

Abstract
Background & Aims
In addition to its role as the primary mediator of the enteroinsular axis, glucose-dependent insulinotropic polypeptide (GIP) may play a critical role in the development of obesity. The purpose of these studies was to characterize the effects of GIP and its receptor (GIPR) in adipocyte development and signaling.
Methods
Effects of GIP and GIPR on differentiated 3T3-L1 cells were analyzed using Western blot analysis, Oil-Red-O staining, cAMP radioimmunoassay, immunofluorescence microscopy, and glucose uptake measurements.
Results
To determine whether GIP and GIPR are important components in adipocyte development, the expression profile of GIPR during differentiation was examined. GIPR protein expression was enhanced during the differentiation process, and co-incubation with its ligand GIP augmented the expression of aP2, a fat cell marker. Conversely, the suppression of GIPR expression by a specific shRNA attenuated Oil-Red-O staining and aP2 expression, suggesting that the GIPR may play a critical role in adipocyte development. To investigate specific signaling components that may mediate the effects of GIP, we analyzed Akt, GLUT-4, and glucose uptake, all of which are modulated by insulin in fat cells. Like insulin, GIP induced the activation of Akt in a concentration-dependent manner, promoted membrane GLUT-4 accumulation, and enhanced [3H]-2-deoxyglucose uptake.
Conclusions
These studies provide further evidence for an important physiological role for GIP in lipid homeostasis and possibly in the pathogenesis of obesity. Furthermore, our data indicate that the GIPR might represent a suitable target for the treatment of obesity.
Keywords: GIP, Akt, GLUT-4, adipocytes, obesity
Introduction
Greater than 60% of the adult population in the United States is considered overweight or obese.1 Despite widespread recognition of the serious complications associated with this chronic condition, the incidence of obesity has continued to increase progressively during the past two decades.1 Several gastrointestinal (GI) regulatory peptides are known to possess important metabolic properties and appear to play an etiologic role in the pathogenesis of obesity and nutrition-related disorders. One peptide of particular interest is glucose-dependent insulinotropic polypeptide (GIP, also known as gastric inhibitory polypeptide), a 42-amino acid hormone synthesized in, and released from, K-cells of the duodenum and jejunum. Upon its postprandial release, GIP induces pancreatic insulin release,2 thus acting as the principal mediator of the enteroinsular axis.
Miyawaki et al. reported that inhibition of GIP signaling could prevent obesity in mice.3 When fed a high-fat diet, GIP receptor-deficient mice (GIPR-/-) maintained normal weight, while the body weight of wild-type littermates increased by 35%. Furthermore, subcutaneous and visceral fat mass, as well as the size of the adipocytes isolated from epididymal fat pads, were significantly enhanced in GIPR+/+ mice when compared to GIPR-/- mice.3 In addition, Clements et al. have reported that patients who undergo Roux-en-Y gastric bypass surgery (RYGB), which bypasses portions of small intestine that produce and secrete GIP, not only lose weight, but also significantly improve their glucose tolerance.4 Moreover, they found that fasting plasma concentrations of GIP, insulin, and C-peptide all steadily declined following the surgery, starting before significant weight loss was evident. These observations support the hypothesis that in addition to its insulinotropic role, GIP/GIPR signaling may represent an important factor in obesity.
Despite these studies indicating the importance of GIP in nutrient deposition and its potential etiological role in obesity, the precise mechanism by which GIP exerts its metabolic properties and in particular, its effects on adipocytes, have not been elucidated. In contrast, the effects of insulin on adipocyte function have been well characterized. Insulin has been shown to be a potent stimulator of lipogenesis,5 and overnutrition causes secondary hyperinsulinemia and subsequent adipocyte hypertrophy.6 Previous studies have shown that the selective disruption of the insulin receptor (IR) gene in fat cells resulted in mice with a low fat mass and that the mice were protected against obesity and obesity-related glucose intolerance.7
Past studies performed in this laboratory have sought to define the physiological properties of GIP and to determine its precise contribution to the pathogenesis of obesity. Previously, we determined that oral administration of glucose potently stimulated GIP expression in the rat,8 and we have demonstrated the presence of functional GIPR on rat adipocytes.9 More recently, we reported that GIP possesses insulin-mimetic properties by virtue of its ability to inhibit isoproterenol-induced lipolysis in perifused rat adipocytes.10 In the present study, we demonstrate that GIPR plays an important role in adipocyte differentiation, and, in addition to stimulating insulin release from pancreatic β-cells, GIP possesses insulin-mimetic signaling properties in adipocytes. Like insulin, GIP induces the activation of the serine/threonine kinase Protein Kinase B (Akt/PKB) via a wortmannin-sensitive pathway. The activation of Akt, in turn, promotes the membrane translocation of glucose transporter-4 (GLUT-4), leading to enhanced adipocyte uptake of glucose.
Materials and Methods
Cell Culture and Treatments
3T3-L1 preadipocytes were maintained in Dulbecco's modified Eagle's Medium (DMEM; Gibco Laboratories, Grand Island, NY), supplemented with 10% calf serum (Gibco, Carlsbad, CA) and 1% penicillin/streptomycin (DMEM/CS/PS). Parental plates were kept below 80% confluency to reduce spontaneous differentiation. On experimental plates, preadipocytes were grown until confluent and further cultured for an additional 2-3 days in DMEM supplemented with 10% fetal calf serum and 1% penicillin/streptomycin (DMEM/FCS/PS). Differentiation was initiated (day 0) by incubating confluent cells with DMEM/FCS/PS containing 10 μg/mL insulin (Sigma, St. Louis, MO), 10 μg/mL dexamethasone (Fujusawa USA, Inc., Deerfield, IL), and 0.5 mM 3-isobutyl-1-methylxanthine (IBMX, Sigma, St. Louis, MO). On day 2, the induction medium was replaced with DMEM/FCS/PS containing 2.5 μg/mL insulin. On day 4, cells were further incubated with DMEM/FCS/PS for full maturation into adipocytes. Unless otherwise noted, differentiated mature adipocytes were harvested at days 8-10 for various experiments. When GIP was co-incubated with the standard induction protocol, the cells were subjected to the peptide for the first 4 days of the aforementioned differentiation process.
Primary human preadipocytes (kindly provided by Dr. James Kirkland, Boston, MA) were normally grown in α-MEM, and after confluence, they were incubated in an enriched medium containing α-MEM, 2 μM insulin, 10 mM glucose, 5 μl/ml 10% Liposyn III (Abbott Laboratories, Abbott Park, IL), and 20% Nuserum (designated DM3) or α-MEM, 2 μM insulin, 10 mM glucose, 125 μM indomethacin, and 10% FBS (designated DM1).11 Continuous incubation in these media induces human preadipocytes to undergo differentiation. Approximately 2 weeks post-induction, human adipocytes were harvested for various experiments.
Porcine GIP (2×10-13 – 2×10-7 M) (1-30 amide, Bachem, Belmont, CA) and insulin (0.0001 – 100 nM) (Sigma) were used to treat adipocytes to examine various signaling components. 1 μM Wortmannin (Roche, Indianapolis, IN) was used to inhibit PI3 kinase (PI3K) activity. A previously characterized GIPR-specific antagonist,12, 13 designated ANTGIP (AG), was used at a concentration of 7.5 – 10 μM.
Short Hairpin RNA (shRNA)
A particular region corresponding to 469-451 of the human GIPR sequence was selected upon validating that this region was capable of reducing GIPR expression. The sequence from this region was inserted into pSilencer 4.1 CMV vectors (Ambion Inc., Austin, TX) prior to transfection into 3T3-L1 preadipocytes. Negative control scrambled siRNA was purchased from Ambion Inc.
Oil Red-O staining
Differentiated adipocytes were rinsed in PBS prior to fixing with 10% formaldehyde. Approximately 15 min after the fixation, Oil-Red O stain was used to stain for lipid accumulation (2-3 h). After rinsing with the distilled water, photomicrographs were taken with a digital camera to document staining.
Western Blot Aanalysis
Total protein was extracted, as previously described.14, 15 For membrane protein extraction, a protocol previously described was utilized.16 Protein quantification was performed using a BCA protein assay kit (Pierce, Rockford, IL) following the manufacturer's instructions. Western blot hybridization analysis was performed, as previously described,14, 15 using the following antibodies: polyclonal aP2 (kindly provided by Dr. Stephen Farmer, Boston University School of Medicine), polyclonal phospho-Akt Ser473 (Cell Signaling, Danvers, MA), monoclonal PPARγ (Santa Cruz, Santa Cruz, CA), monoclonal phospho-ERK1/2 (Cell Signaling), polyclonal GIPR (kindly provided by Dr. Joel Habener, Harvard Medical School), monoclonal GLUT-4 (IF8, kindly provided by Dr. Paul Pilch, Boston University School of Medicine) and monoclonal β-actin (Sigma).
Cyclic Adenosine Monophosphate (cAMP) Measurement
3T3-L1 cells were differentiated on a 12-well plate. On day 8, cells were serum starved overnight before 20 μM forskolin, insulin, or GIP was added in the presence of 1 mM IBMX for an additional 10 min at 37 °C. After aspirating the media, 500 μL of 100% ethanol were added to each well, and the samples were dried under a vacuum. When the wells were completely dry, lysates were prepared in the 1X cAMP buffer. Isolated rat adipocytes were prepared, as previously described,17 and the accumulation of cyclic AMP (cAMP) was measured by radioimmunoassay using the manufacturer's protocol (NEN Life Science Products, Boston, MA). Media with and without IBMX were used as negative controls for background.
GLUT-4 Immunofluorescence
3T3-L1 cells were differentiated in a slide chamber (LabTek, Campbell, CA). On day 8 or 9, adipocytes were washed twice with Krebs Ringer Biocarbonate (KRB) buffer 0.12 M NaCl, 0.025 M NaHCO3, 5 mM KCl, 12 mM KH2PO4, 12 mM MgSO4, 20 mM MOPS, 25 mM CaCl2, and 2 mM glucose) containing 0.1% bovine serum albumin (BSA). The cells were treated with either insulin or GIP in the absence and presence of ANTGIP for 1 h and washed twice with 1X PBS. The cells were fixed with Histofix (Sigma) for 15 min at room temperature. After washing the cells twice with 1X PBS, 0.1% Triton X-100 was used for 10 min to permeabilize the cells. The cells were washed again in 1X PBS and blocked with 5% BSA for 1 h at room temperature. An overnight incubation with polyclonal GLUT-4 antibodies (Novus Biologicals, Littleton, CO) at 4 °C yielded optimal results for detecting GLUT-4 protein. Cells were washed three times with 1X PBS and further incubated with anti-rabbit FITC secondary antibodies (Jackson Immunoresearch, West Grove, PA) for 1 h at room temperature, and they were visualized using confocal laser scanning microscopy (Fluoview 500, Olympus America Inc., Melville, NY). RG-7G was excited at 488 nm via an argon/krypton laser; emitted fluorescence was detected through a 515-540 nm band pass filter.
[3H]-2-deoxyglucose Uptake
On day 8 or 9 of differentiation, cells were washed twice with warm serum-free DMEM/PS, and further serum starved for 2 h at 37 °C. Adipocytes were incubated with either insulin or GIP in the absence and presence of ANTGIP for 15 min. After aspirating media, cells were washed twice with warm KRH buffer, pH 7.4 (121 mM NaCl, 4.9 mM KCl, 1.2 mM MgSO4, 0.33 mM CaCl2, and 12 mM HEPES). A mixture of KRH buffer containing 2 mM 2-deoxyglucose and 1 μCi/mL [3H]-2-deoxyglucose was made and added to the cells for 10 min at room temperature. The reaction was stopped by washing three times with ice-cold KRH buffer supplemented with 25 mM D-glucose. The cells were solubilized on ice with Triton X-100 lysis buffer (1% Triton X-100, 20 mM Tris, and 150 mM NaCl) and vortexed. Equal amounts of lysed cell were mixed with scintillation fluid, and specific activity was counted in a liquid scintillation counter (Beckman Instruments, Fullerton, CA) in duplicate. [3H]-2-deoxyglucose uptake, expressed as nmol/mL per min, was normalized by using a BCA protein quantification of each sample.
Statistical analysis
Data analysis was performed using two-way Student's T-test for paired comparisons and ANOVA in which significant inter-group differences were defined with all-pairwise comparison with Bonferroni correction. Statistical significance was assigned if P < 0.05.
Results
GIPR Protein Expression is Rapidly Induced During the Early Stages of the Differentiation Process
We initially demonstrated the presence of the GIPR in mouse 3T3-L1 cells, rat fat, and in human adipocytes upon differentiation from preadipocytes (Figure 1A). To determine whether GIPR induction represents an early event during differentiation, whole cell lysates were harvested at different time points and probed. GIPR was induced as early as 30 min following the induction in 3T3-L1 cells (Figure 1B). Densitometry readings normalized to β-actin suggested an approximate two-fold increase in the GIPR at this time point. To compare the relative induction of GIPR to other known protein expression patterns during the differentiation process, we also assessed the expression of phospho-ERK1/2 and peroxisome proliferator-activated receptor gamma (PPARγ) in these protein lysates. As depicted in Figure 1B, phospho-ERK1/2 expression peaked at 10 min, and PPARγ expression was detected on day 3, a profile similar to that detected by Prusty et al.23. These results indicate that the GIPR possesses a distinctive expression profile, and that similar to ERK1/2, it is induced early, but unlike ERK1/2, its induction is sustained or even enhanced throughout the process of adipocyte maturation.
Figure 1.
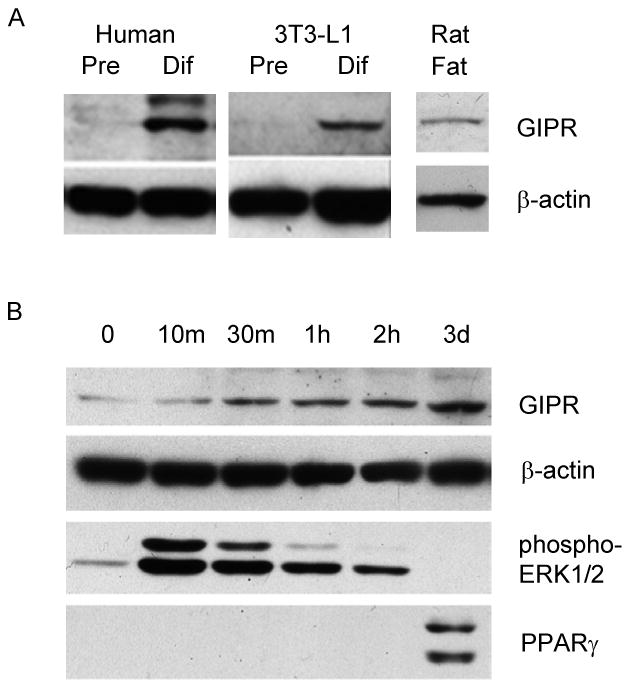
GIPR differentiation. (A) Human and 3T3-L1 preadipocytes (Pre) were converted to differentiated fat cells (Dif). These samples, as well as isolated rat fat, were probed for GIPR protein using a polyclonal antibody. (B) 3T3-L1 preadipocytes were induced, and protein was extracted at the indicated time points for Western anaylsis of GIPR, phosphorylated-ERK1/2, PPARγ, and β-actin. Western blots representative of at least four independent experiments are depicted.
Co-incubation of GIP During the Differentiation Process Enhances the Expression of aP2, a Fat Cell Marker
Because we observed that the GIPR is induced early in the differentiation process, we examined the possibility that the addition of its ligand (GIP) during this process might enhance lipogenesis. GIP was added to the differentiation medium for the first 4 days, however the concentration of insulin was decreased (1 – 0.01 μg/mL) to attenuate the effect of insulin on adipocyte development. When cells were harvested on day 9 of differentiation, we observed that co-incubation with GIP enhanced aP2 expression (lanes 5, 7, 9, and 10) compared to cells that were differentiated only with insulin, dexamethasone, and IBMX (lanes 4, 6, and 8) (Figure 2). This finding suggests that GIP may increase adipocyte development by potentiating the effects of insulin.
Figure 2.
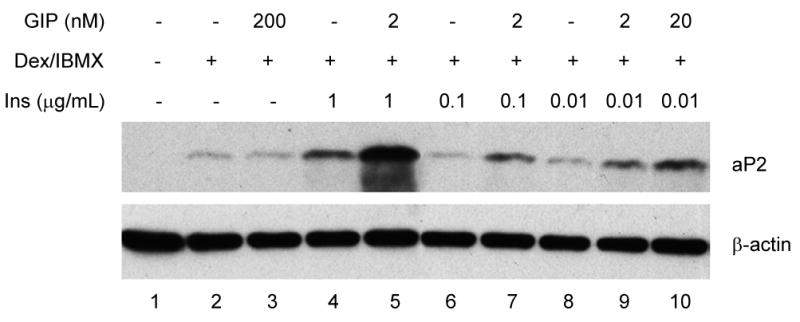
GIP enhancement of aP2 expression. 3T3-L1 cells were incubated with GIP, IBMX, dexamethasone, and insulin (1 – 0.01 μg/mL) for the first 4 days of the differentiation process. Cells were harvested on day 9 and protein extracted. Western blot analyses were performed with polyclonal aP2 and β-actin antibodies.
GIPR shRNA Inhibits Differentiation of Preadipocytes into Adipocytes
Compared to parental 3T3-L1 cells or control cells that have retained the expression of GIPR during the differentiation process, GIPR repressed cells failed to mature into adipocytes by day 8, as evident by the aP2 protein expression pattern (Figure 3A) and Oil-Red-O staining (Figure 3B). These observations suggest that GIPR appears to constitute a necessary component in the differentiation process.
Figure 3.
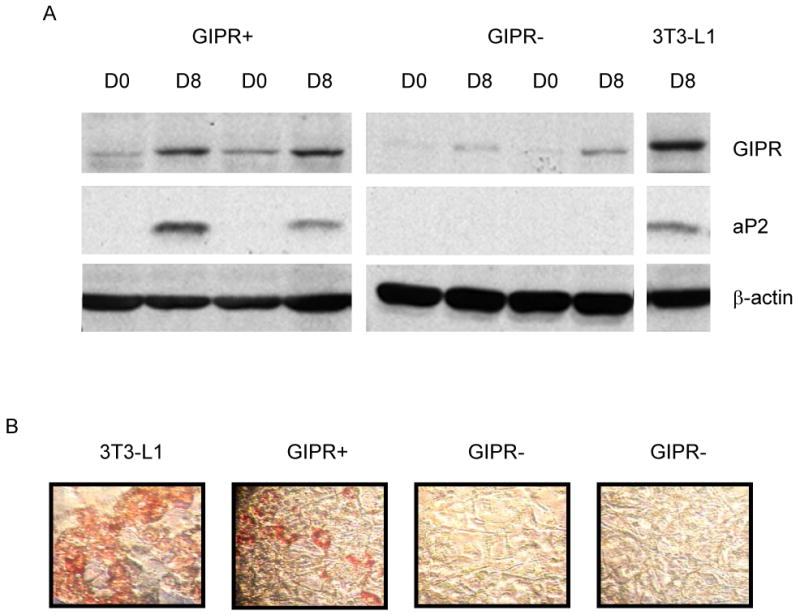
Effect of GIPR on preadipocyte differentiation. GIPR expression was repressed (GIPR-) by stably transfecting GIPR shRNA into 3T3-L1 preadipocytes. (A) Cells were harvested on day 0 (D0) and day 8 (D8), and the expression of GIPR, aP2, and β-actin protein was analyzed. (B) Oil-Red-O staining was performed on day 8 for parental 3T3-L1, negative control (GIPR+), and two GIPR shRNA (GIPR-) clones.
GIP Does Not Signal Through Adenylyl Cyclase in Rat Fat and Differentiated 3T3-L1 Adipocyte
We have previously reported that GIP, like insulin, attenuates isopreteronol induced lipolysis, suggesting an anti-lipolytic role for this peptide.18 However, it has been previously reported in pancreatic islet β-cells that GIP signals through adenylyl cyclase and induces cAMP accumulation,19 a process that, in adipocytes, has long been associated with active lipolysis.20 Thus, we performed experiments to determine whether GIP might likewise modulate cAMP in rat fat and differentiated 3T3-L1 adipocytes. As seen in Figure 4, unlike its effect on pancreatic β-cells, GIP did not appear to signal through adenylyl cyclase in fat cells, suggesting that the effect of GIP is cell-specific. Furthermore, co-incubation of GIP with isoproterenol did not alter the effects of isoproterenol (Data not shown), consistent with a proposed non-lipolytic role in adipocytes.
Figure 4.

Effect of GIP on cAMP accumulation of in fat cells. Following a 10-min incubation under different conditions, cAMP extracted from both rat fat and differentiated 3T3-L1 cells was measured by radioimmunoassay. Basal signifies cAMP levels in the untreated sample sets. 1 μM isoproterenol (Iso) and 20 μM forskolin (Forsk) were used as positive controls. Similar results were obtained from at least two independent experiments. Data are expressed as the mean ±SE in pmol/mL.
GIP Enhances Phosphorylation of Akt in Pancreatic Beta Cells and Adipocytes
We next sought to examine whether GIP might also signal through downstream targets of insulin. One particularly well-recognized insulin-stimulated metabolic pathway involves the activation of PI3K through insulin receptor substrate (IRS),21 which, in turn, promotes Akt phosphorylation and activation. As seen in Figure 5A, both GIP and insulin independently activated Akt in pancreatic islet β-cells. Similarly, when 3T3-L1 and human preadipocytes were transformed into adipocytes and were incubated in the presence of GIP, Akt was activated in a concentration-dependent manner (Figure 5B). These results indicate that GIP, like insulin, appears to exert its effects by activating Akt in both pancreatic islet β and fat cells.
Figure 5.
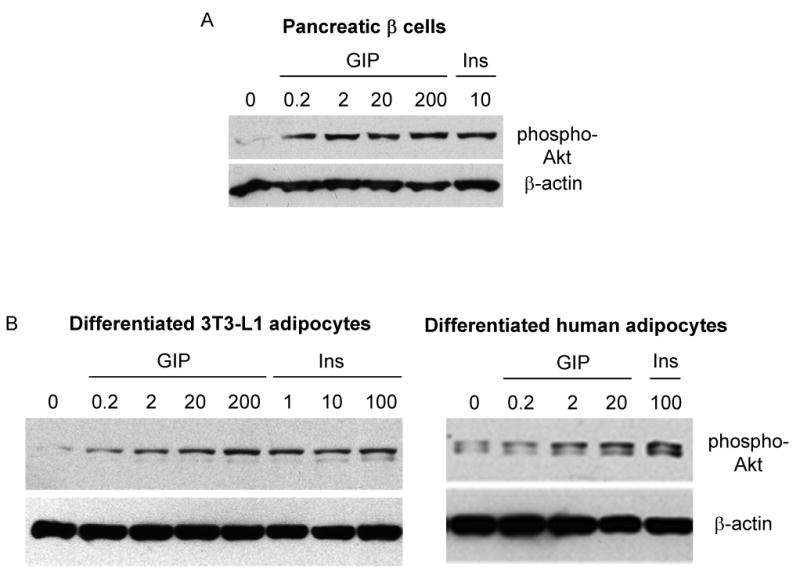
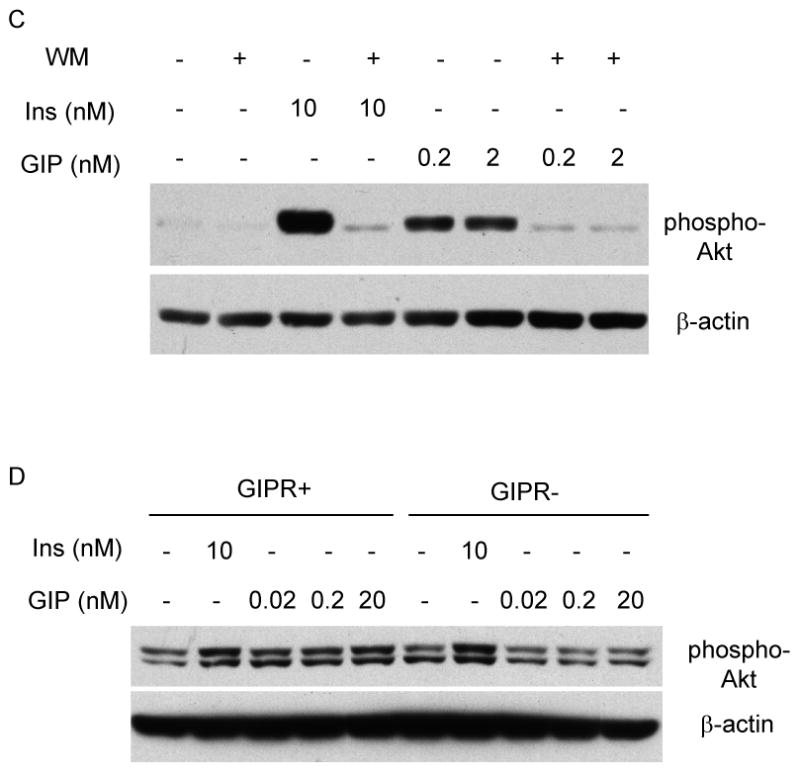
Effects of GIP and insulin on Akt phosphorylation. (A) Rin 5AH cells (pancreatic islet β-cell line) were incubated in the presence of GIP (0.2 – 200 nM) and 10 nM insulin (Ins) for 10 min. Total protein lysates were extracted and probed for expression of the Ser 473 phosphorylated form of Akt (pAkt) and total β-actin. (B) Differentiated 3T3-L1 and human adipocytes were incubated with GIP (0.2 – 200 nM) and insulin (1 – 100 nM) for 10 min. Total protein lysates were extracted and probed for pAkt expression. (C) Differentiated 3T3-L1 adipocytes were initially treated for 30 min with 1 μM Wortmannin (Wort), followed by an additional 10-min incubation with 0.2 nM GIP, 2 nM GIP, or 10 nM insulin. Total protein lysates were extracted and probed for pAkt. Representative Western blots are depicted, and similar results were obtained from at least three independent experiments. (D) Negative control and GIPR repressed cells were treated with either 10 nM or insulin or GIP (0.02 – 20 nM) for 10 min and probed for pAkt and β-actin expression.
To examine whether GIP, like insulin, activates Akt through its upstream regulator PI3K, differentiated 3T3-L1 adipocytes were pre-treated with PI3K inhibitor wortmannin for 30 min and further incubated with either GIP or insulin for an additional 10 min. As depicted in Figure 5C, the effect of GIP on activation of Akt was attenuated by wortmannin, suggesting that both GIP and insulin regulation of Akt is wortmannin-sensitive. To confirm that GIP specifically signals through its receptor in activating Akt, we compared its effects in GIPR repressed 3T3-L1 cells. Compared to control cells, which responded to both insulin and GIP in activating Akt, GIPR repressed cells retained only their response to insulin (Figure 5D).
GIP Augmented Membrane Accumulation of GLUT-4
A downstream consequence of PI3K/Akt signaling is GLUT-4 cell membrane translocation and the subsequent cellular accumulation of glucose. Fat cells were incubated in the presence of 20 nM of GIP or 10 nM of insulin with or without the GIPR-specific antagonist ANTGIP, and membrane fractions were harvested for Western analysis. Both GIP and insulin independently augmented the accumulation of GLUT-4 in the cell membrane (Figure 6A). Furthermore, ANTGIP selectively inhibited the effect of GIP, but not that of insulin. To confirm these findings, immunocytochemistry was performed in differentiated 3T3-L1 adipocytes. Compared to untreated cells, which demonstrated diffuse cytoplasmic staining of GLUT-4, both insulin and GIP induced the accumulation of GLUT-4 in the membrane, as indicated by border around the differentiated fat cells (Figure 6B, red arrows). ANTGIP alone had no effect on the fat cells, but did selectively inhibit the effect of GIP, but not insulin, suggesting that GIP independently enhances the membrane accumulation of GLUT-4.
Figure 6.
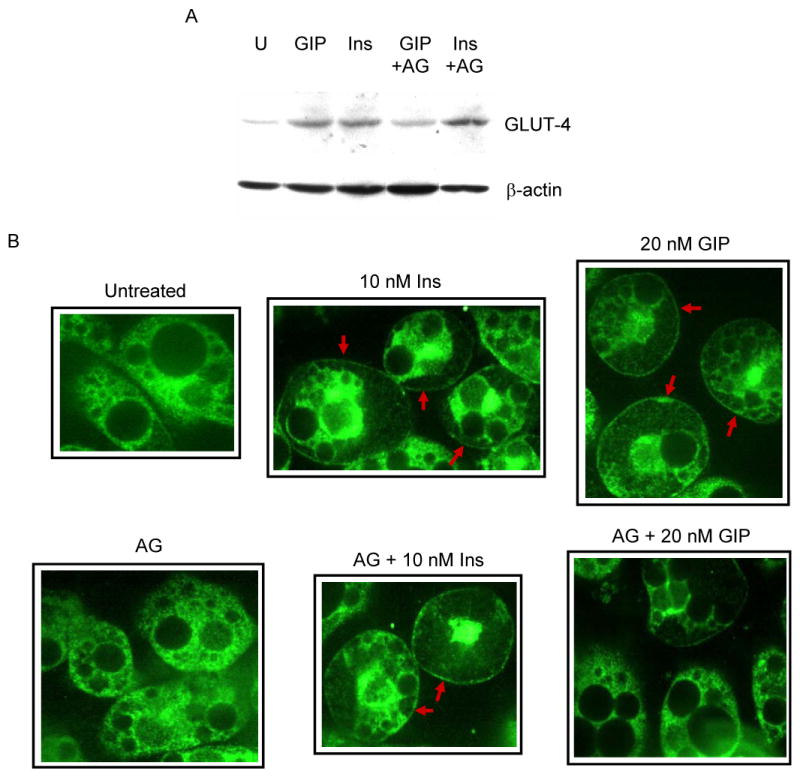
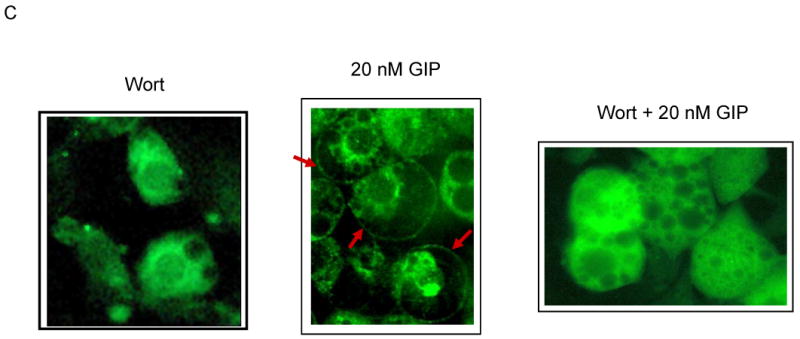
Effects of GIP and insulin on the accumulation of GLUT-4 in the plasma membrane. (A) Differentiated 3T3-L1 adipocytes were incubated in the presence of either 20 nM GIP or 10 nM insulin with or without the GIP-specific antagonist ANTGIP (AG) for 1 h. Membrane fractions were extracted and probed for the accumulation of GLUT-4 protein. (B) 3T3-L1 preadipocytes were grown in chamber slides and differentiated, and probed for the accumulation of GLUT-4 protein. Cells were visualized using confocal laser scanning microscopy. Representative images are shown, and similar images were visualized in three additional independent experiments. (C) Differentiated 3T3-L1 adipocytes were pre-treated with 1 μM wortmannin for 30 min prior to additional incubation in the presence or absence of GIP. Fat cells were stained with polyclonal GLUT-4 primary and counterstained with FITC conjugated secondary antibodies.
To determine whether GIP-dependent membrane accumulation of GLUT-4 is mediated through PI3K, cells were pretreated with wortmannin before co-incubation with GIP. Wortmannin blocked the effects of GIP on GLUT-4 translocation (Figure 6C), confirming that like Akt activation, GIP signals this process in a wortmannin-sensitive manner.
GIP Enhanced Glucose Uptake
To further investigate the insulin-mimetic effects of GIP via activation of Akt and membrane accumulation of GLUT-4, glucose uptake was measured on differentiated 3T3-L1 adipocytes. Compared to untreated cells, 20 nM of GIP enhanced [3H]-2-deoxyglucose uptake nearly 3-fold (Figure 7). The addition of the GIPR-specific antagonist ANTGIP attenuated the effect of GIP on glucose uptake, confirming that glucose uptake elicited by GIP was specific in 3T3-L1 adipocytes.
Figure 7.

Effects of GIP and insulin on [3H]-2-deoxyglucose uptake. Differentiated 3T3-L1 adipocytes were incubated under control conditions (U) or with 100 nM insulin, 20 nM GIP, or 20 nM GIP in the presence of ANTGIP (AG). Cells were lysed after the incorporation of [3H]-2-deoxyglucose, and equal amounts were counted in a liquid scintillation counter. Samples were assayed in duplicate. Data are expressed as nmol/mL per min (mean ±SE). * P ≤ 0.02 to U. ** P ≤ 0.02 to GIP.
Discussion
Earlier studies support the hypothesis that the marked increase in the prevalence of obesity might be due, at least in part, to optimal energy homeostasis. For example, insulin, the most potent mammalian anabolic hormone,22 appears to have evolved from the need to maximize energy efficiency, obviating the requirement to continuously forage for food. Organisms expressing this important peptide hormone possessed a distinct survival advantage and flourished. During evolution, insulin biosynthesis translocated to the pancreas, possibly to protect β-cells from enteric organisms, which then necessitated a messenger from the intestine to complete an enteroinsular axis. The eventual development of GIP and other incretins fulfilled this requirement.13, 23 The additional survival benefit offered by GIP may have been the ability of this regulatory peptide to not only stimulate intestinal glucose absorption and maximally release insulin12, but to also possess insulin mimetic properties, including lipid deposition in adipocytes, as demonstrated in the present study, as well as in recent studies in our laboratory (Figure 8).10 This physiological redundancy ensured the survival of organisms during times when food was scarce.
Figure 8.

Summary diagram. Overnutrition appears to increase GIP expression, which in turn enhances intestinal glucose absorption and insulin release. Our data suggest that GIP independently modulates signaling components that serve to further promote the storage of fat, thereby augmenting nutrient efficiency and potentially promoting obesity.
The consideration of GIP as an efficiency hormone can be further corroborated through its direct link with food ingestion. It has been demonstrated that ingestion of nutrients increases GIP gene expression,8 while the deprivation of food decreases GIP synthesis and secretion.24 Furthermore, a diet chronically high in fat caused K-cell hyperplasia.25 Compared to lean controls, ob/ob mice had higher basal concentrations of intestinal mucosal GIP,26 and the oral administration of glucose and particularly fat, caused additional increases in GIP concentrations.27 As we have moved into a period in time in which food is no longer scarce, at least in most Western nations, this survival advantage, characterized by optimal intestinal nutrient absorption and storage capacity, may be contributing to the obesity epidemic we are now witnessing. Our study is the first to demonstrate that in adipocytes, GIP independently modulates signaling components similarly involved in the insulin signaling pathway, thereby promoting storage, both directly and indirectly through increasing the release of insulin.
Earlier studies had shown that GIP regulates the release of lipoprotein lipase (LPL), which hydrolyzes chylomicrons and very low-density lipoproteins, thereby liberating fatty acids for uptake and storage within the adipocyte. Murphy et al.28 demonstrated that LPL and plasma GIP increased in parallel in vivo after a series of meals in which lipid content was increased. These studies indicate that GIP may represent a major hormonal signal linking meal content and size to postprandial LPL activity 29, 30 and modulating circulating lipoprotein homeostasis.28 In addition, GIP has been found to promote the incorporation of glucose into extractable lipids.31 GIP, like insulin, stimulates fatty acid synthesis in explants of rat adipose tissue, measured by the incorporation of [14C]acetate into saponifiable fat.32 Thus, GIP may function to increase the postprandial uptake of glucose and thereby may enhance the synthesis of fatty acid from glucose.
To further define the mechanism by which GIP may modulate metabolic properties in adipocytes, we examined various signaling components associated with insulin because of its insulin-mimetic properties in adipocytes. Previous studies have shown that insulin-stimulated uptake of glucose in striated muscle and adipose tissue is mediated by the translocation of GLUT-4 from an intracellular pool to the plasma membrane.33, 34 Upon binding to the receptor, insulin activates its receptor's intrinsic kinase, leading to autophosphorylation and tyrosine phosphorylation of several substrates, including members of the insulin receptor substrate (IRS) family.35 IRS phosphorylation, in turn, recruits other signaling molecules, including PI3K.36 One of the downstream targets of PI3K is Akt, whose activation leads to increased glucose transport in 3T3-L1 adipocytes37 and in isolated rat adipocytes.38 Our data suggest that GIP, like insulin, activates Akt and appears to signal through this intermediate component. Although others have reported activation of Akt by GIP in pancreatic islet β-cells39, 40 as well as activation of GIP in the presence of insulin in adipocytes,41 this report is the first to demonstrate the capacity of GIP to independently activate Akt in adipocytes in a wortmannin-sensitive manner. Although Akt is not the only factor responsible for activating glucose transport, GIP appeared to elicit its signaling effects through PI3K/Akt and modulate subsequent accumulation of GLUT-4 and enhanced glucose uptake, well-recognized downstream targets of Akt in fat cells.
One of the major diseases related to obesity is type 2 diabetes mellitus, in which the insulinotropic effects of GIP may be diminished.42 Previous reports have suggested that by restoring its function, GIP may potentially provide therapeutic benefit in diabetic patients 42. However, the results of our studies suggest that GIP may actually promote, rather than prevent, the development of obesity. Previous studies43 have indicated that, similar to insulin, GIP may be overexpressed in obese individuals, further exacerbating the insulin resistance manifested by patients with type 2 diabetes mellitus. The results of the present study support the hypothesis that by antagonizing the effects of GIP and indirectly, the effects of insulin, greater benefit can ultimately be achieved, including the prevention of the development of obesity and related disorders. This hypothesis is supported by a recently published study by Zhou et al.,44 who demonstrated that insulin resistance was ameliorated in mice harboring disruptions of both IRS-1 (IRS-1-/-) and the GIPR (GIPR-/-). Furthermore, compared to IRS-1-/- mice, both the visceral and subcutaneous fat mass, as well as plasma leptin levels, were markedly diminished in the double knockout mice.45 Finally, Gault et al. recently demonstrated that by antagonizing the effects of endogenous GIP with (Pro3)GIP, pancreatic insulin content was reduced, glucose tolerance was improved, and islet β-cell hyperplasia was attenuated.46
In conclusion, the results of the present studies demonstrate that, in addition to stimulating insulin release from pancreatic β-cells, GIP possesses insulin-mimetic properties in adipocytes. Like insulin, GIP induces the activation of Akt via a wortmannin-sensitive pathway, thereby promoting the membrane translocation of GLUT-4, which leads to enhanced adipocyte glucose uptake. Although additional studies will be necessary to fully delineate and characterize the complex nature and the biological significance of GIP, the present studies provide further evidence that GIP plays a central role in the development of nutrition-induced obesity. Finally, our findings, as well as the recent observations of other investigators cited above, suggest that the GIPR may potentially represent a suitable target for the treatment of obesity and obesity-related disorders.
Acknowledgments
We would like to acknowledge the valuable technical assistance of Dr. Danielle Gross and Drs. Satish Singh and Selvi Krishnan during the performance of glucose uptake experiments and confocal microscopy, respectively. We thank Dr. Stephen Farmer for general discussion and a critical review of the manuscript.
Grant Support: This study was supported by grant R01 DK53158 (M.M. Wolfe) and by grant F32 DK67812 and a pilot grant from the Boston Obesity and Nutrition Research Center (BONRC) from the National Institutes of Health (D.H. Song).
Abbreviations used in this paper
- GIP
Glucose-dependent insulinotropic polypeptide
- GIPR
GIP receptor
- ANTGIP
GIPR antagonist
- GI
gastrointestinal
- PKB/Akt
Protein Kinase B
- PI3K
phosphatidylinositol 3-kinase
- PPARγ
peroxisome proliferator-activated receptor gamma
- GLUT-4
glucose transporter-4
- IRS
insulin receptor substrate
- RYGB
Roux-en-Y gastric bypass surgery
- IBMX
3-isobutyl-1-methylxanthine
- cAMP
cyclic Adenosine Monophosphate
- shRNA
short hairpin RNA
Footnotes
Financial Disclosure: M. Michael Wolfe is the founder and president CEO of EnteroMed Inc. Barbara E. Corkey is the founder and chair of the board of AdipoGenix. The remaining authors have no disclosure to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960-1994. Int J Obes Relat Metab Disord. 1998;22:39–47. doi: 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- 2.Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37:826–8. doi: 10.1210/jcem-37-5-826. [DOI] [PubMed] [Google Scholar]
- 3.Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, Fujimoto S, Oku A, Tsuda K, Toyokuni S, Hiai H, Mizunoya W, Fushiki T, Holst JJ, Makino M, Tashita A, Kobara Y, Tsubamoto Y, Jinnouchi T, Jomori T, Seino Y. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. 2002;8:738–42. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- 4.Clements RH, Gonzalez QH, Long CI, Wittert G, Laws HL. Hormonal changes after Roux-en Y gastric bypass for morbid obesity and the control of type-II diabetes mellitus. Am Surg. 2004;70:1–4. discussion 4-5. [PubMed] [Google Scholar]
- 5.Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001;2:282–6. doi: 10.1093/embo-reports/kve071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. Faseb J. 2001;15:312–21. doi: 10.1096/fj.00-0590. [DOI] [PubMed] [Google Scholar]
- 7.Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, Kahn CR. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002;3:25–38. doi: 10.1016/s1534-5807(02)00199-5. [DOI] [PubMed] [Google Scholar]
- 8.Tseng CC, Jarboe LA, Wolfe MM. Regulation of glucose-dependent insulinotropic peptide gene expression by a glucose meal. Am J Physiol. 1994;266:G887–91. doi: 10.1152/ajpgi.1994.266.5.G887. [DOI] [PubMed] [Google Scholar]
- 9.Yip RG, Boylan MO, Kieffer TJ, Wolfe MM. Functional GIP receptors are present on adipocytes. Endocrinology. 1998;139:4004–7. doi: 10.1210/endo.139.9.6288. [DOI] [PubMed] [Google Scholar]
- 10.Getty-Kaushik L, Song DH, Boylan MO, Corkey BE, Wolfe MM. Glucose-dependent insulinotropic polypeptide modulates adipocyte lipolysis and reesterification. Obesity (Silver Spring) 2006;14:1124–31. doi: 10.1038/oby.2006.129. [DOI] [PubMed] [Google Scholar]
- 11.Caserta F, Tchkonia T, Civelek VN, Prentki M, Brown NF, McGarry JD, Forse RA, Corkey BE, Hamilton JA, Kirkland JL. Fat depot origin affects fatty acid handling in cultured rat and human preadipocytes. Am J Physiol Endocrinol Metab. 2001;280:E238–47. doi: 10.1152/ajpendo.2001.280.2.E238. [DOI] [PubMed] [Google Scholar]
- 12.Tseng CC, Zhang XY, Wolfe MM. Effect of GIP and GLP-1 antagonists on insulin release in the rat. Am J Physiol. 1999;276:E1049–54. doi: 10.1152/ajpendo.1999.276.6.E1049. [DOI] [PubMed] [Google Scholar]
- 13.Tseng CC, Kieffer TJ, Jarboe LA, Usdin TB, Wolfe MM. Postprandial stimulation of insulin release by glucose-dependent insulinotropic polypeptide (GIP). Effect of a specific glucose-dependent insulinotropic polypeptide receptor antagonist in the rat. J Clin Invest. 1996;98:2440–5. doi: 10.1172/JCI119060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song DH, Dominguez I, Mizuno J, Kaut M, Mohr SC, Seldin DC. CK2 phosphorylation of the armadillo repeat region of beta-catenin potentiates Wnt signaling. J Biol Chem. 2003;278:24018–25. doi: 10.1074/jbc.M212260200. [DOI] [PubMed] [Google Scholar]
- 15.Song DH, Sussman DJ, Seldin DC. Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells. J Biol Chem. 2000;275:23790–7. doi: 10.1074/jbc.M909107199. [DOI] [PubMed] [Google Scholar]
- 16.Ausubel F, Brent R, Kingston E, Moore DD, Seidman JG, Smith JA, Struhl K. Short protocols in molecular biology. 3rd. John Wiley and Sons; 1995. [Google Scholar]
- 17.Kelly KL, Deeney JT, Corkey BE. Cytosolic free calcium in adipocytes. Distinct mechanisms of regulation and effects on insulin action. J Biol Chem. 1989;264:12754–7. [PubMed] [Google Scholar]
- 18.Wolfe MM, Getty L, Boylan MO, Corkey BE. Glucose-dependent insulinotropic polypeptide (GIP) promotes triglyceride synthesis in adipocytes. Gastroenterology. 2003;124:A-30. [Google Scholar]
- 19.Szecowka J, Grill V, Sandberg E, Efendic S. Effect of GIP on the secretion of insulin and somatostatin and the accumulation of cyclic AMP in vitro in the rat. Acta Endocrinol (Copenh) 1982;99:416–21. doi: 10.1530/acta.0.0990416. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg D. Hormonal control of lipolysis in adipose tissue. Adv Exp Med Biol. 1972;26:77–88. doi: 10.1007/978-1-4684-7547-0_6. [DOI] [PubMed] [Google Scholar]
- 21.White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- 22.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Wang RM, Owji AA, Smith DM, Ghatei MA, Bloom SR. Glucagon-like peptide-1 is a physiological incretin in rat. J Clin Invest. 1995;95:417–21. doi: 10.1172/JCI117671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashimoto Y, Opara EC, Liddle RA. Dietary regulation of glucose-dependent insulinotropic peptide (GIP) gene expression in rat small intestine. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 1995;110:207–14. doi: 10.1016/0742-8413(94)00087-q. [DOI] [PubMed] [Google Scholar]
- 25.Bailey CJ, Flatt PR, Kwasowski P, Powell CJ, Marks V. Immunoreactive gastric inhibitory polypeptide and K cell hyperplasia in obese hyperglycaemic (ob/ob) mice fed high fat and high carbohydrate cafeteria diets. Acta Endocrinol (Copenh) 1986;112:224–9. doi: 10.1530/acta.0.1120224. [DOI] [PubMed] [Google Scholar]
- 26.Mooney MH, Abdel-Wahab YH, Morgan LM, O'Harte FP, Flatt PR. Detection of glycated gastric inhibitory polypeptide within the intestines of diabetic obese (ob/ob) mice. Endocrine. 2001;16:167–71. doi: 10.1385/ENDO:16:3:167. [DOI] [PubMed] [Google Scholar]
- 27.Flatt PR, Bailey CJ, Kwasowski P, Page T, Marks V. Plasma immunoreactive gastric inhibitory polypeptide in obese hyperglycaemic (ob/ob) mice. J Endocrinol. 1984;101:249–56. doi: 10.1677/joe.0.1010249. [DOI] [PubMed] [Google Scholar]
- 28.Murphy MC, Isherwood SG, Sethi S, Gould BJ, Wright JW, Knapper JA, Williams CM. Postprandial lipid and hormone responses to meals of varying fat contents: modulatory role of lipoprotein lipase? Eur J Clin Nutr. 1995;49:578–88. [PubMed] [Google Scholar]
- 29.Eckel RH, Fujimoto WY, Brunzell JD. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes. 1979;28:1141–2. doi: 10.2337/diab.28.12.1141. [DOI] [PubMed] [Google Scholar]
- 30.Knapper JM, Puddicombe SM, Morgan LM, Fletcher JM. Investigations into the actions of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1(7-36)amide on lipoprotein lipase activity in explants of rat adipose tissue. J Nutr. 1995;125:183–8. doi: 10.1093/jn/125.2.183. [DOI] [PubMed] [Google Scholar]
- 31.Hauner H, Glatting G, Kaminska D, Pfeiffer EF. Effects of gastric inhibitory polypeptide on glucose and lipid metabolism of isolated rat adipocytes. Ann Nutr Metab. 1988;32:282–8. doi: 10.1159/000177467. [DOI] [PubMed] [Google Scholar]
- 32.Oben J, Morgan L, Fletcher J, Marks V. Effect of the entero-pancreatic hormones, gastric inhibitory polypeptide and glucagon-like polypeptide-1(7-36) amide, on fatty acid synthesis in explants of rat adipose tissue. J Endocrinol. 1991;130:267–72. doi: 10.1677/joe.0.1300267. [DOI] [PubMed] [Google Scholar]
- 33.Nugent C, Prins JB, Whitehead JP, Wentworth JM, Chatterjee VK, O'Rahilly S. Arachidonic acid stimulates glucose uptake in 3T3-L1 adipocytes by increasing GLUT1 and GLUT4 levels at the plasma membrane. Evidence for involvement of lipoxygenase metabolites and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2001;276:9149–57. doi: 10.1074/jbc.M009817200. [DOI] [PubMed] [Google Scholar]
- 34.Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem. 1980;255:4758–62. [PubMed] [Google Scholar]
- 35.White MF, Maron R, Kahn CR. Insulin rapidly stimulates tyrosine phosphorylation of a Mr-185,000 protein in intact cells. Nature. 1985;318:183–6. doi: 10.1038/318183a0. [DOI] [PubMed] [Google Scholar]
- 36.Nadler ST, Stoehr JP, Rabaglia ME, Schueler KL, Birnbaum MJ, Attie AD. Normal Akt/PKB with reduced PI3K activation in insulin-resistant mice. Am J Physiol Endocrinol Metab. 2001;281:E1249–54. doi: 10.1152/ajpendo.2001.281.6.E1249. [DOI] [PubMed] [Google Scholar]
- 37.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–8. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 38.Cong LN, Chen H, Li Y, Zhou L, McGibbon MA, Taylor SI, Quon MJ. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol. 1997;11:1881–90. doi: 10.1210/mend.11.13.0027. [DOI] [PubMed] [Google Scholar]
- 39.Trumper A, Trumper K, Horsch D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J Endocrinol. 2002;174:233–46. doi: 10.1677/joe.0.1740233. [DOI] [PubMed] [Google Scholar]
- 40.Kim SJ, Winter K, Nian C, Tsuneoka M, Koda Y, McIntosh CH. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J Biol Chem. 2005;280:22297–307. doi: 10.1074/jbc.M500540200. Epub 2005 Apr 6. [DOI] [PubMed] [Google Scholar]
- 41.Kim SJ, Nian C, McIntosh CH. Activation of lipoprotein lipase by glucose-dependent insulinotropic polypeptide in adipocytes. A role for a protein kinase B, LKB1, and AMP-activated protein kinase cascade. J Biol Chem. 2007;282:8557–67. doi: 10.1074/jbc.M609088200. [DOI] [PubMed] [Google Scholar]
- 42.Meier JJ, Nauck MA, Schmidt WE, Gallwitz B. Gastric inhibitory polypeptide: the neglected incretin revisited. Regul Pept. 2002;107:1–13. doi: 10.1016/s0167-0115(02)00039-3. [DOI] [PubMed] [Google Scholar]
- 43.Crockett SE, Mazzaferri EL, Cataland S. Gastric inhibitory polypeptide (GIP) in maturity-onset diabetes mellitus. Diabetes. 1976;25:931–5. doi: 10.2337/diab.25.10.931. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437:569–73. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- 45.Zhou H, Yamada Y, Tsukiyama K, Miyawaki K, Hosokawa M, Nagashima K, Toyoda K, Naitoh R, Mizunoya W, Fushiki T, Kadowaki T, Seino Y. Gastric inhibitory polypeptide modulates adiposity and fat oxidation under diminished insulin action. Biochem Biophys Res Commun. 2005;335:937–42. doi: 10.1016/j.bbrc.2005.07.164. [DOI] [PubMed] [Google Scholar]
- 46.Gault VA, Irwin N, Green BD, McCluskey JT, Greer B, Bailey CJ, Harriott P, O'Harte FP, Flatt PR. Chemical ablation of gastric inhibitory polypeptide receptor action by daily (Pro3)GIP administration improves glucose tolerance and ameliorates insulin resistance and abnormalities of islet structure in obesity-related diabetes. Diabetes. 2005;54:2436–46. doi: 10.2337/diabetes.54.8.2436. [DOI] [PubMed] [Google Scholar]