

Abstract
Nerve growth factor (NGF) deprivation induces a Bax-dependent, caspase-dependent programmed cell death in sympathetic neurons. We examined whether the release of cytochrome c was accompanied by the loss of mitochondrial membrane potential during sympathetic neuronal death. NGF- deprived, caspase inhibitor–treated mouse sympathetic neurons maintained mitochondrial membrane poten-tial for 25–30 h after releasing cytochrome c. NGF- deprived sympathetic neurons became committed to die, as measured by the inability of cells to be rescued by NGF readdition, at the time of cytochrome c release. In the presence of caspase inhibitor, however, this commitment to death was extended beyond the point of cytochrome c release, but only up to the subsequent point of mitochondrial membrane potential loss. Caspase-9 deficiency also arrested NGF-deprived sympathetic neurons after release of cytochrome c, and permitted these neurons to be rescued with NGF readdition. Commitment to death in the NGF-deprived, caspase- 9–deficient sympathetic neurons was also coincident with the loss of mitochondrial membrane potential. Thus, caspase inhibition extended commitment to death in trophic factor–deprived sympathetic neurons and allowed recovery of neurons arrested after the loss of cytochrome c, but not beyond the subsequent loss of mitochondrial membrane potential.
Keywords: apoptosis, caspase-9, bax, sympathetic neurons, NGF
Introduction
Extensive programmed cell death (PCD) occurs during neuronal development. Much of this apoptotic death occurs during the developmental period of target innervation when many neuronal populations are acutely dependent on target-derived trophic factors (Oppenheim 1991). Apoptotic cell death is also observed in pathological situations such as stroke, spinal cord injury, and in certain neurodegenerative diseases (Deshmukh 1998; Schulz et al. 1999). Therefore, understanding how neurons undergo PCD is important for developing rational therapies that may prevent neuronal death after injury or disease.
The mechanism by which trophic factor deprivation induces PCD in neurons has been extensively studied in sympathetic neurons that are dependent on NGF for survival (Deshmukh and Johnson 1997). In culture, sympathetic neurons deprived of NGF undergo apoptosis within 24–48 h after NGF removal (Martin et al. 1988; Edwards et al. 1991; Deckwerth and Johnson 1993; Edwards and Tolkovsky 1994). This cell death is dependent on macromolecular synthesis (Martin et al. 1988) and Bax function (Deckwerth et al. 1996), and is prevented by caspase inhibitors such as boc-aspartyl(OMe)-fluoromethylketone (BAF; Deshmukh et al. 1996; Stefanis et al. 1996; Troy et al. 1996; M.J. McCarthy et al. 1997).
Recent studies have focused on the importance of mitochondria during apoptosis because experiments with cell-free systems have indicated that the release of cytochrome c from mitochondria represents a critical event during apoptosis (Liu et al. 1996; Reed 1997). Data from these studies are consistent with a model in which Bcl-2 family proteins modulate the release of cytochrome c from the mitochondria in response to an apoptotic stimulus. Once in the cytosol, cytochrome c presumably binds to Apaf-1 and procaspase-9 and forms a functional apoptosome; the binding of cytochrome c to Apaf-1 and procaspase-9 in the presence of dATP/ATP is sufficient to promote caspase activation in cell-free extracts (Liu et al. 1996; Ellerby et al. 1997; Li et al. 1997). Consistent with this model, Bax translocates to the mitochondria (Putcha et al. 1999) and is required for cytochrome c release from the mitochondria (Deshmukh and Johnson 1998) before caspase activation during NGF deprivation–induced sympathetic neuronal death. The cytoplasmic accumulation of cytochrome c is necessary, but not sufficient, to mediate sympathetic neuronal death (Neame et al. 1998; Deshmukh and Johnson 1998). NGF deprivation also induces sympathetic neurons to develop competence-to-die, a macromolecular synthesis-independent, Bax-independent event, which is required along with the cytosolic cytochrome c to induce apoptosis in these neurons (Deshmukh and Johnson 1998).
The exact mechanism by which cytochrome c is released from mitochondria during apoptosis remains unknown. Bcl-2 family proteins, in particular Bax, may regulate the formation of a channel in the outer mitochondrial membrane, allowing components of the intermembrane space, including cytochrome c, to be released (Schendel et al. 1998). Alternatively, the release of cytochrome c may occur as a consequence of mitochondrial permeability transition (MPT), an event in which the mitochondrial inner membrane becomes permeable to molecules and, as a consequence, the mitochondria become rapidly depolarized. Mitochondrial matrix swelling and rupture of the outer mitochondrial membrane would then release cytochrome c (Crompton 1999). Indeed, mitochondrial depolarization, a consequence of MPT, is observed in several models of apoptosis (Lemasters et al. 1998; Marzo et al. 1998; Wadia et al. 1998). However, whether the release of cytochrome c precedes or follows mitochondrial depolarization remains controversial, and may depend on the cell type and the apoptotic stimulus. For example, mitochondrial depolarization precedes, or occurs simultaneously, with cytochrome c release in hepatocytes treated with tumor necrosis factor α (Bradham et al. 1998) and in PC6 cells treated with 5 μM staurosporine (Heiskanen et al. 1999). In contrast, the loss of mitochondrial membrane potential occurs after cytochrome c release in 1-μM staurosporine- or UV-treated HeLa cells (Yang et al. 1997; Bossy-Wetzel et al. 1998; Goldstein et al. 2000) and in etoposide-treated monocytic THP.1 cells (Zhuang et al. 1998). Similarly, cortical neurons treated with DNA-damaging agents (Stefanis et al. 1999) and hippocampal neurons treated with 300 nM staurosporine (Krohn et al. 1999) release cytochrome c before changes in the mitochondrial membrane potential.
Finally, the events that determine when a cell becomes committed to die during apoptosis remain unclear. Since the mitochondrial events precede caspase activation in many models of apoptosis, cells saved by caspase inhibition may not be able to recover, and may eventually go on to die because mitochondrial oxidative phosphorylation is presumably compromised after cytochrome c loss (Green and Kroemer 1998). Indeed, in mitotic cells induced to undergo apoptosis, caspase inhibitors block the characteristic features of apoptosis but not the commitment to death; the caspase inhibitor–treated cells cannot be rescued (lose their ability to form colonies) even if serum is added back to the cultures (N.J. McCarthy et al. 1997; Ohta et al. 1997; Amarante et al. 1998; Brunet et al. 1998; Gibson 1999). In contrast, NGF-deprived, sympathetic neurons that are saved with caspase inhibitors are capable of resuming somal growth (Deshmukh et al. 1996), restoring mitochondrial cytochrome c (Martinou et al. 1999), and regaining electrophysiological function (Werth et al. 2000) if NGF is added back to the cultures. However, the window of time afforded by caspase inhibition, during which rescue with NGF readdition is possible, has not been examined, and the molecular events that permit rescue in these neurons are not known. Answers to both these points are important to assess the clinical usefulness of caspase inhibition in a therapeutic context.
In this paper, we examined the events that permit the NGF-deprived, caspase inhibitor–treated sympathetic neurons to be rescued by readdition of NGF. Specifically, we asked the following questions. How long can a cell survive and be capable of rescue with NGF readdition after cytochrome c is released? What events ultimately determine when a cell, treated with caspase inhibitor, becomes committed to death? We also examined the temporal relationship between the loss of cytochrome c and the loss of mitochondrial membrane potential after NGF removal. We found that cytochrome c was released before any significant changes in mitochondrial membrane potential during sympathetic neuronal death. Furthermore, the NGF-deprived sympathetic neurons became committed to die at the time of cytochrome c loss. However, in BAF-treated or caspase-9–deficient sympathetic neurons, this commitment to death was extended beyond the time of cytochrome c release, but only until the point at which the mitochondrial membrane potential was lost. Thus, our data indicate that caspase inhibition extends the commitment to death of mouse sympathetic neurons by 25–30 h after the loss of cytochrome c, but not beyond the subsequent loss of mitochondrial membrane potential.
Materials and Methods
Reagents
Mitotracker orange CM-H2TMRos was purchased from Molecular Probes. Caspase inhibitor BAF was purchased from Enzyme Systems Products. Collagenase and trypsin were purchased from Worthington Biochemical Corp. All other reagents were purchased from Sigma Chemical Co., unless otherwise stated. Untimed, pregnant ICR mice were purchased from Harlan Sprague Dawley.
Sympathetic Neuronal Cultures
Primary cultures of sympathetic neurons from the superior cervical ganglion (SCG) neurons were prepared from postnatal-day-1–old (P1) mice, essentially as described previously for rats (Johnson and Argiro 1983; Deshmukh et al. 1996). In brief, the dissected ganglia were treated with collagenase (1 mg/ml) and trypsin (2.5 mg/ml) for 30 min each at 37°C. The ganglia were triturated, and the dissociated cells were plated on collagen-coated dishes in NGF-containing medium (AM50). This medium contained Eagle's minimum essential medium with Earle's salts (Life Technologies Inc.), with the addition of 50 ng/ml 2.5S NGF, 10% FCS, 2 mM glutamine, 100 μg/ml penicillin, and 100 μg/ml streptomycin; 20 μM fluorodeoxyuridine, 20 μM uridine, and 3.3 μg/ml aphidicolin were also included to reduce the number of nonneuronal cells. ICR outbred mice (Harlan Sprague Dawley) were used for all experiments except those involving Bax-deficient and caspase-9–deficient sympathetic neurons. The genetic background of Bax-deficient and caspase-9–deficient mice was C57BL/6; wild-type littermates were used as controls in these experiments.
Culture Conditions and Assessment of Cell Viability after NGF Readdition
Sympathetic neuronal cultures were grown in NGF-containing medium (AM50) for 4–5 d, and then either maintained in AM50 or treated with various conditions. For NGF deprivation, cultures were rinsed twice with medium lacking NGF (AM0: AM50 medium without NGF), followed by addition of AM0 containing goat anti–NGF neutralizing antibody (Ruit et al. 1990). For NGF deprivation in the presence of cycloheximide or the caspase inhibitor, 1 μg/ml cycloheximide or 50 μM BAF was added, respectively, to the anti-NGF–containing medium. For rescue experiments in which NGF was readded to NGF-deprived cultures at various times after NGF deprivation (maintained in the absence or presence of caspase inhibitor BAF), cultures were rinsed three times and incubated in the NGF-containing medium (AM50) for seven additional days. After 7 d of NGF readdition, the rescued neurons were clearly identifiable with large and phase-bright cell bodies, whereas the nonrescued neurons became atrophic and appeared degenerated. The number of rescued cells was quantitated after fixing the cultures with 4% paraformaldehyde and staining with crystal violet as described previously (Deckwerth and Johnson 1993). Neurons were scored as viable if the crystal violet–positive cells had large, well-defined cellular outlines. Dead neurons and debris stain faintly or show no staining with crystal violet.
Mitochondrial Membrane Potential and Cytochrome c Staining
To assess the status of the mitochondrial membrane potential and cytochrome c in individual cells, sympathetic neurons were loaded with Mitotracker orange and immunostained with cytochrome c antibodies. Sympathetic neurons that were maintained for 4–5 d in NGF-containing medium were first treated with various conditions and loaded with 1 μM Mitotracker orange (freshly prepared in DMSO) in the appropriate culture medium for 1 h at 37°C in the dark. We found that loading cells with Mitotracker orange before subjecting cells to various experimental conditions was not a reliable method of assessing the mitochondrial membrane potential in sympathetic neurons. For the NGF deprivation experiments, 50 μM BAF was included in the culture medium to eliminate any cell loss that would otherwise affect the quantitation; no BAF was added when the caspase-9–deficient cells were analyzed in this assay. Cells were washed four times with PBS and fixed in freshly prepared 4% paraformaldehyde for 30 min at 4°C. After three washes in TBS (100 mM Tris-HCl, pH 7.6, and 0.9% NaCl), the cultures were processed for cytochrome c immunohistochemistry as described previously (Deshmukh and Johnson 1998). Anti–cytochrome c antibody (PharMingen) was used at a final concentration of 0.5 μg/ml and an anti-mouse Alexa 488–conjugated secondary antibody (Molecular Probes) was used at a final concentration of 2 μg/ml. Nuclei were stained with Hoechst 33258 (Molecular Probes) as described previously (Deshmukh and Johnson 1998). The status of the mitochondrial membrane potential and cytochrome c staining in sympathetic neurons was quantitated by visualizing the cells with a Zeiss Axiophot microscope. Cells were first visualized on the basis of their nuclear staining, and then scored as positive or negative for Mitotracker orange and cytochrome c staining by a naive observer. Only cells that had completely lost all punctate staining were scored as negative.
Mitotracker Staining and NGF Rescue in Sympathetic Neurons
The status of Mitotracker orange staining and rescue with NGF readdition in cultures of sympathetic neurons was examined as follows. Sympathetic neuronal cultures were maintained in NGF-containing medium for 5 d, and were deprived of NGF in the presence of 50 μM BAF for 60 h. This time corresponded to the point at which almost all cells had lost cytochrome c staining, and about half the cells had lost their Mitotracker staining. Cells were loaded with Mitotracker by incubating in the same medium containing 250 nM Mitotracker orange (prepared freshly in DMSO) for 1 h at 37°C. Care was taken to ensure minimum exposure of cultures to light in all subsequent manipulations (particularly while taking photographs) because exposure to intense light was found to be toxic to the Mitotracker-labeled cells. The Mitotracker concentration that was used in these rescue experiments (250 nM) was lower than that used in the previous immunostaining experiments (1 μM) to minimize phototoxicity. Since no immunostaining steps with washes were involved in these rescue experiments, the lower Mitotracker concentration was found to be sufficient to provide an adequate signal. After the labeling period, cells were washed four times and incubated in NGF-containing medium. Representative fields of cells (marked from a reference point; day 0 [d0] after NGF rescue) were photographed under phase-contrast microscopy to visualize the cells and under fluorescence microscopy to visualize Mitotracker staining (Fuji Provia film, ASA 1600). Cells were returned to the 37°C incubator, and phase-contrast photographs of the same fields of cells were taken at d2, d4, and d7 after the NGF readdition. For quantitation, the phase-contrast photographs of cells at d0 after NGF rescue were first projected onto a screen and individual cells were numbered. The Mitotracker staining status of these cells was scored as positive or negative. The phase-contrast photographs of the same field of cells at d2, d4, and d7 after NGF rescue were compared to determine which cells were rescued; rescue was defined by the ability of these cells to increase somal diameter after NGF readdition. Only cells whose rescue fate was clearly identifiable were counted. Approximately 200 cells were scored per experiment.
Bax−/− and Caspase-9−/− Mice
Breeding and genotyping of Bax-deficient mice has been described previously (Knudson et al. 1995; Deckwerth et al. 1996). Bax-deficient sympathetic neurons were isolated from postnatal-1-d-old mice. Breeding and genotyping of caspase-9–deficient mice was performed as described previously (Kuida et al. 1998). Caspase-9–deficient sympathetic neurons were isolated from embryonic 17-d-old mice; these cultures were maintained in NGF-containing medium for 6–7 d (instead of the normal 4–5 d) before subjecting them to various experimental conditions.
Results
Use of Mitotracker Orange as an Indicator of Mitochondrial Membrane Potential in Sympathetic Neurons
To examine the temporal relationship between cytochrome c release and the mitochondrial membrane potential loss in sympathetic neurons after NGF deprivation, cells were loaded with Mitotracker orange and subjected to cytochrome c immunocytochemistry. Mitotracker orange is a mitochondrial membrane potential–sensitive dye that is aldehyde fixable and, therefore, compatible with subsequent immunocytochemical analysis (Poot et al. 1996; Matylevitch et al. 1998). This allowed us to assess the status of mitochondrial membrane potential and localization of cytochrome c simultaneously in individual cells undergoing apoptosis.
NGF-maintained sympathetic neurons loaded with Mitotracker orange showed a punctate staining pattern identical to that of the mitochondria-localized cytochrome c (Fig. 1). To confirm that Mitotracker orange was an indicator of mitochondrial membrane potential in sympathetic neurons, we examined Mitotracker staining after treatment with agents that disrupt the mitochondrial membrane potential. NGF-maintained sympathetic neurons were treated with carbonyl cyanide m-chlorophenylhydrazone (CCCP) or valinomycin, uncouplers of oxidative phosphorylation (Chen 1988), for 2 h, and then were loaded with Mitotracker orange. Treatment with CCCP or valinomycin reduced dramatically or eliminated Mitotracker staining in sympathetic neurons in a dose-dependent manner (Fig. 1 and Table ). The Mitotracker-loaded cells were also immunostained for cytochrome c to assess the localization of cytochrome c after such treatments. Cytochrome c immunostaining remained largely unaffected with CCCP or valinomycin despite the loss of Mitotracker staining, at least during the period examined (Fig. 1 and Table ). Treatment of sympathetic neurons for 2 h with agents that disrupt the plasma membrane potential such as KCl or ouabain (Chen 1988) did not affect either Mitotracker or cytochrome c staining (Fig. 1 and Table ). Thus, Mitotracker orange appeared to be a reliable indicator of mitochondrial membrane potential in sympathetic neurons.
Figure 1.

Validation of Mitotracker orange staining as an indicator of mitochondrial membrane potential in sympathetic neurons. Shown are representative photomicrographs of sympathetic neurons that were either untreated (NGF) or treated with CCCP (100 μM), KCl (100 mM), or ouabain (100 μM) for 2 h in the presence of NGF. Neurons were loaded with Mitotracker orange (1 μM for 1 h), and subsequently processed for cytochrome c immunostaining. Cytochrome c immunostaining and Mitotracker signal for the same cells is shown; staining with Hoechst 33258 shows the corresponding nuclei of these cells. Bar, 15 μm.
Table 1.
Quantitation of Cytochrome c and Mitotracker Staining in Sympathetic Neurons Treated with Various Conditions
| Percent total cytochrome c + | Percent total Mitotracker+ | |
|---|---|---|
| +NGF (untreated) | 100 | 100 |
| +CCCP 10 μM 100 μM | 100 100 | 98 ± 2 0 |
| +Valinomycin 10 μM 100 μM | 100 100 | 0 0 |
| +KCl 100 mM | 100 | 100 |
| +Ouabain 10 μM 100 μM | 100 100 | 100 99 ± 1 |
The number of cells showing intact mitochondrial cytochrome c and Mitotracker staining after treatment with various condition (Fig. 1) is shown as a percentage of total number of cells examined. Cells were considered to be negative for cytochrome c or Mitotracker only if the characteristic punctate staining pattern was lost completely. Results are mean ± range of two independent experiments with 100–200 cells counted for each condition.
An important point to note is that, in these and subsequent experiments, sympathetic neurons were loaded with Mitotracker orange after treatment with various conditions. Loading cells with Mitotracker orange before treatment with CCCP did not result in decreased Mitotracker staining after disruption of the mitochondrial membrane potential (data not shown). Thus, although the uptake of the Mitotracker dye was sensitive to mitochondrial membrane potential, once the Mitotracker dye was taken up by the mitochondria, its signal did not decrease reliably under conditions that disrupted the mitochondrial membrane potential.
Loss of the Mitochondrial Membrane Potential during Sympathetic Neuronal Death Requires Upstream Events that Are Dependent on Macromolecular Synthesis and Bax Function
NGF deprivation induces a series of events that result in the loss of cytochrome c from mitochondria during sympathetic neuronal death. Inhibition of protein synthesis with cycloheximide addition or Bax deficiency blocks the pathway leading to the release of cytochrome c, whereas caspase inhibition does not (Neame et al. 1998; Deshmukh and Johnson 1998; Martinou et al. 1999; Putcha et al. 1999). To determine whether sympathetic neurons also lose mitochondrial membrane potential after NGF deprivation, we examined the status of the mitochondrial membrane potential in NGF-deprived sympathetic neurons; BAF was added to the medium in these experiments to prevent any cell death after NGF deprivation that would otherwise complicate quantitation of results caused by a loss of neurons from the population. The status of the mitochondrial membrane potential and cytochrome c staining in individual sympathetic neurons undergoing apoptosis was assessed by using Mitotracker orange and cytochrome c immunostaining as described above.
Staining for both cytochrome c and Mitotracker in NGF-maintained sympathetic neurons appeared punctate, as expected for their mitochondrial localization (Fig. 2; +NGF). NGF deprivation for 72 h resulted in the loss of both cytochrome c and Mitotracker staining in most cells, indicating that cells had released cytochrome c and lost their mitochondrial membrane potential (Fig. 2; −NGF/+BAF). We examined whether the loss of mitochondrial membrane potential seen with NGF deprivation was dependent on events requiring macromolecular synthesis or Bax expression. NGF-deprived, cycloheximide-treated sympathetic neurons maintained both cytochrome c and Mitotracker staining even after 72 h of NGF deprivation (Fig. 2; −NGF/+CHX). Likewise, sympathetic neurons from Bax-deficient mice showed intact cytochrome c and Mitotracker staining even after 72 h of NGF deprivation (Fig. 2; −NGF/Bax−/−). These results suggest that a common, macromolecular synthesis–dependent, Bax-dependent pathway leads to the release of cytochrome c and loss of mitochondrial membrane potential during NGF deprivation–induced sympathetic neuronal death.
Figure 2.

Loss of mitochondrial membrane potential during sympathetic neuronal death requires upstream events that are dependent on macromolecular synthesis and Bax expression. Shown are representative photomicrographs of sympathetic neurons either maintained in NGF (+NGF) or deprived of NGF for 72 h either in the presence of 1 μg/ml cycloheximide (−NGF/+CHX) or 50 μM BAF (−NGF/+BAF). Also shown are Bax-deficient sympathetic neurons that were deprived of NGF for 72 h (−NGF/ Bax−/−). Neurons were loaded with 1 μM Mitotracker orange for 1 h and processed for cytochrome c immunostaining. Cytochrome c immunostaining and Mitotracker signal for the same cells, and the corresponding nuclei labeled with Hoechst 33258, are shown. Bar, 15 μm.
Release of Cytochrome c Precedes the Loss of Mitochondrial Membrane Potential during Sympathetic Neuronal Apoptosis
To understand the mechanism of cytochrome c release during sympathetic neuronal death, we examined whether the loss of cytochrome c preceded or followed the loss of mitochondrial membrane potential in individual sympathetic neurons undergoing apoptosis after NGF deprivation. Sympathetic neurons were deprived of NGF, and at various times after NGF deprivation, the cells were loaded with Mitotracker orange and processed for cytochrome c immunostaining. The status of cytochrome c and Mitotracker staining in individual sympathetic neurons was determined, and the results were grouped into four possible categories: (1) cells that maintained both cytochrome c and Mitotracker staining (CytC+ MitoT+); (2) cells that lost cytochrome c but maintained Mitotracker staining (CytC− MitoT+); (3) cells that maintained cytochrome c but lost Mitotracker staining (CytC+ MitoT−); and (4) cells that lost both cytochrome c and Mitotracker staining (CytC− MitoT−). Examples of cells that were seen in these categories are shown in Fig. 3 A. Note that no CytC+ MitoT− cells were observed during NGF deprivation–induced cell death (see below).
Figure 3.
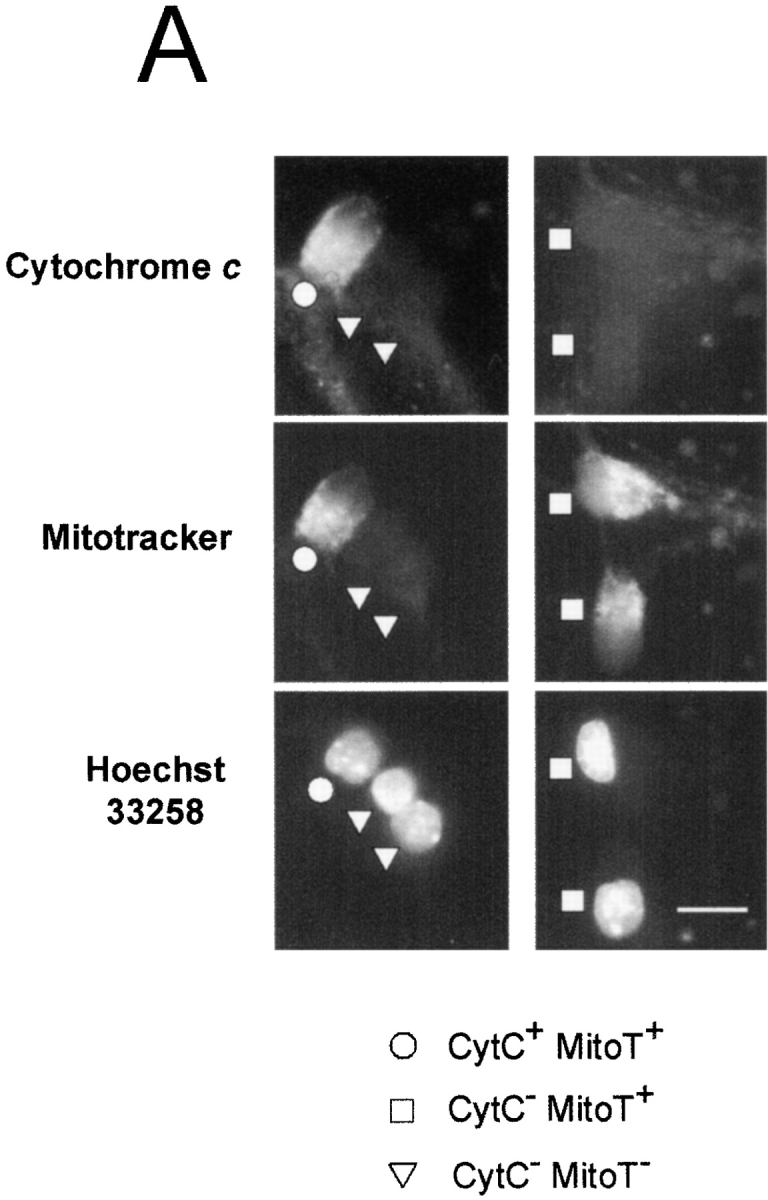

Time course of loss of cytochrome c and mitochondrial membrane potential during NGF deprivation–induced sympathetic neuronal death. NGF-maintained sympathetic neurons (5 d in vitro) were deprived of NGF in the presence of 50 μM BAF. At various times after NGF removal, cultures were loaded with Mitotracker orange (1 μM for 1 h) and immunostained for cytochrome c. The cytochrome c and Mitotracker status of individual cells were quantitated by a naive observer. Results are grouped in four possible outcomes: cells maintaining cytochrome c and Mitotracker staining (CytC+ MitoT+); cells that lost cytochrome c but maintained Mitotracker staining (CytC– MitoT+); cells that maintained cytochrome c but lost Mitotracker staining (CytC+ MitoT–); or cells that lost both cytochrome c and Mitotracker staining (CytC– MitoT–). (A) Examples of cells found in these states at various times after NGF removal are shown. Each column of pictures represents one field of neurons. CytC+ MitoT+ (circles); CytC– MitoT+ (squares); and CytC– MitoT– (triangles). No CytC+ MitoT– cells were found at any time after NGF deprivation. (B) Quantitation of data showing the percentage of cells displaying the various states of cytochrome c and Mitotracker staining at the indicated times after NGF deprivation. (C) Data from B are replotted to show the time course of loss of cytochrome c and Mitotracker staining at the indicated times after NGF deprivation. Results are mean ± SEM from at least three independent experiments with ∼100–200 cells counted for each time point. Bar, 20 μm.
All NGF-maintained sympathetic neurons had intact cytochrome c and Mitotracker staining (Fig. 3 B; 0 h). Upon NGF deprivation, this population of cells decreased gradually, such that by 24 h after NGF removal, only 50% of cells were CytC+ MitoT+ and, by 120 h after NGF removal, none of the cells had intact cytochrome c or Mitotracker staining (Fig. 3 B). Conversely, the population of cells that had lost both cytochrome c and Mitotracker staining increased gradually after NGF removal and by 120 h after NGF removal, all cells were CytC− MitoT−. We also observed a transient increase in the population of cells that had lost cytochrome c but still maintained Mitotracker staining (CytC− MitoT+) after NGF removal. More important however, we did not observe any cells that maintained cytochrome c, but lost Mitotracker staining (CytC+ MitoT−) at any time point after NGF deprivation (Fig. 3 B). Since we observed cells that were CytC− MitoT+ but never cells that were CytC+ MitoT− after NGF deprivation, our data indicate that individual sympathetic neurons first released cytochrome c and subsequently lost mitochondrial membrane potential.
The time course of the loss of cytochrome c and the loss of mitochondrial membrane potential in NGF-deprived, BAF-treated sympathetic neurons (data taken from Fig. 3 B) is shown in Fig. 3 C. Consistent with our previous results, 50% percent of the mouse (ICR) sympathetic neurons released cytochrome c by 24 h after NGF removal (Deshmukh and Johnson 1998). In contrast, Mitotracker staining was not lost in 50% of the NGF-deprived, BAF-saved sympathetic neurons until 55–60 h after NGF removal (Fig. 3 C). Thus, the NGF-deprived, BAF-saved sympathetic neurons were able to maintain a mitochondrial membrane potential for ∼30 h after release of cytochrome c. As discussed above, these experiments were done in the presence of caspase inhibitor. Without caspase inhibition, mitochondrial depolarization presumably follows the loss of cytochrome c relatively rapidly, since cells activate caspases and become committed to die soon after releasing cytochrome c (see below).
Caspase Inhibition Allows Sympathetic Neurons to Recover after Cytochrome c Release, but Not Beyond the Subsequent Loss of Mitochondrial Membrane Potential
Recent studies indicate that NGF-deprived, BAF-treated sympathetic neurons that have released cytochrome c are capable of recovering with subsequent NGF readdition (Deshmukh et al. 1996; Martinou et al. 1999; Werth et al. 2000). However, whether caspase inhibition permits this neuronal rescue with NGF readdition indefinitely has not been examined. Since the recovery of cells that have been saved by caspase inhibition is important from a therapeutic perspective, we wanted to determine how long caspase inhibition could extend the commitment to death in these cells.
NGF-maintained sympathetic neurons were deprived of NGF either in the absence or presence of the caspase inhibitor BAF. At various times after NGF deprivation, NGF was added back to the cultures and the neurons were allowed to recover for 7 d. The number of surviving cells after this 7-d rescue period was quantitated. This long-term rescue assay was needed to determine unambiguously whether caspase inhibition extended the commitment to death. Examples of BAF-saved sympathetic neurons that were rescued with NGF readdition are shown in Fig. 4 A. Control cells deprived of NGF for 72 h without any BAF show extensive cell death (Fig. 4 A; −NGF 72 hr). Caspase inhibition with BAF addition blocks cell death in these neurons. Note that all seven cells marked in the −NGF + BAF condition appear saved at 72 h by the criterion of being phase-bright. However, after 7 d of NGF readdition, only three of the seven cells in that field were able to be recovered with NGF (Fig. 4 A; NGF rescue 7 d). Thus, after 7 d of NGF rescue, it was possible to distinguish unequivocally between those cells that were rescued with NGF readdition (cells 2, 5, and 6), and those cells that had already become committed to die and were not able to recover with NGF readdition (cells 1, 3, 4, and 7).
Figure 4.
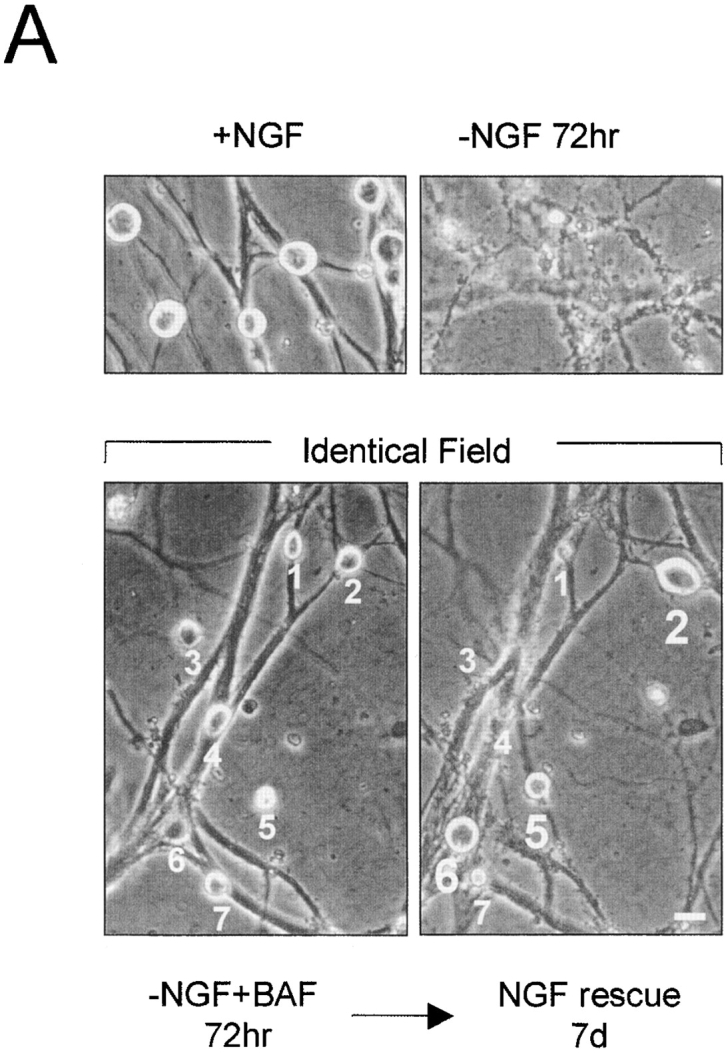

Time course of commitment to death in NGF-deprived sympathetic neurons in the absence or presence of caspase inhibitor. (A) Phase-contrast photographs showing sympathetic neurons rescued with NGF readdition. Representative field of NGF-deprived, BAF-treated cells (72 h of NGF deprivation, 50 μM BAF) showing seven neurons (numbered 1–7) that are prevented from undergoing apoptosis (left; −NGF + BAF 72 h). The same field of cells is shown 7 d after NGF readdition (right; NGF rescue 7d). Cells numbered 2, 5, and 6 were rescued with NGF, whereas cells numbered 1, 3, 4, and 7 were not. Top panels show photographs of control cells that were either maintained in NGF (+NGF) or deprived of NGF without BAF for 72 h (−NGF 72 h). (B) Data showing the percentage of sympathetic neurons capable of recovery with NGF readdition. NGF-maintained mouse sympathetic neurons (5 d in vitro) were deprived of NGF either in the absence (open circles) or presence of 50 μM BAF (open squares). At the indicated times after NGF deprivation, NGF was added back to the cultures for 7 d. The number of NGF-rescued cells was quantitated by crystal violet staining and counting all stained cells in the culture dish. Data showing time course of loss of cytochrome c and Mitotracker staining are taken from Fig. 3 C. Results are mean ± SEM of at least three independent experiments. Bar, 20 μm.
Using this NGF rescue assay, we determined the percentage of cells that became committed to die at various times after NGF deprivation, either in the absence or presence of BAF. The rescue data (Fig. 4 B) were compared with the time course of loss of cytochrome c and Mitotracker staining (from Fig. 3 C) since internally controlled parallel cultures that were deprived of NGF at the same time were used in both these assays. Without BAF addition, 50% of mouse sympathetic neurons became committed to die by ∼25 h after NGF deprivation (Fig. 4 B). The time course of commitment after NGF deprivation is almost identical to the time course of the loss of cytochrome c (Fig. 4 B). BAF addition extended the commitment of sympathetic neuronal death by ∼25–30 h. 50% of the NGF-deprived, BAF-saved sympathetic neurons became committed to die by 55–60 h of NGF deprivation (Fig. 4 B). Remarkably, the time course of commitment to death in the presence of caspase inhibitor was indistinguishable from the time course of loss of Mitotracker staining, which marks the loss of mitochondrial membrane potential in these neurons (Fig. 4 B). We define commitment to death in the absence of caspase inhibitor as Commitment 1 (coincident with cytochrome c release) and commitment to death in the presence of caspase inhibitor as Commitment 2 (coincident with mitochondrial membrane potential loss).
Mitotracker Staining Predicts Rescue with NGF Readdition in Individual Sympathetic Neurons Undergoing Apoptosis
The population-based study shown above demonstrates a good correlation between loss of Mitotracker staining and failure to be rescued with NGF readdition (Fig. 4). To test further the hypothesis that loss of mitochondrial membrane potential marks the commitment to death in caspase inhibitor–treated neurons (Commitment 2), we examined whether the correlation between the maintenance of mitochondrial membrane potential and rescue with NGF readdition could also be seen in individual sympathetic neurons undergoing apoptosis. Sympathetic neurons were deprived of NGF in the presence of BAF for 60 h (2.5 d). NGF deprivation for 60 h corresponds to a time when >90% of cells had lost cytochrome c and ∼50% of cells had lost Mitotracker staining (Fig. 3 C). Cells were loaded with Mitotracker orange (250 nM for 1 h) and photographed to document which cells had lost or maintained Mitotracker staining (Fig. 5 A, −NGF + BAF 2.5 d). As expected, ∼50% of cells had lost Mitotracker staining at that time (data not shown). NGF was added to the cultures, and the neurons were allowed to recover for 7 d. Phase-contrast photographs of the identical field were taken at 2, 4, and 7 d after NGF readdition to follow the status of individual sympathetic neurons (Fig. 5 A; +NGF rescue d2, d4, and d7). More than 90% of the cells that were Mitotracker-positive were rescued with NGF readdition (Fig. 5 B), in contrast to <15% of the Mitotracker-negative cells that were capable of rescue with NGF readdition (Fig. 5 B). The photograph in Fig. 5 A show examples of the correlation seen between Mitotracker staining and rescue with NGF addition. In the field shown, all Mitotracker-positive cells (4 out of 10 cells, numbered 1–4) were rescued with NGF readdition, whereas the Mitotracker-negative cells were not (Fig. 5 A).
Figure 5.


Mitotracker staining predicts rescue with NGF readdition. (A) NGF-maintained sympathetic neurons were deprived of NGF in the presence of 50 μM BAF for 60 h (2.5 d). Cells were loaded with Mitotracker orange (250 nM for 1 h) and photographed to document the status of Mitotracker staining in cells. A representative field shows 4 cells out of 10 that were Mitotracker positive (numbered 1–4; arrow points to one MitoT+ cell). Cells were then treated with NGF, and the same field of cells were photographed after 2, 4, and 7 d of NGF readdition. Photographs show that the Mitotracker positive cells were rescued with NGF readdition, whereas the Mitotracker negative cells were not (arrowhead shows one MitoT– cell). (B) Data showing the correlation between Mitotracker positivity and the ability of cells to be rescued with NGF readdition. Cells were treated as described in A. Results are mean (± range) of two independent experiments with ∼200 cells counted per experiment. Bar, 20 μm.
Caspase-9–deficient Sympathetic Neurons Are Arrested after the Loss of Cytochrome c and Can Be Rescued with NGF Readdition
One prediction from our results is that any genetic manipulation that arrests NGF-deprived sympathetic neurons after the release of cytochrome c would allow these neurons to be rescued with NGF readdition but only until the point of loss of mitochondrial membrane potential. We tested this prediction on sympathetic neurons from caspase-9–deficient mice. We chose to examine caspase-9–deficient sympathetic neurons because caspase-9 is thought to be the apical caspase that becomes activated once cytochrome c is released from mitochondria (Li et al. 1997). Moreover, caspase-9 appears important for neuronal death in vivo since most caspase-9–deficient mice, which die perinatally, have a markedly enlarged brain that is caused by reduced apoptosis during development (Hakem et al. 1998; Kuida et al. 1998).
Sympathetic neurons from caspase-9–deficient mice were deprived of NGF for 72 h to determine first the effect of caspase-9 deletion on sympathetic neuronal death. Cultures from the wild-type littermates were examined in parallel. Caspase-9–deficient neurons appeared phase-bright and had intact neurites even after 72 h of NGF deprivation, whereas those from wild-type littermates appeared degenerated and dead at this time (Fig. 6 A). These results indicate that caspase-9 deficiency prevented sympathetic neurons from undergoing apoptosis after NGF deprivation. The NGF-deprived, caspase-9−/− neurons appeared atrophic presumably because these neurons, like the NGF-deprived, BAF-saved neurons are arrested after the metabolic changes that occur after NGF removal (Deshmukh et al. 1996). Similar neuroprotection was also observed in caspase-3–deficient sympathetic neurons deprived of NGF (data not shown).
Figure 6.
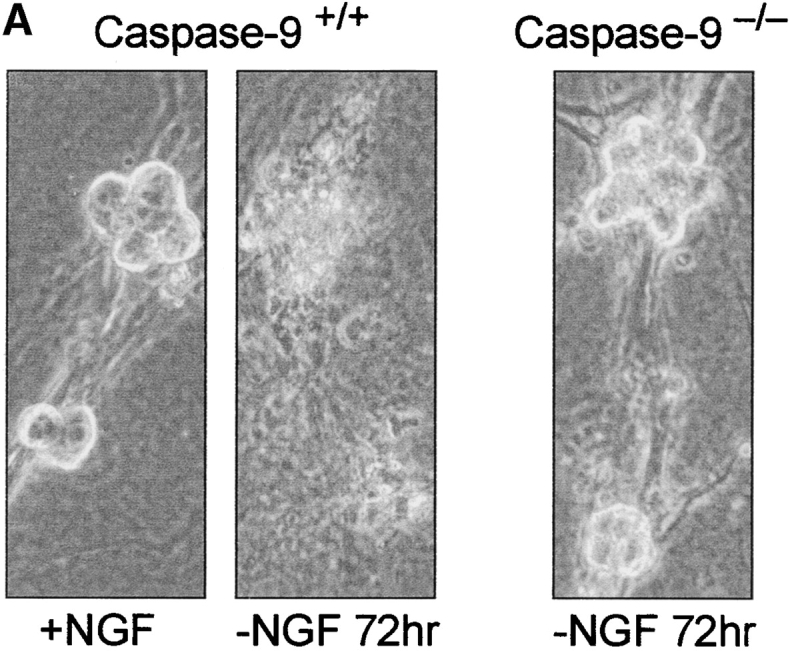

Caspase-9 is required for sympathetic neuronal death after NGF deprivation. (A) Sympathetic neurons from caspase-9+/+ (wild-type) and caspase-9−/− (knockout) mice (littermates) were deprived of NGF for 72 h. Photographs show that, whereas wild-type neurons degenerated and died by this time (Caspase-9+/+, −NGF 72 hr), the caspase-9–deficient neurons were alive and appeared phase-bright (Caspase-9−/−, −NGF 72 hr). (B) Time course of loss of cytochrome c (closed circles) and mitochondrial membrane potential (closed squares) in caspase-9−/− sympathetic neuronal cultures at the indicated times after NGF deprivation. Also shown are survival data after NGF rescue for caspase-9+/+ (open circles) or caspase-9−/− (open squares) sympathetic neurons. Sympathetic neurons from caspase-9+/+ (wild-type) and caspase-9−/− (knockout) mice (littermates) were deprived of NGF and, at the indicated times, NGF was readded to the cultures for 7 d. Rescue experiments were done in parallel with the cytochrome c and Mitotracker experiments. Results for the cytochrome c and Mitotracker experiments are mean ± SEM of two to three independent experiments with ∼100–200 cells counted for each time point. Results for the rescue experiments are mean ± SEM of two to three independent experiments with all surviving cells counted for each time point.
We next examined whether the NGF-deprived, caspase-9–deficient sympathetic neurons were capable of being rescued with NGF readdition. Caspase-9−/− neurons were deprived of NGF and, at various times after NGF deprivation, NGF was added back for 7 d to assess the number of rescued neurons. Cultures from wild-type littermates were examined in parallel. The status of cytochrome c and Mitotracker staining in caspase-9−/− neurons was also examined in parallel at identical times after NGF deprivation. Caspase-9 deficiency inhibited sympathetic neuronal death after the point of cytochrome c release; virtually all caspase-9–deficient cells had lost cytochrome c by 24 h after NGF deprivation (Fig. 6 B). Cytochrome c release in these cells is complete by 24 h, as reported previously for this genetic background (C57BL/6) (Putcha et al. 1999). NGF-deprived, BAF-saved sympathetic neurons from wild-type littermates of caspase-9–deficient mice also released cytochrome c by 24 h after NGF deprivation (data not shown).
Consistent with the time course of cytochrome c loss, >95% of wild-type sympathetic neurons were committed to die (i.e., could not be rescued with NGF readdition) by 24 h after NGF deprivation (Fig. 6 B). In contrast, at that time, >80% of caspase-9–deficient sympathetic neurons were capable of rescue with NGF readdition (Fig. 6 B). Thus, caspase-9−/− sympathetic neurons were arrested at a point after the loss of cytochrome c and could be rescued be with NGF readdition. Longer periods of NGF deprivation resulted in the rescue of fewer caspase-9–deficient cells. 50% of caspase-9–deficient sympathetic neurons were not capable of recovery by 50 h of NGF deprivation, and none were capable of rescue after 5 d of NGF deprivation (Fig. 6 B). As predicted from our data with BAF (Fig. 4), the time course of commitment to death of caspase-9–deficient sympathetic neurons was found to be similar to the time course of loss of Mitotracker staining in these neurons (Fig. 6 B).
Discussion
In this study, we examined the molecular events that determine when sympathetic neurons become committed to die after trophic factor withdrawal. We asked whether caspase inhibition, which inhibits NGF deprivation–induced apoptosis after release of cytochrome c, extends the commitment to death and examined what events might ultimately commit the caspase inhibitor-treated neurons to die. Three main conclusions from this study are as follows. First, NGF-deprived sympathetic neurons released cytochrome c before loss of mitochondrial membrane potential. The NGF-deprived, BAF-treated sympathetic neurons maintained the mitochondrial membrane potential 25–30 h after release of cytochrome c. Second, NGF-deprived sympathetic neurons became committed to die at the time of cytochrome c loss (defined as Commitment 1). Third, in the presence of a caspase inhibitor or in caspase-9–deficient sympathetic neurons, this commitment point of death was extended beyond cytochrome c release but only until the later point of loss of mitochondrial membrane potential (defined as Commitment 2). Once cells lost mitochondrial membrane potential, they were no longer capable of recovery with NGF readdition. Thus, caspase inhibition allowed NGF-deprived sympathetic neurons to recover with NGF readdition after cytochrome c release but not beyond the subsequent loss of mitochondrial membrane potential.
Cytochrome c Release Precedes the Loss of the Mitochondrial Membrane Potential during Sympathetic Neuronal Apoptosis
NGF deprivation induces the loss of cytochrome c, before caspase activation, during sympathetic neuronal death (Deshmukh and Johnson 1998; Neame et al. 1998; Martinou et al. 1999; Putcha et al. 1999). We examined the temporal relationship between cytochrome c release and loss of mitochondrial membrane potential during sympathetic neuronal apoptosis. 50% of the NGF-deprived, BAF-saved sympathetic neurons lost cytochrome c by 24 h after NGF removal, whereas 50% of these neurons did not lose Mitotracker staining nearly 55 h after NGF removal (Fig. 3 C). Our observation that a loss of cytochrome c preceded mitochondrial depolarization in NGF-deprived sympathetic neurons is consistent with similar reports in other cell types (Kluck et al. 1997; Yang et al. 1997; Bossy-Wetzel et al. 1998; Zhuang et al. 1998; Goldstein et al. 2000), including hippocampal (Krohn et al. 1999) and cortical (Stefanis et al. 1999) neurons undergoing apoptosis. Neame et al. 1998 also report NGF-deprived sympathetic neurons release cytochrome c but still maintain Mitotracker staining. These results argue against the involvement of MPT as a mechanism for releasing cytochrome c during NGF deprivation–induced sympathetic neuronal death. Consistent with this, mitochondrial swelling, expected with MPT, is not observed in sympathetic neurons undergoing apoptosis after NGF deprivation (Martin et al. 1988; Martinou et al. 1999). Also, mitochondria isolated from the brain are more resistant to Ca2+-induced MPT than are mitochondria isolated from the liver (Andreyev and Fiskum 1999). Therefore, whether MPT is required for releasing cytochrome c during apoptosis may depend on the specific cell type and the apoptotic signal.
We cannot exclude the possibility that subtle changes in mitochondrial membrane potential undetectable with Mitotracker may have occurred during the release of cytochrome c in sympathetic neurons. Although several reagents are available for measuring mitochondrial membrane potential, a lack of consensus exists regarding which is the best reagent to use on intact cells (Bernardi et al. 1999). We used Mitotracker orange in our studies because it allowed us to assess mitochondrial membrane potential and cytochrome c localization simultaneously in individual cells undergoing apoptosis. Control experiments indicated that the uptake of Mitotracker was dependent on the mitochondrial membrane potential in sympathetic neurons (Fig. 1 and Table ). Also, the time course of the loss of Mitotracker staining was similar to that of the loss of tetramethylrhodamine methyl ester staining, another mitochondrial membrane potential–sensitive dye, in NGF-deprived sympathetic neurons (Deshmukh, M., unpublished observations).
How are NGF-deprived sympathetic neurons able to maintain their mitochondrial membrane potential after cytochrome c release? One possibility is that glycolytic pathways may generate enough ATP, such that mitochondrial membrane potential can be maintained by reversal of the F0F1-ATPase. For example, ρ− cells that lack mitochondrial DNA use glycolytic ATP to maintain mitochondrial membrane potential (Skowronek et al. 1992; Marchetti et al. 1996). Alternatively, a small amount of cytochrome c, which is undetectable by immunocytochemistry, may still be present in the mitochondria of the NGF-deprived, BAF-saved neurons and may be sufficient to maintain electron transport. HeLa cells can maintain a mitochondrial membrane potential even after the loss of cytochrome c because of residual electron transport activity (Goldstein et al. 2000). Nevertheless, the ability of sympathetic neurons to maintain mitochondrial membrane potential for 25–30 h with no detectable cytochrome c and without any trophic support is remarkable. Whether postmitotic cells like sympathetic neurons engage novel ATP-generating pathways to survive during periods of cellular stress such as those described here is unknown.
Loss of Mitochondrial Membrane Potential Marks the Commitment to Death in the Caspase Inhibitor–saved Sympathetic Neurons
We have used the criterion of NGF rescue (the ability to respond to NGF readdition with an increase in somal diameter) to demonstrate that caspase inhibition extended the commitment to death in NGF-deprived sympathetic neurons from the point of cytochrome c release to the later point of mitochondrial depolarization (Fig. 4). This study extends previous observations showing that NGF-deprived, BAF-saved sympathetic neurons are capable of increasing somal diameter (Deshmukh et al. 1996) and restoring cytochrome c in mitochondria (Martinou et al. 1999) with NGF readdition. BAF-saved neurons that are rescued with NGF readdition appear to have normal function since their electrophysiological properties are indistinguishable from those of NGF-maintained sympathetic neurons (Werth et al. 2000).
The ability of NGF-deprived, BAF-saved sympathetic neurons to be rescued with NGF readdition correlated well with their ability to maintain a mitochondrial membrane potential after cytochrome c release (Fig. 4). This correlation held remarkably well, even in individual sympathetic neurons (Fig. 5). These results are consistent with the hypothesis that once cells lose their ability to generate sufficient ATP to maintain their mitochondrial membrane potential, they can no longer be rescued with NGF readdition.
The NGF-deprived, BAF-treated sympathetic neurons that could not be rescued with NGF readdition eventually died without exhibiting any characteristics of apoptosis. For example, dying cells did not immunostain with CM1 antibodies that label active caspase-3 (Srinivasan et al. 1998), nor did they display the typical pattern of annexin V or TUNEL staining (data not shown). Since most apoptotic characteristics in dying cells are likely a consequence of caspase activation (Samali et al. 1999), death occurring in the presence of caspase inhibitors is not expected to display the same apoptotic characteristics. For example, cerebellar granule neurons or cortical neurons, which normally undergo an apoptotic death with caspase activation in response to various insults, undergo a delayed nonapoptotic cell death when caspase inhibitors are added (Miller et al. 1997; Stefanis et al. 1999). Likewise, caspase-9– or caspase-3–deficient cells exhibit few apoptotic features when undergoing cell death (Hakem et al. 1998; Kuida et al. 1998; Woo et al. 1998; Cregan et al. 1999; D'Mello et al. 2000).
Caspase-9 Is Required for Sympathetic Neuronal Death
Our results show that caspase-9 is important in mediating sympathetic neuronal death (Fig. 6). NGF-deprived, caspase-9–deficient sympathetic neurons were capable of rescue with NGF readdition even after the point of cytochrome c loss. As with the BAF-treated cells, the eventual commitment to death in the NGF-deprived, caspase-9–deficient sympathetic neurons also correlated with the loss of mitochondrial membrane potential (Fig. 6 B). The observation that caspase-9 deficiency or BAF addition provided similar neuroprotection in NGF-deprived sympathetic neurons suggests that caspase-9 is the critical apical caspase activated in these cells once cytochrome c is released from the mitochondria. Fig. 7 depicts our current model of the sympathetic neuronal death pathway after NGF deprivation.
Figure 7.

Sympathetic neuronal death induced by NGF deprivation. Sequence of events occurring after NGF deprivation in sympathetic neurons. Cycloheximide (CHX) inhibits an upstream event that leads to the translocation of BAX to the mitochondria and the subsequent release of cytochrome c (Cyt c) from the mitochondria. Cytosolic cytochrome c presumably activates caspase-9 and induces cell death. Cytochrome c–mediated activation of caspases also requires the NGF deprivation–induced development of competence. Loss of mitochondrial membrane potential (Δψm) occurs subsequent to loss of cytochrome c. If caspases are inhibited, rescue of the NGF-deprived, caspase-inhibited neurons is possible with NGF readdition even after the point of cytochrome c release, but not beyond the point of loss of mitochondrial membrane potential.
What Criteria May Permit Rescue of Cells Arrested after Release of Cytochrome c
The ability of caspase inhibitors to keep postmitotic sympathetic neurons alive and capable of rescue with NGF readdition contrasts with the results in primary fibroblasts and several cell lines in which caspase inhibition merely changes the phenotype of death but ultimately has no effect on the commitment to death. Specifically, caspase inhibition did not allow these mitotic cells to maintain clonogenecity even with serum readdition (N.J. McCarthy et al. 1997; Ohta et al. 1997; Amarante et al. 1998; Brunet et al. 1998; Gibson 1999). One explanation for this difference may be that mitotic cells may not be able to generate sufficient ATP and maintain their mitochondrial membrane potential for the period after cytochrome c release that may be required for recovery. Rescue in mitotic cells, as assessed by their ability to divide and form colonies, may arguably require more ATP than rescue in postmitotic sympathetic neurons. However, recovery after cytochrome c release may not depend solely on whether a cell is mitotic or postmitotic. Hsp70 overexpression blocks tumor necrosis factor–induced cell death after cytochrome c release in murine fibrosarcoma cells, yet the Hsp70-overexpressing cells appear capable of normal growth in culture in the presence of the tumor necrosis factor (Jaattela et al. 1998). However, whether individual cells that have released cytochrome c are able to recover and resume growth in these Hsp70-overexpressing cells is unclear.
Alternatively, mitochondrial sequestered, death-promoting proteins, such as AIF, that are released along with cytochrome c (Lorenzo et al. 1999) may mediate caspase-independent death in some cells. Since Commitment 2 in NGF-deprived, BAF-treated sympathetic neurons correlated remarkably well with the loss of Mitotracker staining, factors such as AIF either may not contribute to sympathetic neuronal death or may become activated only with complete loss of mitochondrial membrane potential in these cells. Kluck et al. 1999 report identifying a cytosolic activity in Xenopus extracts that causes permeabilization of the outer mitochondrial membrane. Whether such an activity contributes to the eventual loss of mitochondrial membrane potential in NGF-deprived, BAF-treated sympathetic neurons is not known.
The time by which commitment to death is extended with caspase inhibition (time between Commitment 1 and Commitment 2) varies not only among different cell types, but also among different species for the same cell type. For example, rat sympathetic neurons lose cytochrome c in 50% of neurons by 22–24 h after NGF removal. Yet, >50% of NGF-deprived, BAF-treated rat sympathetic neurons are rescued with NGF readdition, even after 4 d of NGF removal (Deshmukh and Johnson 2000). Thus, rat sympathetic neurons are capable of rescue with NGF readdition for ∼72 h after cytochrome c release, in contrast to a period of ∼30 h in mouse sympathetic neurons (Fig. 4). Whether differences in cell sizes of sympathetic neurons from rat versus mouse contribute to the window of time between Commitment 1 and Commitment 2 is unclear. Consistent with this hypothesis, the survival of mouse cerebellar granule neurons, which are much smaller than sympathetic neurons, is extended for only a few hours after potassium deprivation with caspase inhibition (Miller et al. 1997; D'Mello et al. 2000).
From a therapeutic perspective, the ability of caspase inhibition to promote survival (Fig. 4 and Fig. 6) and functional recovery (Werth et al. 2000) of NGF-deprived sympathetic neurons is very encouraging. Strategies aimed at increasing the time between Commitment 1 and Commitment 2 in neurons may represent means to enhance the neuroprotective effect of caspase inhibitors. Ultimately, whether a caspase inhibitor–treated cell survives the release of cytochrome c and is capable of being rescued may depend on several factors. First, its ability to withstand potentially toxic factors, which may be released along with cytochrome c from the mitochondria. Second, its ability to generate ATP, even after cytochrome c loss and in the continued presence of an apoptotic stimulus; and third, its energetic requirements for recovering and maintaining normal physiological functions.
Acknowledgments
We thank Kevin A. Roth and Barbara Klocke for supplying us with the caspase-9 knockout mice and Stanley Korsmeyer for the Bax-deficient mice. We also thank Louis Chang, Charles Harris, Krista Moulder, Patricia A. Osborne, Girish Putcha, and Brian Tsui-Pierchala for critical review of this manuscript.
This work was supported by the National Institutes of Health grants AG 12947 and NS 38651 (to E.M. Johnson Jr.) and Paralyzed Veterans of America Spinal Cord Research Foundation grant 1786 (to M. Deshmukh).
Footnotes
Abbreviations used in this paper: BAF, boc-aspartyl(OMe)-fluoromethylketone; CCCP, carbonyl cyanide m-chlorophenylhydrazone; MPT, mitochondrial permeability transition; PCD, programmed cell death.
References
- Amarante M.G., Finucane D.M., Martin S.J., Cotter T.G., Salvesen G.S., Green D.R. Anti-apoptotic oncogenes prevent caspase-dependent and independent commitment for cell death. Cell Death Differ. 1998;5:298–306. doi: 10.1038/sj.cdd.4400354. [DOI] [PubMed] [Google Scholar]
- Andreyev A., Fiskum G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999;6:825–832. doi: 10.1038/sj.cdd.4400565. [DOI] [PubMed] [Google Scholar]
- Bernardi P., Scorrano L., Colonna R., Petronilli V., Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer D.D., Green D.R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradham C.A., Qian T., Streetz K., Trautwein C., Brenner D.A., Lemasters J.J. The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Mol. Cell. Biol. 1998;18:6353–6364. doi: 10.1128/mcb.18.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet C.L., Gunby R.H., Benson R.S., Hickman J.A., Watson A.J., Brady G. Commitment to cell death measured by loss of clonogenicity is separable from the appearance of apoptotic markers. Cell Death Differ. 1998;5:107–115. doi: 10.1038/sj.cdd.4400334. [DOI] [PubMed] [Google Scholar]
- Chen L.B. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Cregan S.P., MacLaurin J.G., Craig C.G., Robertson G.S., Nicholson D.W., Park D.S., Slack R.S. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- D'Mello S.R., Kuan C.Y., Flavell R.A., Rakic P. Caspase-3 is required for apoptosis-associated DNA fragmentation but not for cell death in neurons deprived of potassium. J. Neurosci. Res. 2000;59:24–31. [PubMed] [Google Scholar]
- Deckwerth T.L., Johnson E.M., Jr. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth T.L., Elliott J.L., Knudson C.M., Johnson E.M., Jr., Snider W.D., Korsmeyer S.J. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deshmukh M. Caspases in ischaemic brain injury and neurodegenerative disease. Apoptosis. 1998;3:387–394. doi: 10.1023/a:1009602401251. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M., Jr. Programmed cell death in neuronsfocus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol. Pharm. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M., Jr. Evidence of a novel event during neuronal deathdevelopment of competence-to-die in response to cytoplasmic cytochrome c . Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M., Jr. Staurosporine-induced neuronal deathmultiple mechanisms and methodological implications. Cell Death Differ. 2000;7:250–261. doi: 10.1038/sj.cdd.4400641. [DOI] [PubMed] [Google Scholar]
- Deshmukh M., Vasilakos J., Deckwerth T.L., Lampe P.A., Shivers B.D., Johnson E.M., Jr. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J. Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.N., Tolkovsky A.M. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J. Cell Biol. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.N., Buckmaster A.E., Tolkovsky A.M. The death programme in cultured sympathetic neurones can be suppressed at the posttranslational level by nerve growth factor, cyclic AMP, and depolarization. J. Neurochem. 1991;57:2140–2143. doi: 10.1111/j.1471-4159.1991.tb06434.x. [DOI] [PubMed] [Google Scholar]
- Ellerby H.M., Martin S.J., Ellerby L.M., Naiem S.S., Rabizadeh S., Salvesen G.S., Casiano C.A., Cashman N.R., Green D.R., Bredesen D.E. Establishment of a cell-free system of neuronal apoptosis—comparison of premitochondrial, mitochondrial, and postmitochondrial phases. J. Neurosci. 1997;17:6165–6178. [PMC free article] [PubMed] [Google Scholar]
- Gibson R.M. Caspase activation is downstream of commitment to apoptosis of Ntera-2 neuronal cells. Exp. Cell Res. 1999;251:203–212. doi: 10.1006/excr.1999.4563. [DOI] [PubMed] [Google Scholar]
- Goldstein J.C., Waterhouse N.J., Juin P., Evan G.I., Green D.R. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Green D.R., Kroemer G. The central executioners of apoptosiscaspases or mitochondria? Trend Cell Biol. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- Hakem R., Hakem A., Duncan G.S., Henderson J.T., Woo M., Soengas M.S., Elia A., de la Pompa J.L., Kagi D., Khoo W. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Heiskanen K.M., Bhat M.B., Wang H.W., Ma J., Nieminen A.L. Mitochondrial depolarization accompanies cytochrome c release during apoptosis in PC6 cells. J. Biol. Chem. 1999;274:5654–5658. doi: 10.1074/jbc.274.9.5654. [DOI] [PubMed] [Google Scholar]
- Jaattela M., Wissing D., Kokholm K., Kallunki T., Egeblad M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6124–6134. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.I., Argiro V. Techniques in the tissue culture of rat sympathetic neurons. Methods Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Esposti M.D., Perkins G., Renken C., Kuwana T., Bossy-Wetzel E., Goldberg M., Allen T., Barber M.J., Green D.R., Newmeyer D.D. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J. Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson C.M., Tung K.S., Tourtellotte W.G., Brown G.A., Korsmeyer S.J. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Krohn A.J., Wahlbrink T., Prehn J.H. Mitochondrial depolarization is not required for neuronal apoptosis. J. Neurosci. 1999;19:7394–7404. doi: 10.1523/JNEUROSCI.19-17-07394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K., Zheng T.S., Na S.Q., Kuan C.Y., Yang D., Karasuyama H., Rakic P., Flavell R.A. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K., Haydar T.F., Kuan C.Y., Gu Y., Taya C., Karasuyama H., Su M.S., Rakic P., Flavell R.A. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Lemasters J.J., Nieminen A.L., Qian T., Trost L.C., Elmore S.P., Nishimura Y., Crowe R.A., Cascio W.E., Bradham C.A., Brenner D.A., Herman B. The mitochondrial permeability transition in cell deatha common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c . Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lorenzo H.K., Susin S.A., Penninger J., Kroemer G. Apoptosis inducing factor (AIF)a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6:516–524. doi: 10.1038/sj.cdd.4400527. [DOI] [PubMed] [Google Scholar]
- Marchetti P., Susin S.A., Decaudin D., Gamen S., Castedo M., Hirsch T., Zamzami N., Naval J., Senik A., Kroemer G. Apoptosis-associated derangement of mitochondrial function in cells lacking mitochondrial DNA. Cancer Res. 1996;56:2033–2038. [PubMed] [Google Scholar]
- Martin D.P., Schmidt R.E., DiStefano P.S., Lowry O.H., Carter J.G., Johnson E.M., Jr. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J. Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou I., Desagher S., Eskes R., Antonsson B., Andre E., Fakan S., Martinou J.-C. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Kroemer G. The central role of the mitochondrial megachannel in apoptosisevidence obtained with intact cells, isolated mitochondria, and purified protein complexes. Biomed. Pharmacother. 1998;52:248–251. doi: 10.1016/S0753-3322(98)80009-7. [DOI] [PubMed] [Google Scholar]
- Matylevitch N.P., Schuschereba S.T., Mata J.R., Gilligan G.R., Lawlor D.F., Goodwin C.W., Bowman P.D. Apoptosis and accidental cell death in cultured human keratinocytes after thermal injury. Am. J. Path. 1998;153:567–577. doi: 10.1016/S0002-9440(10)65599-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M.J., Rubin L.L., Philpott K.L. Involvement of caspases in sympathetic neuron apoptosis. J. Cell Sci. 1997;110:2165–2173. doi: 10.1242/jcs.110.18.2165. [DOI] [PubMed] [Google Scholar]
- McCarthy N.J., Whyte M.K., Gilbert C.S., Evan G.I. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol. 1997;136:215–227. doi: 10.1083/jcb.136.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T.M., Moulder K.L., Knudson C.M., Creedon D.J., Deshmukh M., Korsmeyer S.J., Johnson E.M., Jr. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame S.J., Rubin L.L., Philpott K.L. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T., Kinoshita T., Naito M., Nozaki T., Masutani M., Tsuruo T., Miyajima A. Requirement of the caspase-3/CPP32 protease cascade for apoptotic death following cytokine deprivation in hematopoietic cells. J. Biol. Chem. 1997;272:23111–23116. doi: 10.1074/jbc.272.37.23111. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Poot M., Zhang Y.Z., Kramer J.A., Wells K.S., Jones L.J., Hanzel D.K., Lugade A.G., Singer V.L., Haugland R.P. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J. Histochem. Cytochem. 1996;44:1363–1372. doi: 10.1177/44.12.8985128. [DOI] [PubMed] [Google Scholar]
- Putcha G.V., Deshmukh M., Johnson E.M., Jr. BAX translocation is a critical event in neuronal apoptosisregulation by neuroprotectants, BCL-2, and caspases. J. Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed J.C. Cytochrome ccan't live with it, can't live without it. Cell. 1997;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- Ruit K.G., Osborne P.A., Schmidt R.E., Johnson E.M., Jr., Snider W.D. Nerve growth factor regulates sympathetic ganglion cell morphology and survival in the adult mouse. J. Neurosci. 1990;10:2412–2419. doi: 10.1523/JNEUROSCI.10-07-02412.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samali A., Zhivotovsky B., Jones D., Nagata S., Orrenius S. Apoptosiscell death defined by caspase activation. Cell Death Differ. 1999;6:495–496. doi: 10.1038/sj.cdd.4400520. [DOI] [PubMed] [Google Scholar]
- Schendel S.L., Montal M., Reed J.C. Bcl-2 family proteins as ion-channels. Cell Death Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- Schulz J.B., Weller M., Moskowitz M.A. Caspases as treatment targets in stroke and neurodegenerative diseases. Annu. Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Skowronek P., Haferkamp O., Rodel G. A fluorescence-microscopic and flow-cytometric study of HeLa cells with an experimentally induced respiratory deficiency. Biochem. Biophys. Res. Commun. 1992;187:991–998. doi: 10.1016/0006-291x(92)91295-2. [DOI] [PubMed] [Google Scholar]
- Srinivasan A., Roth K.A., Sayers R.O., Shindler K.S., Wong A.M., Fritz L.C., Tomaselli K.J. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Stefanis L., Park D.S., Yan C.Y.I., Farinelli S.E., Troy C.M., Shelanski M.L., Greene L.A. Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support—dissociation from apoptosis. J. Biol. Chem. 1996;271:30663–30671. doi: 10.1074/jbc.271.48.30663. [DOI] [PubMed] [Google Scholar]
- Stefanis L., Park D.S., Friedman W.J., Greene L.A. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J. Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy C.M., Stefanis L., Prochiantz A., Greene L.A., Shelanski M.L. The contrasting roles of ICE family proteases and interleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc. Natl. Acad. Sci. USA. 1996;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia J.S., Chalmers-Redman R.M.E., Ju W.J.H., Charlie G.W., Philips J.L., Fraser A.D., Tatton W.G. Mitochondrial membrane potential and nuclear changes in apoptosis caused by serum and nerve growth factor withdrawaltime course and modification by (–)-deprenyl. J. Neurosci. 1998;18:932–947. doi: 10.1523/JNEUROSCI.18-03-00932.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth J., Deshmukh M., Cocabo J., Johnson E.M., Jr., Rothman S.M. Reversible physiological alterations in sympathetic neurons deprived of NGF but protected by caspase inhibitor or Bax deletion. Exp. Neurol. 2000;161:203–211. doi: 10.1006/exnr.1999.7241. [DOI] [PubMed] [Google Scholar]
- Woo M., Hakem R., Soengas M.S., Duncan G.S., Shahinian A., Kagi D., Hakem A., McCurrach M., Khoo W., Kaufman S.A. Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998;12:806–819. doi: 10.1101/gad.12.6.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T.I., Jones D.P., Wang X. Prevention of apoptosis by Bcl-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zhuang J., Dinsdale D., Cohen G.M. Apoptosis, in human monocytic THP.1 cells, results in the release of cytochrome c from mitochondria prior to their ultracondensation, formation of outer membrane discontinuities and reduction in inner membrane potential. Cell Death Differ. 1998;5:953–962. doi: 10.1038/sj.cdd.4400440. [DOI] [PubMed] [Google Scholar]