplemented with 0.05 mg/ml ascorbic acid (Sigma-Aldrich), 0.5 mg/ml l-glutamine, 10 mg/ml streptomycin, 10 units/ml penicillin (Cat. 11679; GIBCO BRL), 1 mM β-glycerophosphate (Sigma-Aldrich), and 0.2% faction V BSA (A-9647; Sigma-Aldrich). Explants were grown at 37°C in a humidified 5% CO2 incubator. TGF-β1 (10 ng/ml) (240-B; R&D Systems) in 4 mM HCL was added to cultures 16–20 h after dissection. Medium was changed on the second day of culture. The rudiments were observed and photographed under a dissecting microscope (Leica) at 0, 2, and 4 d of treatment. The total length (TL) and the hypertrophic length (HL) of chondrocyte, which includes the zone of mineralized chondrocytes and the light regions flanking it, were measured with an eyepiece scale. Changes in length were expressed as percentage increase relative to the value before the treatment (percentage increase = [length at day 4 − length at day 0]/length at day 0). Data are expressed as mean ± SD, and the significance of differences was evaluated with Student's t test.
Results
Skeletal Defects of the Smad3ex8/ex8 Mice
To study function of Smad3 in skeletal formation and development, we examined Smad3ex8/ex8mice created previously (Yang et al. 1999). Our data showed that these mice were often kyphotic (abnormal rearward curvature of the spine) and moved with difficulties. Using x-ray radiographic analysis, kyphoscoliosis and abnormal calcification of the synovial joints was observed in the Smad3ex8/ex8 mice >6 mo of age (Fig. 1 B). The knee joints were usually enlarged due to osteophytes (extra cartilage and bones) that had developed at the joint margins and within the joint space (Fig. 1 D). The osteophytes were also observed in vertebral bone joints (Fig. 1 F) and sternum joints (Fig. 1 H). As the mutant mice aged, the clinical signs were getting progressively worse and eventually resulted in loss of movement. No similar abnormalities were seen in age-matched wild-type and heterozygous controls (Fig. 1A, Fig. C, Fig. E, and Fig. G).
Figure 1.
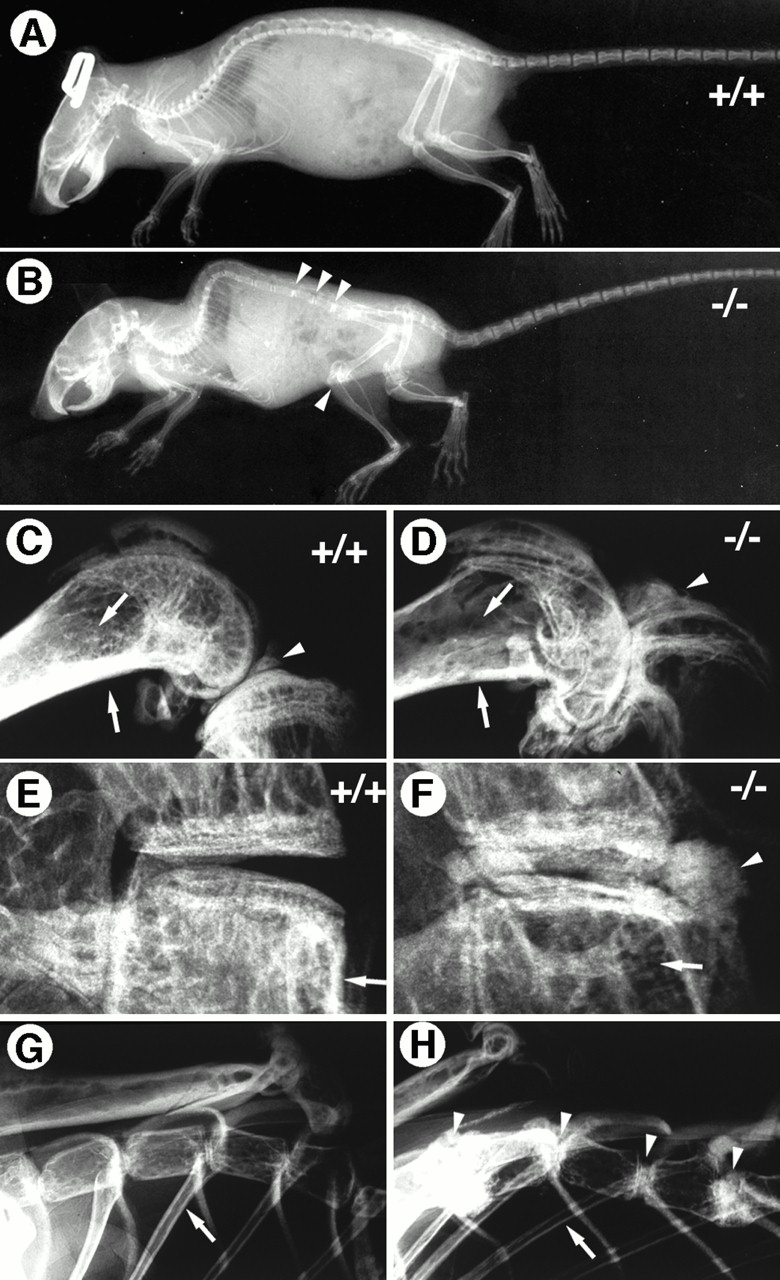
Radiographs of the skeletal abnormalities in 7-mo-old Smad3 ex8/ex8 mice. (A and B) Whole-mount view of control and mutant skeletons. Mutant mice exhibited increased intensities in vertebral and knee joints (arrowheads). (C–H) Enlarged views of knee (C and D), vertebral (E and F), and sternum joints (G and H). The increased density in mutant joints is caused by formation of extra bones (arrowheads). However, decreased bone densities were found in other areas of mutant bones compared with corresponding regions of control mice (arrows).
Next, we examined alizarin red and alcian blue whole mount skeletal preparations of mice at ages from postnatal day (P)1, P21, P30, and 4 and 6 mo to determine the onset of skeletal abnormalities. Smad3ex8/ex8 mice at ≤1 mo of age did not show any apparent abnormalities in synovial joints or rib cages compared with their wild-type and heterozygous littermates (Fig. 2A, Fig. C, and Fig. D). In contrast, all older Smad3ex8/ex8 mice exhibited varying abnormalities in rib cages characterized by the accumulation of bony materials (Fig. 2, E–G). As the mutant mice aged, the symptoms were getting progressively stronger and became obvious in all mutant mice examined at ages ≥6 mo of age (n = 10). Osteophytes were also detected in other joints (Fig. 2 B; data not shown). The ribs of mutant mice were severely distorted due to abnormal ossification (Fig. 2 G). Because these abnormalities were not found in younger mice and they become progressively worsen in aging population, we conclude that Smad3 plays an important role in maintaining skeletal integrity rather than in its formation.
Figure 2.

Abnormal skeletal development in Smad3 ex8/ex8 mice. (A and B) Alizarin red S and Alcian blue staining of hindlimbs of P21 (A) and 7-mo-old (B) mice. Genotypes as indicated. (C–H) Rib cages of P21 (C and D), 5- (E and F), and 7-mo-old (G and H) mice. F is an enlarged view of E showing accumulation of bony material in sternum joints. Arrows point to abnormally formed bones of mutant mice. (I and J) Angular defects of tarsal bones in P21 mouse.
Although the majority of bones in younger mice were normal, we found that ∼30% of the Smad3ex8/ex8 mice exhibited unilateral or bilateral angular distortion in their forelimbs due to abnormal formation in tarsal bones (Fig. 2 I). Since this defect is seen in P1 mice and does not show progressive nature compared with the degenerative skeletal defects found in older mice, it may therefore represent a patterning defect in embryonic skeletal development caused by the Smad3 deficiency. The mechanism underlying this defect is currently unknown and will be addressed in future studies.
Histologic Analysis of Skeletal Tissues of the Smad3ex8/ex8 Mice
To characterize the degenerative abnormalities in Smad3ex8/ex8 mice in more detail, synovial joints were sectioned for histologic analysis. There were no apparent differences in bone mass between Smad3ex8/ex8 and control mice ∼30 d of age as revealed by x-ray and whole mount skeletal preparation (Fig. 2A and Fig. C; data not shown). However, an abnormal increase in the number of hypertrophic chondrocytes was seen in the articular cartilage of mutant mice at this stage (Fig. 3 B), whereas the majority of articular cartilage cells in the control mice remained as resting chondrocytes (Fig. 3 A). The histology of knee joints from mutant mice at 4 mo of age revealed progressive loss of the smooth surface of articular cartilage. The articular surface of mutant joints with mild degeneration was covered with abnormally differentiated chondrocytes (Fig. 3 D) instead of a thin layer of resting chondrocytes found in control mice (Fig. 3 C). Surface fibrillation (vertical cleft development) and osteophytes with varying sizes were seen in the synovial cavities of most 6-mo-old mutant mice (Fig. 3F and Fig. G). Although these osteophytes were positive for type II collagen (Fig. 3 H), a marker for chondrocytes, type I collagen, and osteocalcin markers for osteoblast cells were also detected (Fig. 3I and Fig. J), revealing the presence of both chondrocytes and osteoblasts in the outgrowth of endochondral tissues. Similar abnormalities were also detected in vertebral joints of mutant mice (data not shown). These observations indicate that Smad3 deficiency does not interfere with synovial joint formation; however, it causes progressive articular cartilage degeneration resembling osteoarthritis in humans.
Figure 3.
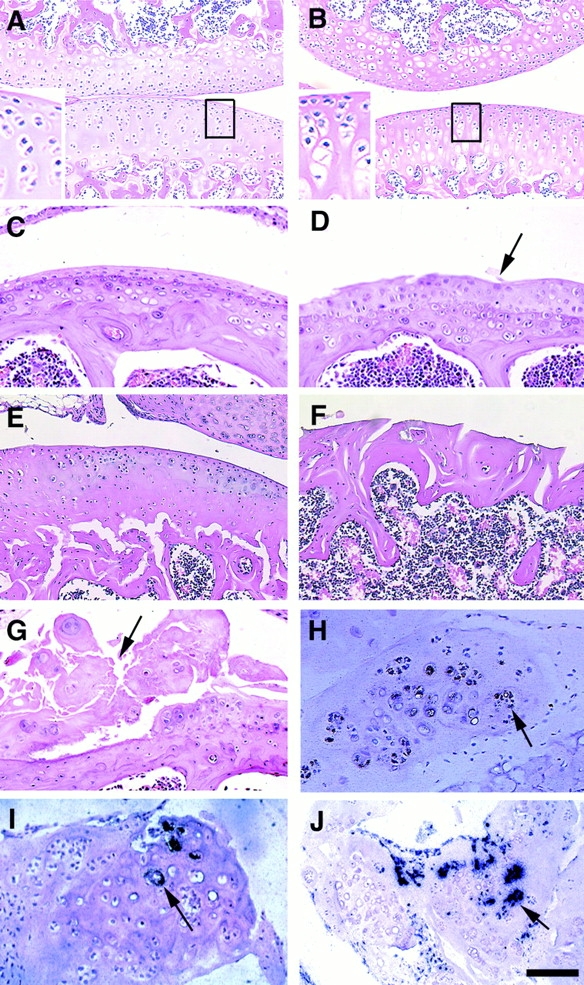
Histologic analysis of articular cartilage of Smad3 ex8/ex8 mice. (A and B) Knee joints of P30 wild-type (A) and mutant (B) mice. Notice the increased number of hypertrophic chondrocytes in mutant articular cartilage. (C–F) Knee joints of 4- (C and D) and 7-mo-old (E and F) wild-type (C and E) and mutant (D and F) mice. Arrow points to abnormally accumulated layer of chondrocytes (D). (G–J) Osteophytes found in synovium of 7-mo-old mice. Arrows point to osteophytes, hematoxylin and eosin (G), collagen type II (H), collagen type I (I), and osteocalcin (J) expression in osteophytes. Bar: (A and B) 150 μm; (C–J) 100 μm.
Increased Hypertrophic Differentiation of Epiphyseal Growth Plate Chondrocytes in Smad3ex8/ex8 Mice
We next examined the epiphyseal growth plates to determine if Smad3 deficiency could cause similar defects. Chondrocytes in wild-type growth plates can be divided into four different cell types: resting, proliferating, maturing, and hypertrophic chondrocytes (Fig. 4A and Fig. B). Examination of mutant mice at multiple early developmental stages, including P1, P9, and P12, did not reveal obvious abnormalities compared with their littermate controls (Fig. 4A and Fig. B). This observation indicates that loss of Smad3 does not interfere with chondrogenesis at these developmental stages. However, the height of the hypertrophic zone gradually increased in some 3–4-wk-old Smad3ex8/ex8 mice (Fig. 4C and Fig. D, arrows). This phenotype became more obvious in 6–8-wk-old mutant mice (Fig. 4, E–H). This observation suggests that the Smad3 deficiency caused increased hypertrophic differentiation of mutant growth plate chondrocytes. Furthermore, growth plates of P21 or older mutant animals also contained fewer proliferating chondrocytes than their controls (Fig. 4, C–H). In this case, mutant chondrocyte columns were not only shorter but also were arranged irregularly (Fig. 4, C–F, arrows), suggesting that Smad3-mediated signals also play a role in chondrocyte proliferation. Because the defects were not observed in younger mice, we suggest that Smad3 is required for maintenance, rather than formation of the epiphyseal growth plate. Similar abnormalities were also detected in vertebrae and sternum (data not shown).
Figure 4.
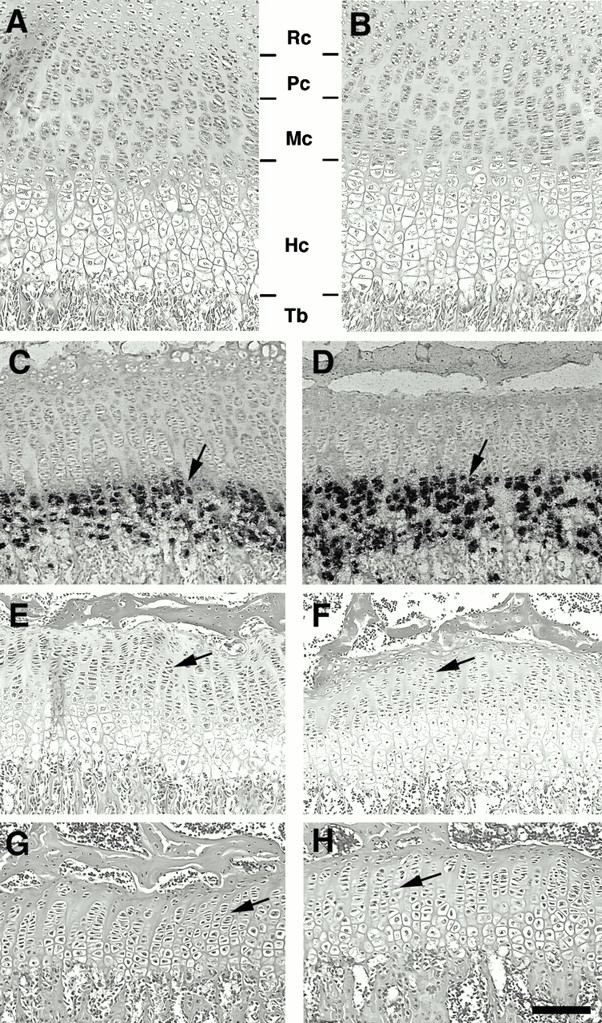
Histologic analysis of epiphyseal growth plates of Smad3 ex8/ex8 mice. (A and B) Sections of wild-type (A) and mutant (B) growth plates from P12 tibiae. Chondrocytes are divided into four distinct zones: resting (Rc), proliferation (Pc), maturing (Mc), and hypertrophic (Hc) chondrocytes. No apparent difference was detected. (C–H) Sections of wild-type (C, E, and G) and mutant (D, F, and H) growth plates isolated from P21, (C and D), P40 (E and F), and P60 (G and H) mice. Arrows in C and D point to hypertrophic chondrocytes positive for type X collagen. Arrows in E and H point to chondrocyte columns. Tb, trabecular bone. Bar: (A, B, G, and H) 80 μm; (C, D, E, and F) 120 μm.
Increased Expression of Type X Collagen and Decreased Proteoglycan Content in Smad3ex8/ex8 Mice
To characterize the skeletal abnormalities of Smad3ex8/ex8 mutant mice at molecular level, we checked the expression of type X collagen, a marker of chondrocyte differentiation. More intense type X collagen staining was found in the Smad3ex8/ex8 growth plate (Fig. 5 B) and articular cartilage (Fig. 5 D), as compared with controls (Fig. 5A and Fig. C). The type X staining also marked the osteophytes (Fig. 5 E).
Figure 5.
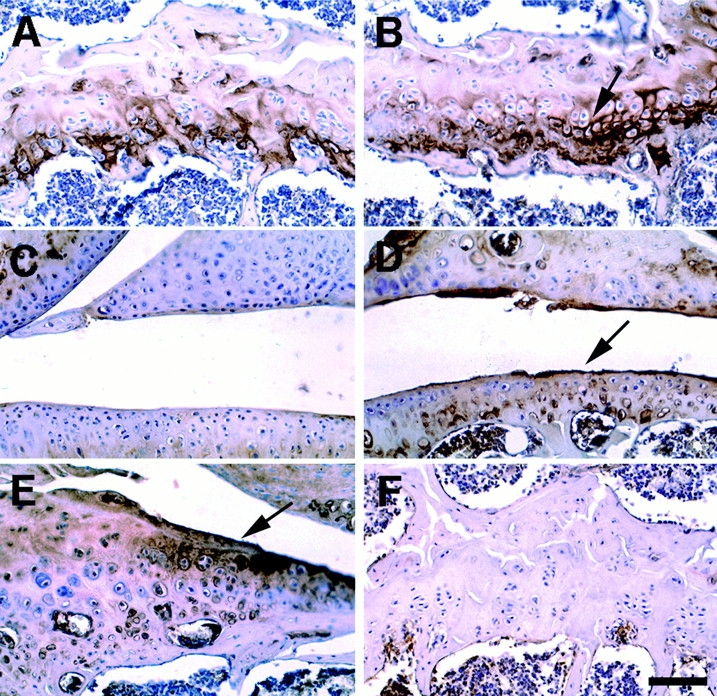
Immunohistochemical detection of collagen type X expression in 6-mo-old wild-type (A and C) and mutant (B, D, and E) mice. A and B, growth plates; C and D, knee joints; and E, osteophyte. F is an image without the first antibody serving as a negative control. Bar, 100 μm.
Abnormal differentiation of articular cartilage may lead to changes of matrix components. Therefore, we examined expression of proteoglycans, a class of matrix proteins that are produced by chondrocytes and interact with hyaluronic acid, link proteins, and collagen fibers. No apparent differences in safranine O, which stains proteoglycans, were found in P12 mutant and control mice (Fig. 6A and Fig. B). However, P40 Smad3 ex8/ex8 mice showed significantly reduced safranine O staining in both the articular cartilage and growth plate compared with wild-type mice (Fig. 6C and Fig. D). Decreased safranine O staining was often found in articular joint of 6-mo-old Smad3 ex8/ex8 mice as more cartilage turned into bone (Fig. 6, E–H). Small regions of enhanced safranine O staining were also observed in some abnormal outgrowths of the articular cavity (not shown), indicating fibrocartilaginous repair occurred. These results suggest that the lack of Smad3-mediated TGF-β signals promotes the terminal differentiation of articular cartilage and may affect the rate of cartilage matrix turnover, eventually leading to articular cartilage degeneration.
Figure 6.
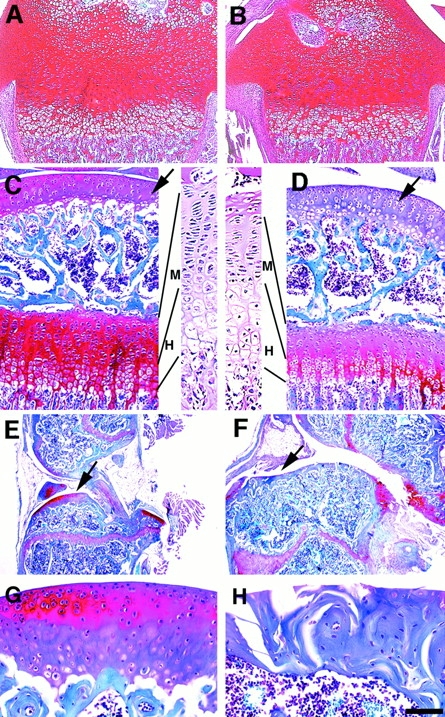
Expression of proteoglycans in wild-type (A, C, E, and G) and mutant (B, D, F, and H) skeletal tissues. Safranine O staining of epiphyseal growth plates and articular cartilage of P12 (A and B), P40 (C and D), and 6-mo-old (E–H) mice. Arrows point to articular cartilage. Enlarged images in C and D show hematoxylin and eosin sections. H, hypertrophic zone; and M, all other zones including Rc, Pc, and Mc. Bar: (A and B) 250 μm; (C, D, G, and H) 150 μm; (E and F) 600 μm.
Expression of Smad3, FGFR3, and Indian Hedgehog in Skeletal Tissues
Previous studies showed that Smad3 is expressed in multiple organs and/or tissues (Yang et al. 1999). Smad3 protein is also detected in the maturing zone of rat epiphyseal growth plates at a level higher than other zones (Sakou et al. 1999). However, its expression in the mouse articular cartilage and synovium has not been studied. Therefore, we checked expression of Smad3 during endochondral ossification in the rib cage and knee joints using in situ hybridization. Our data showed that Smad3 is expressed in the perichondrium of the ribs and the sternebrae joints with higher levels (Fig. 7, A–C). Smad3 transcripts were also detected in articular cartilage and trabecular bones (Fig. 7, D–F). This expression pattern correlates with sites of abnormalities found in the Smad3 ex8/ex8 mice, suggesting that loss of Smad3 signals in Smad3 ex8/ex8 mice could be primary for these defects.
Figure 7.
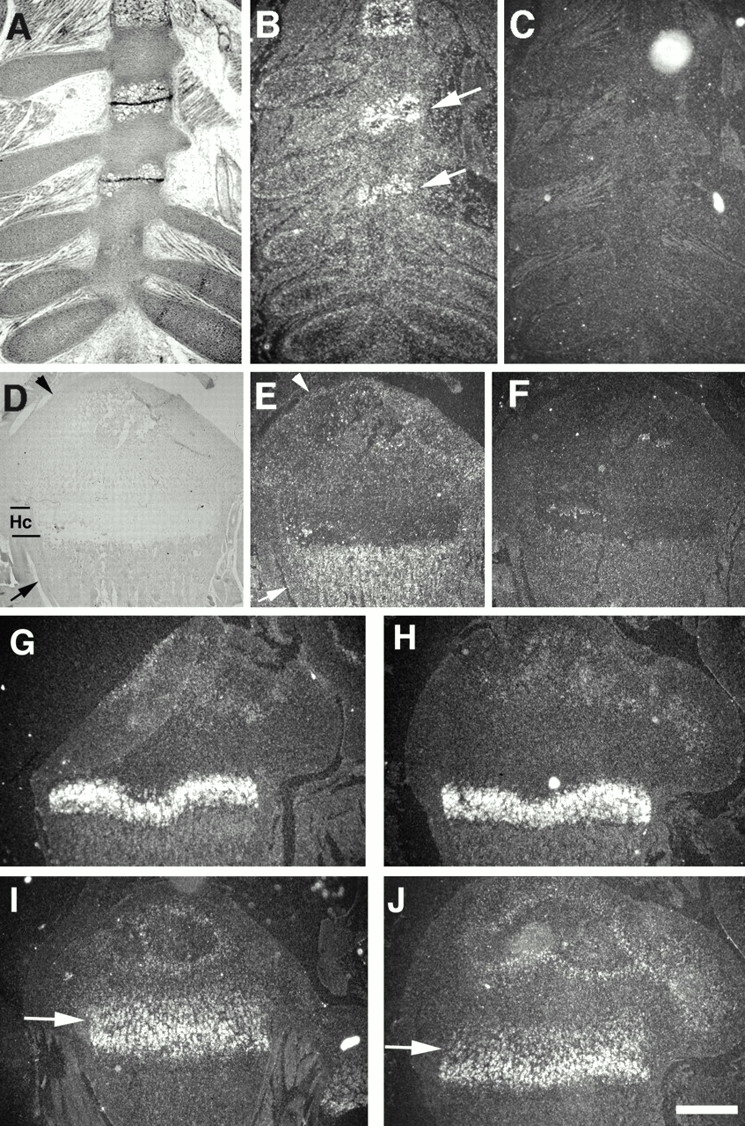
Analysis of gene expression by in situ hybridization. (A–F) Smad3 expression in ribs of embryonic day 16 (A–C) and P12 growth plate (D–F) of wild-type mice. A and D are bright field view, and B, C, E, and F are dark field view. B and E are anti-sense, and C and F are sense controls. Arrow and arrowhead in E point to perichondrium and articular cartilage, respectively. (G and H) Ihh expression in wild-type (G) and mutant (H) mice. (I and J) FGFR3 expression in wild-type (I) and mutant (J) mice. Hc, hypertrophic chondrocytes. Bar: (A and B) 700 μm; (D–F) 600 μm; (G–J) 500 μm.
Notably, expression pattern of Smad3 overlaps with that of several growth factors and signaling molecules, including Indian hedgehog (Ihh) and FGFR3, both of which are important for long bone growth (Deng et al. 1996; Chen et al. 1999; Li et al. 1999; St-Jacques et al. 1999; Iwata et al. 2000). In situ hybridization analysis revealed no significant change of Ihh (Fig. 7G and Fig. H). However, FGFR3 expression in proliferating chondrocytes was slightly decreased (Fig. 7I and Fig. J, arrows), whereas no change in the maturing zone of chondrocytes was detected. This observation suggests that Smad3 signals may regulate FGFR3 expression in chondrocytes. However, the significance of this regulation is currently unknown and should be addressed in future studies. Next, we checked expression of Smad2, which shares a transduction pathway with Smad3 in mediating TGF-β and activin signals. Our data showed that Smad2 expression in wild-type growth plates is similar to Smad3 (data not shown). We detected no change in expression of Smad2 in Smad3 ex8/ex8 chondrocytes, indicating that the loss of Smad3 did not cause a complementary alteration of this highly related gene.
Diminished Response of Smad3ex8/ex8 Bone Rudiment to TGF-β1 Inhibition
Previous studies have shown that TGF-β1 can inhibit both chondrocyte proliferation and hypertrophic differentiation (Ballock et al. 1993; Tschan et al. 1993; Bohme et al. 1995; Serra et al. 1997). TGF-β1, 2, 3 and their receptors are expressed in the perichondrium, articular cartilage, and growth plate chondrocytes (Fukumura et al. 1998; Horner et al. 1998; Kabasawa et al. 1998; Sakou et al. 1999) where Smad3 is expressed. It is conceivable that the diminished chondrocyte response to TGF-β at these sites is a primary cause for the observed abnormalities. To test this, we used an in vitro embryonic bone culture system where bone growth could be maintained in defined conditions to monitor chondrocyte proliferation and differentiation. For these experiments, we cultured embryonic metatarsals from E17.5 mice. Bone rudiments at this stage have already initiated endochondral ossification as illustrated in Fig. 8 A. Bone growth was determined by increased whole bone length and hypertrophic zone expansion toward the rudiments' ends 4 d after culture (Fig. 8A and Fig. B). In the presence of TGF-β1 (10 ng/ml), however, the expansion of hypertrophic zone was repressed in comparison to untreated controls (Fig. 8B and Fig. F). Quantitative measurement of nine pairs of cultured bones indicated that the average length of hypertrophic zones (HLs) of the treated bones is ∼70% that of wild-type bones (Fig. 8 M). In the histologic sections, bones treated with TGF-β1 exhibited a narrower region containing hypertrophic chondrocytes of smaller sizes (Fig. 8 K, arrow) compared with corresponding region in untreated controls (Fig. 8 I). These observations indicate that TGF-β1 significantly inhibits chondrocyte differentiation.
Figure 8.
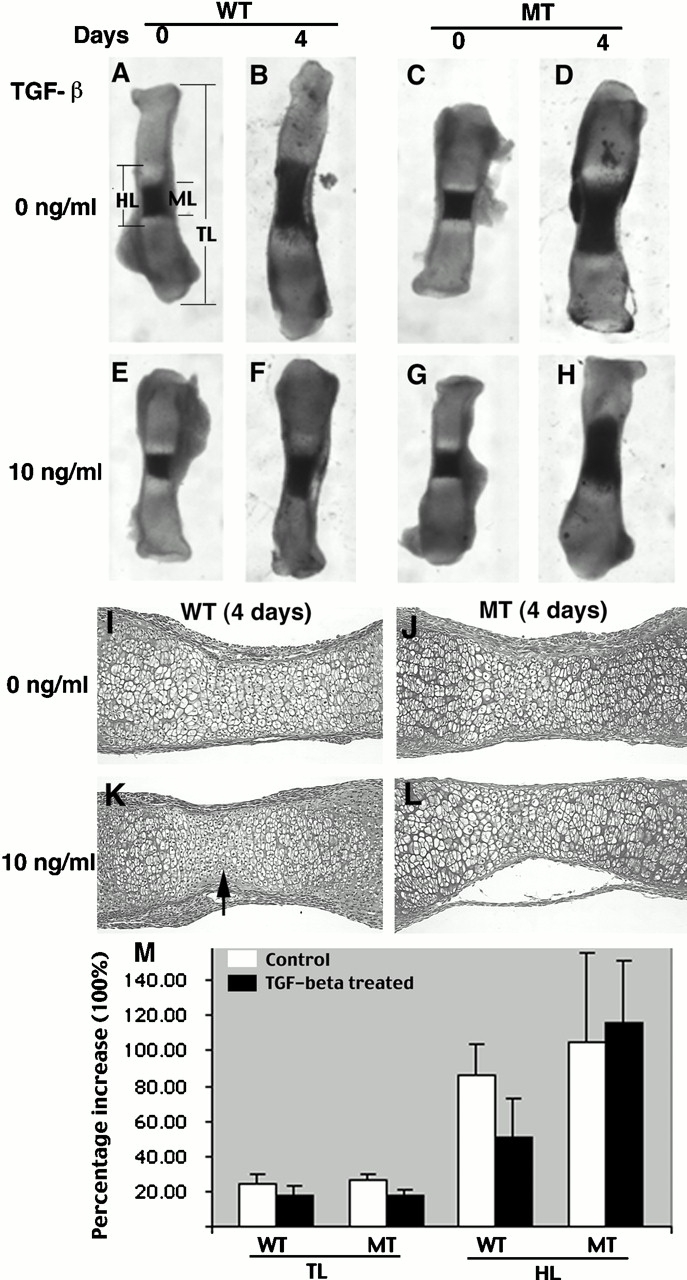
Effects of TGF-β1 treatment on cultured wild-type (A, B, E, F, I, and K) and Smad3 mutant (C, D, G, H, J, and L) embryonic (E17.5) metatarsal bones. (ML) The length of the mineralized hypertrophic chondrocytes (dark area). The light areas flanking the length of the mineralized hypertrophic chondrocytes (ML) are the nonmineralized hypertrophic chondrocytes. The regions outside the light zones are proliferating chondrocytes. Hypertrophic zone (HL) includes both mineralized and unmineralized hypertrophic zones. TL, total bone length. (I–L) Histologic sections of wild-type and mutant bones cultured for 4 d in the absence (I and J) and presence (K and L) of TGF-β1. The arrow in K points to a region containing hypertrophic chondrocytes with smaller sizes. (M) Percentage increases in TL and HL of wild-type and mutant bones cultured for 4 d in the absence (0 ng/ml) or presence of TGF-β1 (10 ng/ml). WT, wild type; MT, mutant bones. (Percentage increase = [day 4 length − day 0 length]/day 0 length). Data represent mean ± SD of values obtained from nine pairs of bones.
We then examined bones isolated from Smad3 ex8/ex8 mice. Without TGF-β1, there is no significant difference between mutant and wild-type bones (Fig. 8B and Fig. D). This is consistent with our observation that Smad3 mutation does not affect bone growth at early stages. Notably, the TGF-β1 treatment could not repress the expansion of the hypertrophic zone of mutant explants (Fig. 8D and Fig. H, and Fig. 8 M for quantitative comparison). Histologic sections revealed no apparent morphological changes between treated and untreated bones (Fig. 8J and Fig. L). These observations indicate that the inhibition of TGF-β1 on chondrocyte hypertrophic differentiation is largely Smad3 dependent.
Our data also showed that the TLs of treated bones were slightly reduced compared with that of the untreated controls (Fig. 8 M), suggesting that TGF-β1 treatment decreased proliferation of chondrocytes in the culture conditions. This function is Smad3 independent since both wild-type and Smad3 mutant bones showed similar reductions.
Discussion
Osteoarthritis occurs at a very high frequency affecting the majority of the aging population (Fujita 1997; Simon 1999). Although multiple factors have been shown to affect articular cartilage in osteoarthritis, the molecular mechanism(s) underlying the cartilage degeneration is largely unknown. In this study, we have investigated a role of TGF-β/Smad3 signals in bone formation and cartilage development in Smad3 ex8/ex8 mice generated by gene targeting. We showed that Smad3 deficiency results in a skeletal condition that exhibits some features mimicking human osteoarthritis, including progressive loss of articular cartilage, formation of large osteophytes, decreased production of proteoglycan, and abnormally increased number of type X collagen–expressing chondrocytes in synovial joints. We further showed that the articular cartilage degeneration in Smad3 ex8/ex8 mice is associated with increased chondrocyte differentiation, which is normally repressed by TGF-β/Smad3 signals. Thus, our study establishes an essential role of Smad3 in maintaining articular cartilage integrity, which is pivotally important for normal joint functions.
TGF-βs, especially TGF-β1, 2, and 3 and their receptors, have been implicated in osteoarthritis (van den Berg 1999). When injected into knee joints of mouse or rabbit, TGF-β1 or 2 can stimulate chondrocyte differentiation and induce osteophyte formation at sites characteristic for osteoarthritis (van Beuningen et al. 1994, van Beuningen et al. 2000; van den Berg 1995), suggesting that TGF-βs may serve as pathogens for this disease. On the other hand, it was also showed that TGF-β1 can suppress acute and chronic arthritis in rat, implicating this cytokine as a potentially important therapeutic agent (Brandes et al. 1991). Moreover, it was shown that expression of a dominant negative type II TGF-β receptors in cartilage and the synovium altered chondrocyte differentiation and resulted in degenerative joint abnormalities resembling human osteoarthritis (Serra et al. 1997). These seemingly contradictory data suggest the function of TGF-βs in endochondral ossification is complex. Indeed, as multifunctional growth factors, TGF-βs could exhibit differential effects depending on the differentiation stage of the target cells. Numerous studies in multiple model systems have documented that although TGF-βs stimulate early chondrocyte differentiation, they inhibit the later stages of cartilage formation, especially the terminal differentiation of chondrocytes (Seyedin et al. 1985; Kulyk et al. 1989; Ballock et al. 1993; Lafeber et al. 1993; Tschan et al. 1993; Dieudonne et al. 1994; Bohme et al. 1995; Denker et al. 1995; Serra et al. 1999). Alternatively, it is possible that different doses of TGF-β could cause a biphasic response in same type of cells by activating different downstream effectors with opposite functions.
TGF-β transduces signals from the cell membrane to the nucleus via specific type I and II receptors and Smad proteins. TGF-β1, 2, and 3 and their receptors are expressed in articular cartilage and the epiphyseal growth plate (Fukumura et al. 1998; Horner et al. 1998; Kabasawa et al. 1998; Sakou et al. 1999). TGF-β/activin–restricted Smads also expressed epiphyseal growth plate in a partially overlapping fashion with Smad2 and 3 strongly expressed in proliferating chondrocytes and maturing chondrocytes, respectively. Smad4, which serves as a central mediator for both TGF-βs and BMPs, is broadly expressed in all zones of epiphyseal growth plate (Sakou et al. 1999). The overlapping expressions of TGF-β, receptors, and Smads suggest that TGF-β functions in an autocrine fashion during endochondral ossification.
This brings an essential question that this study aims to address, that is, what is the physiological function of Smad3-mediated TGF-β signals in endochondral ossification? The observations that blocking TGF-β signals at either receptor (Serra et al. 1997) or intracellular level (this study) causes increased chondrocyte terminal differentiation and progressive degeneration of articular cartilage indicate that one of the physiological functions of TGF-β/Smad3 signals must be the inhibition of chondrocyte terminal differentiation. This notion is strongly supported by our in vitro rudiment culture experiments, where TGF-β1 treatment dramatically inhibited chondrocyte hypertrophy in wild-type but not in Smad3 ex8/ex8 bones. These observations indicate that the inhibition of TGF-β1 on chondrocyte differentiation is largely Smad3 dependent. Thus, lacking TGF-β1/Smad3 signals, chondrocytes undergo abnormal differentiation, which eventually leads to osteoarthritis.
Because TGF-β has been known as a cartilage inducer and can stimulate cartilage formation both in vitro and in vivo (Kulyk et al. 1989; Joyce et al. 1990; Lafeber et al. 1993; Denker et al. 1995; Chimal-Monroy and Diaz de Leon 1997), we carefully examined Smad3 ex8/ex8 mice to see if blocking TGF-β responsiveness could affect early stages of chondrogenesis. Our examinations at multiple developmental stages detected no developmental defects in synovial joints and epiphyseal growth plate formation, suggesting that Smad3 is dispensable in the early stages of cartilage formation. In histologic sections, we found that the increased hypertrophic differentiation of chondrocytes started when animals were ∼1 mo of age. This leads to the progressive loss of articular cartilage and formation of osteophytes in synovial joints of older mutant mice. Of note, the severity of cartilage degeneration correlates well with expression levels of Smad3. Sternebrae joints, where Smad3 is expressed at a particularly higher level, showed most severe phenotype (Fig. 2, E–G, and Fig. 7 B).
Cartilage degeneration associated with osteoarthritis is also linked to aberrant cytokine and growth factor expression in affected tissues. One of the best studied factors so far is IL-1, which stimulates chondrocytes to release destructive proteases and at the same time, represses proteoglycan synthesis (Beekman et al. 1998; Hui et al. 1998; Cawston et al. 1999; Flannery et al. 1999; Sandy et al. 1999). Thus, the combined effect of IL-1 action is reduced matrix production and enhanced cartilage degradation. However, the osteoarthritis in Smad3 ex8/ex8 mice is distinct from that caused by IL-1 overexpression. It shows no sign of enzymatic bone destruction rather than the abnormally increased bone production in synovial joints at the expense of increased chondrocyte differentiation, which starts shortly after weaning.
Another feature exhibited by Smad3 ex8/ex8 articular cartilage is significantly reduced levels of proteoglycan. This observation is consistent with previous reports that synthesis of proteoglycan can be stimulated by TGF-β both in vitro and in vivo (Lafeber et al. 1993; Redini et al. 1997; Moller et al. 2000). The reduced proteoglycan production may significantly contribute to osteoarthritis since it may lead to further changes in the components of cartilage matrix and the rate of matrix turnover in Smad3 ex8/ex8 mice. Interestingly, the decreased levels of proteoglycans associated with Smad3 deficiency is similar to that caused by the IL-1 overexpression (van de Loo et al. 1994, van de Loo et al. 1998). This suggests partial overlapping roles between IL-1 and TGF-β/Smad3 in osteoarthritis initiation and progression.
In summary, we show that Smad3-mediated TGF-β signals are important for maintaining articular cartilage in the quiescent state by repressing chondrocyte differentiation and controlling matrix molecule synthesis. Consequently, impairment of TGF-β signals due to Smad3 disruption results in phenotypes resembling human osteoarthritis. These mice should serve as an ideal animal model for performing studies that may eventually lead to the prevention and effective treatment of osteoarthritis.
Acknowledgments
We thank B. Olsen for providing polyclonal antibody and in situ hybridization probe to type X collagen, and G. Karsenty, A. McMahan, and Y. Yamada for probes for mouse osteocalcin, Ihh, and type I collagen, respectively. We are grateful for S.G. Brodie, B. Ding, A. Engle, and C. Tifft for critically reading the manuscript.
X. Yang is supported in part by grants from the National Natural Science Foundation of China (39970413), the National Science Fund for Distinguished Young Scholars (30025028), and the National High-Tech Research and Development program (102-08-08-02).
Footnotes
X. Yang and L. Chen contributed equally to this work.
Abbreviations used in this paper: BMP, bone morphogenetic protein; FGFR, FGF receptor; HL, hypertrophic length; Ihh, Indian hedgehog; P, postnatal day; TL, total length.
References
- Ashcroft G.S., Yang X., Glick A.B., Weinstein M., Letterio J.L., Mizel D.E., Anzano M., Greenwell-Wild T., Wahl S.M., Deng C. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat. Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- Ballock R.T., Heydemann A., Wakefield L.M., Flanders K.C., Roberts A.B., Sporn M.B. TGF-beta 1 prevents hypertrophy of epiphyseal chondrocytesregulation of gene expression for cartilage matrix proteins and metalloproteases. Dev. Biol. 1993;158:414–429. doi: 10.1006/dbio.1993.1200. [DOI] [PubMed] [Google Scholar]
- Beekman B., Verzijl N., de Roos J.A., TeKoppele J.M. Matrix degradation by chondrocytes cultured in alginateIL-1 beta induces proteoglycan degradation and proMMP synthesis but does not result in collagen degradation. Osteoarthritis Cartilage. 1998;6:330–340. doi: 10.1053/joca.1998.0132. [DOI] [PubMed] [Google Scholar]
- Bohme K., Winterhalter K.H., Bruckner P. Terminal differentiation of chondrocytes in culture is a spontaneous process and is arrested by transforming growth factor-beta 2 and basic fibroblast growth factor in synergy. Exp. Cell Res. 1995;216:191–198. doi: 10.1006/excr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Brandes M.E., Allen J.B., Ogawa Y., Wahl S.M. Transforming growth factor beta 1 suppresses acute and chronic arthritis in experimental animals. J. Clin. Invest. 1991;87:1108–1113. doi: 10.1172/JCI115073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawston T., Billington C., Cleaver C., Elliott S., Hui W., Koshy P., Shingleton B., Rowan A. The regulation of MMPs and TIMPs in cartilage turnover. Annu. NY Acad. Sci. 1999;878:120–129. doi: 10.1111/j.1749-6632.1999.tb07678.x. [DOI] [PubMed] [Google Scholar]
- Chen L., Adar R., Yang X., Monsonego E.O., Li C., Hauschka P.V., Yayon A., Deng C.X. Gly369Cys mutation in mouse FGFR3 causes achondroplasia by affecting both chondrogenesis and osteogenesis. J. Clin. Invest. 1999;104:1517–1525. doi: 10.1172/JCI6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimal-Monroy J., Diaz de Leon L. Differential effects of transforming growth factors beta 1, beta 2, beta 3 and beta 5 on chondrogenesis in mouse limb bud mesenchymal cells. Int. J. Dev. Biol. 1997;41:91–102. [PubMed] [Google Scholar]
- Datto M., Wang X.F. The Smadstranscriptional regulation and mouse models. Cytokine Growth Factor Rev. 2000;11:37–48. doi: 10.1016/s1359-6101(99)00027-1. [DOI] [PubMed] [Google Scholar]
- Datto M.B., Frederick J.P., Pan L., Borton A.J., Zhuang Y., Wang X.F. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol. Cell. Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C., Wynshaw-Boris A., Zhou F., Kuo A., Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Denker A.E., Nicoll S.B., Tuan R.S. Formation of cartilage-like spheroids by micromass cultures of murine C3H10T1/2 cells upon treatment with transforming growth factor-beta 1. Differentiation. 1995;59:25–34. doi: 10.1046/j.1432-0436.1995.5910025.x. [DOI] [PubMed] [Google Scholar]
- Dieudonne S.C., Semeins C.M., Goei S.W., Vukicevic S., Nulend J.K., Sampath T.K., Helder M., Burger E.H. Opposite effects of osteogenic protein and transforming growth factor beta on chondrogenesis in cultured long bone rudiments. J. Bone Miner. Res. 1994;9:771–780. doi: 10.1002/jbmr.5650090603. [DOI] [PubMed] [Google Scholar]
- DiLeone R.J., King J.A., Storm E.E., Copeland N.G., Jenkins N.A., Kingsley D.M. The Bmp8 gene is expressed in developing skeletal tissue and maps near the Achondroplasia locus on mouse chromosome 4. Genomics. 1997;40:196–198. doi: 10.1006/geno.1996.4533. [DOI] [PubMed] [Google Scholar]
- Ducy P., Karsenty G. The family of bone morphogenetic proteins. Kidney Int. 2000;57:2207–2214. doi: 10.1046/j.1523-1755.2000.00081.x. [DOI] [PubMed] [Google Scholar]
- Eppert K., Scherer S.W., Ozcelik H., Pirone R., Hoodless P., Kim H., Tsui L.C., Bapat B., Gallinger S., Andrulis I.L. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86:543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- Erlebacher A., Filvaroff E.H., Gitelman S.E., Derynck R. Toward a molecular understanding of skeletal development. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- Flannery C.R., Little C.B., Caterson B., Hughes C.E. Effects of culture conditions and exposure to catabolic stimulators (IL-1 and retinoic acid) on the expression of matrix metalloproteinases (MMPs) and disintegrin metalloproteinases (ADAMs) by articular cartilage chondrocytes. Matrix Biol. 1999;18:225–237. doi: 10.1016/s0945-053x(99)00024-4. [DOI] [PubMed] [Google Scholar]
- Fujita T. Osteoporosispast, present and future Osteoporos. Int 7Suppl.1997. S6 S9 [DOI] [PubMed] [Google Scholar]
- Fukumura K., Matsunaga S., Yamamoto T., Nagamine T., Ishidou Y., Sakou T. Immunolocalization of transforming growth factor-beta s and type I and type II receptors in rat articular cartilage. Anticancer Res. 1998;18:4189–4193. [PubMed] [Google Scholar]
- Gatherer D., Ten Dijke P., Baird D.T., Akhurst R.J. Expression of TGF-beta isoforms during first trimester human embryogenesis. Development. 1990;110:445–460. doi: 10.1242/dev.110.2.445. [DOI] [PubMed] [Google Scholar]
- Goldring S.R. A 55-year-old woman with rheumatoid arthritis. JAMA. 2000;283:524–531. doi: 10.1001/jama.283.4.524. [DOI] [PubMed] [Google Scholar]
- Graff J.M., Bansal A., Melton D.A. Xenopus Mad proteins transduce distinct subsets of signals for the TGF beta superfamily. Cell. 1996;85:479–487. doi: 10.1016/s0092-8674(00)81249-0. [DOI] [PubMed] [Google Scholar]
- Heldin C.H., Miyazono K., ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Horner A., Kemp P., Summers C., Bord S., Bishop N.J., Kelsall A.W., Coleman N., Compston J.E. Expression and distribution of transforming growth factor-beta isoforms and their signaling receptors in growing human bone. Bone. 1998;23:95–102. doi: 10.1016/s8756-3282(98)00080-5. [DOI] [PubMed] [Google Scholar]
- Hui A., Min W.X., Tang J., Cruz T.F. Inhibition of activator protein 1 activity by paclitaxel suppresses interleukin-1-induced collagenase and stromelysin expression by bovine chondrocytes. Arthritis Rheum. 1998;41:869–876. doi: 10.1002/1529-0131(199805)41:5<869::AID-ART15>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Iwata T., Chen L., Li C., Ovchinnikov D.A., Behringer R.R., Francomano C.A., Deng C.X. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum. Mol. Genet. 2000;9:1603–1613. doi: 10.1093/hmg/9.11.1603. [DOI] [PubMed] [Google Scholar]
- Joyce M.E., Roberts A.B., Sporn M.B., Bolander M.E. Transforming growth factor-beta and the initiation of chondrogenesis and osteogenesis in the rat femur. J. Cell Biol. 1990;110:2195–2207. doi: 10.1083/jcb.110.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabasawa Y., Ejiri S., Matsuki Y., Hara K., Ozawa H. Immunoreactive localization of transforming growth factor-beta type II receptor-positive cells in rat tibiae. Bone. 1998;22:93–98. doi: 10.1016/s8756-3282(97)00249-4. [DOI] [PubMed] [Google Scholar]
- Katagiri T., Boorla S., Frendo J.L., Hogan B.L., Karsenty G. Skeletal abnormalities in doubly heterozygous Bmp4 and Bmp7 mice. Dev. Genet. 1998;22:340–348. doi: 10.1002/(SICI)1520-6408(1998)22:4<340::AID-DVG4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Kawabata M., Imamura T., Miyazono K. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998;9:49–61. doi: 10.1016/s1359-6101(97)00036-1. [DOI] [PubMed] [Google Scholar]
- Kingsley D.M., Bland A.E., Grubber J.M., Marker P.C., Russell L.B., Copeland N.G., Jenkins N.A. The mouse short ear skeletal morphogenesis locus is associated with defects in a bone morphogenetic member of the TGF beta superfamily. Cell. 1992;71:399–410. doi: 10.1016/0092-8674(92)90510-j. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M., Liu F., Hata A., Doody J., Massague J. The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev. 1997;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- Kulyk W.M., Rodgers B.J., Greer K., Kosher R.A. Promotion of embryonic chick limb cartilage differentiation by transforming growth factor-beta. Dev. Biol. 1989;135:424–430. doi: 10.1016/0012-1606(89)90191-7. [DOI] [PubMed] [Google Scholar]
- Lafeber F.P., Vander Kraan P.M., Huber-Bruning O., Vanden Berg W.B., Bijlsma J.W. Osteoarthritic human cartilage is more sensitive to transforming growth factor beta than is normal cartilage. Br. J. Rheumatol. 1993;32:281–286. doi: 10.1093/rheumatology/32.4.281. [DOI] [PubMed] [Google Scholar]
- Lagna G., Hata A., Hemmati-Brivanlou A., Massague J. Partnership between DPC4 and SMAD proteins in TGF-beta signalling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- Li C., Chen L., Iwata T., Kitagawa M., Fu X.Y., Deng C.X. A Lys644Glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum. Mol. Genet. 1999;8:35–44. doi: 10.1093/hmg/8.1.35. [DOI] [PubMed] [Google Scholar]
- Liu F., Hata A., Baker J.C., Doody J., Carcamo J., Harland R.M., Massague J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- Luo G., Hofmann C., Bronckers A.L., Sohocki M., Bradley A., Karsenty G. BMP-7 is an inducer of nephrogenesis, and is also required for eye development and skeletal patterning. Genes Dev. 1995;9:2808–2820. doi: 10.1101/gad.9.22.2808. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M., Abdollah S., Hoodless P.A., Pirone R., Attisano L., Wrana J.L. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Massague J. TGF-beta signal transduction. Ann. Rev. Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- McLeod M.J. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- Mikic B., van der Meulen M.C., Kingsley D.M., Carter D.R. Long bone geometry and strength in adult BMP-5 deficient mice. Bone. 1995;16:445–454. [PubMed] [Google Scholar]
- Millan F.A., Denhez F., Kondaiah P., Akhurst R.J. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. doi: 10.1242/dev.111.1.131. [DOI] [PubMed] [Google Scholar]
- Moller H.D., Fu F.H., Niyibizi C., Studer R.K., Georgescu H.J., Robbins P.D., Evans C.H. TGF-beta-1 gene transfer in joint cartilage cells. Stimulating effect in extracellular matrix synthesis. Orthopade. 2000;29:75–79. doi: 10.1007/s001320050012. [DOI] [PubMed] [Google Scholar]
- Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C.H., Miyazono K., ten Dijke P. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T., Snyder M.A., Grewal S.S., Tsuneizumi K., Tabata T., Christian J.L. Xenopus Smad8 acts downstream of BMP-4 to modulate its activity during vertebrate embryonic patterning. Development. 1998;125:857–867. doi: 10.1242/dev.125.5.857. [DOI] [PubMed] [Google Scholar]
- Pelton R.W., Dickinson M.E., Moses H.L., Hogan B.L. In situ hybridization analysis of TGF beta 3 RNA expression during mouse developmentcomparative studies with TGF beta 1 and beta 2. Development. 1990;110:609–620. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- Redini F., Min W., Demoor-Fossard M., Boittin M., Pujol J.P. Differential expression of membrane-anchored proteoglycans in rabbit articular chondrocytes cultured in monolayers and in alginate beads. Effect of transforming growth factor-beta 1. Biochim. Biophys. Acta. 1997;1355:20–32. doi: 10.1016/s0167-4889(96)00115-2. [DOI] [PubMed] [Google Scholar]
- Sakou T., Onishi T., Yamamoto T., Nagamine T., Sampath T., Ten Dijke P. Localization of Smads, the TGF-beta family intracellular signaling components during endochondral ossification. J. Bone Miner. Res. 1999;14:1145–1152. doi: 10.1359/jbmr.1999.14.7.1145. [DOI] [PubMed] [Google Scholar]
- Sandberg M., Autio-Harmainen H., Vuorio E. Localization of the expression of types I, III, and IV collagen, TGF-beta 1 and c-fos genes in developing human calvarial bones. Dev. Biol. 1988;130:324–334. doi: 10.1016/0012-1606(88)90438-1. [DOI] [PubMed] [Google Scholar]
- Sandy J.D., Thompson V., Verscharen C., Gamett D. Chondrocyte-mediated catabolism of aggrecanevidence for a glycosylphosphatidylinositol-linked protein in the aggrecanase response to interleukin-1 or retinoic acid. Arch. Biochem. Biophys. 1999;367:258–264. doi: 10.1006/abbi.1999.1234. [DOI] [PubMed] [Google Scholar]
- Serra R., Johnson M., Filvaroff E.H., LaBorde J., Sheehan D.M., Derynck R., Moses H.L. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J. Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra R., Karaplis A., Sohn P. Parathyroid hormone-related peptide (PTHrP)-dependent and -independent effects of transforming growth factor beta (TGF-beta) on endochondral bone formation. J. Cell Biol. 1999;145:783–794. doi: 10.1083/jcb.145.4.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedin S.M., Thomas T.C., Thompson A.Y., Rosen D.M., Piez K.A. Purification and characterization of two cartilage-inducing factors from bovine demineralized bone. Proc. Natl. Acad. Sci. USA. 1985;82:2267–2271. doi: 10.1073/pnas.82.8.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon L.S. Osteoarthritisa review. Clin. Cornerstone. 1999;2:26–37. doi: 10.1016/s1098-3597(99)90012-1. [DOI] [PubMed] [Google Scholar]
- Solloway M.J., Dudley A.T., Bikoff E.K., Lyons K.M., Hogan B.L., Robertson E.J. Mice lacking Bmp6 function. Dev. Genet. 1998;22:321–339. doi: 10.1002/(SICI)1520-6408(1998)22:4<321::AID-DVG3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- St-Jacques B., Hammerschmidt M., McMahon A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation Genes Dev 13 1999. 2072 2086(erratum published 13:2617). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm E.E., Kingsley D.M. Joint patterning defects caused by single and double mutations in members of the bone morphogenetic protein (BMP) family. Development. 1996;122:3969–3979. doi: 10.1242/dev.122.12.3969. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Chang C., Yingling J.M., Wang X.F., Hemmati-Brivanlou A. Smad5 induces ventral fates in Xenopus embryo. Dev. Biol. 1997;184:402–405. doi: 10.1006/dbio.1997.8548. [DOI] [PubMed] [Google Scholar]
- Tschan T., Bohme K., Conscience-Egli M., Zenke G., Winterhalter K.H., Bruckner P. Autocrine or paracrine transforming growth factor-beta modulates the phenotype of chick embryo sternal chondrocytes in serum-free agarose culture. J. Biol. Chem. 1993;268:5156–5161. [PubMed] [Google Scholar]
- van Beuningen H.M., van der Kraan P.M., Arntz O.J., van den Berg W.B. Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Invest. 1994;71:279–290. [PubMed] [Google Scholar]
- van Beuningen H.M., Glansbeek H.L., van der Kraan P.M., van den Berg W.B. Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-beta injections. Osteoarthritis Cartilage. 2000;8:25–33. doi: 10.1053/joca.1999.0267. [DOI] [PubMed] [Google Scholar]
- van de Loo A.A., Arntz O.J., Otterness I.G., van den Berg W.B. Proteoglycan loss and subsequent replenishment in articular cartilage after a mild arthritic insult by IL-1 in miceimpaired proteoglycan turnover in the recovery phase. Agents Actions. 1994;41:200–208. doi: 10.1007/BF02001917. [DOI] [PubMed] [Google Scholar]
- van de Loo F.A., Arntz O.J., van Enckevort F.H., van Lent P.L., van den Berg W.B. Reduced cartilage proteoglycan loss during zymosan-induced gonarthritis in NOS2-deficient mice and in anti-interleukin-1-treated wild-type mice with unabated joint inflammation. Arthritis Rheum. 1998;41:634–646. doi: 10.1002/1529-0131(199804)41:4<634::AID-ART10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- van den Berg W.B. Growth factors in experimental osteoarthritistransforming growth factor beta pathogenic? J. Rheumatol. Suppl. 1995;43:143–145. [PubMed] [Google Scholar]
- van den Berg W.B. The role of cytokines and growth factors in cartilage destruction in osteoarthritis and rheumatoid arthritis. Z. Rheumatol. 1999;58:136–141. doi: 10.1007/s003930050163. [DOI] [PubMed] [Google Scholar]
- Vukicevic S., Luyten F.P., Reddi A.H. Stimulation of the expression of osteogenic and chondrogenic phenotypes in vitro by osteogenin. Proc. Natl. Acad. Sci. USA. 1989;86:8793–8797. doi: 10.1073/pnas.86.22.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein M., Yang X., Li C., Xu X., Gotay J., Deng C.X. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc. Natl. Acad. Sci. USA. 1998;95:9378–9383. doi: 10.1073/pnas.95.16.9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein M., Yang X., Deng C. Functions of mammalian Smad genes as revealed by targeted gene disruption in mice. Cytokine Growth Factor Rev. 2000;11:49–58. doi: 10.1016/s1359-6101(99)00028-3. [DOI] [PubMed] [Google Scholar]
- Wozney J.M. Bone morphogenetic proteins. Prog. Growth Factor Res. 1989;1:267–280. doi: 10.1016/0955-2235(89)90015-x. [DOI] [PubMed] [Google Scholar]
- Wozney J.M. The bone morphogenetic protein family and osteogenesis. Mol. Reprod. Dev. 1992;32:160–167. doi: 10.1002/mrd.1080320212. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A., Katagiri T., Ikeda T., Wozney J.M., Rosen V., Wang E.A., Kahn A.J., Suda T., Yoshiki S. Recombinant human bone morphogenetic protein-2 stimulates osteoblastic maturation and inhibits myogenic differentiation in vitro. J. Cell Biol. 1991;113:681–687. doi: 10.1083/jcb.113.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Letterio J.J., Lechleider R.J., Chen L., Hayman R., Gu H., Roberts A.B., Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Feng X., We R., Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Richardson J.A., Parada L.F., Graff J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]