

Abstract
Rab escort proteins (REP) 1 and 2 are closely related mammalian proteins required for prenylation of newly synthesized Rab GTPases by the cytosolic heterodimeric Rab geranylgeranyl transferase II complex (RabGG transferase). REP1 in mammalian cells is the product of the choroideremia gene (CHM). CHM/REP1 deficiency in inherited disease leads to degeneration of retinal pigmented epithelium and loss of vision. We now show that amino acid residues required for Rab recognition are critical for function of the yeast REP homologue Mrs6p, an essential protein that shows 50% homology to mammalian REPs. Mutant Mrs6p unable to bind Rabs failed to complement growth of a mrs6Δ null strain and were found to be dominant inhibitors of growth in a wild-type MRS6 strain. Mutants were identified that did not affect Rab binding, yet prevented prenylation in vitro and failed to support growth of the mrs6Δ null strain. These results suggest that in the absence of Rab binding, REP interaction with RabGG transferase is maintained through Rab-independent binding sites, providing a molecular explanation for the kinetic properties of Rab prenylation in vitro. Analysis of the effects of thermoreversible temperature-sensitive (mrs6 ts) mutants on vesicular traffic in vivo showed prenylation activity is only transiently required to maintain normal growth, a result promising for therapeutic approaches to disease.
Keywords: choroideremia, REP1, CHM, vesicle traffic, MRS6
Introduction
Choroideremia (CHM) is a form of X-linked retinal degeneration that falls under the broad classification of hereditary peripheral retinal dystrophies or retinitis pigmentosa (Heckenlively and Bird 1988). The discovery of the function of CHM came with the observation that CHM gene product (Cremers et al. 1990) is identical to that of component A or Rab escort protein 1 (REP1) of the Rab geranylgeranyl transferase II (RabGG transferase) protein complex. RabGG transferase posttranslationally modifies Rab proteins with 20 carbon geranylgeranyl isoprene lipids (Seabra et al. 1992a), a modification essential for Rab function (reviewed in Seabra 1998). The function of Rab GTPases is to regulate the docking and fusion of vesicle carriers mediating the transport of protein between compartments of the exocytic and endocytic pathways (Schimmoller et al. 1998). A second, highly related isoform, REP2, was subsequently discovered (Cremers et al. 1994). The sequence between the rat, mouse, and human homologues is highly conserved (87–91%). A yeast homologue, MSI4/MRS6, is an essential gene and shows ∼50% identity with mammalian CHM (Fujimura et al. 1994; Jiang and Ferro-Novick 1994).
Current evidence suggests that CHM/REP1 and REP2 function by binding newly synthesized Rab proteins for delivery to the catalytic component B of RabGG transferase (Anant et al. 1998; Alexandrov et al. 1999) that has closely related homologues in yeast (Jiang et al. 1993; Li et al. 1993). Component B is a heterodimer composed of α- and β-subunits (Seabra et al. 1992a). REP has been demonstrated to form a 1:1 protein complex with all newly synthesized Rab proteins examined to date (Andres et al. 1993; Anant et al. 1998; Alexandrov et al. 1999). Residues involved in binding Rab by REP or those dictating recognition of the Rab–REP complex with RabGG transferase are unknown. After prenylation, the CHM/REP1–Rab complex delivers the newly prenylated protein to the membrane for use in vesicular traffic, presumably through receptors used by the cytosolic Rab–GDI complex (Soldati et al. 1994; Ullrich et al. 1994; Luan et al. 1999). Thus, REP proteins can associate with both the unprenylated and prenylated forms of Rab, and function in the first step of delivery of Rab to membranes (Andres et al. 1993; Alexandrov et al. 1994; Cremers et al. 1994). REP1, but not REP2, is missing from lymphoblasts prepared from CHM patients (Seabra et al. 1993), and loss of REP1 serves as a practical diagnostic test for CHM (MacDonald et al. 1998). Only truncations leading to complete loss of protein are correlative with disease. The underlying defect in response to REP1 loss in CHM leading to retinal degeneration remains to be established. Given the essential role of REP1 in the geranylgeranylation of Rab proteins, a leading hypothesis is that defective prenylation of one or more isoforms of Rab that are substrates for REP1 results in defects in vesicular trafficking in cells comprising the choriocapillaris and retinal pigmented epithelium (RPE), initiating retinal degeneration.
Further insight into the function of CHM came with the realization that it shows sequence similarity (∼30%) to guanine nucleotide dissociation inhibitor (GDI) involved in the recycling of small GTPases belonging to the Rab gene family (Fodor and Lee, 1991). GDI is largely specific for only the prenylated form and, therefore, does not participate in delivery of nascent Rab to the catalytic subunits of RabGG transferase (Sasaki et al. 1990; Alexandrov et al. 1994; Wu et al. 1998). Whereas regions of homology are observed throughout the primary sequence, three regions of high identity have been noted (Waldherr et al. 1993). One of these, an NH2-terminal sequence conserved region (SCR1), has a 23/46 identical match for conserved amino acids, whereas a second region found towards the middle of the protein (SCR3B) has a 21/42 match. Other regions, including SCR2 and SCR3A, are less conserved. We have demonstrated using X-ray crystallography that SCR1 and SCR3B interact to form a Rab-binding domain at the apex of α-GDI (Schalk et al. 1996; Luan et al. 2000).
The critical role of REP in the function of Rab GTPases involved in membrane transport throughout the endocytic and exocytic pathways stresses the importance of understanding the contribution of individual amino acid residues in directing the interaction of CHM/REP1, not only with different Rab species, but importantly, the catalytic subunits of RabGG transferase. To gain insight into the underlying molecular basis for these interactions, we have turned to the yeast homologue, MRS6 (Waldherr et al. 1993; Jiang and Ferro-Novick 1994). Saccharomyces cerevisiae contains only a single copy of the gene and it is essential for cell growth. We find that point mutations encoding homologous residues of α-GDI involved in Rab binding in vitro (Wu et al. 1998; Luan et al. 1999) exhibit deficiency in Rab binding and loss of prenylation in vitro, yet supported partial growth when present as the only copy in a mrs6Δ null strain in vivo. When combined in double mutants, complete loss of Mrs6p function was observed in the mrs6Δ null strain, yet dominant effects on growth were observed in wild-type cells. The important implications of these results on the general mechanism of the REP-regulated prenylation cycle in the general biology of the vesicular traffic and in eye dysfunction in CHM are discussed.
Materials and Methods
Strains, Plasmids, and Media
Yeast strains used in this study were SEY6210a/α (MAT a /MATα ura3 leu2 his3 trp1 lys2 suc2Δ9; obtained from S. Emr, Howard Hughes Medical Institute, University of California San Diego), SEY6210 Δmrs6, and SEY6210 MRS6ts. Yeast strains were grown in standard media containing yeast extract, peptone, and dextrose (YPD; Sherman et al. 1979) or synthetic media (SM) supplemented with essential amino acid supplements (Sherman et al. 1979) as required for maintenance of plasmids. An mrs6::LEU2 disruption plasmid used for generating the mrs6 null strain was constructed by replacing the 1,285-bp DNA fragment encoding the amino acids 2–570 of Mrs6p with a 1,812-bp fragment containing the HIS3 gene. An mrs6Δ strain was constructed by transformation of a wild-type diploid strain (SEY 6210a/α) with a CEN URA3 plasmid containing MRS6, followed by transformation of this strain with the mrs6Δ::HIS3 deletion-disruption construct. After confirming by PCR that this strain contained one intact and one disrupted copy of MRS6, it was sporulated, and colonies derived from Ura+ His+ spores were identified.
Random Mutagenesis of the MRS6 Gene
The MRS6 gene was subjected to the random PCR mutagenesis using an in vivo gap-repair method. Using an MRS6-containing plasmid as a template, a 2,123-bp PCR fragment was amplified under mutagenic PCR conditions (Cadwell and Joyce 1992). This fragment corresponds to the positions −196 to +1,927 of the MRS6 gene, where the first nucleotide in the open reading frame (ORF) is defined as the +1 nucleotide. A 2μ LEU2 plasmid containing the wild-type MRS6 gene was gapped to remove the region corresponding to the ORF. The gapped plasmid and the PCR fragment were then cotransformed into the yeast strain SEY6210a/α. Leu− transformants containing circular plasmids generated via homologous recombination were identified by growth on 5-fluoroorotic acid. Transformants were screened at the permissive temperature, 30°C, and at the nonpermissive temperature, 37°C, resulting in yeast strain SEY6210 MRS6 ts.
DNA Methods
Standard DNA manipulation (Maniatis et al. 1982) was used with restriction endonucleases and modification enzymes from Boehringer, New England Biolabs, and Promega. Point mutations were introduced by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All PCR-amplified DNAs were sequenced to confirm that only the desired mutations were present.
Yeast and Bacterial Methods
Transformation of S. cerevisiae strains was done according to the lithium acetate method (Ito et al. 1983) with single-stranded DNA used as carrier (Schiestl and Gietz 1989). Standard bacterial medium (Miller 1972) was used for E. coli cultures. E. coli transformations were done as described (Hanahan 1983).
Protein Expression, Purification, and Antibody Production
NdeI (position +1) and BamHI (position + 1,818) sites, respectively, were created in the MRS6 ORF. After PCR amplification, the 1.8-kb NdeI–BamHI fragment was cloned into the pET11d vector (Novagen) containing a 5′ six-histidine (6xHis) tag. The 6xHis-tagged Mrs6p was expressed and purified from E. coli extracts by native purification on Ni2+-agarose as described by the manufacturer (Qiagen), followed by gel filtration and concentration. The anti-Mrs6p polyclonal antibody was raised in rabbits and the antibody purified using affinity column containing Mrs6p according to the manufacturer's directions (Affi-Gel, BioRad). The YPT1 expression plasmid, pET11dYPT1, was a gift from Dr. S. Ferro-Novick (Yale University, New Haven, CT). The Ypt1p protein was purified from E. coli extracts as described (Wagner et al. 1987). The anti-Ypt1p mouse monoclonal and anti-Ypt1p rabbit polyclonal antibodies were a gift from Dr. D. Gallwitz (MPI, Gottingen, Germany). His6-tagged Rab3A expressed from E. coli and prenylated His6-tagged Rab3A expressed from Spodoptera frugiperda (Sf9) were purified as described (Nuoffer et al. 1995; Schalk et al. 1996).
Growth Analysis
Cells were grown overnight in selective medium. The cells were then diluted and sonicated to separate them into individual cells. Subsequently, the cells were counted and diluted to 1,000, 400, 100, and 10 cells/μl. 5 μl of each dilution was spotted onto selective plates. The plates were incubated at 30°C and growth was assessed after 2–4 d.
Metabolic Labeling and Immunoprecipitation
Yeast cultures were radiolabeled using previously published procedures (Horazdovsky and Emr 1993; Horazdovsky et al. 1994). Immunoprecipitations of carboxypeptidase Y (CPY) were done as described (Klionsky et al. 1988). Immunoprecipitations of HA epitope-tagged Mrs6p proteins were done with an mAb to the HA tag (12CA5; Amersham Pharmacia Biotech). Cells were washed and resuspended (50 OD600 U/ml) in standard buffer (20 mM Hepes, pH 6.2, 200 mM sorbitol, 100 mM potassium acetate, 2 mM MgCl2, 1 mM dithiothreitol), and were then broken by vortexing with glass beads on ice. The homogenates were centrifuged at 100,000 g for 1 h to yield membrane and cytosolic fractions. Anti-HA antibody was added (20 μg/ml), incubated with rocking at 4°C for 2 h, and HA-tagged antibody containing Mrs6p recovered using G–Sepharose as described by the manufacturer (Amersham Pharmacia Biotech).
Subcellular Fractionation
Spheroplasts made from cells were labeled and processed as described (Gaynor et al. 1994). After clearing extracts of unbroken cells, lysates were centrifuged at 100,000 g for 1 h to yield P100 particulate (membrane) and S100 soluble (cytosolic) fractions. Proteins were precipitated with chloroform/methanol (Wessel and Flugge 1984), separated by SDS-PAGE, and immunoblotted using a polyclonal rabbit antisera raised against the Ypt1p protein.
Distribution of Ypt1p
Cells were grown to early logarithmic phase at 30°C in selection medium. Then one half of the culture was maintained at the permissive temperature (30°C) and the other half was incubated at the restrictive temperature (37°C). At each time point, 5 OD of cells was washed and resuspended in standard buffer, and permeabilized by vortexing with 1 vol of glass beads on ice. After removing the cell debris by low-speed centrifugation, the supernatant was subjected to centrifugation at 100,000 g for 1 h to separate the membrane from the cytosolic fraction. After precipitation of the cytosolic proteins with chloroform/methanol (Wessel and Flugge 1984), the proteins from both fractions were separated by SDS-PAGE and immunoblotted using anti-Ypt1p antibodies. The distribution of Ypt1p was quantitated using densitometry.
Geranylgeranylation Assay
Rab geranylgeranyltransferase activity was assayed by incubating E. coli-produced Ypt1p with [3H]geranylgeranyl pyrophosphate (Seabra and James 1998). A 60-μl reaction contains 50 mM Tris/HCl, pH 7.5, 5 mM dithiothreitol, 10 mM MgCl2, 0.5 μl [3H]geranylgeranyl pyrophosphate (20 Ci/mmol), 10 μg of 6xHis-Ypt1p, and 250 μg of yeast crude extract. The reaction mixture was incubated at 30°C. After incubation, the reaction mixture was applied onto Whatman GF/A paper and washed twice with ethanol. The filters were dried and radioactivity was counted in a scintillation counter.
Fluorescence Assay
An assay to measure methylanthraniloyl guanosine diphosphate (mGDP; Molecular Probes) binding to Rab GTPases was performed as described. In brief, recombinant His6-Rab3A from E. coli and Sf9 cells was loaded with the fluorescent GDP analogue, mGDP, by incubating mGDP and His6-Rab3A at a 100:1 molar ratio in 50 mM Tris/HCl, pH 7.2, 10 mM EDTA, 1 mM dithiothreitol, 100 μM mGDP at 32°C for 45 min. At the end of the incubation, the mixture was adjusted to 20 mM MgCl2 (to trap mGDP in Rab) and incubated 15 min more at 32°C. The mixture can be kept on ice for 1 h before use. The free mGDP was removed by using MicroSpin™ G-25 columns (Amersham Pharmacia Biotech). The dissociation of mGDP from His6-Rab3A was assessed by measuring the decrease in relative fluorescence (λ excitation 360 nm, λ emission 440 nm) using a Perkin-Elmer LS50B Fluorescence Spectrometer as described. The dissociation was measured using 100 nM Rab3A mGDP incubated with increasing concentration of Mrs6p in 300 μl of buffer A (25 mM Tris-HCl, pH 7.2, 0.3 mM GDP, 0.5 mM MgCl2, 0.6 mM EDTA). Dissociation constants (K d) for His6-Rab3A binding to His6-Mr6p wild-type and mutants were determined as previously described for His6-Rab3A and GDI (Wu et al. 1998).
Electron Microscopy
Cells were grown to early logarithmic phase at 30°C in YPD or selection medium. Then, each culture was divided and one half was maintained at the permissive temperature (30°C), and the other half was incubated at the restrictive temperature (37°C). After 3 h of incubation, the cells were fixed with 3% glutaraldehyde and processed for EM as described (Rieder et al. 1996).
Results
A Structural Basis to Characterize Residues in Mrs6p Required for Function
As a basis to begin to explore the crucial residues involved in REP function in Rab prenylation, we focused on the key features derived from the molecular and structural alignment of CHM/REP1 and GDI family members (Wu et al. 1996). Such an alignment shows four sequence-conserved regions (SCRs) located in the NH2-terminal and central areas with an insert of variable length separating SCR1 and SCR2 (Wu et al. 1996; Fig. 1). Residues comprising motifs SCR1A and 1B (Fig. 1, yellow), and SCR3B (Fig. 1, red) are particularly diagnostic of REP/GDI-related proteins, due to their striking composition of invariant di- and tripeptides (Waldherr et al. 1993). These particular motifs, defined by the consensus sequences DVxxxGTGxxExx (SCR1A), GxxVLHxDxxxYYGxY (SCR1B), and GExxQGFxRxxAxxG (SCR3B) define members of the REP/GDI superfamily (Waldherr et al. 1993). These motifs reveal that SCR1 and 3B form a compact region found at the apex of the protein in domain I (Fig. 1, red and yellow; Schalk et al. 1996). Residues Tyr39, Glu233, Gln236, and Arg240, which are strongly conserved residues in the SCRs, 1B (GxxVLHxDxxxYYGxY) and 3B (GExxQGFxRxxAxxG), between GDI and REP family members, and are required for α-GDI binding to Rab (Schalk et al. 1996; Wu et al. 1998) and, when mutated, lead to loss of function in yeast (Luan et al. 1999). In contrast, conserved residues common to both Mrs6p and GDI that are found in SCR2 (Fig. 1, green) and 3A (Fig. 1, purple) extend the full-length along one face of GDI (Schalk et al. 1996) and contribute to the organization of a lower structurally defined region, referred to as domain II (Fig. 1; Schalk et al. 1996). We have recently provided direct evidence that in the case of GDI, domain II is involved in the interaction of GDI with Rab recycling factors (RRF), receptors mediating Rab retrieval after vesicle targeting and fusion (Luan et al. 1999, Luan et al. 2000). The conserved residues in common between GDI and Mrs6p are likely to correspond to similar structural arrangements, providing a basis to explore Mrs6p at the molecular level.
Figure 1.

Potential location of substitutions (yeast numbering) based on the crystal structure of bovine α-GDI. The tentative location of each of the residues (Mrs6p residue numbering) mutated in this study based on their homologous residue in α-GDI (Table ) are indicated (Schalk et al. 1996). The insert region (blue) and the Rab-binding region containing SCRs 1B (yellow) and 3B (red) are highlighted at the top of GDI and form domain I. The conserved face of GDI containing SCRs 2 (green) and 3A (purple) link domain I with a second domain II, and are oriented to the front of the image (Schalk et al. 1996). Bolded residues are those that have prominent phenotype in altering REP function.
A Model System to Study REP Function in Yeast
The currently recognized biological function common to members of the GDI and REP families is to bind a diverse group of Rab proteins. Because the highly conserved SCR1 and 3B motifs fold to generate a compact region at the apex of GDI in domain I, it was a strong candidate for direct participation in Rab association by CHM/REP1. The yeast S. cerevisiae contains a single REP homologue, MRS6 (Waldherr et al. 1993; Jiang and Ferro-Novick 1994), that is essential for cell growth. Therefore, we carried out a systemic screen of mutants in Mrs6p for function in Rab binding and prenylation in vitro, and cell growth and transport in vivo.
To map amino acid residues of Mrs6p that are important for function, a plasmid shuffle-based complementation assay was developed that allowed rapid screening of site-directed point mutants for their ability to complement an mrs6Δ null mutation (Table ). mrs6 plasmids (CEN LEU2) containing site-directed mutations were used to transform a mrs6Δ::HIS3 null strain containing a CEN URA3 MRS6 plasmid as the sole source of wild-type Mrs6p. Transformed strains were streaked to 5-flouroorotic acid (5-FOA) plates to select for loss of the URA3 MRS6 plasmid leaving the mutant mrs6 gene as the only source of Mrs6p. Growth of the resultant strains was assayed after restreaking to rich medium. This method allowed us to assess if any mrs6 mutants constructed were able to supply the essential function of MRS6. As a test to confirm the feasibility of this approach, a double mutant, G53A-G55A, was introduced into MRS6. Based on the crystal structure of GDI, the conserved nature of these residues, and their buried location in the protein (Schalk et al. 1996), we predicted that this double mutant would affect the structural integrity of Mrs6p, as it does for GDI (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999), and lead to a complete loss of function through misfolding. Indeed, the double mutant failed to yield Leu+ Ura− strains after streaking to 5-FOA plates, indicating noncomplementation of the mrs6Δ null mutation and thus confirming the validity of this assay (see below, Table ).
Table 1.
Effect of Single Point Mutations in Mrs6p on Growth of the 6Δ Null Strain
| Region | Location | Residue mutated | Growth at 30°C | Yeast GDI residue | Mammalian GDI residue |
|---|---|---|---|---|---|
| Wild-type | − | − | +++ | − | − |
| SCR1A | a1 strand | D48N | +++ | 11 | 5 |
| SCR1A | a1 loop A | G53A | +++ | 16 | 11 |
| SCR1A | a1 loop A | G55A | +++ | 18 | 13 |
| SCR1B | a2 loop f1 | D76N | ++ | 39 | 34 |
| SCR1B | a2 loop f1 | Y81V | +++ | 44 | 39 |
| − | B helix | K99D | +++ | 59 | 55 |
| SCR2 | f2 strand | R127A | +++ | 78 | 70 |
| SCR2 | b2 strand | E174Y | +++ | 125 | 117 |
| SCR2 | D helix | T185D | +++ | 136 | 128 |
| SCR2 | E helix | R195A | + | 146 | 138 |
| SCR2 | E helix | K199D | ++ | 150 | 142 |
| SCR2 | E loop F | E208Q | +++ | 160 | 152 |
| − | F helix | L226Y | +++ | 180 | 172 |
| SCR3A | H loop e3 | D269A | +++ | 225 | 217 |
| SCR3A | H loop e3 | V270R | +++ | 226 | 218 |
| SCR3A | H loop e3 | Y271F | +++ | 227 | 219 |
| SCR3B | I helix | E286S | + | 241 | 233 |
| SCR3B | I helix | Q289L | +++ | 244 | 236 |
| SCR3B | I helix | R293A | + | 248 | 240 |
| SCR3B | a3 strand | T301P | +++ | 256 | 248 |
−, Dead; +, slow; ++, slower than wild-type; +++, like wild-type.
Table 2.
Effect of Double Mutants on the Growth of mrs6Δ Null and Wild-type Strains
| Growth at 30°C in: | |||
|---|---|---|---|
| Mutants | yrepΔ strain | wt strain | Mammalian GDI residue |
| Wild-type | +++ | NE | − |
| G53A-G55A | − | NE | 11–13 |
| Y81V-G53A | +++ | NE | 11–39 |
| Y81V-293A | + | NE | 39–240 |
| R127A-R293A | − | NE | 70–240 |
| R195A-R293A | − | Dominant negative | 138–240 |
| L226Y-R293A | − | NE | 172–240 |
| D269A-R293A | + | NE | 217–240 |
| V270R-R293A | + | NE | 218–240 |
| Y271F-R293A | + | NE | 219–240 |
| E286S-R293A | − | Dominant negative | 233–240 |
| T301P-R293A | + | NE | 240–248 |
+, Dead; +, very slow; +++, like wild-type; NE, no effect.
Generation of Single Point Mutants Partially Defective in Mrs6p Function
Mutation of conserved residues in SCRs 1B and 3B found at the apex of GDI affect the ability of the α-GDI to bind Rab3A in vitro (Schalk et al. 1996) and for function in yeast in vivo (Luan et al. 1999). Therefore, we used the crystallographic structure (Schalk et al. 1996) and the aligned sequences of all GDIs and REPs comprising the GDI superfamily as a guide to generate point mutations changing conserved, surface-exposed amino acids of the various SCRs that would not affect protein folding or stability, but that were likely candidate residues for interactions with Rabs and RabGG transferase. The mutants examined are shown with respect to their corresponding location in GDI (Fig. 1, residue numbers are those of Mrs6p). Each was tested in the complementation assay.
The results of our analysis is shown in Table . Despite the extensive collection of point mutants tested, no single amino acid change was found that resulted in a complete loss of Mrs6p function. However, several point mutations, notably R195A, E286S, and R293A (yeast Mrs6p numbering) affected growth as is evident by the slower growth of these strains (Table ; Fig. 2A and Fig. c). R195A is located in domain II, whereas E286S and R293A are found in SCR3B (Fig. 1). In particular, R293A (Table ) was previously shown to be important in mammalian and yeast GDI function and is crucial for Rab binding in vitro and in vivo (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999). From these results, and by analogy to the effect of similar mutants for binding of Rab in vitro by GDI (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999, Luan et al. 2000), potentially even reduced levels of Rab recognition by REP are sufficient to maintain a functional Rab pool in vivo for cell survival. We suggest that the prenylation activity of wild-type REP is likely to be in functional excess in living cells.
Figure 2.
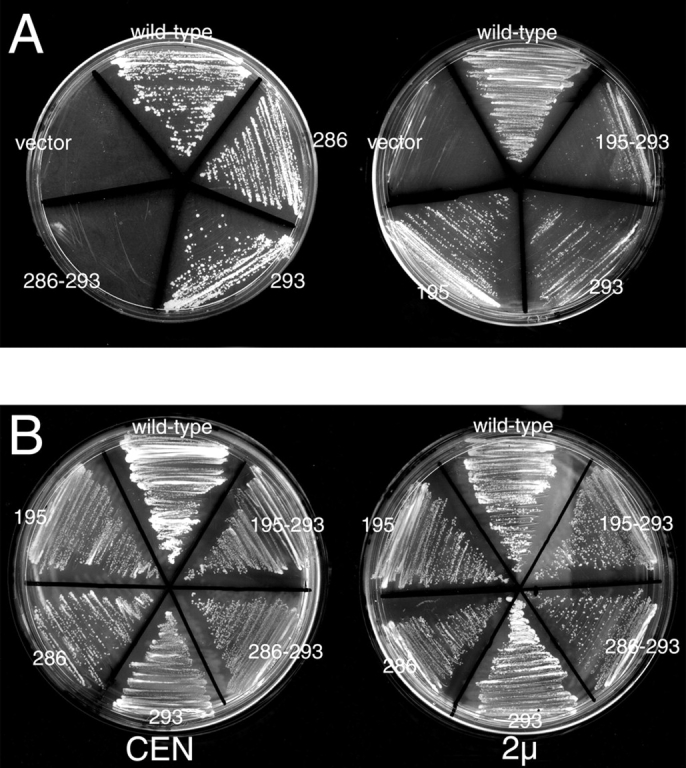

Ability of selected single and double point mutants to complement growth of mrs6Δ null strain. A, Indicated mutants were expressed in the mrs6Δ null strain using the single copy CEN plasmid as described in Materials and Methods. B, Indicated mutants were expressed using the single copy CEN plasmid, or multicopy 2μ plasmid in wild-type cells. The ability of overexpressed Mrs6p double mutants to partially inhibit growth is illustrated by reduced level of colonies appearing on the plate. C, Indicated mutants were expressed in the mrs6Δ null strain using the single copy CEN plasmid as described in Materials and Methods. 5,000, 2,000, 500, and 50 cells were spotted onto a plate. The growth was assessed after a 2–4-d incubation at 30°C.
Double Mutants Demonstrate the Essential Role of the Rab-binding Platform in Mrs6p Function
Because the E286S and R293A mutants showed comparable slower growth phenotypes (Table ), we chose the R293A mutation as a starting point to construct a set of double mutants. Additional amino acid substitutions were made in either the same SCR containing R293 (SCR3B; Fig. 1, red) or in different SCRs, and each of these was tested in combination with R293A in the complementation assay (Table ). We also generated double mutants within and between SCRs that did not include the R293 residue, to test whether multiple mutations in regions flanking the putative Rab-binding platform would yield defective function.
While many of the double mutants did complement growth (Table and not shown), five double mutant combinations with R293A complemented the mrs6Δ null strain with a similar phenotype to the single R293A mutant, whereas four had additional more-lethal phenotypes (Table II; Fig. 2 A and not shown). Like single mutants, all double mutants (with the exception of G53A-G55A discussed above) were found to be expressed at levels similar to wild-type Mrs6p when examined by immunoblotting (see Fig. 4 A and not shown), suggesting that instability or proteolysis was not responsible for loss of function of double mutants in the mrs6Δ null strain. Significantly, one of the strong double mutant combinations, E286S-R293A (Table II; Fig. 2 A), contained residues that, when mutated separately, previously have been implicated directly in Rab-binding in vitro and in vivo by GDI (Schalk et al. 1996; Wu et al. 1998). Thus, full disruption of the physiological function of Mrs6p in vivo may require mutation of at least two surface residues in the putative Rab-binding region. Notably, when the E286S-R293A double mutant was transformed into the MRS6 wild-type strain on a 2μ vector to promote overexpression, we observed significant inhibition of growth compared with a similar level of overexpression of the wild-type Mrs6p, which shows no effect (Fig. 2 B; Table ). These results raise the possibility that this Mrs6p double mutant can act as a potent competitive inhibitor of prenylation (not observed by overexpression of wild-type Mrs6p) by either inappropriately competing for binding to endogenous unprenylated Rab species, or, more likely, by binding to the catalytic components of RabGG transferase or membrane-associated receptors involved in Rab recycling.
Figure 4.

Coimmunoprecipitation of Ypt1p and Sec4p with wild-type and mutant HA-Mrs6p proteins. A and B, Identical amounts of cell homogenates of mrs6 ts cells (grown at 30°C [not shown] or 37°C) expressing HA epitope-tagged wild-type or mutant Mrs6p proteins (indicated at the top of each lane) were cleared of membranes by centrifugation at 100,000 g, and HA-Mrs6p was immunoprecipitated from each supernatant fraction (S100) under native conditions. In A, HA-Mrs6p was detected using immunoblotting with an HA-specific antibody in cell homogenates (before immunoprecipitation) to show that identical amounts of HA-Mrs6p are present in each sample. In B, antibodies to Ypt1p were used to detect Ypt1p in the immunoprecipitates using immunoblotting. All blots were treated in an identical fashion to allow quantitative comparison of levels of recovered Ypt1p.
An additional residue in domain I was found to be required for Mrs6p function in combination with R293 was R127 (Table ). R127 is found in the insert region in α-GDI (Fig. 1, blue). Loss of function in support of growth of the mrs6Δ null strain was similar to that observed for the E286S-R293A mutant (Table ). This region is greatly expanded in mammalian REP (Wu et al. 1998). Moreover, a similar residue in GDI was also found to be essential for specific aspects of α-GDI function (Wu et al. 1998). These results imply a role for the insert region adjacent to the Rab-binding platform in GDI/REP superfamily function. In the case of REP, one possibility is that this region may provide additional sites for interaction with unprenylated, nascent forms of Rab to insure efficient delivery to RabGG transferase.
We also noted that the R195A mutant showed a slower growth phenotype, particularly in combination with R293A (Fig. 2 A; Table and Table ). Moreover, R195A-R293A, like E286A-R293A, had a partial dominant suppressing effect on growth when overexpressed in wild-type cells (Fig. 2 B, Table ). R195 is present in domain II, a region that we have now implicated in interaction with membrane receptors (Luan et al. 2000). These results suggest an important role for residues outside the Rab-binding region in REP function, leading us to conclude that both the domain I and domain II (Fig. 1) play a key role in REP activity.
Double Mutants Will Not Complement the Growth Defect in Temperature-sensitive mrs6ts Cells
To further define the function of the mutant Mrs6p proteins that failed to complement the mrs6Δ null strain, we examined if they could complement the growth defect of yeast strains expressing a temperature-sensitive (ts) Mrs6 protein (Mrs6pts). For this purpose, MRS6 was treated to random PCR mutagenesis and subsequently used to replace wild-type MRS6 by the gapped plasmid repair transformation method. Ts-variants were identified by replica plating and incubation at 30°C (permissive) or 37°C (nonpermissive) temperature. Five ts-variants that failed to show colonies on agar plates at the restrictive temperature, but which grew normally at the permissive temperature (Fig. 3 A), were identified and the mutations mapped. All showed single point mutations in key structural elements (α helices and β strands) found in analogous residues in GDI (not shown). Thus, these structural elements are likely to be destabilized by incubation at elevated temperature.
Figure 3.
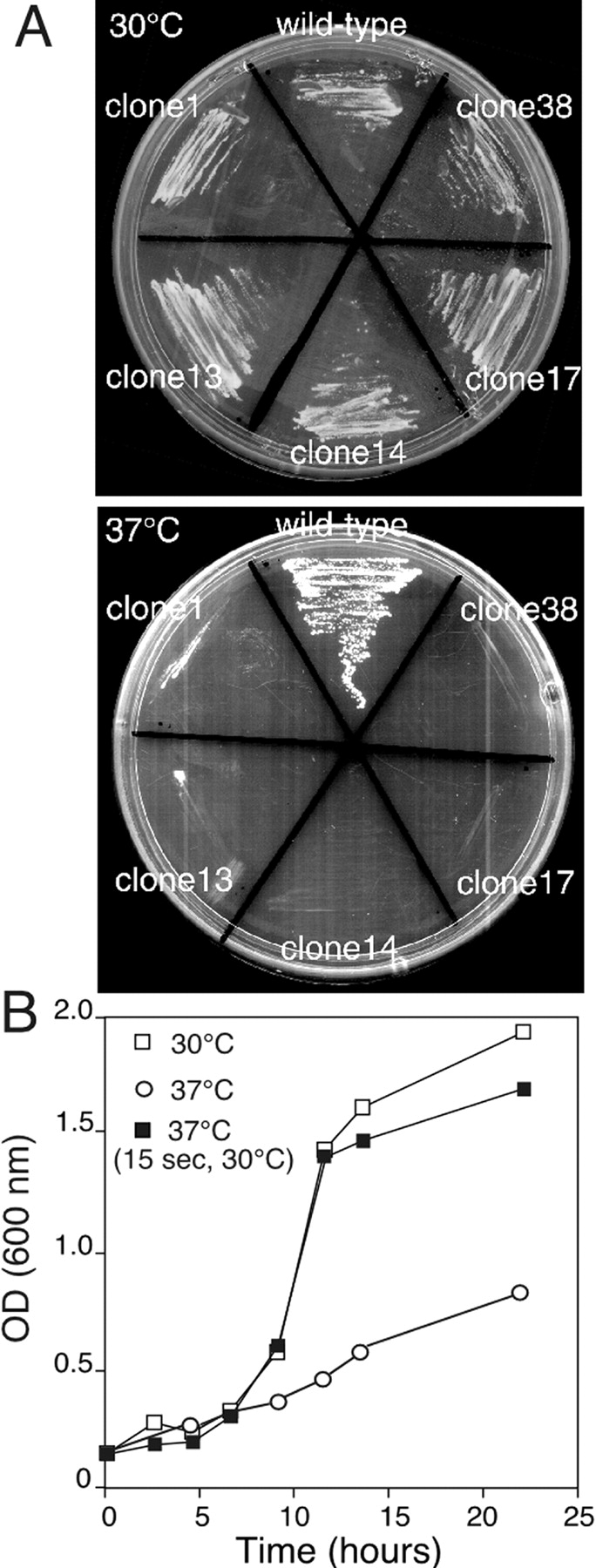
Growth of the mrs6 ts strain. A, The indicated temperature-sensitive strains (clones 1, 13, 14, 17, and 38) were incubated at the permissive (30°C; left) or the restrictive (37°C; right) temperature. B, Growth of clone 14 at the permissive (open squares) or restrictive (open circles) temperatures. In the curve shown by the closed squares, the culture was exposed to the permissive temperature for 15 s before reequilibration to the restrictive temperature (∼2 min total time at the permissive temperature).
A typical growth curve for one of the ts-variants (clone 14) is shown in Fig. 3 B. Growth within the first generation was markedly reduced after shift to the restrictive temperature. Given that prenylated Rabs are relatively stable molecules, requiring at least a single generation for turnover, the decreased rate of growth is likely to reflect progressively reduced pools of prenylated Rab proteins available to promote membrane traffic and cell division. The immediate onset of a reduced rate of growth suggests that Mrs6pts may be rapidly inactivated after shift to the restrictive temperature. Remarkably, when cells grown at the restrictive temperature (37°C) are exposed to 30°C for less than two minutes at hourly intervals, growth was normal (Fig. 3 B). This illustrates that clone 14 phenotype is thermoreversible in vivo. Moreover, the ability of the mutant Mrs6pts to rapidly refold and burst prenylate, an unprenylated Rab pool illustrates that REP function is only transiently required to support normal growth (about two minutes per generation). This may have important implications for understanding the basis of disease and therapeutic approaches to CHM (see Discussion).
To assess whether single or double mutants will complement loss of Mrs6pts function at the restrictive temperature, mutant genes on CEN vectors were transformed into the mrs6 ts strain, and transformants were selected and maintained at 30°C. Transformants were then restreaked and shifted to 37°C for three days and the growth of each of the strains was assessed. Consistent with the above results, single mrs6 mutations suppressed the ts growth defect in a comparable fashion to that observed in the mrs6Δ null strain (not shown). In contrast, each of the double mutations, such as R195A-R293A and E286S-R293A, that did not complement the mrs6Δ null mutation (Table ) showed a considerably reduced ability to complement the mrs6 ts mutation at 37°C (not shown).
Single and Double Mutants Show Deficiencies in Rab Binding In Vivo
Mutant mrs6 alleles that failed to complement the mrs6Δ null mutation or the mrs6 ts phenotype could produce proteins that were defective for folding and therefore unstable and degraded. Alternatively, they could be deficient in any essential aspect of REP function, including Rab binding, interactions with the RabGG transferase catalytic subunits, or interaction of Rab with membrane receptors involved in vesicular transport (Soldati et al. 1994; Ullrich et al. 1994; Luan et al. 1999, Luan et al. 2000).
To distinguish between Rab binding and prenylation, a vector was constructed that allowed us to tag mutant and wild-type Mrs6p proteins with the influenza hemagglutinin (HA) epitope at the COOH terminus (HA-Mrs6p). This region in bovine GDI is disordered in the crystal structure (Schalk et al. 1996). By analogy, the epitope tag would not be expected to interfere with Mrs6p function. Consistent with this conclusion, all HA-tagged constructs generated behaved in an identical fashion to their equivalent untagged versions in growth and in binding to Rab (not shown), as has been observed of HA-tagged Gdi1p (Luan et al. 1999, Luan et al. 2000). The double mutant genes and their corresponding single mutant genes cloned into the HA-vector were used to transform the mrs6 ts strain. Immunoblotting of total cell lysates derived from these strains (incubated at either the permissive or restrictive temperatures) with an HA-specific antibody revealed that a single band of ∼66 kD was present (Fig. 4 A and not shown).
To determine if the single or double mutants would bind Rab, we used a coimmunoprecipitation assay to examine their interaction in vivo with the Rab GTPase Ypt1p that is required for ER/Golgi transport (Segev et al. 1988). Extracts were prepared from strains grown at permissive temperature (30°C) and centrifuged at 100,000 g for one hour to yield P100 particulate fractions containing membranes and S100 cytosolic fractions. HA-Mrs6p was immunoprecipitated from the S100 fractions under native conditions and the immunoprecipitates were probed by immunoblotting with specific antibody against Ypt1p.
Whereas Ypt1p was efficiently coimmunoprecipitated with wild-type HA-Mrs6p (Fig. 4 B), it was only poorly coimmunoprecipitated in extracts from strains expressing each of the double mutants containing modified residues in the Rab-binding platform (Fig. 4 B and not shown). Moreover, significant but reduced and variable amounts of Ypt1p, with the exception of the R195A mutant strain, were coprecipitated by each of the corresponding Rab-binding platform single mutants (E286S and R293A; Fig. 4 B and not shown). This result is consistent with the observation that these single mutants partially affect binding of GDI to Rab3A in vitro and in vivo, yet still support cell growth (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999).
The Domain II Mutant Binds Rab Normally In Vivo
In contrast to the effects of mutations in the Rab-binding platform on interaction of REP with Rab, the single point mutation R195A in domain II, which poorly complemented growth of the mrs6Δ null strain (Table ), showed normal binding to Rab (Fig. 4 B). However, this binding was markedly reduced in the double R195A-R293A mutant (Fig. 4 B). These results now raise the intriguing possibility that domain II is specifically involved in recognition of the RabGG transferase catalytic complex.
Mrs6p Wild-type and Mutant Interactions with Rab In Vitro
We recently reported a novel GDI-Rab binding assay to measure the strength of Rab and GDI interactions based on the property of GDI to prevent the intrinsic dissociation of GDP from Rab3A in combination with mGDP using fluorescence spectroscopy (Wu et al. 1998). The dissociation of mGDP from Rab is determined by measuring the decrease in relative fluorescence (λ excitation 360 nm, λ emission 440 nm) that accompanies the normal release of mGDP. As shown in Fig. 5, in a fashion similar to that reported for GDI (Wu et al. 1998), addition of increasing concentrations of recombinant, purified Mrs6p to prenylated Rab3A led to a decrease in the loss of intrinsic fluorescence through formation of a Rab3A-Mrs6p-mGDP ternary complex. Thus, Mrs6p functions as a GDI. Interestingly, binding Mrs6p to the prenylated form of Rab3A (Fig. 5 A) was stronger than that observed for unprenylated forms (Fig. 5 B) based on the level of inhibition of mGDP dissociation observed in the presence of increasing Mrs6p (K d = 650 nM for unprenylated Rab3A; K d = 320 nM for prenylated Rab). The approximate twofold relative increase in binding is consistent with the observation that REP is likely to function as a transient carrier for delivery of Rab to membranes after prenylation. While the ability of Mrs6p to bind and suppress nucleotide dissociation of mammalian Rab3A confirms the evolutionary conservation of the Rab-binding domain in recognizing Rabs as divergent as those found in yeast and mammals, it was not possible to measure the K d of interaction of REP with the yeast Rab proteins Ypt1p or Sec4p, as they could not be stably loaded with mGDP (our unpublished observations).
Figure 5.
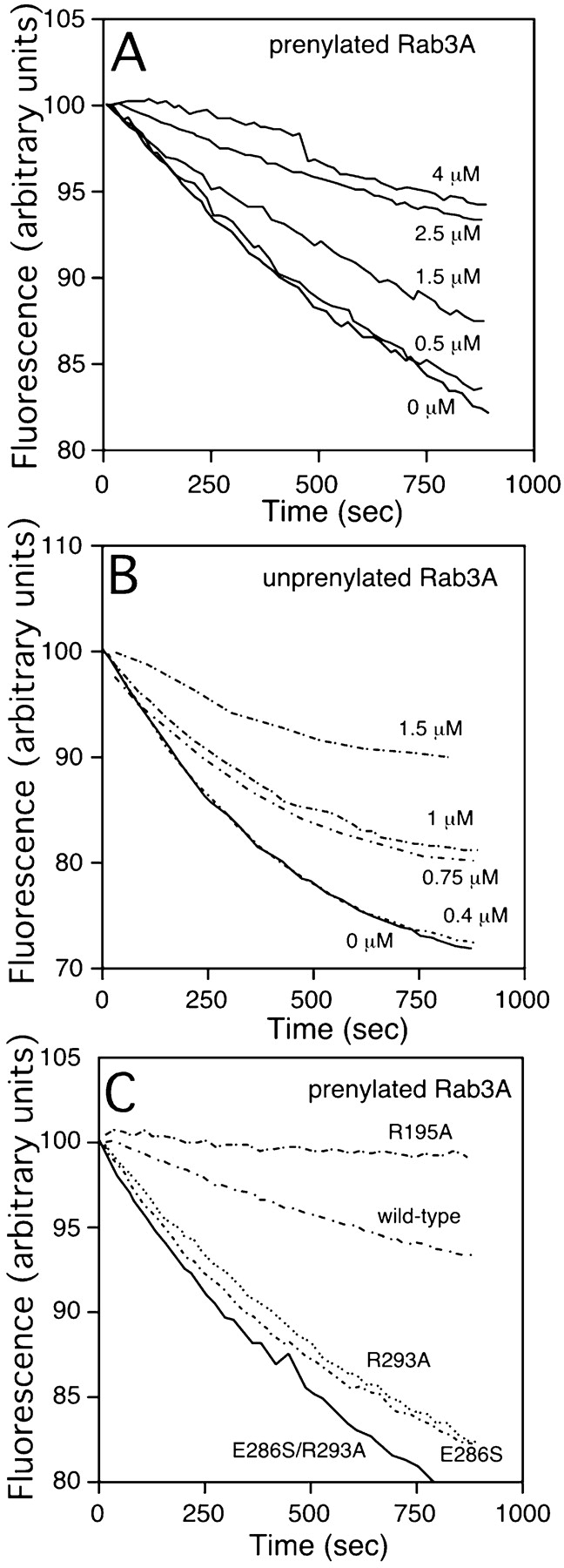
Fluorescence assay to measure Mrs6p-Rab3A interactions in vitro. A and B, Effect of Mrs6p wild-type and mutants on the intrinsic dissociation of mant-GDP from Rab3A. Mrs6p was added at the indicated final concentration (μM). The interaction of Mrs6p with prenylated (A) and unprenylated (B) Rab3A was determined by measuring the decrease in relative fluorescence (λ excitation 360 nm, λ emission 440 nm) that accompanies the release of mant-GDP from Rab3A as described in Materials and Methods. C, The interaction of Mrs6p mutants with Rab3A was determined by mixing 100 nM Rab3A with 2.5 μM Mrs6p wild-type or mutant protein as indicated.
Using this assay, we also tested the effects of mutation of selected residues on the ability of Mrs6p to interact with Rab3A. As shown in Fig. 5 C, both the single and double mutants defining the Rab-binding region of GDI (E286S-R293A) completely interfered with the ability of Mrs6p to prevent mGDP dissociation. Given that both single point mutations partially supported growth in vivo, these results emphasize the importance of these conserved residues in both GDI and REP binding to Rab GTPases, and the fact that even reduced activity of REP in vivo can support growth.
In contrast to the effects of mutants in the Rab-binding region on binding to Mrs6p, R195A, a substitution in domain II that poorly supported growth (Table ; Fig. 2A and Fig. c) had no effect on Rab binding in vitro. Because the double mutant (R195A-R293A) that had reduced Rab binding had a dominant negative effect on growth in cells expressing wild-type Mrs6p, these results provide an additional line of evidence that the single mutant may affect the functional interaction of REP with RabGG transferase.
The Absence of Mrs6p Function Results in the Accumulation of Unprenylated Rab in the Cytosol
To determine if in the absence of Mrs6p function in the clone 14 Mrs6pts strain grown at the restrictive temperature, Rab proteins showed altered distributions between the cytosol or membranes in vivo, the P100 and S100 fractions from the experiments described above were probed by immunoblotting with a Rab-specific antibody (Fig. 6). Interestingly, we found that compared with wild-type cells in which the ratio of cytosol to membrane bound forms was 0.6 at the permissive temperature, the ratio in ts cells grown at the permissive temperature was 3, indicating an accumulation of unprenylated Rab in the cytosol, even under conditions that support normal growth. These results are consistent with the observation that the prenylation by the ts strain is defective in vitro and suggests that only 10–20% of the normal membrane-associated pool found in wild-type cells in sufficient to sustain growth. Interestingly, when either the wild-type or ts cells were shifted to the restrictive temperature for five hours, only a modest shift in the respective cytosol to membrane ratios was observed (Fig. 6). Moreover, the level of total Rab in both the wild-type or ts strain remained the same (Fig. 6, inset). These results suggest that in the case of the ts strain, further misfolding of Mrs6pts at the higher temperature completely inactivated function and was responsible for the markedly reduced rate of growth as shown previously (Fig. 3 B). These results illustrate that newly synthesized Rabs that fail to be prenylated accumulate in the cytosol. Thus, burst prenylation of Rabs by temporal stabilization of the folding of clone 14 Mrs6pts during transient shift to the permissive temperature (Fig. 3 B) may occur on either a newly synthesized pool of Rab or the preexisting pool.
Figure 6.

Distribution of wild-type and mutant Rab proteins between cytosol and membranes. Wild-type and clone 14 mrs6 ts strain were incubated at the indicated temperature. After 5 h, the same OD of cells was lysed and subjected to centrifugation at 100,000 g to generate a P100 membrane fraction (p) and an S100 cytosol fraction (s). The amount of Ypt1p in each fraction was quantitated by immunoblotting with specific antibody. The inset illustrates that the amount of total Rab at the 5-h time point that was detected using immunoblotting was the same in both the wild-type and ts strains at either 30°C or 37°C.
Effect of Single Point Substitutions on Prenylation In Vitro
Because selected single and double mutations in domains I and II of Mrs6p will either partially complement or fail to complement growth of the mrs6 ts or mrs6Δ null strains (Table and Table ), respectively, a prediction is that they should be deficient to some degree in prenylation of Rab proteins in vitro. Prenylation activity in vitro is measured by incorporation of 3H-GGPP to recombinant Rab added to S100 fractions prepared from wild-type or mutant protein-expressing strains.
Single and double mutants that partially or fully complemented growth were expressed in the mrs6Δ null strain and extracts prepared for assay in the presence or absence of recombinant Rab. K99D, T185D, Q289L, and Y271F, among others, had no defect in prenylation relative to that found for wild-type Mrs6p (Fig. 7 A), consistent with the ability of these mutants to support normal growth (Table ). In contrast, in the mrs6Δ null strain background, the single mutants E286S, R293A, and T301P were completely deficient in prenylation in vitro (Fig. 7 B). These domain I residues were implicated in Rab binding in vitro and in vivo (Fig. 4 B and 5 C). Therefore, efficient prenylation of Rab requires binding of Rab to the Rab-binding platform.
Figure 7.

Analysis of prenylation activity of Mrs6p single and double mutants in vitro. A, Single and double mutants that partially or fully complemented growth were expressed in the mrs6Δ strain and extracts prepared for assay in the presence or absence of recombinant Ypt1p as described in Materials and Materials. B, To assess the prenylation activity of inviable double mutants, these were expressed in the mrs6 ts strain and extracts prepared for assay as indicated. The assay profile of clone 14 is typical for that observed for clones 13, 17, and 38. Double mutants were assayed in the clone 14 strain at 37°C.
Interestingly, the R195A mutant found in helix E of domain II, which showed a weak growth phenotype in vivo in the mrs6Δ null strain (Table ), but showed normal Rab binding (Fig. 4 B and 5 C), was completely deficient in prenylation (Fig. 7 A). These results are consistent with the interpretation that domain II function is necessary for delivery of Rab to the catalytic subunits of RabGG transferase. In addition, a second residue, L226, found in the adjacent helix (Fig. 1), while having no effect on growth as a single point mutant (Table ) showed a striking effect on growth as double mutant in combination with R293A. In vitro, the L226A was defective in prenylation, suggesting that this substitution contributes instability to helix F and, therefore, loss of domain II function in vitro. We conclude that at least two residues, one in helix E and a second in helix F, contribute to the structure–function relationships within the domain II–helical tripod, providing strong evidence for its role in prenylation.
In general, the inability of Rab-binding platform and domain II mutants to support prenylation in vitro, yet support growth in vivo, suggests that prenylation in vitro is a sensitive measure of potential effects of mutants on REP function. The ability of single point mutants to support growth must indicate that reduced activity of REP in vivo is sufficient to generate a minimal, but functional, pool of prenylated Rab proteins necessary for normal cell function. This observation could now account for why only complete loss of CHM/REP1 gives rise to the hereditary disease CHM.
Mobile Effector Domain Found in GDI Is Not Required for Prenylation by Mrs6p
We have recently shown that two highly conserved residues found in a mobile effector loop of GDI found in domain II are required for recycling of Rabs after the targeting and fusion of vesicles (Luan et al. 2000). Therefore, we tested the potentially homologous residues in Mrs6p. As shown in Fig. 7 A, mutations D269A, V270R, and Y271F had no effect on prenylation in vitro, consistent with their inability to support normal growth of the mrs6Δ null strain and to have no effect on the growth phenotype of the R293A mutant when expressed as double mutants (Table and Table ). Thus, the mobile effector loop found in GDI responsible for Rab recycling after vesicle targeting and fusion is not required for binding of REP to RabGG transferase, a result consistent with the observation that REP is involved in the delivery, but not retrieval, of Rabs from membranes.
Effect of Double Mutants on Prenylation Activity In Vitro
To assess the prenylation activity of inviable double mutants, these were expressed in the mrs6 ts strain. For this purpose, we first screened each of the Mrs6pts clones for thermosensitive prenylation in vitro. As shown in Fig. 7 B, three classes of activity were observed. The first group included extracts prepared from wild-type cells grown at 30°C that showed efficient prenylation at all temperatures tested (23, 30, and 37°C). The second group, including clone 1 extracts, while efficiently prenylating Rab at 23 and 30°C, showed markedly reduced levels of prenylation upon incubation at 37°C. In contrast, group 3, characterized by extracts prepared from clones 14 (Fig. 7 B) and 13, 17, and 38 (not shown), were unable to prenylate Rabs at any temperature (23, 30, or 37°C), suggesting that although the folding of these variants was sufficiently stable to support growth in vivo, they were not stable under our current in vitro incubation conditions, a typical characteristic of ts variants. As expected, double mutants that were stably expressed, but deficient in growth in vivo in the mrs6 ts strain, were deficient in prenylation in vitro in a mrs6 ts (clone 14) background at both the permissive and restrictive temperature (Fig. 7 B).
Intracellular Membrane Trafficking in mrs6 Mutants
Rab GTPases are required for numerous membrane trafficking steps in the endocytic and exocytic pathways. These involve at least 11 different Rab proteins in yeast and >40 proteins in mammalian cells (Martinez and Goud 1998). Therefore, we examined our collection of single point mutants for secretory transport function. We monitored Mrs6p function in the early secretory pathway (ER/Golgi complex) and the vacuolar protein sorting/endocytic (VPS) pathway by examining the processing of CPY. In pulse-chase immunoprecipitation assays, the conversion of the ER p1 precursor form (67 kD) to the Golgi complex-modified p2 (69 kD) precursor, reflects rapid transport through the ER and Golgi complex, and conversion of p2 form to mature CPY (61 kD) reflects transport from the Golgi complex to the vacuole. CPY biosynthesis was assayed at 5- and 30-min chase at 30°C, time points that allowed us to visualize potential reduced rates of transport through each of these CPY biosynthetic transport steps.
For the viable Mrs6p mutants, pulse-chase assays were conducted with strains in which the mutant mrs6 alleles were the sole source of Mrs6p. As shown in Fig. 8, the R195A mutant and the R127A (that showed normal growth in the mrs6Δ null strain; Table ) transported CPY at a rate characteristic of that observed in wild-type cells. Transport was rapid at 30°C and nearly complete within the 5-min chase period as indicated by the appearance of the mature form (Fig. 8 A, m, compare a, b, and c); results quantitated in Fig. 8 B). In contrast, two of the single mutants, E286S and R293A, clearly exhibited defects in CPY biosynthesis that reflected partial kinetic delays in transport at different steps of the secretory pathway. The R293A mutant, which showed markedly reduced prenylation of the ER to Golgi complex-specific Rab (Ypt1p) in vitro (Fig. 7), demonstrated a clear reduction in ER to Golgi complex transport leading to a decrease in the kinetics of processing of the P1 (ER) to the P2 (Golgi complex) form (Fig. 8). The fact that the p2 form was subsequently rapidly processed to the mature vacuolar form indicates that the major kinetic delay in the presence of this mutant is ER to Golgi complex transport. However, the E286S mutation showed also a potent block in transport of CPY from the p2 (Golgi form) to the vacuole as indicated by the transient accumulation of the p2 form at the 5-min point. Moreover, the p2 form only partially matured to the vacuolar form by 30 min (Fig. 8d). These results suggest that another yeast Rab involved in vacuolar targeting may not be efficiently recognized by this mutant and that different residues in the Rab-binding region of REP are required for efficient binding of different Rab proteins. A similar result has been observed for GDI (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999, Luan et al. 2000). As vacuolar protein transport is not required for growth of yeast, while ER to Golgi complex transport is essential, the kinetic delay of CPY transport to the Golgi complex observed in the presence of the R293A mutant is consistent with the reduced rate of growth of this mutant in the mrs6Δ null strain (Table ).
Figure 8.
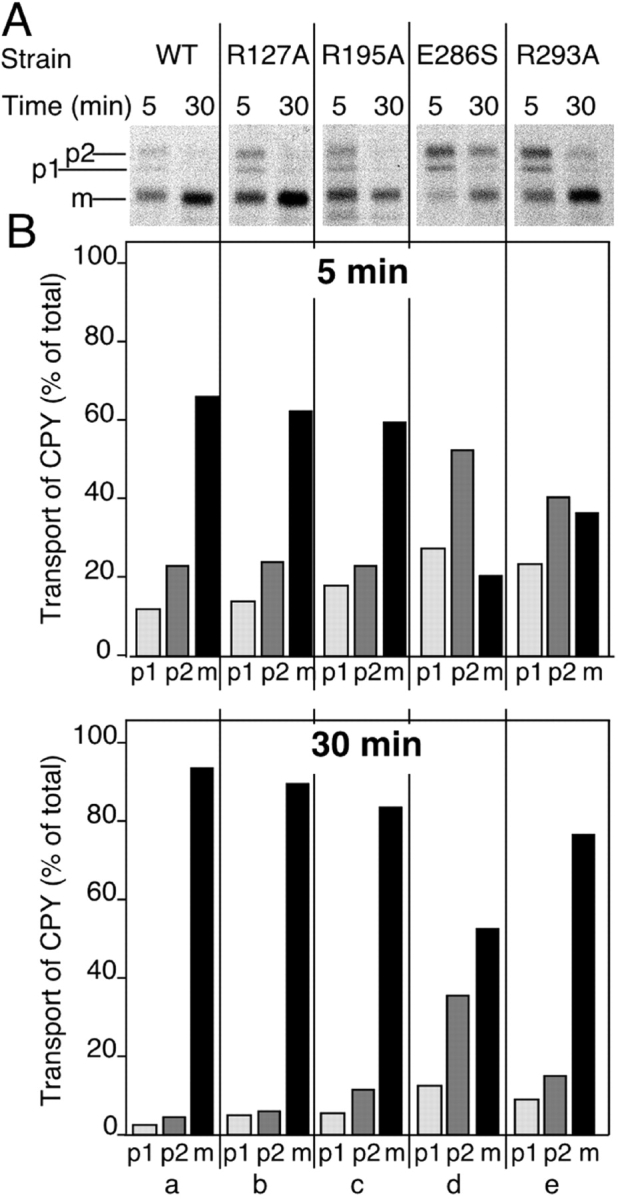
Analysis of vesicle-mediated trafficking of mrs6 single mutants. The indicated strains in mrs6Δ null cells were prepared and the cultures were labeled with [35S]methionine/cysteine for 20 min, then chased for 5 or 30 min. CPY was immunoprecipitated from extracts derived from each culture. The positions of p1-CPY (ER), p2-CPY (Golgi), and mature (m) CPY (vacuole) are indicated to the left. In wild-type cells incubated at 37°C transport is very rapid. Here, CPY is largely recovered in the mature form, even after a 5-min chase period. B, The distribution of CPY in the p1, p2, and m forms at the 5-min (top) and 30-min (bottom) time points for each mutant were quantitated using densitometry.
These results provide direct support for the interpretation that REP interacts differentially with Rabs to modulate the availability of the active, prenylated form Rab for distinct transport steps in the exocytic pathway. This is consistent with the need for specialized REP isoforms in mammalian cells that have cell lineage–specific defects in function such as found in CHM.
EM Reveals the Accumulation of Numerous Vesicular Structures in the mrs6ts Strain Incubated at the Restrictive Temperature
EM was used in an effort to visualize membrane trafficking intermediates that might accumulate in mrs6 ts mutants. Clone 14 was prepared for analysis by growing at the permissive temperature (30°C), and then the culture was divided and one half was transferred to 37°C and incubated for three hours while the other half remained at 30°C. Cells were then fixed and prepared for EM.
Fig. 9 shows that the mrs6 ts cells grown at the permissive temperature (Fig. 9 B) were similar in appearance to wild-type cells (Fig. 9 A; Vida and Emr 1995). They contained a dense, ribosome-rich cytoplasm dominated by a large circular vacuole. In contrast, numerous aberrant membrane structures were observed in the mrs6 ts strain incubated at the restrictive temperature (Fig. 9 C). Not only were numerous vesicular structures evident throughout the cytoplasm (Fig. 9 C, arrowheads), but the vacuole was surrounded by large electron-lucent structures giving the vacuole a punched-in type of appearance (Fig. 9, arrows). This phenotype is very similar to the phenotype observed in the sec19-1 or sec19-1 (R248A-Y44V) strains in which a temperature-sensitive mutant of yeast GDI (sec19-1) can be rapidly inactivated by shift to the restrictive temperature. These results illustrate that striking morphological alterations accompany loss of general REP function. In CHM, where the defect is due to loss of the CHM/REP1 isoform function, early morphological phenotypes will be expected to be highly diagnostic of the specific pathway regulated by Rab protein(s) uniquely prenylated by CHM/REP1.
Figure 9.
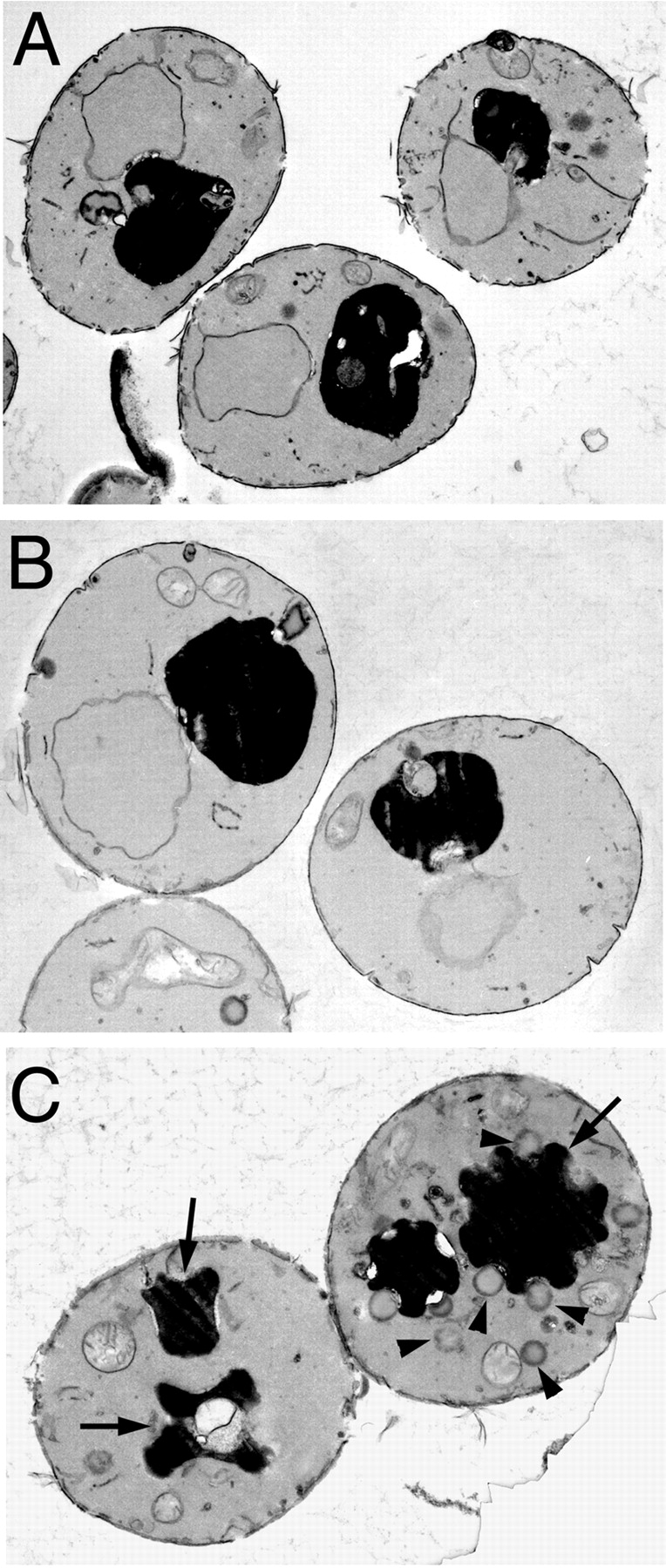
EM of a mrs6 ts mutant strain. The clone 14 mrs6 ts mutant was grown overnight at 30°C, the culture was divided, and one half was incubated at 37°C for 3 h (C) while the other half was maintained at 30°C (B). In C, note the numerous large vesicles that accumulate (arrowheads), and the punched-in appearance of vacuoles (arrows) as a consequence of inactivation of Mrs6p. A typical wild-type cell is shown in A.
Discussion
Our genetic and biochemical analyses provide new mechanistic insight into the role of REP in prenylation of Rab GTPases. Using site-directed mutagenesis to inactivate Mrs6p function in yeast we have now established the physiological importance of residues found in domain I of REP, a region previously implicated in Rab binding in vitro and in vivo to GDI (Schalk et al. 1996; Wu et al. 1998) and, the potential role of domain II in recognition of the RabGG transferase catalytic subunits. The importance of these findings and their implication for the dysfunction of the CHM/REP1 in disease are discussed below.
The Rab-binding Region Plays a Physiological Role in REP Function In Vivo
Previous biochemical studies based on the structure of bovine α-GDI demonstrated that residues present in SCRs 1 and 3B fold to form a platform at the apex of GDI that binds Rab (Schalk et al. 1996; Wu et al. 1998; Luan et al. 2000). We have now shown that mutation of conserved residues found in the homologous Rab-binding platform of MRS6, while having only a partial effect on growth when expressed as single point mutants in the mrs6Δ null or mrs6 ts strain, can markedly affect their ability to bind Rab and to support prenylation in vitro. Thus, we now conclude that the Rab-binding platform is a universal functional feature of the REP-GDI superfamily (Fig. 10). Moreover, the phenotype observed under conditions in which Rab binding is clearly reduced in vivo lead us to conclude that REP activity is normally in functional excess over that required to maintain a minimal pool of Rab GTPases required for cell growth. This was particularly evident when we allowed a temperature-sensitive variant of Mrs6p to function only transiently by rapid shift of cells from the restrictive to the permissive temperature before reincubation at the restrictive temperature. Consistent with a rapid inactivation, growth was promptly inhibited and prenylation in vitro was completely defective at the restrictive temperature. Because it is apparent that unprenylated Rabs appear to be stable during incubation of ts cells at the restrictive temperature, transient prenylation of this pool is sufficient to support growth, consistent with the fact that only a small recycling pool of Rab is sufficient to support growth in yeast strains harboring less active, mutant forms of Gdi1p (Luan et al. 1999). The ability of cells to grow with marginal functional levels of REP is likely to explain why only a complete loss of CHM/REP1 function gives rise to disease.
Figure 10.
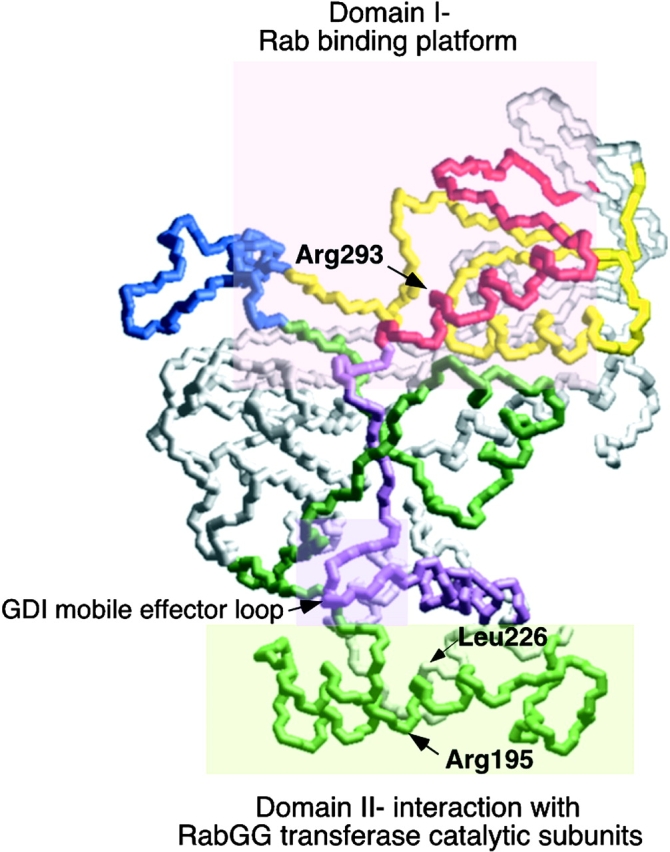
Domain organization of REP for Rab-binding and prenylation. The figure highlights principle regions involved in REP function based on the homologous residues in GDI through sequence alignment. Residues shaded by the pink box in upper region (Domain I) directs Rab interaction; residues shaded by the light green box in the lower region (Domain II) participate in recognition of the catalytic subunits of GG Tr II. The mobile effector loop involved in recognition of recycling factors by GDI (Luan et al. 2000), but not REP (this study), are indicated by the shaded purple box at the interface of domains I and II.
In contrast to the marginal effects of most single mrs6 point mutations on complementation of growth of the mrs6Δ null strain, selected combinations of Rab-binding platform mutations rendered Mrs6p inactive in vivo as these mutants could not complement growth of mrs6 ts strain at the restrictive temperature. This is consistent with the effect of similar double mutants in Gdi1p function in Rab recognition in vitro and Rab recycling in vivo (Wu et al. 1998; Luan et al. 1999). Our results now provide strong physiological evidence that this Arg residue plays a pivotal role, along with other residues found at the apex of GDI in the activity of the Rab-binding platform for the REP-GDI superfamily (Fig. 1 and Fig. 10). This conclusion is consistent with the observation that a double mutant containing the R293A mutation and a mutation in a residue localized to the insert region that forms a boundary on one side of the Rab-binding platform (R127 in Mrs6p) renders both proteins inactive. These results raise the possibility that the variable insert region also contributes to superfamily function.
Function of Rabs in Membrane Traffic Is Differentially Sensitive to Residues in the Rab-binding Platform Mrs6p
Our studies have now demonstrated that the function of REP proteins in prenylation is differentially sensitive to single point mutations. In particular, the R293A mutant clearly affected ER to Golgi complex transport, a result consistent with its reduced ability to prenylate the ER to Golgi complex-specific Rab, Ypt1p. In contrast, a different point mutation, E286S, affected in addition post-Golgi complex trafficking to the vacuole. One explanation for this result is that residues in the Rab-binding pocket of Mrs6p contribute in different ways to the strength of interaction between REP and distinct Rab species through multiple polar and/or hydrophobic interactions. Such an interpretation would be consistent with in vitro binding studies with GDI where we (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999) and others (Shisheva et al. 1999) have shown that various mutations in the Rab-binding platform differentially affect Rab binding in vitro and recycling in vivo.
Role of Domain II in REP Function
We now provide evidence for the role of the domain II region (Fig. 10) in REP recognition of the catalytic subunits of RabGG transferase. Mutation of amino acid residue R195 in helix E had no effect on Rab binding or stability in vivo. However, it led to reduced ability of Mrs6p to support growth of mrs6Δ null strain and a striking loss of function in prenylation of Ypt1p in vitro, particularly in combination with an R293A substitution which weakens Rab binding. These results suggest that this region (Fig. 10) is involved in Rab prenylation. A similar result was observed for the role of the adjacent helix F. We found that L226 is crucial for maintaining the activity of CHM in Rab prenylation in vitro.
The importance of domain II in interacting with other proteins is consistent with our recent studies based on the 1.04-Å structure of α-GDI (Luan et al. 2000), where we demonstrated that residues found in a mobile effector loop (residues surrounding Tyr271 [Mrs6p numbering], as shown in Fig. 10) were important for interaction with downstream receptors involved in the recycling of Rab through RRF after vesicle targeting and fusion. Thus, domain II provides additional function to the members of the GDI/REP superfamily.
Interestingly, the highly conserved residues comprising the mobile effector loop in GDI family members are not strictly conserved in REP. Indeed, mutation of these residues (269–271 Mrs6p numbering) failed to inhibit prenylation. Because current evidence suggests that REP is involved in delivery to membranes after prenylation, but not retrieval (Wu et al. 1996), these results provide further evidence that the mobile effector loop in domain II function of GDI is restricted to a late Rab recycling step (Luan et al. 2000). Thus, REP, like GDI, contains two distinct regions required for function: an upper platform found at the apex required for binding of Rab; and a second domain composed of tightly packed helices that appears to play a role in the recognition of the α/β complex of RabGG transferase (Fig. 10).
Molecular Explanation for Kinetic Models of REP Interaction with Rab and RabGG Transferase
Recent biochemical studies (Anant et al. 1998; Alexandrov et al. 1999) have suggested the following mechanistic model for REP in promoting Rab prenylation by RabGG transferase. In the first step, REP and unprenylated Rab form a binary complex that involves residues in the switch 2 region of Rab (Overmeyer et al. 1998) that are sensitive to the nucleotide bound state, but insensitive to hypervariable COOH terminus. In a second step, the binding of the Rab–REP complex to RabGG transferase occurs by a two-stage kinetic mechanism that is independent of prenyl substrate. The first stage is fast and concentration (Rab)-dependent, whereas the second stage is concentration-independent and rate-limiting. Finally, after prenylation, the REP-prenylated Rab complex rapidly dissociates and delivers prenylated Rab to membranes.
Our evidence now suggests that in step 1, Rab binding principally relies on residues comprising the Rab-binding platform of domain I (Fig. 10). In the case of REP, this may also involve the large insert motif flanking the Rab-binding region (Fig. 10, blue). Once bound, both domain I containing bound Rab and domain II residues contribute to recognition of RabGG transferase. One possibility is that this recognition is first achieved by Rab. However, this would be inconsistent with the fact that Rab, independent of REP, cannot bind RabGG transferase, and that excess wild-type REP is a competitive inhibitor of Rab prenylation. A second, favored model, is that the first rapid and concentration-dependent stage reflects residues in domain II that direct the docking the Rab–REP complex to RabGGTase. Support for this model comes the fact that prenylation can be readily competed, at least in the case of the mammalian REP protein in vitro, with unbound REP (Anant et al. 1998; Alexandrov et al. 1999). Moreover, while we found that overexpression of wild-type Mrs6p did not inhibit growth reflecting the ability of a modest pool of prenylated Rab to function efficiently, we found the striking result that mutation of residue 195 found in domain II showed reduced growth in the mrs6Δ null background or mrs6 ts background, and rendered REP defective in prenylation activity in vitro. The second slower and concentration-independent stage we propose to be the presentation of hypervariable COOH terminus of Rab to the catalytic subunit of RabGG transferase. Rapid dissociation after prenylation would be expected to modulate the affinity of domain II for RabGG transferase, consistent with the fact that binding of the prenyl groups on Rab may occur at a hydrophobic pocket situated at the interface between domain I and II (D'Adamo et al. 1998). This model is consistent with our studies that showed a higher affinity of REP for prenylated Rab than for unprenylated Rab. Further crystallographic and molecular studies are currently in progress to address this model.
Implications for Physiological Function of the CHM/REP Family in CHM
REP2 appears to be the major housekeeping form distributed throughout all tissues (Cremers et al. 1994). In contrast, REP1 is confined to a more limited spectrum of tissues, such as the RPE (Seabra et al. 1992b; Andres et al. 1993). The differential tissue distribution of this REP isoform combined with our observations that residues in the Rab-binding region contribute to selective activity in Rab recognition and recycling (Schalk et al. 1996; Wu et al. 1998; Luan et al. 1999) indicates a specialized role in handling subsets of RPE-specific Rabs. Such specialization may contribute to maintenance of specific types of Rab-dependent vesicle traffic in these tissues (Seabra et al. 1995). By analogy to the effects of various mutants in Mrs6p, our results now demonstrate that the deficiency in CHM may be overcome by only a modest level of increased expression of REP1 or through selective augmentation of REP2 function to facilitate prenylation of REP1-specific Rabs.
Acknowledgments
We are grateful to Dieter Gallwitz and Susan Ferro-Novick for generously providing reagents, and Matthias Jost and Scott Emr for advice. Electron Microscopy Core Facility was conducted by Core B of CA58689.
This work was supported by a National Institutes of Health grant to W.E. Balch (EY11606).
Footnotes
Abbreviations used in this paper: CHM, choroideremia; CPY, carboxypeptidase Y; GDI, guanine nucleotide dissociation inhibitor; mGDP, methylanthraniloyl guanosine diphosphate; RabGG transferase, Rab geranylgeranyl transferase II; REP, Rab escort protein; RPE, retinal pigmented epithelium; SCR, sequence-conserved region.
References
- Alexandrov K., Horiuchi H., Steele-Mortimer O., Seabra M.C., Zerial M. Rab escort protein-1 is a multifunctional protein that accompanies newly prenylated rab proteins to their target membranes. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:5262–5273. doi: 10.1002/j.1460-2075.1994.tb06860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov K., Simon I., Yurchenko V., Iakovenko A., Rostkova E., Scheidig A.J., Goody R.S. Characterization of the ternary complex between Rab7, REP-1 and Rab geranylgeranyl transferase. Eur. J. Biochem. 1999;265:160–170. doi: 10.1046/j.1432-1327.1999.00699.x. [DOI] [PubMed] [Google Scholar]
- Anant J.S., Desnoyers L., Machius M., Demeler B., Hansen J.C., Westover K.D., Deisenhofer J., Seabra M.C. Mechanism of Rab geranylgeranylationformation of the catalytic ternary complex. Biochemistry. 1998;37:12559–12568. doi: 10.1021/bi980881a. [DOI] [PubMed] [Google Scholar]
- Andres D.A., Seabra M.C., Brown M.S., Armstrong S.A., Smeland T.E., Cremers F.P.M., Goldstein J.L. cDNA cloning of component A of rab geranylgeranyl transferase and demonstration of its role as a Rab escort protein. Cell. 1993;73:1091–1099. doi: 10.1016/0092-8674(93)90639-8. [DOI] [PubMed] [Google Scholar]
- Cadwell R.C., Joyce G.F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Cremers F.P.M., van de Pol D.J.R., van Kerkhoff L.P.M., Wieringa B., Ropers H.-H. Cloning of a gene that is rearranged in patients with choroideraemia. Nature. 1990;347:674–677. doi: 10.1038/347674a0. [DOI] [PubMed] [Google Scholar]
- Cremers F.P.M., Armstrong S.A., Seabra M.C., Brown M.S., Goldstein J.L. REP-2, a rab escort protein encoded by the choroideremia-like gene. J. Biol. Chem. 1994;269:2111–2117. [PubMed] [Google Scholar]
- D'Adamo P., Menegon A., Lo Nigro C., Grasso M., Gulisano M., Tamanini F., Bienvenu T., Gedeon A.K., Oostra B., Wu S.K. Mutations in GDI1 are responsible for X-linked non-specific mental retardation [published erratum appears in Nat. Genet. 1998. 19:303] Nat. Genet. 1998;19:134–139. doi: 10.1038/487. [DOI] [PubMed] [Google Scholar]
- Fodor E., Lee R.T., O'Donnell J.J. Analysis of choroideraemia gene. Nature. 1991;351:614. doi: 10.1038/351614b0. [DOI] [PubMed] [Google Scholar]
- Fujimura K., Tanaka K., Nakano A., Toh-e A. The Saccharomyces cerevisiae MS14 gene encodes the yeast counterpart of component A of rab geranylgeranyltransferase. J. Biol. Chem. 1994;269:9205–9212. [PubMed] [Google Scholar]
- Gaynor E.C., te Heesen S., Graham T.R., Aebi M., Emr S.D. Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J. Cell Biol. 1994;127:653–665. doi: 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Heckenlively, J.R., and A.C. Bird. 1988. Retinitis Pigmentosa. Heckenlively, J.R., editor. J.B. Lippincott & Co., New York, NY. 176–187.
- Horazdovsky B.F., Emr S.D. The VPS16 gene product associates with a sedimentable protein complex and is essential for vacuolar protein sorting in yeast. J. Biol. Chem. 1993;268:4953–4962. [PubMed] [Google Scholar]
- Horazdovsky B.F., Busch G.R., Emr S.D. VPS21 encodes a rab5-like GTP binding protein that is required for the sorting of yeast vacuolar proteins. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:1297–1309. doi: 10.1002/j.1460-2075.1994.tb06382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H., Fukuda Y., Murata K., Kimura A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Ferro-Novick S. Identification of yeast component Areconstitution of the geranylgeranyltransferase that modifies Ypt1p and Sec4p. Proc. Natl. Acad. Sci. USA. 1994;91:4377–4381. doi: 10.1073/pnas.91.10.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Rossi G., Ferro-Novick S. Bet2p and Mad2p are components of a prenyltransferase that adds geranylgeranyl onto Ypt1p and Sec4p. Nature. 1993;366:84–86. doi: 10.1038/366084a0. [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Banta L.M., Emr S.D. Intracellular sorting and processing of a yeast vacuolar hydrolaseproteinase A propeptide contains vacuolar targeting information. Mol. Cell. Biol. 1988;8:2105–2116. doi: 10.1128/mcb.8.5.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Havel C., Watson J.A., Murray A.W. The mitotic feedback control gene MAD2 encodes the alpha-subunit of a prenyltransferase [published erratum appears in Nature. 1994. 371:438] Nature. 1993;366:82–84. doi: 10.1038/366082a0. [DOI] [PubMed] [Google Scholar]
- Luan P., Balch W.E., Emr S.D., Burd C.G. Molecular dissection of guanine nucleotide dissociation inhibitor function in vivo. J. Biol. Chem. 1999;274:14806–14817. doi: 10.1074/jbc.274.21.14806. [DOI] [PubMed] [Google Scholar]
- Luan P., Heine A., Zeng K., Moyer B., Greasely S.E., Kuhn P., Balch W.E., Wilson I.A. A new functional domain of guanine nucleotide dissociation inhibitor (α-GDI) involved in Rab recycling. Traffic. 2000;1:270–281. doi: 10.1034/j.1600-0854.2000.010309.x. [DOI] [PubMed] [Google Scholar]
- MacDonald I.M., Mah D.Y., Ho Y.K., Lewis R.A., Seabra M.C. A practical diagnostic test for choroideremia. Ophthalmology. 1998;105:1637–1640. doi: 10.1016/S0161-6420(98)99031-5. [DOI] [PubMed] [Google Scholar]
- Maniatis, T., E.F. Fritsch, and J. Sambrook. 1982. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Martinez O., Goud B. Rab proteins. Biochim. Biophys. Acta. 1998;1404:101–112. doi: 10.1016/s0167-4889(98)00050-0. [DOI] [PubMed] [Google Scholar]
- Miller J. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. [Google Scholar]
- Nuoffer C., Peter F., Balch W.E. Purification of His6-tagged Rab1 proteins using bacterial and insect cell expression systems. Meth. Enzymol. 1995;257:3–8. doi: 10.1016/s0076-6879(95)57003-9. [DOI] [PubMed] [Google Scholar]
- Overmeyer J.H., Wilson A.L., Erdman R.A., Maltese W.A. The putative “switch 2” domain of the Ras-related GTPase, Rab1B, plays an essential role in the interaction with Rab escort protein. Mol. Biol. Cell. 1998;9:223–235. doi: 10.1091/mbc.9.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder S.E., Banta L.M., Kohrer K., McCaffery J.M., Emr S.D. Multilamellar endosome-like compartment accumulates in the yeast vps28 vacuolar protein sorting mutant. Mol. Biol. Cell. 1996;7:985–999. doi: 10.1091/mbc.7.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T., Kikuchi A., Araki S., Hata Y., Isomura M., Kuroda S., Takai Y. Purification and characterization from bovine brain cytosol of a protein that inhibits the dissociation of GDP from and the subsequent binding of GTP to smg p25A, a ras p21-like GTP-binding protein. J. Biol. Chem. 1990;265:2333–2337. [PubMed] [Google Scholar]
- Schalk I., Zeng K., Wu S.K., Stura E.A., Matteson J., Huang M., Tandon A., Wilson I.A., Balch W.E. Structure and mutational analysis of Rab GDP-dissociation inhibitor. Nature. 1996;381:42–48. doi: 10.1038/381042a0. [DOI] [PubMed] [Google Scholar]
- Schiestl R.H., Gietz R.D. High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet. 1989;16:339–346. doi: 10.1007/BF00340712. [DOI] [PubMed] [Google Scholar]
- Schimmoller F., Simon I., Pfeffer S.R. Rab GTPases, directors of vesicle docking. J. Biol. Chem. 1998;273:22161–22164. doi: 10.1074/jbc.273.35.22161. [DOI] [PubMed] [Google Scholar]
- Seabra M.C. Membrane association and targeting of prenylated Ras-like GTPases. Cell. Signal. 1998;10:167–172. doi: 10.1016/s0898-6568(97)00120-4. [DOI] [PubMed] [Google Scholar]
- Seabra M.C., James G.L. Prenylation assays for small GTPases. Methods Mol. Biol. 1998;84:251–260. doi: 10.1385/0-89603-488-7:251. [DOI] [PubMed] [Google Scholar]
- Seabra M.C., Goldstein J.L., Sudhof T.C., Brown M.S. Rab geranylgeranyl transferasea multisubunit enzyme that prenylates GTP-binding proteins terminating in Cys-X-Cys or Cys-Cys J. Biol. Chem. 267 1992. 14497 14503a [PubMed] [Google Scholar]
- Seabra M.C., Brown M.S., Slaughter C.A., Sudhof T.C., Goldstein J.L. Purification of component A of Rab geranylgeranyl transferasepossible identity with the choroideremia gene product Cell 70 1992. 1049 1057b [DOI] [PubMed] [Google Scholar]
- Seabra M.C., Brown M.S., Goldstein J.L. Retinal degeneration in choroideremiadeficiency of Rab geranylgeranyl tranferase. Science. 1993;259:377–381. doi: 10.1126/science.8380507. [DOI] [PubMed] [Google Scholar]
- Seabra M.C., Ho Y.K., Anant J.S. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J. Biol. Chem. 1995;270:24420–24427. doi: 10.1074/jbc.270.41.24420. [DOI] [PubMed] [Google Scholar]
- Segev N., Mulholland J., Botstein D. The yeast GTP-binding YPT1 protein and a mammalian counterpart are associated with the secretion machinery. Cell. 1988;52:915–924. doi: 10.1016/0092-8674(88)90433-3. [DOI] [PubMed] [Google Scholar]
- Sherman, F., G.R. Fink, and L.W. Lawrence. 1979. Methods in yeast genetics. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Shisheva A., Chinni S.R., DeMarco C. General role of GDP dissociation inhibitor 2 in membrane release of Rab proteinsmodulations of its functional interactions by in vitro and in vivo structural modifications. Biochemistry. 1999;38:11711–11721. doi: 10.1021/bi990200r. [DOI] [PubMed] [Google Scholar]
- Soldati T., Shapiro A.D., Svejstrup A.B.D., Pfeffer S.R. Membrane targeting of the small GTPase rab9 is accompanied by nucleotide exchange. Nature. 1994;369:76–78. doi: 10.1038/369076a0. [DOI] [PubMed] [Google Scholar]
- Ullrich O., Horiuchi H., Bucci C., Zerial M. Membrane association of Rab5 mediated by GDP-dissociation inhibitor and accompanied by GDP/GTP exchange. Nature. 1994;368:157–160. doi: 10.1038/368157a0. [DOI] [PubMed] [Google Scholar]
- Vida T.A., Emr S.D. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J. Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner P., Molenaar C.M., Rauh A.J., Brokel R., Schmitt H.D., Gallwitz D. Biochemical properties of the ras-related YPT protein in yeasta mutational analysis. EMBO (Eur. Mol. Biol. Organ.) J. 1987;6:2373–2379. doi: 10.1002/j.1460-2075.1987.tb02514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldherr M., Ragnini A., Schweyen R.J., Boguski M.S. MRS6-yeast homologue of the choroideraemia gene. Nat. Genet. 1993;3:193–194. doi: 10.1038/ng0393-193. [DOI] [PubMed] [Google Scholar]
- Wessel D., Flugge U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Wu S.K., Zeng K., Wilson I.A., Balch W.E. Structural insights into the function of the Rab GDI superfamily. Trends Biochem. Sci. 1996;21:472–476. doi: 10.1016/s0968-0004(96)10062-1. [DOI] [PubMed] [Google Scholar]
- Wu S.K., Luan P., Matteson J., Zeng K., Nishimura N., Balch W.E. Molecular role for the Rab binding platform of guanine nucleotide dissociation inhibitor in endoplasmic reticulum to Golgi transport. J. Biol. Chem. 1998;273:26931–26938. doi: 10.1074/jbc.273.41.26931. [DOI] [PubMed] [Google Scholar]