

Abstract
In the unicellular alga Chlamydomonas, two anterior flagella are positioned with 180° rotational symmetry, such that the flagella beat with the effective strokes in opposite directions (Hoops, H.J., and G.B. Witman. 1983. J. Cell Biol. 97:902–908). The vfl1 mutation results in variable numbers and positioning of flagella and basal bodies (Adams, G.M.W., R.L. Wright, and J.W. Jarvik. 1985. J. Cell Biol. 100:955–964). Using a tagged allele, we cloned the VFL1 gene that encodes a protein of 128 kD with five leucine-rich repeat sequences near the NH2 terminus and a large α-helical–coiled coil domain at the COOH terminus. An epitope-tagged gene construct rescued the mutant phenotype and expressed a tagged protein (Vfl1p) that copurified with basal body flagellar apparatuses. Immunofluorescence experiments showed that Vfl1p localized with basal bodies and probasal bodies. Immunogold labeling localized Vfl1p inside the lumen of the basal body at the distal end. Distribution of gold particles was rotationally asymmetric, with most particles located near the doublet microtubules that face the opposite basal body. The mutant phenotype, together with the localization results, suggest that Vfl1p plays a role in establishing the correct rotational orientation of basal bodies. Vfl1p is the first reported molecular marker of the rotational asymmetry inherent to basal bodies.
Keywords: basal body, flagella, cilia, centriole, Chlamydomonas
Introduction
The formation of flagella and cilia in eukaryotic cells is organized by basal bodies, cylindrical organelles with walls composed of nine triplet microtubules that serve as templates for assembly of doublet microtubules in the axoneme. Basal bodies and centrioles are conserved in structure and the functions of these organelles are often interchangeable. For example, in some cells one of the two centrioles functions as a basal body for assembly of a primary cilium in interphase. The cilium is resorbed before mitosis and the basal body then serves as a centriole at the spindle pole (Rieder et al. 1979; Masuda and Sato 1984; for review see Wheatley et al. 1996). Although centrioles and basal bodies were first observed more than a century ago and their structure and behavior have been studied extensively in numerous cell types, the identification and functional analysis of molecular components of these organelles are just beginning.
Basal bodies possess innate axial polarity as well as rotational polarity (for review see Beisson and Jerka-Dziadosz 1999). A conserved cartwheel structure marks the proximal end of the organelle, where assembly is initiated. The distal end of the basal body docks with the cell surface, where axoneme assembly begins. Rotational polarity (the asymmetry of the nine triplet microtubules) has been inferred from results showing asymmetry of structures associated with the axonemal doublet microtubules (Afzelius 1959; Gibbons 1961; Hoops and Witman 1983). Further evidence of rotational polarity comes from the asymmetric attachment of various appendages to specific triplets in the basal bodies (for review see Lynn 1981; Anstrom 1992; Beech and Melkonian 1993; Geimer et al. 1998) and from the site of daughter basal body formation adjacent to defined triplets in the mature organelles (Dippell 1968; Allen 1969). The direction of the effective stroke in flagellar or ciliary beating is correlated with the rotational orientation of both the axoneme and the basal body appendages (Afzelius 1959; Gibbons 1961; Tamm et al. 1975; Hoops and Witman 1983; Lemullois et al. 1988; Tamm and Tamm 1988). It follows that the precise orientation of basal bodies at the cell surface with respect to their rotational asymmetry is essential for the coordinated beating of flagella and cilia in unicellular organisms and in sheets of ciliated epithelial cells.
The biflagellate, unicellular alga Chlamydomonas provides a model system for genetic analysis of eukaryotic flagella and basal bodies (for review see Mitchell 2000). In Chlamydomonas, the basal bodies function both as organizers of axonemal microtubule assembly and as components of the microtubule organizing center in interphase and mitotic cells. Normal cell division and normal axoneme assembly and motility are dependent on a cycle of basal body duplication and semiconservative segregation to daughter cells, and also on correct positioning of basal bodies and associated fibrous structures at the anterior end of the cell.
Previous studies identified mutations in several Chlamydomonas genes that result in cell populations with a variable number of flagella (vfl mutations), reflecting a variable number of basal bodies capable of flagellar assembly (Wright et al. 1983, Wright et al. 1989; Adams et al. 1985). One recessive mutation, vfl1, was observed to have pleiotropic effects on flagellar number and localization, the timing of flagellar outgrowth during the cell cycle, and the positioning of the cleavage furrow at cytokinesis (Adams et al. 1985). Defects in the number and localization of basal bodies and in their associated striated fibers were observed at the ultrastructural level. Basal bodies and probasal bodies were sometimes found embedded in the cytoplasm and not associated with the plasma membrane. Tam and Lefebvre 1993 recovered a new allele of the vfl1 mutation generated by insertional mutagenesis. They obtained wild-type genomic DNA fragments that rescued the mutant phenotype when transformed into vfl1 mutant cells. We report the structure of the VFL1 gene, the nature of the vfl1-1 mutation, and the predicted structure of the protein product. A chimeric VFL1 gene encoding an epitope tag rescued the mutant phenotype and facilitated the localization of the tagged Vfl1 protein (Vfl1p) using immunofluorescence and immunoelectron microscopy. The localization of Vfl1p, taken together with the motility defects and structural defects observed in vfl1 mutant cells, suggest a role for the VFL1 gene product in the establishment of the rotational orientation of the basal bodies to allow beating of the two flagella in opposite directions.
Materials and Methods
Strains and Culture Conditions
Strain 5E8 (vfl1-2::NIT1) was generated by insertional mutagenesis (Tam and Lefebvre 1993). This allele is designated vfl1-2 throughout the paper. Strains 5E8IV2B (vfl1-2; apm1-19; mt +) and 5E8I4F(vfl1-2; mt −) were derived from backcrosses of the strain 5E8 to strains L5 and L8 (Tam and Lefebvre 1993). Strains CC-1690 (wild-type strain 21 gr, mt +) and CC1388 (vfl1-1; mt +) were obtained from the Chlamydomonas Genetics Center. Strain JB4A2 (vfl1-1; mt +) was derived from backcrosses of strain CC1388 to wild-type strains B26 and B27 (Schnell and Lefebvre 1993). Strain CS977G3 (vfl1-2; arg7) was used for phenotypic rescue experiments. Phenotypically rescued strain VFL1-2-R2 expressed a hemagglutinin (HA)-tagged VFL1 gene; rescued strain VFL1-2-R29 expressed the wild-type untagged gene. Chlamydomonas strains were grown at 24°C in minimal medium I (Sager and Granick 1953) or in tris-acetate-phosphate (TAP) medium (Gorman and Levine 1965) supplemented with 0.005% arginine. Cells were grown on solid agar medium (1.2% agar) or in liquid culture bubbled continuously with filtered air and illuminated with white light. Synchronized cells were grown in liquid minimal medium I on a 14-h light/10-h dark schedule. For mating, cells were suspended in minimal medium I lacking nitrogen in bright light conditions to induce gametogenesis.
Quantitation of Flagellar and Nuclear Number
To determine flagellar number, cells were grown synchronously in liquid minimal medium I to a cell density of 105 cells/ml. Cells were collected 3–4 h into the light period and fixed by adding an equal volume of medium containing 1% glutaraldehyde. Cells were examined by phase–contrast microscopy to determine flagellar number. To determine nuclear number, cells were fixed and stained with DAPI as described by Hirono and Yoda 1997. Cells were examined by epifluorescence microscopy using a UV filter.
DNA Sequencing and Sequence Analysis
Genomic DNA encoding the wild-type VFL1 gene was contained in the clone λ5E-10 described previously (Tam and Lefebvre 1993). Subcloned fragments were sequenced on both DNA strands and the DNA sequences were assembled using Genetics Computer Group software (Devereux et al. 1984). The sequence is available at EMBL/GenBank/DDBJ under accession number AF154916. To predict exons in the genomic sequence, the data were analyzed using the GeneMark program (Borodovsky and McIninch 1993). Parameters for this analysis were determined from a training set of 50 Chlamydomonas gene sequences.
Reverse Transcriptase PCR Amplification of Vfl1 cDNA Fragments
Based on exon positions predicted from the GeneMark analysis of the genomic DNA sequence, specific primers were synthesized and used with the reverse transcriptase (RT)-PCR to amplify cDNA fragments. First-strand cDNA synthesis was carried out with specific 17-mer oligonucleotide primers and SuperScript RT II (Life Technologies). For amplification of double-stranded cDNA, specific primers (22–25 mers, Tm 72°C–74°C) were used, together with purified product from the first-strand cDNA synthesis reaction, in a buffer containing Taq DNA polymerase (Fisher Scientific), buffer A of the manufacturer, 10% glycerol, and 5% formamide. Amplification conditions consisted of a hot start at 94°C for 5 min and 80°C for 10 min, followed by 35 cycles of 94°C for 1 min, 58°C for 45 s, and 72°C for 2 min. Nested primers were used for a second round of amplification. The resulting fragments were subcloned into pCRII using the Original TA Cloning Kit (Invitrogen) for DNA sequencing. The 5′ rapid amplification of cDNA ends (RACE v2.0; Life Technologies) was used to determine the 5′ end of the transcript (Frohman et al. 1988).
Construction of an HA-tagged VFL1 Gene
A plasmid encoding three copies of the 9–amino acid HA-epitope was constructed from the pHA1 cassette plasmid (a gift from Dr. M. Jacobs-Lorena, Case Western Reserve University, Cleveland, OH; Surdej and Jacobs-Lorena 1994) encoding a single copy of the epitope. Two complementary 60-mer oligonucleotides encoding two additional copies of the HA epitope with the Chlamydomonas codon bias: (a) 5′-CGATACCCCTACGACGTGCCCGACTACGCCTACCCCTACGACGTGCCC- GACTACGCCGAT-3′ and (b) 5′-ATCGGCGTAGTCGGGCACGTCGTAGGGGTAGGCGAGTCGGGCACGTCGTAGGGGTATCG-3′ were annealed and ligated into the NruI site of pHA1. One NruI site flanking the HA epitope was reconstructed to allow for excision of the triple HA epitope cassette. The modified region of the resulting p3xHA plasmid was sequenced to confirm that it encoded three copies of the HA tag. Plasmid pW6-10.0 containing the VFL1 gene was cleaved at a PstI site 12-bp upstream of the stop codon and the ends were blunted by treatment with bacteriophage T4 DNA polymerase (Life Technologies). A SmaI fragment of 136 bp encoding the triple HA epitope tag was recovered from the p3xHA plasmid and ligated into the blunted PstI site. The resulting pW6-10.0-3HA plasmid was partially sequenced to confirm that the HA epitope sequences were in the proper orientation and reading frame.
Chlamydomonas Transformation
Strain CS977G3 was cotransformed with linearized plasmids (see Fig. 1), together with an equal amount of the linearized plasmid pARG7.8 DNA (Debuchy et al. 1989), using the protocol of Tam and Lefebvre 1993. Plasmids pW6-10.0 and pW6-10.0-3HA were linearized by digestion with SspI; pARG7.8, with XmnI. Cells were plated on TAP plates and transformant Arg+ colonies were picked into liquid medium to screen for rescue of the vfl1 motility phenotype.
Figure 1.

Genomic region of the VFL1 gene. The top bar represents a 16-kb fragment of genomic DNA cloned in the plasmid pW6 that rescues the phenotype of the vfl1-2 mutation when transformed into mutant cells. SstI sites are indicated. Subclones of the genomic region (represented by solid black bars) were tested for their ability to rescue the mutation. A 9.2-kb region within the complementing subclone pW6-10.0 was sequenced. GeneMark analysis of the sequence and RT-PCR confirmation of the predicted gene structure identified a coding region spanning 7.8 kb. In DNA from the vfl1-2 allele, a deletion of ∼4 kb was mapped to the region indicated by the bottom hatched bar.
Immunoblotting
Protein extracts from intact cells were prepared by pelleting 1.8 × 106 cells (per gel lane) and mixing these with sample buffer (0.0625 M Tris-Cl, pH 6.8, 2.0% SDS, 10% glycerol, 5% 2-mercaptoethanol). The mixture was boiled for 90 s and centrifuged at 14,000 g for 2 min to pellet cell debris before loading the supernatant on the gel. Nucleoflagellar apparatuses (NFAps) were isolated using the method of Wright et al. 1985. Synchronous cells in G1 (4 × 108 cells) were treated with autolysin (Tam and Lefebvre 1993) to remove cell walls and gently lysed. The NFAps were purified on Percoll gradients, dissolved in sample buffer, and boiled for 90 s. Proteins from intact cells and from isolated NFAps were size-fractionated on SDS-PAGE minigels (1.5-mM thick, 7% acrylamide) and transferred to Immobilon-P membranes (Millipore) using a Genie electrophoretic blotter (Idea Scientific Co.) with 0.025 M Tris-Glycine buffer (pH 8.3) in 20% methanol at 24 V for 1 h. The HA-tagged proteins were localized using the rat anti-HA high-affinity antibody and other reagents from Roche Molecular Biochemicals. The antibody was diluted to 200 ng/ml in blocking reagent diluted in TBS. The secondary antibody was sheep IgG anti–rat Fab fragments conjugated with peroxidase at a 1:1,000 dilution. The detection system used BM chemiluminescence blotting substrate (POD) and exposure to x-ray film (X-OMAT AR; Eastman Kodak). The blots were stripped by incubating the membrane in TBS (50 mM Tris-Cl, 150 mM NaCl, pH 7.5) containing 100 mM 2-mercaptoethanol and 2.0% SDS for 30 min at 50°C, and washed twice for 15 min in TBS containing 0.1% Tween 20. To localize β-tubulin, mouse monoclonal antibody 2-10-B6 was used at a 1:75 dilution of the cell culture supernatant (a gift from Dr. G. Piperno, Mount Sinai School of Medicine, New York, NY). Molecular weight standards on the blot were stained for 5–10 min in a solution containing 0.02% ponceau S, 0.3% trichloroacetic acid, and 0.3% sulfosalicylic acid. Proteins in the gel were silver-stained (Wray et al. 1981).
Isolation and Blotting of Nucleic Acids
Chlamydomonas DNA was isolated as described by Schnell and Lefebvre 1993. RNA was isolated using the method of Wilkerson et al. 1994. Polyadenylated RNA was isolated using Magnetight oligo (dT) particles (Novagen). Polyadenylated RNA (∼10 mg/lane) was separated on formaldehyde agarose gels (1.2% agarose, 0.5-cm thick gels; Sambrook et al. 1989) and blotted to Hybond-N1 membranes (Amersham Pharmacia Biotech) using the downward transfer method described in Ambion technical bulletin 169 (Ambion, Inc.). RNA was cross-linked to the membranes by baking at 80°C for 2 h. Hybridization probes were labeled by random-primer labeling with 32P-dCTP using the DECAprime II kit (Ambion, Inc.) and hybridized in ULTRAhyb buffer (Ambion, Inc.).
Immunofluorescence Labeling
Transformant cell lines expressing the wild-type VFL1 gene (strain VFL1-2-R29) or the wild-type gene tagged with the HA-epitope (strain VFL1-2-R2) were grown under synchrony conditions. Cells in the G1 phase of the cell cycle were used for isolation of NFAps for immunofluorescence microscopy as described by Wright et al. 1985. The cells were treated with autolysin to remove cell walls, resuspended in MT buffer (50 mM Pipes, 3 mM EGTA, 3 mM MgSO4, and 25 mM KCl, pH 7.2) and applied to multiwell slides (treated with 0.1% polyethylenimine). The slide was washed in MT buffer, placed in MT buffer containing 1% NP-40, and agitated for 2 min to lyse the cells. The slide was washed briefly in MT buffer and placed in a fixative solution containing 4% paraformaldehyde (EM Sciences) in MT buffer for 30 min at room temperature. The slide was placed in methanol at −20°C for 2 min. Acetone treatment of the fixed complexes was omitted. Fixed apparatuses were incubated with blocking buffer (Sanders and Salisbury 1995) for 1 h followed by anti-HA high affinity antibody (Roche Molecular Biochemicals) diluted to 500 ng/ml in blocking buffer for 16 h at 4°C. The secondary antibody was an FITC-conjugated goat anti–rat IgG fraction (ICN Biomedicals) diluted 1:400 in blocking buffer. Microtubules were labeled with a rabbit polyclonal antibody raised against Chlamydomonas α-tubulin (Silflow and Rosenbaum 1981) using a serum dilution of 1:2,000. The secondary antibody was a Texas red–conjugated goat anti–rabbit IgG diluted 1:400 (ICN Biomedicals). DNA was stained with DAPI at 4 mg/ml in MT buffer. The slides were mounted in Vectashield (Vector Laboratories) antifading medium. Preparations were viewed using a photomicroscope (Eclipse E800; Nikon) equipped with differential interference contrast (DIC) and fluorescence optics, including a 100 W mercury lamp epi-fluorescence illumination with UV (excitation filter 330–380, barrier 420 nm), FITC, and Texas red (excitation filter 510–560 nm, barrier 570–620 nm) filter sets. The samples were viewed using either a 60×, 1.4 NA plan apo or a 100×, 1.3 NA plan apo objective. Digital images were collected using a CoolCam liquid-cooled, three-chip color CCD camera (Cool Camera Company) and captured to a Pentium® II computer using Image Pro Plus v4.0 software (Media Cybernetics). Prints were made using Adobe Photoshop® v5.5.
To compare the localization of HA-tagged Vfl1p with centrin in cells, cells were treated with autolysin, resuspended in buffer containing 10 mM Hepes, pH 7.2, 5 mM EGTA, and 1 mM MgCl2, and allowed to attach to slides. Cells were fixed in 4% paraformaldehyde, 10 mM Hepes for 30 min at room temperature. The slides were incubated in methanol at −20°C for 2 min, and then in acetone at −20°C for 2 min. The cells were incubated with antibodies against the HA epitope as described above and with the MC1 rabbit polyclonal antibody against centrin (diluted 1:200; a gift from Dr. J.L. Salisbury, Mayo Clinic, Rochester, MN).
Preembedding Immunogold Labeling of Nucleoflagellar Complexes
Immunogold electron microscopy was performed as described by McFadden et al. 1987. Pellets of NFAps from VFL1-R29 cells and from VFL1-R2 cells were suspended in 0.5 ml HMT buffer containing 30 mM Hepes, 5 mM MgSO4, 5 mM EGTA, and 25 mM KCl, pH 7.0. The apparatuses were fixed by addition of an equal volume of 6% paraformaldehyde and 0.5% glutaraldehyde in HMT buffer on ice for 45 min. After washing in PBS three times (including one wash with PBS containing 50 mM NH4Cl), the apparatuses were incubated in blocking buffer for 90 min at 4°C and in a 1:150 dilution of the primary anti-HA antibody for 16 h. After three washes in PBS, the apparatuses were suspended in blocking buffer containing a 1:20 dilution of goat anti–rat IgG labeled with 12 nm colloidal gold (Jackson ImmunoResearch Laboratories) for 90 min at 37°C. After three washes in PBS, the apparatuses were post-fixed in 2% glutaraldehyde in PBS overnight at 4°C. The final pellet was osmicated in 1% OsO4 in MT buffer for 30 min on ice. After additional washes, the pellet was embedded in 2% low-melt agarose, dehydrated, and embedded in EM bed-812 resin (EM Sciences). Sections were stained with Reynold's lead citrate for 2 min and with uranyl acetate for 15 min, and were imaged at 60,000× using a transmission electron microscope (CM-12; Philips) at 60 kV. To measure the location of colloidal gold particles at the distal ends of the basal bodies, negatives were printed at a uniform magnification. Data for the graph in Fig. 8 were obtained by drawing a horizontal line across basal body longitudinal sections at the point where C tubules end and where a thin election-dense line crosses the interior of the basal body. The vertical distance of each gold particle from the horizontal line was determined and was normalized for the distance between the horizontal line and the proximal end of the cylinder of the transition region. This procedure allowed us to reduce the effects of distortions in basal body shape that occurred during preparation of the samples. To determine the radial position of gold particles, printed images of basal body cross-sections were analyzed. The distance of each gold particle from the center point of the basal body was compared with the distance from the center point to the ring of doublet/triplet microtubules. Comparison of the two lengths allowed the gold particles to be classed as lying on the inner half or outer half of the basal body radius.
Figure 8.
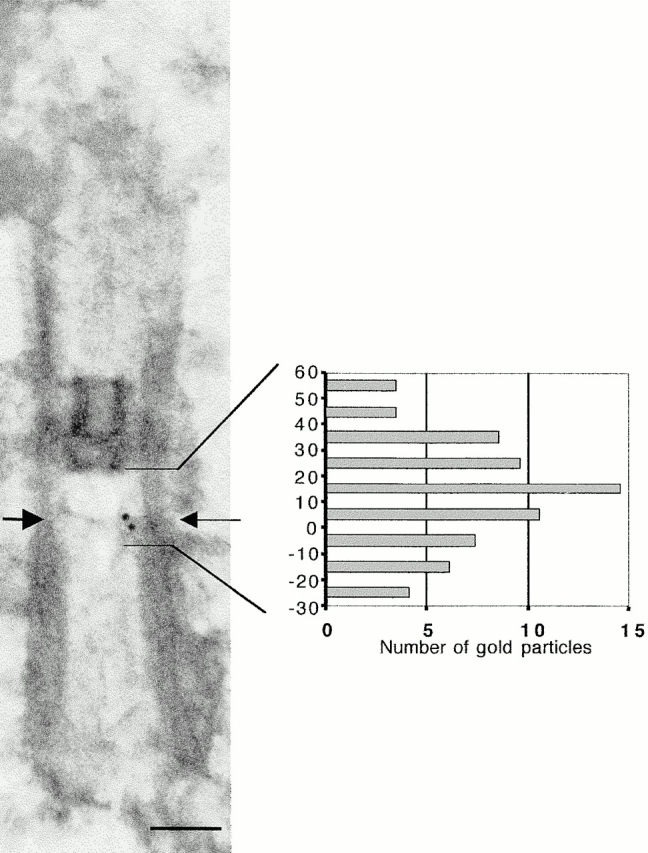
Localization of Vfl1p relative to the distal ends of the triplet microtubules. The positions of 67 gold particles on 49 longitudinal basal body sections were determined with respect to the end of the triplet microtubules of the basal body (arrow). The data were normalized as described in Materials and Methods and plotted to indicate the distribution distal (positive numbers) or proximal (negative numbers) to the end of the triplet microtubules. Bar, 0.2 μm
To analyze the positions of gold particles with respect to individual doublet/triplet microtubules, serial cross-sections (50 nm) of the labeled and embedded apparatuses were examined. For each basal body analyzed, digitized negatives from six to seven consecutive serial cross-sections were aligned using the SliceAlign v1.6 component of a deconvolution software system (Vaytek, Inc.). Cross-sections on which gold particles were located were divided into pie-shaped wedges, each associated with one of the nine doublet microtubules. Each particle was assigned to a specific doublet according to its location within a wedge. The X-Y center of the labeled basal body section was determined and five concentric rings were constructed over the cross-section, with the outermost ring (number 5) containing the microtubule wall of the cylinder. To categorize its distance from the microtubule wall, each gold particle was assigned to one of these rings.
Results
Characterization of a New Vfl1 Allele
We compared some aspects of the phenotypes of the vfl1 insertional allele (vfl1-2::NIT1) described by Tam and Lefebvre 1993 with those of the original allele (vfl1-1; Adams et al. 1985). The distribution of flagellar number per cell was similar for both alleles (Table ). Although approximately one third of the cells are biflagellate, few swim normally. Most biflagellate cells swim with a large helical path, tumble, or swim in tight circles; uniflagellate cells spin in place. Adams et al. 1985 observed the presence of multiple nuclei in some vfl1-1 mutant cells, indicating a possible defect in the placement of the cleavage furrow at cytokinesis. We compared nuclear number in cells with the vfl1-1 and vfl1-2 alleles using DAPI staining and immunofluorescence microscopy of cells in G1 stage of the cell cycle to determine the number of nuclei per cell. From 5 to 7% of cells of vfl1-1 or vfl1-2 cultures contained two nuclei, whereas wild-type cells never contained multiple nuclei (Table ).
Table 1.
Distribution of Flagellar Numbers
| Percentage of cells with various flagellar numbers | ||||||
|---|---|---|---|---|---|---|
| Strains | 0 | 1 | 2 | 3 | ≥4 | |
| n | % | % | % | % | % | |
| CC-1690 (wild-type) | 685 | 10 | 2 | 88 | — | — |
| JB4A2 (vfl1-1) | 1501 | 19 | 32 | 32 | 13 | 4 |
| 5E8IV2B (vfl1-2) | 422 | 25 | 30 | 32 | 9 | 3 |
| VFL1-2-R29 (rescue strain without tag) | 682 | 9 | 2 | 89 | — | — |
| VFL1-2-R2 (rescue strain with tag) | 1097 | 8 | 2 | 90 | — | — |
Table 2.
Number of Nuclei in Wild-type and Mutant Cells
| Binucleate cells | ||
|---|---|---|
| n | % | |
| CC-1690 (wild-type) | 900 | 0 |
| JB4A2 (vfl1-1) | 1601 | 6.9 |
| 5E8IV2B (vfl1-2) | 2268 | 5.3 |
| VFL1-2-R29 (rescue strain without tag) | 2087 | 0.2 |
| VFL1-2-R2 (rescue strain with tag) | 2224 | 0.3 |
Characterization of the VFL1 Gene and Its Product
The wild-type VFL1 gene was contained on a 16-kb genomic DNA fragment that rescued the mutant phenotype upon transformation (Tam and Lefebvre 1993). Using fragments from the cloned DNA as hybridization probes, we mapped a deletion of ∼4 kb of genomic DNA in the vfl1-2 mutant strain (Fig. 1). A 10-kb plasmid subclone (pW6-10.0) rescued the mutant phenotype upon transformation. Subclones truncated at either end of this subclone did not rescue the mutation, indicating that the VFL1 gene spans most of the 10-kb region. DNA sequence from the plasmid insert was obtained (data not shown; EMBL/GenBank/DDBJ under accession number AF154916) and was analyzed using the GeneMark program to predict exon locations (Borodovsky and McIninch 1993). We screened several cDNA libraries using genomic fragments from within the predicted gene region but did not obtain a cDNA clone. To confirm the intron–exon boundaries predicted by the GeneMark program, we used RT-PCR with primers from the predicted exon regions. The sequence from the RT-PCR products was compared with genomic DNA sequence to identify 16 introns and 17 exons that defined a predicted open reading frame of 3,648 bp. The initiation codon of the predicted open reading frame was located 39 bp downstream of a stop codon in the same reading frame, indicating that it is the authentic initiation codon. We used 5′ RACE to determine the transcription start site and obtained products that extended at least 145 bp upstream of the initiation codon. At a position 233 bp downstream of the predicted termination codon of the open reading frame, we observed the sequence TGTAA, the highly conserved polyadenylation signal in Chlamydomonas genes (for review see Silflow 1998). This analysis of the VFL1 gene indicated it should produce a transcript of at least 4.0 kb.
The transcript produced by the VFL1 gene was identified on gel blots of polyadenylated RNA using the first exon (570 bp) of the gene as a hybridization probe. The probe hybridized to a single transcript of ∼4.1 kb in RNA from wild-type cells and from vfl1-1 cells; the transcript was absent in RNA from vfl1-2 cells (Fig. 2). The genomic DNA deletion associated with the vfl1-2 mutation extends well into the 3′ region of the gene (Fig. 1), apparently resulting in an unstable transcript.
Figure 2.

The VFL1 gene encodes a transcript of ∼4.0 kb. Polyadenylated RNA (10 μg/lane) was electrophoresed on a denaturing gel and blotted to a nylon membrane. The RNA was hybridized with a labeled probe consisting of the first exon of the gene. RNA was obtained from: lane 1, wild-type cells; lane 2, vfl1-1 cells; and lane 3, vfl1-2 cells. The lower panel is an RNA loading control showing hybridization with a radioactive probe for the CRY1 gene (Nelson et al. 1994).
The deduced protein encoded by the VFL1 gene contains 1,216 amino acids, with a predicted molecular mass of 127,942 Da and a predicted pI of 5.77 (Fig. 3). Near the NH2 terminus, extending from amino acids 42–159, are five copies of a conserved leucine-rich repeat (LRR) sequence (Fig. 4 a). This motif, found in proteins with diverse intracellular and extracellular functions, facilitates protein–protein interactions (Kobe and Deisenhofer 1995a,Kobe and Deisenhofer 1995b; Buchanan and Gay 1996; Kajava 1998). Each repeat of 22–29 amino acids forms a short β-strand packed against an α-helix; parallel β-sheets interact with the target protein. Comparisons with different classes of LRR repeats showed that the repeats in the Vfl1 protein are conserved in length and sequence with the SD22-like class of repeats (Kajava 1998). Within the COOH-terminal two thirds of the protein, large blocks of coiled coil tertiary structure were predicted by the COILS algorithm (Fig. 4 b; Lupas et al. 1991). Other notable sequence motifs include a high density of glutamine residues in positions 282–433 and a region of nine proline residues at positions 460–470.
Figure 3.

Predicted sequence of the Vfl1 protein. The five LRRs are underlined.
Figure 4.

Summary of the predicted Vfl1 protein structure. (a) The LRR region (vertical bars) is located near the NH2 terminus, and the coiled coil regions (hatched bars) are at the COOH terminus. The position where the amino acid sequence is disrupted in the protein encoded by the vfl1-1 allele is shown by a vertical open arrow. The five LRR sequences are aligned, with the consensus sequence shown below. (b) COILS output for the Vfl1p using a window size of 21 (Lupas et al. 1991).
To determine the lesion associated with the original vfl1-1 mutation, we used RT-PCR to amplify products from polyadenylated RNA of vfl1-1 cells. No differences in sequence between PCR products from wild-type and mutant cells were observed except for an 8-bp deletion in exon 5 of products generated from vfl1-1 cells. Primers specific for sequences in introns 4 and 5 were used to amplify the entire exon 5 from genomic DNA of vfl1-1 cells. DNA sequence of the PCR product confirmed the existence of the 8-bp deletion in genomic DNA of the vfl1-1 cells. The deletion results in a shift in the reading frame at amino acid 639, followed by a termination codon 123–amino acids downstream (Fig. 4 a). The mutant protein of 765 amino acids contains the LRR sequences, but lacks the predicted coiled coil domain of the wild-type protein. Although vfl1-1 mutant cells produce a stable transcript (Fig. 2), the truncated protein is apparently not stable or is not functional because the phenotype of the vfl1-1 allele is similar to that of the null allele vfl1-2.
The Cellular Localization of Vfl1p
The cellular localization of Vfl1p, the protein encoded by the VFL1 gene, was examined using an epitope-tagged version of the protein. Three copies of the sequence encoding the 9–amino acid influenza virus HA epitope (Field et al. 1988) were inserted at a position two codons upstream of the stop codon in the wild-type VFL1 gene. The modified construct rescued the mutant phenotype of vfl1-2 cells as efficiently as the untagged construct did. Flagellar number on the rescued cells was similar to that of wild-type cells, with no cells having more than two flagella (Table ). The frequency of binucleate cells was much lower than that observed in mutant cells (Table ), indicating that binucleate cells are a result of the vfl1 mutations. Protein extracts were prepared from rescued cells transformed with the tagged gene with or without the untagged construct. To determine whether the Vfl1p might be associated with the basal body apparatus, we also isolated NFAps by detergent treatment of cells (Wright et al. 1985). The NFAps include the basal bodies with attached axonemes, the nucleus connected to the basal bodies via centrin-containing fibrous nucleus-basal body connectors, and microtubule rootlets and striated fibers associated with the basal bodies. Proteins from whole cell extracts and from NFAps were fractionated by PAGE and blotted. An antibody raised against the HA-epitope reacted with a protein of 140 kD in whole cell extracts and in NFAps from cells transformed with the HA-tagged gene construct (Fig. 5 a, lanes 1 and 2). This protein migrated at the expected size (137 kD) for Vfl1p with the 27–amino acid tag. The antibody did not react with proteins in cells transformed with the nontagged construct (Fig. 5 a, lanes 3 and 4). The blot was stripped and probed with an antibody raised against β-tubulin (Fig. 5 b), which detected similar levels of β-tubulin in the protein extracts from cells transformed with either construct. The enrichment of the HA-tagged protein in the NFAp proteins compared with that in total cell proteins was similar to the enrichment observed for β-tubulin.
Figure 5.

The Vfl1 protein copurifies with the basal body apparatus. Whole cell proteins (lanes 1 and 3) and proteins from NFAps (lanes 2 and 4) were isolated from VFL1-2-R2 cells expressing the VFL1 HA-tagged gene construct (lanes 1 and 2) and from VFL1-2-R29 cells expressing an untagged VFL1 gene construct (lanes 3 and 4). The proteins were separated by SDS-PAGE and blotted to nylon membrane. (a) The anti-HA high affinity antibody was used to identify the tagged Vfl1p which migrates as a 140,000-M r protein. (b) The blot was stripped and reprobed with an anti–β-tubulin antibody as a control. Silver-stained lanes from the gel are shown in panel c.
Because immunoblot data indicated that the Vfl1p is associated with the basal body apparatus, we prepared NFAps for immunofluorescence localization of the HA tag (Wright et al. 1985). Cells transformed with either the tagged or the nontagged construct were isolated at the G1 phase of the cell cycle. NFAps were isolated by detergent extraction of cells and probed with the antibody raised against the HA epitope. In NFAps from cells expressing the tagged construct, we observed highly reproducible staining localized in two to four dots adjacent to the DAPI-stained nuclei (Fig. 6 a). No dots were observed in NFAps from cells expressing the untagged construct (data not shown). To further localize the Vfl1p staining on NFAps, the structures were colabeled with the anti-HA antibody and with an antibody against α-tubulin (Fig. 6b and Fig. c). The antitubulin antibody labeled the two flagella as well as the cytoplasmic microtubules associated with the basal bodies in the NFAps. Two to four dots labeled by the anti-HA antibody were observed consistently at the base of the flagella in positions close to the basal bodies and probasal bodies. When more than two dots were present, as in Fig. 6 b, the dots were often of unequal intensity, with the most intense signals at the positions expected for mature basal bodies (Fig. 6 b, dots on the right and left) and less intense signals at positions expected for probasal bodies (Fig. 6 b, dots at the top and bottom). The number and positions of the HA dots were not dependent on cytoplasmic microtubules present in an NFAp; the same results were obtained when NFAps were isolated from cold-treated cells in which all the cytoplasmic microtubules were depolymerized except the cold-stable cytoplasmic rootlet microtubules (data not shown). The Vfl1p localization also was not dependent on the presence of a nucleus in the isolated NFAp structure, as the dots were observed in NFAps from which the nucleus was lost during the isolation procedure (Fig. 6b and Fig. c). To localize Vfl1p in intact cells, we colabeled cells with the anti-HA antibody and with an antibody against centrin, a 20-kD calcium-binding protein that labels several different structures in NFAps (Fig. 6, d–g). We observed anticentrin staining in the distal striated fiber extending between the two basal bodies, in the nucleus–basal body connector fibers that extend from the each basal body to the surface of the nucleus, and in a perinuclear array of fimbria (Fig. 6 e) as had been described previously (Huang et al. 1988; Salisbury et al. 1988). The anti-HA staining was restricted to two to four dots (Fig. 6 f). Two dots of anti-HA staining were located at the distal ends of the distal striated fiber at the positions expected for the basal bodies (Fig. 6 g). When additional labeled dots were present, they were positioned orthogonally to the dots at the ends of the distal striated fiber, consistent with their localization on probasal bodies.
Figure 6.
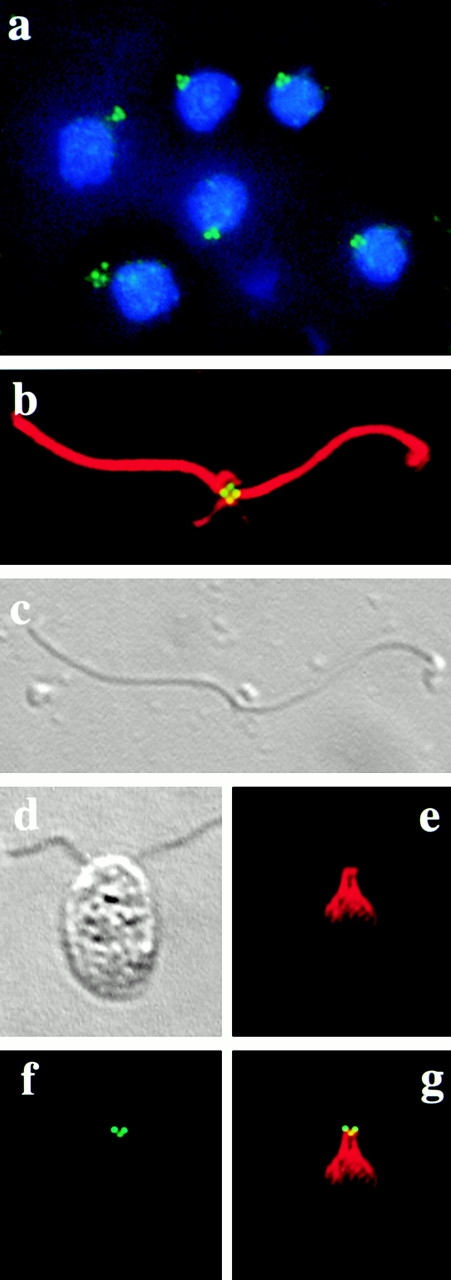
Immunofluorescence microscopy of NFAps and whole cells. (a) NFAps isolated from cells expressing HA-tagged Vfl1p were stained with rat anti-HA antibody, followed by FITC-conjugated goat anti–rat IgG (green). The DNA was labeled with DAPI (blue). (b) A single apparatus was costained with rat anti-HA and rabbit anti–α-tubulin antibodies, followed by FITC-conjugated goat anti–rat (green) and Texas red–conjugated goat anti–rabbit (red) IgGs. (c) DIC microscopy of the apparatus labeled in panel b. The nucleus is missing in this apparatus. (d) DIC microscopy of an intact cell expressing HA-tagged Vfl1p, analyzed by immunofluorescence microscopy (e–g). (e) Texas red channel showing the centrin distribution with the distal striated fiber that connects the two basal bodies. (f) FITC channel showing Vfl1p localization with two intensely stained dots and one weakly stained dot. (g) Superimposed images from panels e and f.
The precise localization of Vfl1p with respect to basal bodies was determined by using immunogold electron microscopy. Isolated NFAps from phenotypically rescued cells expressing HA-tagged Vfl1p were labeled before embedding, using the anti-HA antibody and a second antibody labeled with 12 nm gold particles. Using the densely stained H-shaped transition region lying distal to the basal body as a marker, several hundred longitudinal sections of basal bodies were identified (Fig. 7, a and b). In a subset of basal body sections used to quantify the extent of labeling, gold particles were found on 27% (n = 199) of the sections at the distal end of the basal bodies and within the microtubule walls of the cylinder. Particles were not observed on the proximal region of the basal body, the transition region, or on the axoneme. In control samples prepared from rescued cells expressing an untagged Vfl1p, no gold particles were found on 69 longitudinal sections of basal bodies (data not shown). To estimate the level of nonspecific background labeling in the tagged Vfl1p material, 80 images containing a basal body labeled with a gold particle were printed. On these images, which contained a total background area ∼20 times larger than the area of the basal body sections, 119 particles were observed, 103 (87%) of which localized on a basal body. Some of the 13 gold particles that were not localized on a basal body section were found in positions expected for daughter basal bodies (see below).
Figure 7.

Ultrastructural localization of Vfl1p by immunogold preembedded labeling of isolated NFAps. (a and b) Longitudinal sections of basal bodies (95-nm sections). Arrows indicate the point at the distal end of the basal body where the C tubules end and the doublet microtubules of the axoneme begin. The electron-dense H-shaped structure is the transition region. Dark arrowheads indicate the distal striated fibers. Gold particles are indicated by light arrow heads. (c and d) Cross-sections made at the distal ends of basal bodies are viewed from the proximal end of the basal body (95-nm sections). Arrows indicate transitional fibers connecting the nine triplet microtubules with the plasma membrane. Gold particles are located within the basal body cylinder. (e and f) Cross-sections of the proximal region of the apparatus showing basal body pairs and probasal bodies. Gold particles indicated by arrowheads are found in positions expected for probasal bodies. e, 200-nm section; f, 50-nm section. Bars, 0.2 μm.
The locations of gold particles were determined with respect to structural landmarks in longitudinal basal body sections. The positions of 67 gold particles were measured on images of 49 basal body sections that appeared to be minimally distorted by the isolation and embedding procedures (Fig. 8). The distal end of each basal body was identified as the point of a reduced thickening of the basal body wall where the C tubule ends (Cavalier-Smith 1974), a point that coincided with a thin horizontal line of matrix material in the interior of the basal body (Fig. 7, a and b, arrows; Fig. 8). Most particles were localized in a region between the distal end of the basal body and the proximal end of the H-shaped transition region.
Nine transitional fibers extend from the distal ends of each of the nine triplet microtubules to make contact with the plasma membrane (Ringo 1967; Cavalier-Smith 1974; Weiss et al. 1977). These electron-dense fibers provide a marker for cross-sections of this region of the basal body. The positions of the gold particles in longitudinal sections allowed us to predict that basal body cross-sections decorated with transitional fibers would contain gold particles. The prediction was confirmed for apparatuses from cells expressing the HA-tagged Vfl1p. Of 103 cross-sections containing transitional fibers, 31% were found to contain one or more gold particle (Fig. 7c and Fig. d). Gold particles were not observed on cross-sections of other regions of the basal bodies or of the axonemes. In the control sample from cells expressing an untagged Vfl1p, we examined 31 basal body distal-end cross-sections and found no gold particles (data not shown). To determine the location of the gold particles within the radius of the basal body cross-section, we examined images of 21 basal body cross-sections containing 31 gold particles, 30 of which were located in the outer half of the radius of the basal body.
The distribution of gold particles on longitudinal basal body sections indicated that Vfl1p is distributed in a rotationally asymmetric pattern within the basal bodies. Previous studies have shown that the nine microtubule doublets of the axoneme have structural differences, giving the axoneme an inherent circumferential asymmetry (Hoops and Witman 1983). Doublet microtubule number 1 in each axoneme faces the opposite axoneme, demonstrating 180° rotational symmetry of the basal bodies and axonemes. The two basal bodies are connected by the distal striated fiber associated with triplets 9, 1, and 2 of each basal body. Longitudinal sections of basal bodies with an attached distal striated fiber allowed us to determine the distribution of Vfl1p with respect to the distal fiber. In several hundred such sections, gold particles were almost always located on the half of the basal body facing the distal fiber. To document this unexpected result, we examined printed images of such basal body sections (two are shown in Fig. 7, a and b) and found that 95% (n = 76) of gold particles were localized on the half of the basal body facing the distal striated fiber. This result indicates that the position of Vfl1p is rotationally asymmetric within the basal body cylinder, lying within the half of the basal body that faces the distal striated fiber and contains triplets 9, 1, and 2.
Further evidence for the rotationally asymmetric localization of Vfl1p was obtained from serial cross-section analysis of gold-labeled basal bodies (Fig. 9). The most obvious structural marker for identifying individual triplet microtubules is the distal striated fiber attached to triplets 9, 1, and 2 at a position ∼200 nm proximal to the H-shaped transition zone (Fig. 7). Sets of serial thin sections, including the region from the transition zone to the distal striated fiber, were examined to determine the axial and rotational orientation of six basal bodies, three of which are shown in Fig. 9. Digitized images were processed using alignment software to identify triplets 9, 1, and 2 and to follow them through adjacent sections to the distal end of the basal body where they converted to doublets. To identify the doublet (or triplet) microtubule most closely associated with each gold particle, the labeled section was divided into pie-shaped wedges, each of which bisected adjacent doublets. Of 10 gold particles analyzed, 9 were most closely associated with doublet 1; 1 was associated with doublet 2. The distance of the gold particle from the doublet microtubule was determined by dividing the labeled axonemal sections into five concentric rings, ending with an outer ring that included the microtubule wall of the basal body cylinder. Of the 10 gold particles examined, 1 localized within the outer ring (ring 5, range 0–24.37 nm); 6 localized within the adjacent ring (ring 4, range 25.01–33.26 nm), and 3 localized in ring 3 (range 40.71–58.41 nm). The mean distance from the X-Y center of doublet 1 to the gold particles was 12.79 nm for the particle in ring 5, 28.05 nm for the six particles in ring 4, and 42.86 nm for the three particles in ring 3. The results do not allow us to localize Vfl1p to a specific microtubule doublet due to the small number of particles analyzed and to the length of the rat IgG-goat IgG complex (∼23 nm) used for localization. However, the results clearly support the conclusion that Vfl1p localizes near the microtubules that face the distal striated fiber.
Figure 9.

Rotationally asymmetric localization of Vfl1p. Three series of adjacent serial sections (50-nm sections) of isolated NFAps labeled by the immunogold preembedding method show the positions of the gold particles with respect to doublet microtubules 9, 1, and 2. The images are viewed from the distal end of the basal body. Numbers 9, 1, and 2 identify these triplets based on their association with the distal striated fiber. Gold particles are marked by light arrowheads; the distal striated fiber is marked by a dark arrowhead. In series 1 a, the upper gold particle is nearest to doublet 2 and the lower particle is nearest to doublet 1. In series 1 b, the particle is nearest to doublet 1. In series 2 a, the gold particle is nearest to doublet 1. In series 3 b, the particle is nearest to doublet 1. Bars, 0.2 μm.
Immunofluorescence images of Vfl1p localization frequently showed three to four spots of labeling, suggesting that daughter basal bodies might contain the protein (Fig. 6). Evidence to support this conclusion was obtained from examining semithick and serial thin sections of basal body apparatuses. Cross-sections of basal body pairs in the region between the proximal end of the basal bodies and the distal striated fiber sometimes contained gold particles positioned orthogonally to the mature basal bodies (Fig. 7e and Fig. f). Although the structure of the daughter basal bodies was not well preserved in the NFAp material prepared by preembedding labeling, the reproducibility of the labeling pattern indicated that the gold particles correspond to the dots of immunofluorescence labeling observed at the positions expected for daughter basal bodies.
Discussion
In Chlamydomonas cells, basal bodies serve as the organizing center for the microtubule cytoskeleton in addition to their role in nucleating the growth of flagella. Detailed ultrastructural studies of the basal body apparatus and its cycle of duplication in Chlamydomonas (Ringo 1967; Johnson and Porter 1968; Cavalier-Smith 1974; Gould 1975; Gaffal 1988) have provided a background for interpreting the phenotype of vfl1 mutations (Adams et al. 1985) and the localization of Vfl1p. In interphase cells, the basal body apparatus consists of two mature (flagellated) basal bodies maintained at an angle of ∼90° by distal and proximal striated connecting fibers. One component of the distal striated fiber is centrin, a 20-kD EF-hand protein that also forms descending fibers connecting the basal bodies with the nucleus (Huang et al. 1988; Salisbury et al. 1988). Originating near the basal bodies are four microtubule rootlets that extend into the cell beneath the plasma membrane, where they are involved in positioning cellular organelles and the cleavage furrow (Holmes and Dutcher 1989; Ehler et al. 1995) Before mitosis, the flagella and the flagellar transition regions are disassembled, leaving the basal bodies attached to the plasma membrane via nine transitional fibers that extend from the microtubule triplets to the plasma membrane (Ringo 1967; Weiss et al. 1977). Striated fibers connecting the basal bodies also disassemble, allowing disorientation of the mature basal bodies. In early prophase, the probasal bodies elongate and dock with the plasma membrane; transitional fibers become visible at their distal ends (Gaffal 1988). Two basal body pairs, each consisting of one mature and one recently elongated daughter basal body, separate and move to positions near the spindle poles where they are seen at metaphase (Coss 1974). Probasal bodies develop in the metaphase to anaphase stage adjacent and perpendicular to the proximal ends of each full-length basal body, but do not elongate until the following prophase (Gould 1975; Gaffal 1988).
A key advantage of Chlamydomonas for studying basal bodies is the availability of mutations that affect basal body function. For example, the bld2 mutation results in failure to assemble basal bodies and in defects in the organization of the cytoskeleton (Ehler et al. 1995). Mutations in the VFL2 gene encoding centrin lead to defects in centrin-containing fibrous components of the basal body apparatus and result in abnormal basal body localization and segregation (Wright et al. 1989). Recently, a mutation in the UNI3 gene led to the identification of δ-tubulin and indicated a role for this protein in assembly of triplet microtubules (Dutcher and Trabuco 1998).
Mutations in the VFL1 gene result in pleiotropic effects, including defects in the number and positioning of basal bodies and defects in basal body–associated fibers. The basal bodies are not restricted in location to the surface of the cell; basal bodies and structures that appear to be intermediates in basal body assembly are found at various positions within the interior of vfl1 cells (Adams et al. 1985). In addition, the cells continue to acquire flagella throughout G1 phase (0.05 flagella per cell per hour) in contrast to wild-type cells, which only produce flagella at the beginning of G1 phase. Based on these observations, Adams et al. 1985 suggested that the VFL1 gene plays a role in control of the basal body assembly cycle and that delays in basal body replication or maturation may lead to delays in the appearance of flagella during the cell cycle.
Our results suggest that Vfl1p may help to ensure that the two basal bodies are positioned with the correct 180° rotational symmetry. The rotationally asymmetric localization of Vfl1p is a reflection of the intrinsic circumferential polarity of basal bodies that previously was inferred from the asymmetric attachment of various appendages (for review see Beisson and Jerka-Dziadosz 1999). For example, in Chlamydomonas cells, the distal striated fiber is attached to triplet microtubules 9, 1, and 2 (Hoops and Witman 1983), whereas centrin-containing fibers of the nucleobasal body connector are attached to triplets 7 and 8 (Salisbury et al. 1988). Several aspects of the vfl1 mutant phenotype point to the role of Vfl1p in positioning basal bodies with the correct rotational orientation. Most biflagellate vfl1 cells show aberrant spinning or tumbling motility (Adams et al. 1985; our unpublished observations), indicating that the flagella do not beat in opposite directions. Ultrastructural defects in the assembly and positioning of the distal striated fiber in mutant cells (Adams et al. 1985; our unpublished observations) likely reflect the misorientation of the basal bodies. Other aspects of the vfl1 phenotype, including multinucleate cells, mispositioned cellular organelles, and heterogeneous cell volume, could result from mispositioning of microtubule rootlets that are organized within the basal body apparatus and that play key roles in spatial positioning of the cleavage furrow and cytoplasmic organelles (Holmes and Dutcher 1989; Gaffal and el-Gammal 1990; Ehler et al. 1995).
Studies in numerous systems have suggested that basal body appendages, whose appearance is dictated by inherent asymmetry within the basal body, interact with the cytoskeleton to lock the basal bodies into specific rotational orientations, causing the cilia to beat in specific directions. For example, when small sections of the cortex of Paramecium were grafted to recipient cells such that the axial orientation of the basal bodies and surrounding cortex was reversed 180° from that of the recipient, the cilia in the graft beat with permanently reversed polarity (Tamm et al. 1975). During the differentiation of ciliated epithelial cells, basal bodies dock in random orientation with the plasma membrane before axoneme assembly. Once the cilia begin to beat, the basal foot and striated rootlet appendages on each basal body become aligned with the uniform direction of ciliary beat (for review see Lemullois et al. 1988). Basal body alignment is dependent on numerous interactions of the appendages with the microtubule and actin filament cytoskeleton at the apical end of the cell (for review see Sandoz et al. 1988). In Chlamydomonas, Vfl1p may play a role in establishing the correct spatial organization of basal body appendages. For example, it may specify the microtubule triplets onto which the distal fiber assembles.
The rotational orientation of a basal body dictates the cytoplasmic location where a probasal body will form because the site of basal body replication is determined by radial asymmetry inherent to the mature organelle (Dippell 1968; Allen 1969). Misorientation of basal bodies in vfl1 mutant cells would likely lead to defects in the positioning of probasal bodies. It is possible that mispositioned probasal bodies would develop abnormally or would be delayed in membrane docking during the cell cycle, leading to the observed phenotype of basal bodies apparently stranded in the cytoplasm. This phenotype was observed in vfl3 mutant cells which have defects in the rotational orientation of basal bodies (Hoops et al. 1984). The localization of Vfl1p near the transitional fibers suggests that it could be involved in the process by which basal bodies dock at the plasma membrane after basal body elongation. Vfl1p does not contain predicted transmembrane domains, but it could interact with membrane proteins.
Our results indicate that Vfl1p is associated with the mature basal body at the base of each axoneme. In addition, in both intact cells and in NFAps we consistently observed one or two additional dots of immunofluorescence in positions expected for the two probasal bodies, a result supported by immunogold localization. Because the cells were in G1 phase of the cell cycle, the results suggest that Vfl1p is associated with probasal bodies soon after they form and before their elongation. These results are interesting because previous studies of the centriolar pathway of basal body assembly have shown that basal body assembly begins at the proximal end (Dippell 1968; Cavalier-Smith 1974). Proteins such as Vfl1p located at the distal end of the mature structure might be expected to be added at the stage of basal body elongation. The early association of Vfl1p with probasal bodies has a precedent in a 205-kD protein component of the transition region in Spermatozopsis similis, a related green alga (Lechtreck et al. 1999). The 205-kD protein was found to associate with probasal bodies soon after their assembly. Further studies of the order of assembly of these and other basal body components during the cell cycle will be useful for determining the assembly pathway.
The LRR and coiled coil structural motifs in Vfl1p provide domains for probable interactions with other protein(s). The localization pattern is distinct from those of other proteins, including centrin (Salisbury et al. 1988), γ-tubulin (Silflow et al. 1999), and δ-tubulin (Dutcher and Trabuco 1998), suggesting that these proteins do not interact directly with Vfl1p. The predicted coiled coil domain is required for protein function or targeting to the basal body, as shown by the similarity in phenotype between the null vfl1-2 allele and the vfl1-1 allele. Numerous centrosome-associated proteins contain coiled coil motifs (Andersen 1999); however, database searches did not reveal potential homologues of Vfl1p when either the entire protein or the unique region (amino acids 160–400) between the conserved structural domains was used to search protein databases. Although no homologues for Vfl1p have yet been found in other organisms, it seems likely that proteins with similarly asymmetric localization within basal bodies will be a universal feature. Future studies to define genes that interact with VFL1 in Chlamydomonas should provide further insight into the molecular mechanisms for establishing the radial asymmetry of basal bodies.
Acknowledgments
We thank Darryl Krueger for help with the immunogold localization experiments and the Electron Optic Facility (Minnesota Agricultural Research Station, St. Paul, MN) for use of the electron microscope. We thank the Imaging Center (College of Biological Sciences, University of Minnesota, St. Paul, MN) for use of immunofluorescence microscope facilities. We are grateful to Dr. William Dentler (University of Kansas, Lawrence, KS) and Dr. Richard Linck (University of Minnesota, Minneapolis, MN) for reading the manuscript and for helpful discussions.
S. Tousey was supported by the Undergraduate Research Opportunity Program, University of Minnesota. This work was supported by the National Institutes of Health grant GM51995 and by funding from the Minnesota Medical Foundation and the Office of the Vice President for Research and Dean of the Graduate School of the University of Minnesota to C.D. Silflow.
Footnotes
Abbreviations used in this paper: DIC, differential interference contrast; HA, hemagglutinin; LRR, leucine-rich repeat; NFAp, nucleoflagellar apparatus; RT, reverse transcriptase.
References
- Adams G.M.W., Wright R.L., Jarvik J.W. Defective temporal and spatial control of flagellar assembly in a mutant of Chlamydomonas reinhardtii with variable flagellar number. J. Cell Biol. 1985;100:955–964. doi: 10.1083/jcb.100.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzelius B. Electron microscopy of the sperm tail. J. Biophys. Biochem. Cytol. 1959;2:269–278. doi: 10.1083/jcb.5.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R.D. The morphogenesis of basal bodies and accessory structures of the cortex of the ciliated protozoan Tetrahymena pyriformis . J. Cell Biol. 1969;40:716–733. doi: 10.1083/jcb.40.3.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen S.S. Molecular characteristics of the centrosome. Int. Rev. Cytol. 1999;187:51–109. doi: 10.1016/s0074-7696(08)62416-x. [DOI] [PubMed] [Google Scholar]
- Anstrom J.A. Organization of the ciliary basal apparatus in embryonic cells of the sea urchin, Lytechinus pictus . Cell Tissue Res. 1992;269:305–313. doi: 10.1007/BF00319622. [DOI] [PubMed] [Google Scholar]
- Beech P.L., Melkonian M. The basal apparatus of the quadriflagellate Spermatozopsis exsultans (chlorophyceae)numbering of basal body triplets reveals triplet individuality and developmental modifications. J. Phycol. 1993;29:191–202. [Google Scholar]
- Beisson J., Jerka-Dziadosz M. Polarities of the centriolar structuremorphogenetic consequences. Biol. Cell. 1999;91:367–378. [PubMed] [Google Scholar]
- Borodovsky M., McIninch J. GeneMarkgene prediction of both DNA strands. Comput. Chem. 1993;17:123–133. [Google Scholar]
- Buchanan S.G., Gay N.J. Structural and functional diversity in the leucine-rich repeat family of proteins. Prog. Biophys. Mol. Biol. 1996;65:1–44. doi: 10.1016/s0079-6107(96)00003-x. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. Basal body and flagellar development during the vegetative cell cycle and the sexual cycle of Chlamydomonas reinhardtii . J. Cell Sci. 1974;16:529–556. doi: 10.1242/jcs.16.3.529. [DOI] [PubMed] [Google Scholar]
- Coss R.A. Mitosis in Chlamydomonas reinhardtii basal bodies and the mitotic apparatus. J. Cell Biol. 1974;63:325–329. doi: 10.1083/jcb.63.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debuchy R., Purton S., Rochaix J.-D. The argininosuccinate lyase gene of Chlamydomonas reinhardtiian important tool for nuclear transformation and for correlating the genetic and molecular maps of the ARG7 locus. EMBO (Eur. Mol. Biol. Organ.) J. 1989;8:2803–2809. doi: 10.1002/j.1460-2075.1989.tb08426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux J., Haeberli P., Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippell R.V. The development of basal bodies in Paramecium. Proc. Natl. Acad. Sci. USA. 1968;61:461–468. doi: 10.1073/pnas.61.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutcher S.K., Trabuco E.C. The UNI3 gene is required for assembly of basal bodies of Chlamydomonas and encodes δ-tubulin, a new member of the tubulin superfamily. Mol. Biol. Cell. 1998;9:1293–1308. doi: 10.1091/mbc.9.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler L.L., Holmes J.A., Dutcher S.K. Loss of spatial control of the mitotic spindle apparatus in a Chlamydomonas reinhardtii mutant strain lacking basal bodies. Genetics. 1995;141:945–960. doi: 10.1093/genetics/141.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J., Nikawa J.-I., Broek D., MacDonald B., Rodgers L., Wilson I.A., Lerner R.A., Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman M.A., Dush M.K., Martin G.R. Rapid production of full-length cDNAs from rare transcriptsamplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffal K.P. The basal body-root complex of Chlamydomonas reinhardtii during mitosis. Protoplasma. 1988;143:118–129. [Google Scholar]
- Gaffal K.P., el-Gammal S. Elucidation of the enigma of the “metaphase band” of Chlamydomonas reinhardtii . Protoplasma. 1990;156:139–148. [Google Scholar]
- Geimer S., Lechtreck K.-F., Melkonian M. A novel basal apparatus protein of 90 kD (Bap90) from the flagellate green alga Spermatozopsis similis is a component of the proximal plates and identifies the d-(dexter) surface of the basal body. Protist. 1998;149:173–184. doi: 10.1016/S1434-4610(98)70022-7. [DOI] [PubMed] [Google Scholar]
- Gibbons I.R. The relationship between the fine structure and the direction of beat in gill cilia of a lamellibranch mollusc. J. Biophys. Biochem. Cytol. 1961;11:179–205. doi: 10.1083/jcb.11.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman D.S., Levine R.P. Cytochrome f and plastocyanintheir sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardtii . Proc. Natl. Acad. Sci. USA. 1965;54:1665–1669. doi: 10.1073/pnas.54.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould R.R. The basal bodies of Chlamydomonas reinhardtii. Formation from probasal bodies, isolation, and partial characterization. J. Cell Biol. 1975;65:65–74. doi: 10.1083/jcb.65.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M., Yoda A. Isolation and phenotypic characterization of Chlamydomonas mutants defective in cytokinesis. Cell Struct. Funct. 1997;22:1–5. doi: 10.1247/csf.22.1. [DOI] [PubMed] [Google Scholar]
- Holmes J.A., Dutcher S.K. Cellular asymmetry in Chlamydomonas reinhardtii . J. Cell Sci. 1989;94:273–285. doi: 10.1242/jcs.94.2.273. [DOI] [PubMed] [Google Scholar]
- Hoops H.J., Witman G.B. Outer doublet heterogeneity reveals structural polarity related to beat direction in Chlamydomonas flagella . J. Cell Biol. 1983;97:902–908. doi: 10.1083/jcb.97.3.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoops H.J., Wright R.L., Jarvik J.W., Witman G.B. Flagellar waveform and rotational orientation in a Chlamydomonas mutant lacking normal striated fibers. J. Cell Biol. 1984;98:818–824. doi: 10.1083/jcb.98.3.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Watterson D.M., Lee V.D., Schibler M.J. Purification and characterization of a basal body-associated Ca21-binding protein. J. Cell Biol. 1988;107:121–131. doi: 10.1083/jcb.107.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson U.G., Porter K.R. Fine structure of cell division in Chlamydomonas reinhardi . J. Cell Biol. 1968;38:403–425. doi: 10.1083/jcb.38.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajava A.V. Structural diversity of leucine-rich repeat proteins. J. Mol. Biol. 1998;277:519–527. doi: 10.1006/jmbi.1998.1643. [DOI] [PubMed] [Google Scholar]
- Kobe B., Deisenhofer J. A structural basis of the interactions between leucine-rich repeats and protein ligands Nature. 374 1995. 183 185a [DOI] [PubMed] [Google Scholar]
- Kobe B., Deisenhofer J. Proteins with leucine-rich repeats Curr. Opin. Struct. Biol. 5 1995. 409 416b [DOI] [PubMed] [Google Scholar]
- Lechtreck K.-F., Teltenkotter A., Grunow A. A 210 kDa protein is located in a membrane-microtubule linker at the distal end of mature and nascent basal bodies. J. Cell Sci. 1999;112:1633–1644. doi: 10.1242/jcs.112.11.1633. [DOI] [PubMed] [Google Scholar]
- Lemullois M., Boisvieux-Ulrich E., Laine M.-C., Chailley B., Sandoz D. Development and functions of the cytoskeleton during ciliogenesis in metazoa. Biol. Cell. 1988;63:195–208. doi: 10.1016/0248-4900(88)90058-5. [DOI] [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Lynn D. The organization and evolution of microtubular organelles in ciliated protozoa. Biol. Rev. 1981;56:243–292. [Google Scholar]
- Masuda M., Sato H. Reversible resorption of cilia and the centriole cycle in dividing cells of sea urchin blastulae. Zool. Sci. 1984;1:455–462. [Google Scholar]
- McFadden D., Schulze B., Surek J.L., Salisbury, Melkonian M. Basal body reorientation mediated by a Ca21-modulated contractile protein. J. Cell Biol. 1987;105:903–912. doi: 10.1083/jcb.105.2.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D.R. Chlamydomonas flagella. J. Phycol. 2000;36:261–273. [Google Scholar]
- Nelson J.A.E., Savereide P.B., Lefebvre P.A. The CRY1 gene in Chlamydomonas reinhardtiistructure and use as a dominant selectable marker for nuclear transformation. Mol. Cell. Biol. 1994;14:4011–4019. doi: 10.1128/mcb.14.6.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder C.L., Jensen C.G., Jensen L.C.W. The resorption of primary cilia during mitosis in a vertebrate (PtK1) cell line. J. Ultrastruct. Res. 1979;68:173–185. doi: 10.1016/s0022-5320(79)90152-7. [DOI] [PubMed] [Google Scholar]
- Ringo D.L. Flagellar motion and fine structure of the flagellar apparatus in Chlamydomonas . J. Cell Biol. 1967;33:543–571. doi: 10.1083/jcb.33.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager R., Granick S. Nutritional studies in Chlamydomonas reinhardtii . Ann. NY Acad. Sci. 1953;56:831–838. doi: 10.1111/j.1749-6632.1953.tb30261.x. [DOI] [PubMed] [Google Scholar]
- Salisbury J.L., Baron A.T., Sanders M.A. The centrin-based cytoskeleton of Chlamydomonas reinhardtiidistribution in interphase and mitotic cells. J. Cell Biol. 1988;107:635–641. doi: 10.1083/jcb.107.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular CloningA Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Sanders M.A., Salisbury J.L. Immunofluorescence microscopy of cilia and flagella. Methods Cell Biol. 1995;47:163–169. doi: 10.1016/s0091-679x(08)60805-5. [DOI] [PubMed] [Google Scholar]
- Sandoz D., Chailley B., Boisvieux-Ulrich E., Lemullois M., Laine M.-C., Bautista-Harris G. Organization and functions of cytoskeleton in metazoan ciliated cells. Biol. Cell. 1988;63:183–193. doi: 10.1016/0248-4900(88)90057-3. [DOI] [PubMed] [Google Scholar]
- Schnell R.A., Lefebvre P.A. Isolation of the Chlamydomonas regulatory gene NIT2 by transposon tagging. Genetics. 1993;134:737–747. doi: 10.1093/genetics/134.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silflow C.D. Organization of the nuclear genome. In: Rochaix J.-D., Goldschmidt-Clermont M., Merchant S., editors. The Molecular Biology of Chloroplasts and Mitochondria in Chlamydomonas. Kluwer Academic Publishers; Dordrecht, The Netherlands: 1998. pp. 25–40. [Google Scholar]
- Silflow C.D., Rosenbaum J.L. Multiple α- and β-tubulin genes in Chlamydomonas and regulation of tubulin mRNA levels after deflagellation. Cell. 1981;24:81–88. doi: 10.1016/0092-8674(81)90503-1. [DOI] [PubMed] [Google Scholar]
- Silflow C.D., Liu B., LaVoie M., Richardson E.A., Palevitz B.A. γ-tubulin in Chlamydomonascharacterization of the gene and localization of the gene product in cells. Cell Motil. Cytoskeleton. 1999;42:285–297. doi: 10.1002/(SICI)1097-0169(1999)42:4<285::AID-CM3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Surdej P., Jacobs-Lorena M. Strategy for epitope tagging the protein-coding region of any gene. Biotechniques. 1994;17:560–565. [PubMed] [Google Scholar]
- Tam L.-W., Lefebvre P.A. Cloning of flagellar genes in Chlamydomonas reinhardtii by DNA insertional mutagenesis. Genetics. 1993;135:375–384. doi: 10.1093/genetics/135.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm S., Tamm S.L. Development of macrociliary cells in Beroe. I. Actin bundles and centriole migration. J. Cell Sci. 1988;89:67–80. doi: 10.1242/jcs.89.1.67. [DOI] [PubMed] [Google Scholar]
- Tamm S., Sonneborn T.M., Dippel R.V. The role of cortical orientation in the control of the direction of ciliary beat in Paramecium . J. Cell Biol. 1975;64:98–112. doi: 10.1083/jcb.64.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R.L., Goodenough D.A., Goodenough U.W. Membrane particle arrays associated with the basal body and with contractile vacuole secretion in Chlamydomonas . J. Cell Biol. 1977;72:133–143. doi: 10.1083/jcb.72.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley D.N., Wang A.M., Strugnell G.E. Expression of primary cilia in mammalian cells. Cell Biol. Int. Rep. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- Wilkerson C.G., King S.M., Witman G.B. Molecular analysis of the gamma-heavy chain of Chlamydomonas flagellar outer arm dynein. J. Cell Sci. 1994;107:497–506. doi: 10.1242/jcs.107.3.497. [DOI] [PubMed] [Google Scholar]
- Wray W., Boulikas T., Wray V.P., Hancock P. Silver staining of proteins in polyacrylamide gels. Anal. Biochem. 1981;118:197–203. doi: 10.1016/0003-2697(81)90179-2. [DOI] [PubMed] [Google Scholar]
- Wright R.L., Chojnacki B., Jarvik J.W. Abnormal basal-body number, location, and orientation in a striated fiber-defective mutant of Chlamydomonas reinhardtii . J. Cell Biol. 1983;96:1697–1707. doi: 10.1083/jcb.96.6.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright R.L., Salisbury J., Jarvik J.W. A nucleus-basal body connector in Chlamydomonas reinhardtii that may function in basal body localization or segregation. J. Cell Biol. 1985;101:1903–1912. doi: 10.1083/jcb.101.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright R.L., Adler S.A., Spanier J.G., Jarvik J.W. Nucleus-basal body connector in Chlamydomonasevidence for a role in basal body segregation and against essential roles in mitosis or in determining cell polarity. Cell Motil. Cytoskel. 1989;14:516–526. doi: 10.1002/cm.970140409. [DOI] [PubMed] [Google Scholar]