

Abstract
Phosphoinositide 3 kinase/Akt pathway plays an essential role in neuronal survival. However, the cellular mechanisms by which Akt suppresses cell death and protects neurons from apoptosis remain unclear. We previously showed that transient expression of constitutively active Akt inhibits ceramide-induced death of hybrid motor neuron 1 cells. Here we show that stable expression of either constitutively active Akt or Bcl-2 inhibits apoptosis, but only Bcl-2 prevents the release of cytochrome c from mitochondria, suggesting that Akt regulates apoptosis at a postmitochondrial level. Consistent with this, overexpressing active Akt rescues cells from apoptosis without altering expression levels of endogenous Bcl-2, Bcl-x, or Bax. Akt inhibits apoptosis induced by microinjection of cytochrome c and lysates from cells expressing active Akt inhibit cytochrome c induced caspase activation in a cell-free assay while lysates from Bcl-2–expressing cells have no effect. Addition of cytochrome c and dATP to lysates from cells expressing active Akt do not activate caspase-9 or -3 and immunoprecipitated Akt added to control lysates blocks cytochrome c–induced activation of the caspase cascade. Taken together, these data suggest that Akt inhibits activation of caspase-9 and -3 by posttranslational modification of a cytosolic factor downstream of cytochrome c and before activation of caspase-9.
Keywords: protein serine-threonine kinase, cytochrome c, apoptosis, neuron, mitochondria
Introduction
Apoptosis plays an important role in the development and homeostasis of multicellular organisms (Vaux and Korsmeyer 1999). In the nervous system, apoptosis is required for normal development (Oppenheim 1991; Jacobson et al. 1997) and has been implicated in the pathogenesis of various neurodegenerative conditions, including acute insults to the central nervous system such as head trauma and spinal cord injury, and chronic degeneration such as Alzheimer's disease and Huntington's disease (Thompson 1995).
Genetic studies in the nematode Caenorhabditis elegans have provided an understanding of apoptosis at the molecular level that show that three genes, ced-3, ced-4, and ced-9, play critical roles in regulating the cell death program. ced-3 and ced-4 were identified as proapoptotic genes, whereas ced-9 was identified as an antiapoptotic gene (Hengartner and Horvitz 1994). Mammalian homologues of CED-3 have been identified as the caspase family. Caspases are activated in response to apoptotic stimuli and subsequently cleave cellular proteins to cause cell death (Salvesen and Dixit 1997; Thornberry and Lazebnik 1998). The mammalian homologue of CED-4 is Apaf-1 (apoptotic protease activating factor 1), which is important for initiating a cytochrome c–dependent caspase activation cascade (Zou et al. 1997; Slee et al. 1999). Mammalian homologues of CED-9 are members of the Bcl-2 family, which includes both positive and negative regulators of cell survival (Adams and Cory 1998; Gross et al. 1999).
Recent biochemical studies have revealed that caspase activation during apoptosis is a tightly regulated process (Salvesen and Dixit 1997; Thornberry and Lazebnik 1998; Budihardjo et al. 1999). Apoptotic stimuli such as activation of cell surface receptors or environmental stress can induce cytochrome c release from mitochondria (Green and Reed 1998). Once in the cytosol, cytochrome c binds to Apaf-1 and induces its oligomerization. Oligomerization of Apaf-1 recruits procaspase-9 and results in subsequent caspase-9 activation (Srinivasula et al. 1998; Zou et al. 1999). Active caspase-9 cleaves procaspase-3 and generates active caspase-3. Active caspase-3 cleaves a number of important cellular proteins to execute cell death and activate additional downstream caspases (Slee et al. 1999). Phenotypes of caspase-3–, caspase-9–, or Apaf-1–deficient mice are very similar to each other in that all these mice manifest brain overgrowth due to reduced apoptosis during brain development (Cecconi et al. 1998; Hakem et al. 1998; Kuida et al. 1998; Yoshida et al. 1998). On the other hand, caspase-1–, caspase-2–, caspase-8–, caspase-11–, Bid-, or FADD-deficient mice do not show obvious defects in brain development (Los et al. 1999; Yin et al. 1999). These genetic studies confirm the critical roles for Apaf-1, caspase-9, and caspase-3 in regulating neuronal apoptosis.
Neurons are dependent on neurotrophic factors for survival, and removal of such factors results in apoptosis. Among the growth factor signaling molecules, phosphoinositide 3 (PI-3) kinase and mitogen-activated protein (MAP) kinase have been shown to be important for neuronal survival (Pettmann and Henderson 1998). Recent studies indicate that the protective effects of PI 3-kinase are mediated primarily by one of its downstream targets—Akt (Franke et al. 1997). Upon activation by PI 3-kinase, Akt phosphorylates Bad at Ser136. This decreases the binding of Bad to Bcl-xL at the mitochondrial membrane and increases its binding to 14-3-3 in the cytosol (Zha et al. 1996; Datta et al. 1997). It has been speculated that Akt inhibits apoptosis by maintaining Bcl-x function and preventing cytochrome c release from mitochondria. However, a direct effect of Akt in regulating cytochrome c translocation during apoptosis has not been shown. Moreover, it remains to be explored whether Akt may also inhibit apoptosis independent of cytochrome c release.
In this report, we examine the cellular mechanism by which Akt inhibits apoptosis in hybrid motor neuron 1 (HMN1) cells, a neuronal cell line that requires PI 3-kinase but not MAP kinase for survival. By generating stable HMN1 lines overexpressing constitutively active Akt, subcellular fractionation, cell-free assays of apoptosis, and microinjection, we investigated the effects of Akt on several critical apoptotic events, with particular focus on its effect on cytochrome c redistribution. Our data indicate that Akt inhibits apoptosis downstream of cytochrome c release based on the following observations: (a) Akt inhibits cell death but does not block release of cytochrome c, (b) Akt inhibits cytochrome c–induced caspase activation in a cell free assay, and (c) HMN1 cells expressing active Akt are resistant to apoptosis induced by microinjection of cytochrome c. In sum, our study indicates that Akt plays an important role in suppressing neural apoptosis at a postmitochondrial stage, downstream of cytochrome c release and before activating caspase-9.
Materials and Methods
Materials
C2-ceramide, C2-dihydroceramide, PD 98059, and LY 294002 were obtained from Calbiochem. LipofectAMINE was obtained from Life Technologies. Hygromycin B was from Boehringer. Fetal bovine serum was from HyClone. Polyvinylidene difluoride transfer membrane, ECL substrate, and [gamma-32P] ATP were from NEN Life Science Products. pCR3.1 vector was from Invitrogen. Caspase-9 colorimetric assay kit was from R&D Systems. Hoechst 33342 and antibody to cytochrome c oxidase (COX) subunit IV (clone 20E8-C12) were from Molecular Probes, Inc. Antibody to cytochrome c (clone 7H8.2C12), antibody to mouse BCL-2 (clone 3F11), and antibody to human BCL-2 (clone 6C8) were from PharMingen. Antibodies to caspase-9 were from Santa Cruz Biotechnology, Inc. and R&D Systems. Rabbit polyclonal antibody to Akt and phosphospecific antibody for Akt Serine-473 were from New England Biolabs, Inc., and a rabbit polyclonal phosphospecific antibody for MAP kinase threonine-202/tyrosine-204 was from Cell Signaling Technology. Fluorescein isothiocyanate–conjugated secondary antibody was from Jackson ImmunoResearch Laboratories. Antibody to hemagglutinin (12CA5) was a generous gift from Dr. Jeffery Field (University of Pennsylvania, Philadelphia, PA). Antibody to activated caspase-3 was a generous gift from Dr. Robert Siman (University of Pennsylvania, Philadelphia, PA). All other reagents were obtained from Sigma-Aldrich.
Cell Viability, Internucleosomal DNA Fragmentation, and Nuclear Condensation
HMN1 cells were washed with serum-free DMEM. LY 294002, PD 98059, or vehicle was diluted into serum-free DMEM at the indicated concentrations. Cell viability and DNA fragmentation were analyzed as described previously (Zhou et al. 1998). For C2-ceramide–induced apoptosis, HMN1 cells were maintained in serum-free DMEM for 24 h before experiments. Staining nuclei with Hoechst 33342 was performed as described previously (Zhou et al. 1998).
Cell Transfection and Generation of Stable Cell Lines
HMN1 cells were plated in six-well plates and cotransfected with 0.5 μg of pREP-4 encoding a hygromycin resistance gene and 4.5 μg of pCMV, pCMVhBcl-2, pCR3.1, or pCR3.1myr-AktΔ4-129 (a constitutively active Akt containing a consensus myristoylation site and lacking the PH domain; Kohn et al. 1996; Zhou et al. 1998). 36 h after transfection, cells were replated into 10-cm plates and selected in medium containing 350 μg/ml hygromycin for 4 wk to select stable clones. Clones expressing human Bcl-2 were screened with monoclonal antibody, 6C8, for human Bcl-2 (PharMingen). Clones expressing constitutively active Akt were screened with anti-hemagglutinin (anti–HA) monoclonal antibody (12CA5) and verified by anti–Akt antibody as well as anti–Akt (Ser473) phosphospecific antibody (New England Biolabs, Inc.).
SDS-PAGE and Western Blot Analysis
Proteins were separated by SDS-PAGE and transferred onto PVDF membranes, followed by probing with various antibodies. Bound antibodies were detected using appropriate peroxidase-coupled secondary antibodies, followed by detection using the Enhanced Chemiluminescence system (NEN Life Science Products).
Subcellular Fractionation
The cytosolic fraction for detecting cytochrome c was prepared as described by Erhardt et al. 1999. In brief, HMN1 cells were harvested by centrifugation at 600 g for 5 min at 4°C. The cell pellet was washed once with ice-cold phosphate-buffered saline and resuspended in 5 vol of buffer (20 mM Hepes, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM sodium EDTA, 1 mM sodium EGTA, 1 mM dithiothreitol, 10 μg/ml leupeptin, 2 μg/ml aprotinin, and 0.1 mM PMSF) containing 250 mM sucrose. Cells were homogenized with 20 strokes of a Dounce homogenizer, and the homogenates were centrifuged at 900 g for 5 min at 4°C to remove nuclei. Supernatants were centrifuged at 10,000 g for 15 min at 4°C. The resulting supernatants were used as the cytosolic fraction for detecting cytochrome c and the resulting pellets were used as a positive control for mitochondrial protein cytochrome c oxidase (COX) subunit IV.
Preparation of Cell-free Extracts and Cell-free Reactions
Cell-free extracts were generated from HMN1 cells as described by Slee et al. 1999 with some modifications. Cells were pelleted and washed twice with PBS, pH 7.2, followed by washing once with 5 ml of ice-cold cell extract buffer (CEB; 20 mM Hepes-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 μM okadaic acid, 1 mM dithiothreitol, 100 μM PMSF, 10 μg/ml leupeptin, 2 μg/ml aprotinin). The cell pellet was resuspended in 2 vol of ice-cold CEB and incubated on ice for 15 min. Cells in CEB were then transferred to a 2-ml Dounce homogenizer and disrupted with 20 strokes of a B-type pestle. Lysates were transferred to Eppendorf tubes and centrifuged at 15,000 g for 15 min at 4°C. The resulting supernatants were obtained as postnuclear extracts and frozen in aliquots at −80°C until required. Cell-free reactions were set up in a 120-μl reaction volume. Cell extracts were brought to a final volume of 120 μl in CEB with protein concentration of 2.5 mg/ml. Apoptosis was induced by addition of bovine heart cytochrome c and dATP to extracts at a final concentration of 50 μg/ml and 1 mM, respectively. The reactions were then incubated at 37°C. At the indicated time points, 20 μl of reactions were removed and boiled with 10 μl 3× sample buffer for 5 min at 95°C. Samples were frozen at −20°C for subsequent SDS-PAGE/Western blot analysis. Addition of cytochrome c and dATP to nonapoptotic HMN1 cell extracts induces rapid cleavage of poly (ADP-ribose) polymerase (PARP) by activated caspase-3. On the other hand, addition of BSA and dATP does not lead to PARP cleavage (data not shown). These observations suggest that, in this cell free system, caspase activation is strictly dependent on the presence of cytochrome c.
Colorimetric Caspase-9 Activity Assay
Cell extracts were incubated in the presence or absence of cytochrome c and dATP at 37°C for 15 min as described above. Then the lysates were incubated with caspase-9 colorimetric substrate (LEHD-pNA) to measure caspase-9 activity according to the protocol suggested by the manufacturer. The cleavage of the peptide was quantitated spectrophotometrically at a wavelength of 405 nm. The results are expressed as fold increase in caspase-9 activity of apoptotic reactions (cytochrome c and dATP added) compared with that of control reactions (BSA and dATP added).
Immunoprecipitation and Akt Kinase Reaction
HMN1 clones expressing constitutively active Akt were lysed in ice-cold lysis buffer (20 mM Tris-HCL, pH 7.4, 10 mM MgCl2, 10 mM NaCl, 1 mM EGTA, 5 mM Na3VO4, 1 mM dithiothreitol, 1 mM PMSF, 1 μg/ml leupeptin, 10 μg/ml aprotinin) as described previously (Zhou et al. 1998). The cell lysates were then incubated with either monoclonal anti–HA antibody (12CA5) or control mouse immunoglobulin for 2 h at 4°C. Protein A sepharose was then added and incubated for another hour. The beads were then washed three times with cold lysis buffer, and then incubated with 120 μl vector lysates (CEB, 20 μM ATP) at 30°C. After 30 min, bovine heart cytochrome c and dATP were added and the reactions were incubated at 37°C.
Microinjection
Cell microinjection was performed as previously described (Cass et al. 1999). HMN1 or HMN1/myr-Akt cells were plated on 35-mm plates 24 h before injection. Rabbit IgG alone or rabbit IgG plus 5 mg/ml cytochrome c (Sigma-Aldrich; bovine heart cytochrome c, diluted in H2O and freshly prepared for each experiment) were injected into the cytoplasm. Injected cells were stained with FITC-conjugated anti–rabbit IgG, and then incubated with Hoechst 33258 (4 μg/ml) for nuclear staining. The intracellular concentrations of microinjected proteins are estimated to represent a 10–100-fold dilution of the pipette concentration based on an estimated HMN1 cell volume of 4–5 pl. We estimate that 0.2–2 pg cytochrome c per cell is delivered after injection of cytochrome c at 5 mg/ml. At various times after injection, injected cells were scored for apoptosis based on condensed chromatin or fragmented nuclei. The percentage of apoptotic cells was calculated as the number of apoptotic nuclei versus total number of injected cells. Data represent the mean ± SEM from three or more independent experiments.
Results
PI 3-kinase Is Required for HMN1 Cell Survival
Depending on the type of neuron, either PI 3-kinase or MAP kinase can promote cell survival (Pettmann and Henderson 1998). To determine which pathway (or both pathways) is required for HMN1 cell survival, HMN1 cells were treated with the PI 3-kinase inhibitor, LY 294002, or with the MEK1 inhibitor PD 98059. In HMN1 cells, PD 98059 blocked serum-stimulated ERK phosphorylation at threonine-202/tyrosine-204, indicating effective inhibition of MEK1, and LY 294002 blocked serum- and insulin-stimulated Akt phosphorylation at serine-473, indicating effective inhibition of PI 3-kinase (Fig. 1 A). After 18 h, LY 294002 induced ∼50% cell death, whereas PD 98059 had no obvious effect on cell survival (Fig. 1 B). Consistent with LY 294002 but not PD 98059–inducing apoptosis in HMN1 cells, DNA laddering was observed only in LY 294002-treated cells (Fig. 1 C). Other features characteristic of apoptosis including cell shrinkage, membrane blebbing, and chromatin condensation were also present in cells treated with LY 294002 but not in cells treated with PD 98059 (data not shown), indicating that PI 3-kinase but not MAP kinase plays an essential role in HMN1 cell survival.
Figure 1.
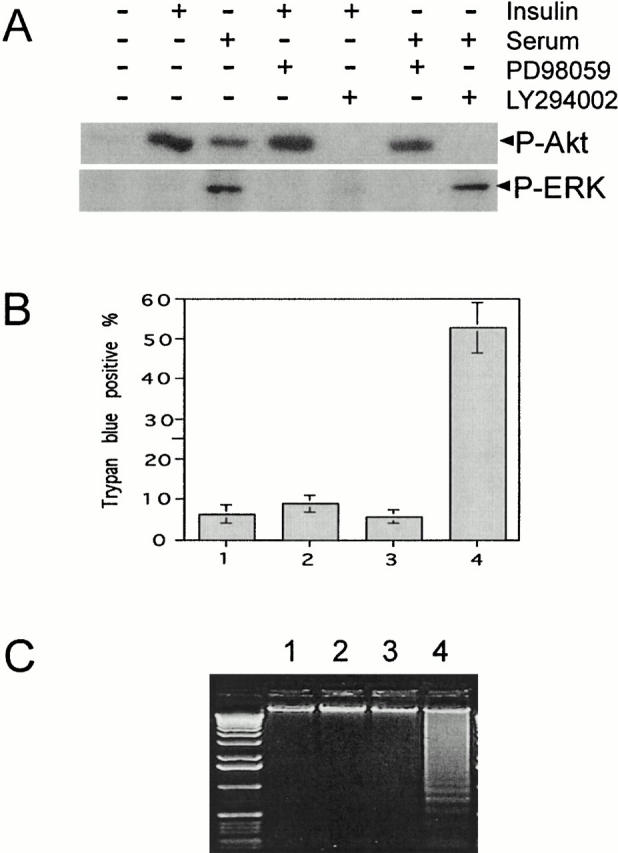
Inhibition of PI 3-kinase signaling but not MAPK signaling induces apoptosis of HMN1 cells. (A) PD 98059 inhibits MAPK signaling (ERK phosphorylation at threonine-202/tyrosine-204) and LY 294002 inhibits PI 3-kinase signaling (Akt phosphorylation at serine-473). HMN1 cells were grown in serum-free medium for 16 h, followed by stimulation with 10 μg/ml insulin or 20% fetal bovine serum in the absence or presence of 30 μM PD 98059 or LY 294002. Samples were blotted with antibodies against phospho-ERK or phospho-Akt. (B) LY 204002, but not PD98059, induces cell death. HMN1 cells were treated with 30 μM LY 294002 (dissolved in ethanol) or 30 μM PD 98059 (dissolved in DMSO), and cell viability was determined after 18 h. 1, DMSO; 2, PD 98059; 3, ethanol; 4, LY 294002. LY 294002 causes a significant decrease in viability. Data represent mean ± SEM of three independent experiments. (C) HMN1 cells were treated as described above, and after 18 h soluble DNA was extracted for DNA fragmentation assay. 1, DMSO; 2, PD 98059; 3, ethanol; 4, LY 294002. Note prominent DNA laddering after treatment with LY 294002. Data are from a representative experiment performed three times.
Caspase-3 Activation, PARP Cleavage and Cytochrome c Release from Mitochondria during C2-Ceramide Induced HMN1 Cell Death
A variety of apoptotic stimuli have been shown to generate ceramide that can serve as a lipid messenger to induce apoptosis (Hannun 1996). We previously reported that cell-permeable C2-ceramide induces HMN1 cell death and inhibits Akt activation that may represent one mechanism by which ceramide promotes apoptosis (Zhou et al. 1998). However, the cellular events involved in ceramide-induced apoptosis remain undetermined. Since caspase-3 activation plays a central role in neuronal apoptosis, we examined caspase-3 activation and cleavage of its substrate, PARP, during ceramide-induced apoptosis. Cells treated with C2-ceramide, but not with an inactive ceramide analogue C2-dihydroceramide exhibited activation of caspase-3 after 3 h based on processing of procaspase-3 into active caspase-3 and the appearance of the p17 subunit. Caspase-3 activation was inhibited by the broad caspase inhibitor zVAD-fmk (benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) (Fig. 2 A). Consistent with the observed caspase-3 activation, PARP, a known substrate for caspase-3, was cleaved with a similar time course, and zVAD-fmk effectively blocked PARP cleavage (Fig. 2 B). Another important cellular event in the progression of apoptosis is the release of cytochrome c from mitochondria; therefore, we examined the distribution of cytochrome c in HMN 1 cells treated with C2-ceramide. Cell fractionation studies revealed that C2-ceramide treatment leads to cytochrome c redistribution into the cytosolic fraction, which can be detected as early as 1 h (Fig. 2 C, top). This observation is consistent with the notion that cytochrome c release occurs before caspase-3 activation. Interestingly, zVAD-fmk did not block cytochrome c release from the mitochondria (Fig. 2 C), even though it inhibited caspase-3 activation and PARP cleavage (Fig. 2A and Fig. B). These results suggest that cytochrome c release from mitochondria during ceramide-induced apoptosis in HMN1 cells is independent of caspase activation.
Figure 2.
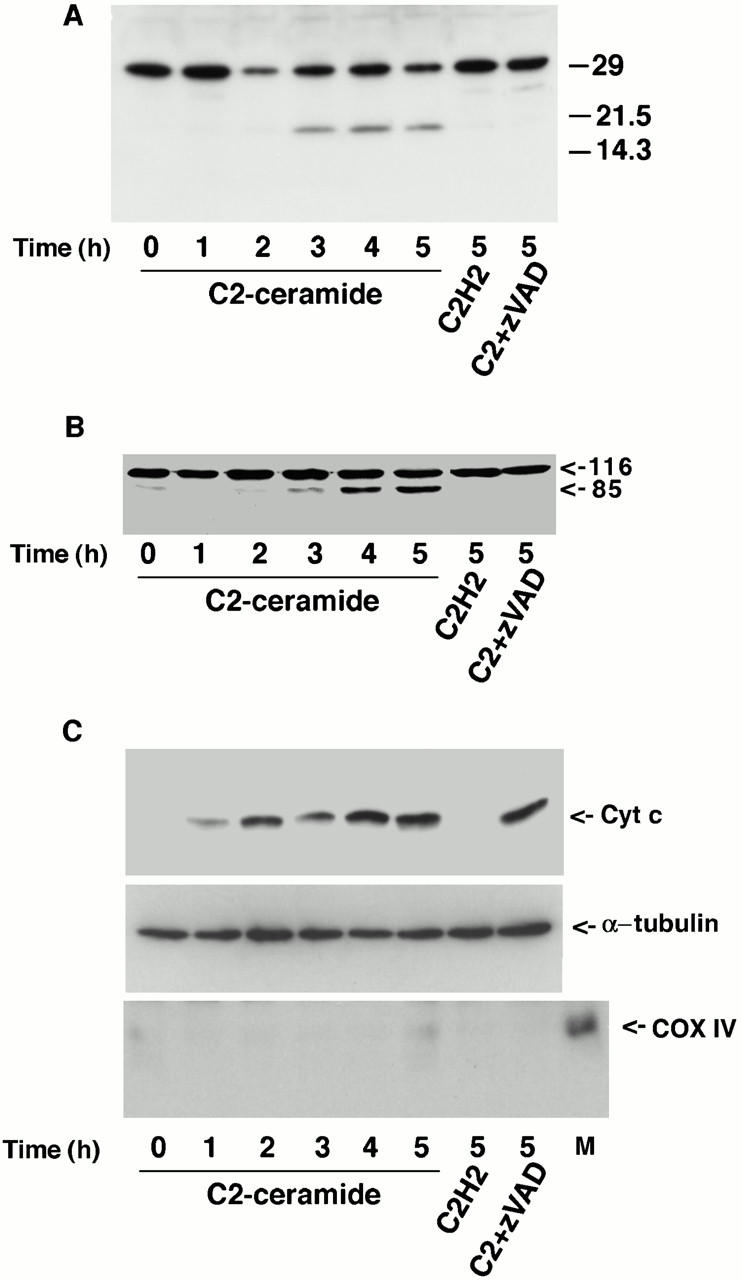
Caspase-3 activation, PARP cleavage and cytochrome c release from mitochondria during C2-ceramide–induced HMN1 cell death. (A) C2-ceramide induces caspase-3 activation. HMN1 cells were treated with 50 μM C2-ceramide, 50 μM C2-dihydroceramide, or 50 μM C2-ceramide plus 100 μM zVAD-fmk for the indicated period. The blot was probed with a polyclonal antibody to caspase-3. Note that zVAD-fmk inhibits caspase-3 processing. (B) C2-ceramide induces PARP cleavage. HMN1 cells were treated as described above for the indicated period. The blot was probed with an mAb to PARP. Note that zVAD-fmk blocks PARP cleavage. (C) C2-ceramide induces cytochrome c release into the cytosol. HMN1 cells were treated as described above for the indicated period. At each time point, cells were fractionated and equal amounts of cytosolic protein were loaded into each lane. The blot was probed with an mAb to cytochrome c (top), or an mAb to α-tubulin (middle). an mAb to cytochrome c oxidase subunit IV (COX IV) was used to show the cytosolic fraction is not contaminated with mitochondria (bottom; M represents mitochondria as a positive control for COX IV). Note that zVAD-fmk does not block cytochrome c release.
Establishing Stable HMN1 Lines Overexpressing Constitutively Active Akt or Human Bcl-2
One mechanism by which active Akt protects BAF/3 cells from interleukin-3 withdrawal–induced apoptosis is to increase the expression level of Bcl-2 (Ahmed et al. 1997). We previously showed that transiently overexpressing constitutively active Akt in HMN1 cells inhibits C2-ceramide–induced apoptosis; however, the expression levels of Bcl-2, Bcl-x, or Bax were not changed (Zhou et al. 1998; data not shown). The lack of an effect in HMN1 cells may have been due to the low transfection efficiency in our transiently transfected cells or possibly that active Akt may protect neurons from apoptosis via a mechanism different from that in BAF/3 cells. To elucidate the cellular mechanisms by which Akt inhibits apoptosis, stable HMN1 lines overexpressing constitutively active Akt were generated. The expression of constitutively active myr-AktΔ4-129 in these clones was confirmed using anti–Akt antibody (Fig. 3 A) and antiphosphospecific Akt (Ser473) antibody (B). Note that myr-AktΔ4-129 has a much higher degree of Ser473 phosphorylation than endogenous Akt despite its relative lower expression level. HMN1 lines overexpressing human Bcl-2 were also generated to help dissect the role of Akt in regulating apoptosis (Fig. 3 C).
Figure 3.
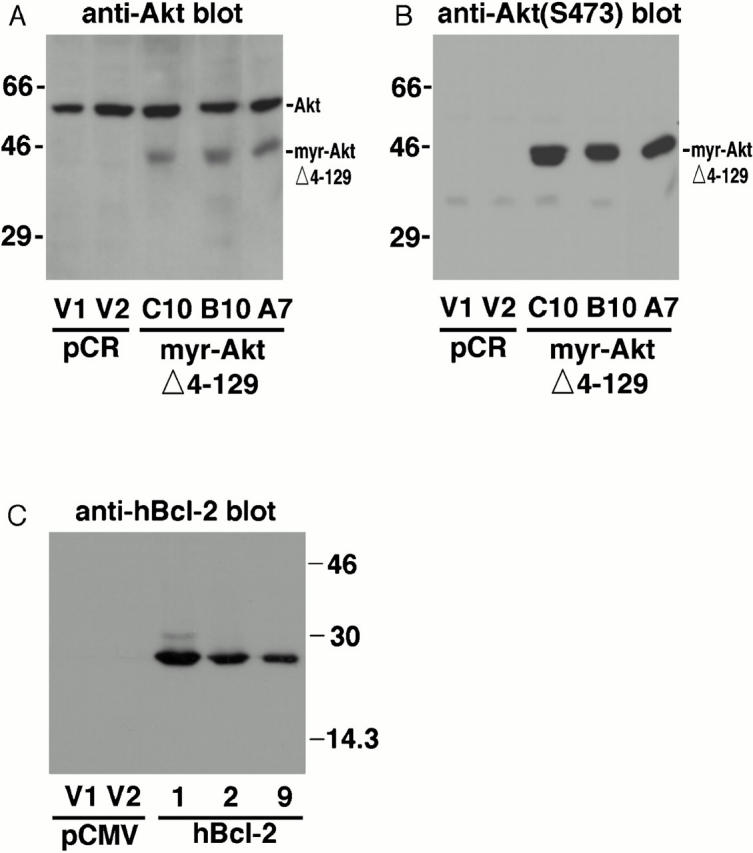
Establishing stable HMN1 lines overexpressing constitutively active Akt or human Bcl-2. (A) Detection of endogenous and transfected Akt in stable HMN1 clones expressing active Akt. HMN1 cells were transfected with either control vector (pCR3.1) or myr-AktΔ4-129 and selected with hygromycin B (350 μg/ml) for 4 wk. V1 and V2 represent control clones transfected with vector. A7, B10, and C10 represent stable HMN1 cell lines expressing myr-AktΔ4-129. Endogenous Akt runs at ∼60 kD and myr-AktΔ4-129 runs at ∼46 kD. The clones were analyzed using a polyclonal antibody against Akt (A) and an mAb against the HA epitope tag (not shown). (B) Detection of activated/phosphorylated Akt in stable HMN1 clones expressing active Akt. Blots were probed with a phosphospecific antibody against Akt phosphorylated on Ser-473. (C) Detection of stable HMN1 clones expressing human Bcl-2. HMN1 cells were transfected with either control vector (pCMV) or human Bcl-2 and selected with hygromycin B (350 μg/ml) for 4 wk. V1 and V2 represent control clones transfected with vector. 1, 2, and 9 represent the stable HMN1 cells expressing human Bcl-2. The clones were analyzed using an mAb specific for human Bcl-2.
Inhibition of Apoptosis by Constitutively Active Akt and Bcl-2
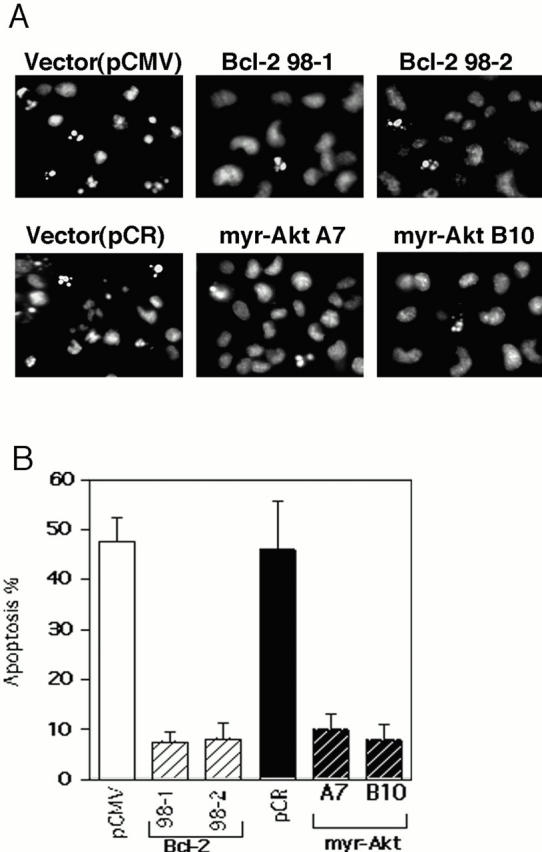
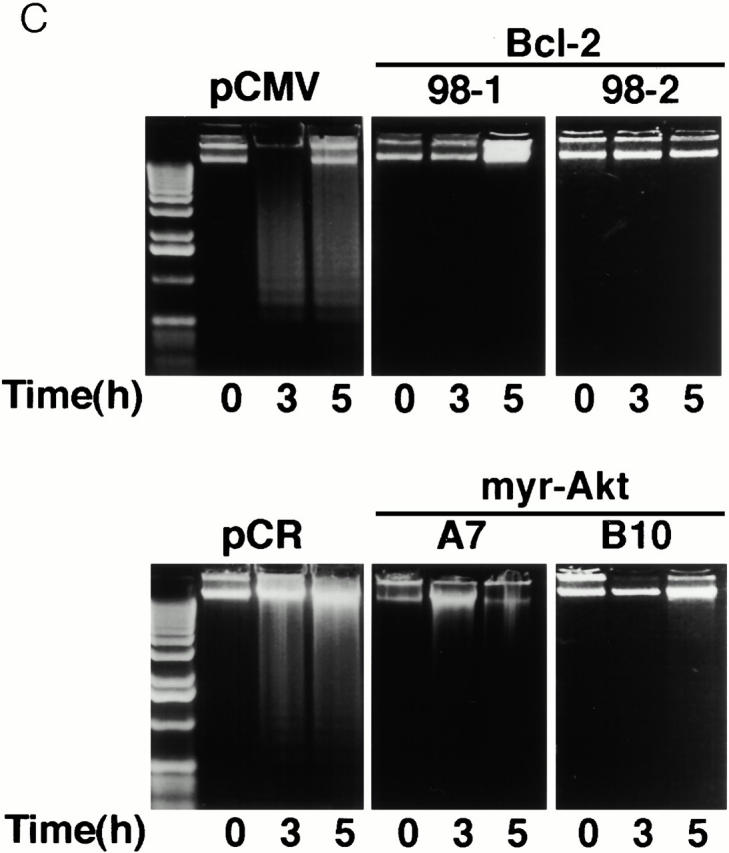
Both constitutively active Akt and Bcl-2 significantly protected HMN1 cells from apoptosis induced by C2-ceramide as determined by morphological changes and the extent of nuclear condensation (Fig. 4 A). When treated with 30 μM C2-ceramide for 5 h, most cells overexpressing constitutively active Akt kinase or Bcl-2 remained flat and exhibited normal nuclear morphology, whereas vector-transfected cells showed extensive cell shrinkage, membrane blebbing, and nuclear condensation (Fig. 4 A, and data not shown). Quantitation of apoptosis by counting Hoechst-stained condensed nuclei revealed that active Akt and Bcl-2 were approximately equally effective at inhibiting apoptosis (Fig. 4 B). This is consistent with data showing that C2-ceramide induced DNA fragmentation in the two vector control lines, whereas DNA fragmentation was inhibited in myr-AktΔ4-129 and Bcl-2–expressing stable cell lines (Fig. 4 C).
Figure 4.
Inhibition of apoptosis by constitutively active Akt and Bcl-2. (A) Fluorescence photomicrographs of Hoechst 33342 staining of HMN1 cells transfected with pCMV, human Bcl-2, pCR3.1, or myr-AktΔ4-129. Cells were treated with 30 μM C2-ceramide for 5 h. (B) Constitutively active Akt and Bcl-2 inhibit ceramide-induced HMN1 cell apoptosis. Cells were treated with 30 μM C2-ceramide for 5 h and apoptosis was assayed by Hoechst 33342 staining. Data represent mean ± SEM for three independent experiments. (C) Constitutively active Akt and Bcl-2 inhibit DNA fragmentation in HMN1 cells. Cells were treated with 30 μM C2-ceramide for 0, 3, or 5 h, and soluble DNA was extracted for DNA fragmentation assay. Data are from a representative experiment performed three times.
Overexpressing Constitutively Active Akt Does Not Alter Bcl-2, Bcl-x, or Bax Expression Levels
Bcl-2 family proteins play an important role in regulating apoptosis and a growing number of signaling pathways are known to regulate apoptosis by modulating expression of Bcl-2 family members (Gross et al. 1999). We therefore explored the possibility that Akt may inhibit apoptosis in HMN1 cells by regulating the expression levels of three key members of the Bcl-2 family: Bcl-2, Bcl-x, and Bax. Western blots of vector-transfected cells and active myr-AktΔ4-129–expressing cells indicated that stable expression of activated Akt did not alter Bcl-2, Bcl-x, or Bax expression to inhibit apoptosis (Fig. 5).
Figure 5.
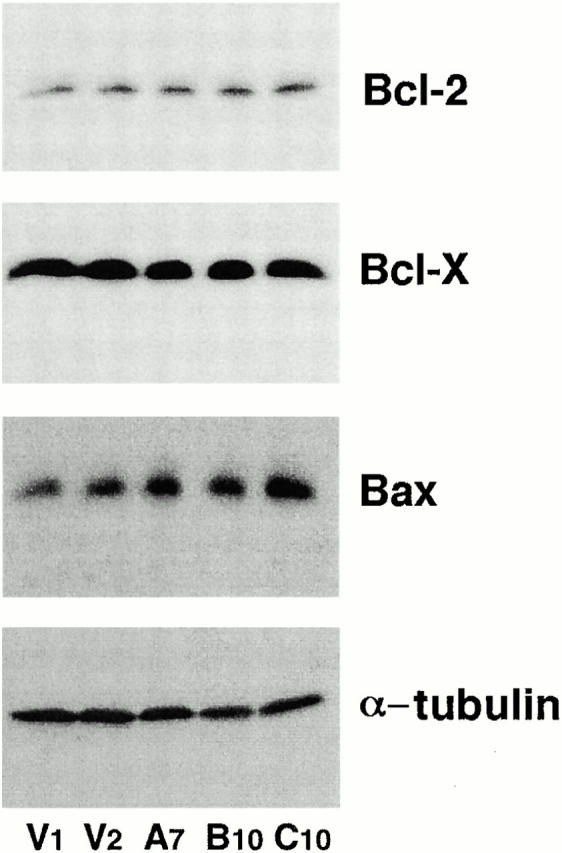
Overexpressing constitutive Akt does not change Bcl-2, Bcl-x, or Bax expression in HMN1 cells. Equal amounts of cell lysates from two control vector lines (V1 and V2) or three active Akt lines (A7, B10, and C10) were separated by SDS-PAGE, followed by Western blot with an anti–mouse Bcl-2 antibody, an anti–Bcl-x antibody, or an anti–Bax antibody. For the internal control, blots were stripped and reprobed with an mAb specific for α-tubulin. Data are from a representative experiment performed three times.
Bcl-2, but Not Active Akt, Blocks Cytochrome c Release
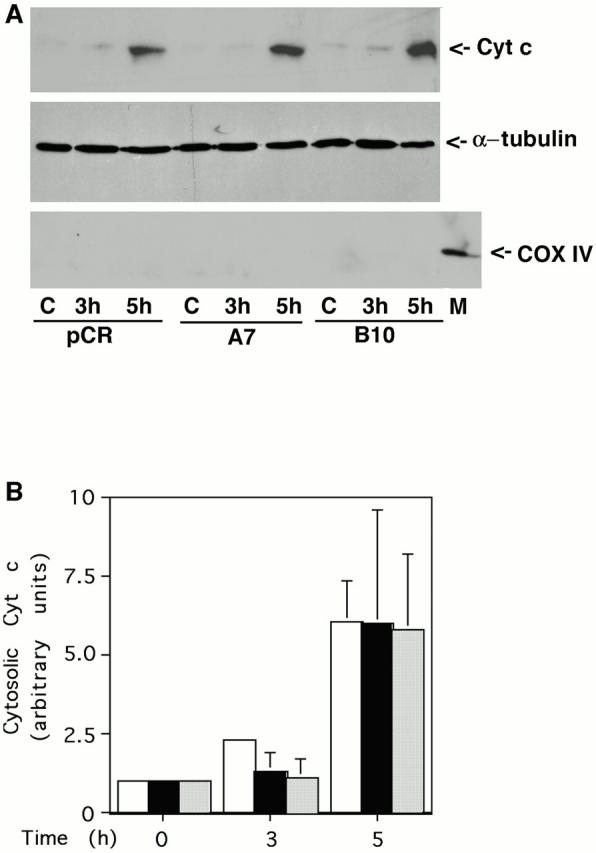
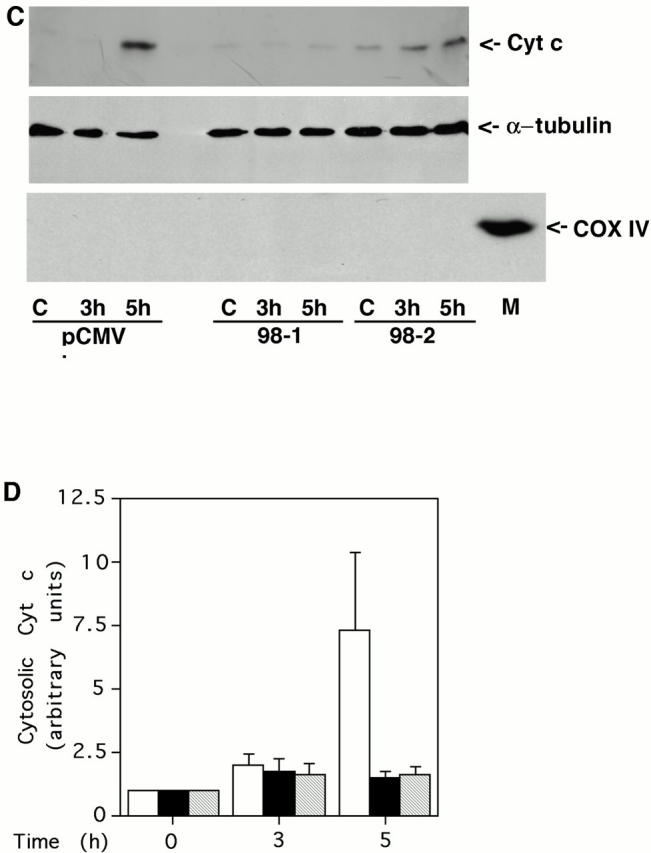
Bad, a proapoptotic member of the Bcl-2 family, has been identified as one of the downstream targets of Akt. Activated Akt increases Bad phosphorylation at Ser136, which shifts binding of Bad from Bcl-XL to 14-3-3 protein (Zha et al. 1996; Datta et al. 1997). Based on this observation, it has been proposed that Akt inhibits apoptosis mainly through maintaining the prosurvival function of Bcl-XL, which regulates cytochrome c release from mitochondria. We tested this model by examining whether active Akt can inhibit cytochrome c redistribution in C2-ceramide induced apoptosis. Surprisingly, Akt did not block cytochrome c release into the cytosol (Fig. 6A and Fig. B), whereas Bcl-2 suppressed cytochrome c release (C and D). This observation is in agreement with the well established role for Bcl-2 to inhibit apoptosis by blocking cytochrome c release from mitochondria (Kluck et al. 1997; Yang et al. 1997). Moreover, it suggests that active Akt inhibits apoptosis at a step after release of cytochrome c into the cytoplasm.
Figure 6.
Bcl-2 but not active Akt blocks cytochrome c release. (A) Active Akt fails to inhibit cytochrome c release induced by C2-ceramide. Vector control line or two independent active Akt lines were treated with 30 μM C2-ceramide for the indicated period. Cells were then fractionated and equal amounts of cytosolic protein were loaded into each lane. The blot was probed with an mAb to cytochrome c (top). For the internal control, the blots were stripped and reprobed with antibody to α-tubulin (middle). The cytosolic fraction was confirmed to be without mitochondria with an mAb to COX IV (bottom; M represents mitochondria as a positive control for COX IV). Data are from a representative experiment performed three times. Note the delay in cytochrome c release is due to cells being treated with 30 μM C2-ceramide in this experiment rather than 50 μM C2-ceramide as used in experiments shown in Fig. 2 C. (B) Quantification of cytochrome c release. Bars: white, pCR vector; black, myr-Akt clone A7; stippled, myr-Akt clone B10. Data represent mean ± SEM of three independent experiments. (C) Bcl-2 inhibits cytochrome c release induced by C2-ceramide. Vector control line or two independent Bcl-2 lines were treated with 30 μM C2-ceramide for the indicated period. Cells were then fractionated and equal amounts of cytosolic protein were loaded into each lane. The blot was probed with an mAb to cytochrome c (top). For the internal control, the blots were stripped and reprobed with antibody to α-tubulin (middle). The cytosolic fraction was confirmed to be without mitochondria with an mAb to COX IV (bottom; M represents mitochondria as a positive control for COX IV). Data are from a representative experiment performed three times. (D) Quantification of cytochrome c release. Bars: white, pCMV vector; black, hBcl-2 clone 98-1; stripped, hBcl-2 clone 98-2. Data represent mean ± SEM of three independent experiments.
Akt, but Not Bcl-2, Inhibits Caspase Activation Induced by Cytochrome c in a Cell-free System
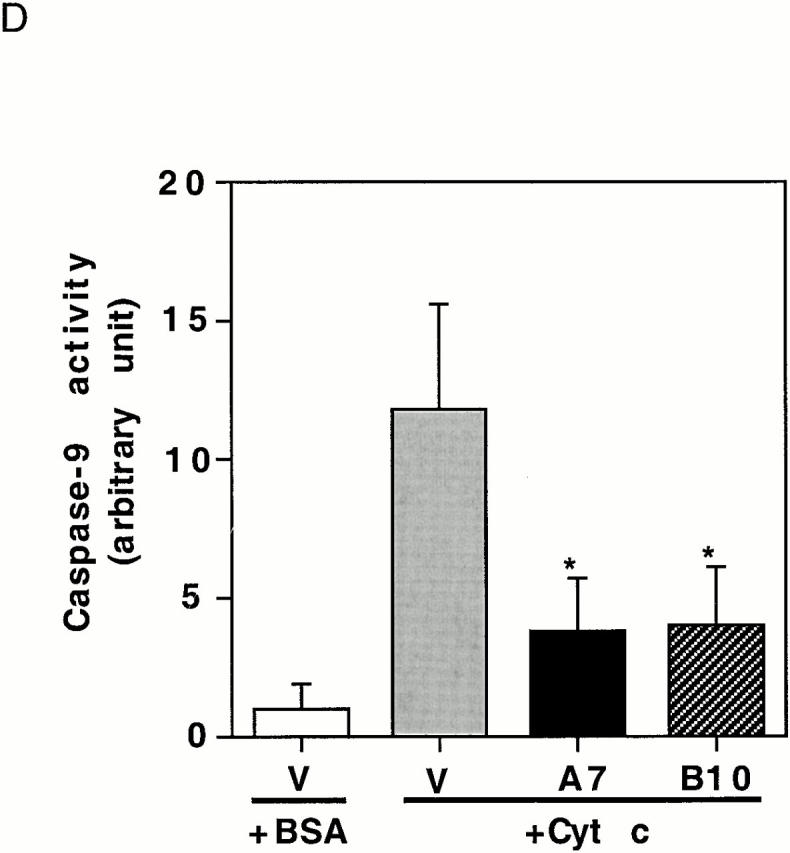
To confirm that expressing active Akt inhibits the apoptotic cascade downstream of cytochrome c release, a cell-free system was used to examine PARP cleavage and caspase-3 activation. In extracts prepared from vector-transfected cells, PARP was rapidly cleaved. After a 15-min incubation with cytochrome c and dATP, only a small fraction of intact PARP remained and, by 30 min, all intact PARP disappeared (Fig. 7 A). In contrast, in extracts prepared from Akt stable cell lines, cytochrome c–induced PARP cleavage was markedly inhibited and delayed. The cleaved PARP fragment was detectable only after 60–90 min and a large percentage of intact PARP was still present after 120 min (Fig. 7 A). These data confirm that active Akt inhibits caspase activation downstream of cytochrome c. On the other hand, PARP cleavage induced by cytochrome c in extracts prepared from Bcl-2–stable lines proceeded similarly to cleavage in vector-control cell extracts (Fig. 7 B). Taken together, these cell-free studies suggest that Akt can regulate apoptosis at a postmitochondrial level, whereas Bcl-2 regulates apoptosis primarily at the mitochondrial level. Using an antibody that recognizes cleaved/activated caspase-3, we further analyzed caspase-3 processing induced by cytochrome c in the cell-free assay. Consistent with results for PARP cleavage, caspase-3 cleavage/activation was also inhibited in extracts prepared from Akt-stable lines compared with caspase-3 cleavage/activation in vector-control cell extracts (Fig. 7 C). Since caspase-9 is the initiator caspase for cleaving and activating caspase-3, we examined whether caspase-9 activation by cytochrome c and dATP was inhibited in extracts prepared from Akt-stable lines (Fig. 7 D). Caspase-9 activity assays showed that activation of caspase-9 by cytochrome c and dATP was inhibited in extracts prepared from Akt-stable lines compared with those in vector-control cell extracts, indicating that expressing active Akt effectively inhibits the cytochrome c–initiated caspase activation cascade.
Figure 7.
Active Akt but not Bcl-2 inhibits caspase activation induced by cytochrome c in a cell-free system. (A) Extracts from active Akt stable cell lines but not a vector control line inhibit cytochrome c–induced PARP cleavage. Cell-free assays were prepared as described in Materials and Methods and samples were taken at the indicated time. The blot was probed with an mAb against PARP. Note that the addition of cytochrome c induced rapid cleavage of intact PARP (116 kD) into the 85-kD fragment, whereas addition of BSA did not. PARP cleavage was markedly inhibited in lysates from two active Akt clones (A7, middle, and B10, bottom) compared with vector control (top). (B) Extracts from Bcl-2 stable cell lines do not inhibit cytochrome c–induced PARP cleavage. Cell-free assays were performed as described above for the indicated period. The blot was probed with an mAb against PARP. Note that addition of cytochrome c induced a similar degree of PARP cleavage in both vector control cells (top) and two Bcl-2 stable lines (98-1, middle, and 98-2, bottom). (C) Extracts from active Akt stable cell lines but not a vector control line inhibit cytochrome c–induced caspase-3 cleavage. Cell-free assays were performed as described above. The blots were probed with a polyclonal antibody selectively recognizing cleaved/activated caspase-3. Note that caspase-3 cleavage/activation was markedly inhibited in two active Akt clones (middle and bottom) compared with vector control (top). Cleaved products represent the 20- and 17-kD processed caspase-3. (D) Caspase-9 activity induced by cytochrome c is inhibited in cell extracts from two active Akt clones compared with vector control. Data represent mean ± SEM of three independent experiments. *P < 0.01 by Student's t test.



Constitutively Active Akt Inhibits Apoptosis After Microinjection of Cytochrome c
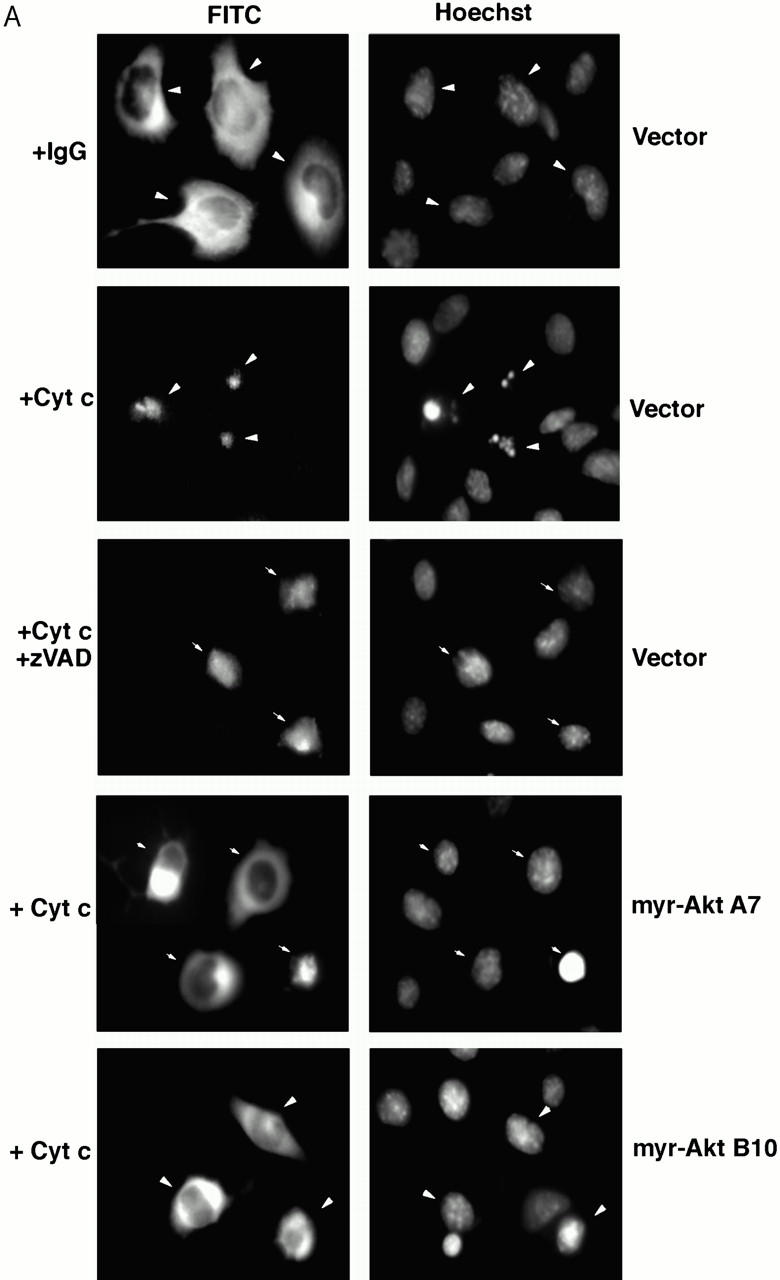
To determine whether Akt inhibits cytochrome c–induced apoptosis in vivo, purified cytochrome c was microinjected into the cytoplasm of Akt-expressing and vector-transfected cells. Control injections did not induce apoptosis, whereas microinjection of cytochrome c was sufficient to induce rapid apoptosis in HMN1 cells. Cytochrome c but not control-injected cells exhibited condensed and/or fragmented nuclei (Fig. 8 A). Moreover, constitutively active Akt inhibited and delayed cytochrome c–induced apoptosis (Fig. 8 B), which is consistent with results obtained from the cell-free system in which expressing active Akt-inhibited apoptotic events such as PARP cleavage and activation of caspase-9 and caspase-3 in response to cytochrome c (Fig. 7a, Fig. c, and Fig. d).
Figure 8.
Overexpressing constitutively active Akt suppresses apoptosis after microinjection of cytochrome c. (A) Fluorescent photomicrographs of FITC-labeled injected cells (left) and Hoechst 33342 nuclear staining (right). Note that cytochrome c but not rabbit IgG induces chromatin condensation in injected cells. Both zVAD pretreatment and expressing active Akt prevented cytochrome c–induced chromatin condensation in injected cells. (B) Quantification of the protective effects of Akt on inhibiting cytochrome c–induced apoptosis. Cells were injected with cytochrome c and, after 1, 2, or 3 h, apoptosis of injected cells was analyzed as described in Materials and Methods. Data represent the mean ± SEM from three or more independent experiments.

Immunoprecipitated Active Akt Added to Vector Cell Extracts Inhibits Caspase Activation Induced by Cytochrome c
To test the role of posttranslational modification of proteins by active Akt in inhibiting cytochrome c–induced caspase activation, active Akt was immunoprecipitated from cells expressing active Akt and incubated with vector cell extracts. Subsequently, cytochrome c and dATP were added to initiate caspase activation. PARP cleavage was markedly inhibited and delayed in vector-cell extracts preincubated with active Akt (Fig. 9). In contrast, PARP cleavage was not altered in vector cell extracts preincubated with control mouse immunoglobulin immunoprecipitate (Fig. 9). These results indicate that active Akt has a direct effect on inhibiting cytochrome c–induced caspase activation, and suggest that the inhibition by Akt is through posttranslational modification of cytosolic factor(s) involved in activating caspases.
Figure 9.

Addition of active Akt to vector cell extracts inhibits cytochrome c–induced PARP cleavage in a cell-free assay. Cell extracts from the vector line were incubated with either control mouse immunoglobulin immunoprecipitation (− myr-Akt) or immunoprecipitated active Akt [+ myr-Akt (A7) or (B10)] from either the A7 or B10 line that expresses constitutively active Akt. Cytochrome c and dATP were then added to initiate caspase activation and PARP cleavage. Data are from a representative experiment performed three times.
Discussion
Apoptotic death of neurons occurs at many different developmental stages. The most frequently studied and modeled neuronal death is that occurring during the period of synaptogenesis (Oppenheim 1991). Neurons are produced in excess, and then compete for specific neurotrophic factors produced by their target neurons or organs. Neurons that do not get sufficient neurotrophic factor die; this appears to be important for matching the extent of innervation with the size of the target field. Compelling evidence supporting this hypothesis comes from a number of studies showing that neurotrophic factors regulate target-dependent apoptotic neuronal death during development in vivo and in vitro (Snider 1994). The two major pathways implicated in supporting neuronal survival by neurotrophic factors are the PI 3-kinase and MAP kinase pathways (Pettmann and Henderson 1998).
Activation of PI 3-kinase generates the lipid products phosphatidylinositol 3,4-biphosphate (PIP2) and phosphatidylinositol 3,4,5-triphosphate (PIP3). These products subsequently activate Akt, a serine-threonine protein kinase believed to be the major effector for PI 3-kinase to promote cell survival (Franke et al. 1997). Pharmacological studies with PI 3-kinase and/or MEK inhibitors indicate that only the PI 3-kinase pathway is essential for HMN1 cell survival (Fig. 1). We previously showed that C2-ceramide inhibited insulin-stimulated Akt activation but did not affect the activity of a membrane-targeted, constitutively active form of Akt, indicating that membrane association activates Akt and bypasses the inhibitory effect of ceramide (Zhou et al. 1998). In this study, we generated stable HMN1 cell lines expressing constitutively active Akt to explore the cellular mechanisms through which Akt regulates cell survival. Overexpression of active Akt inhibits apoptosis, as exemplified by its inhibitory effects on caspase-3 activation, PARP cleavage, and DNA fragmentation. However, unlike the observation that Akt upregulates Bcl-2 expression in BAF/3 cells (Ahmed et al. 1997), Akt promotes HMN1 cell survival in the absence of altering Bcl-2, Bcl-X, and Bax expression, which is consistent with data from hippocampal H19-7 neuronal cells and epithelial BRK cells (Eves et al. 1998; Sabbatini and McCormick 1999). Nevertheless, given the pivotal role of BCL-2 family proteins in regulating apoptosis and a growing number of signaling pathways shown to regulate apoptosis by modulating expression of BCL-2 family members (Gross et al. 1999), these results do not rule out the possibility that Akt may regulate the expression of other members of the BCL-2 family to inhibit apoptosis.
Apoptosis induced by a variety of initiators such as activation of p75 low affinity NGF receptor and growth factor withdrawal is associated with increased ceramide production. In addition, a strong correlation exists between production of ceramide and subsequent cell death (Hannun 1996; Mathias et al. 1998). Moreover, cell-permeable ceramide analogues induce apoptosis in many different cell types including neurons (Hannun 1996; Mathias et al. 1998). These observations are consistent with ceramide acting as a lipid second messenger that functions to link cell-surface receptors and environmental stresses to the cellular apoptotic machinery. However, the cellular mechanisms for ceramide-induced apoptosis remain unclear. Our current study as well as work by others suggests that caspase activation is required for ceramide-induced apoptosis and that ceramide-induced apoptosis can be suppressed by either constitutively active Akt or Bcl-2, probably by different cellular mechanisms. Embryonic fibroblasts lacking Apaf-1 are less susceptible to C6-ceramide–induced apoptosis than wide-type embryonic fibroblasts, suggesting that Apaf-1 is involved in ceramide-induced apoptosis (Cecconi et al. 1998). Further studies are needed to determine whether members of the Bcl-2 family, such as Bax are involved in ceramide-induced apoptosis.
Cytochrome c release from mitochondria represents an important control point during apoptosis, although the mechanisms causing cytochrome c release remain unclear (Green and Reed 1998). It has been proposed that at least two different mechanisms exist for cytochrome c release from mitochondria, one that is caspase dependent and the other that is caspase independent. In Fas-induced apoptosis, oligomerization of the Fas receptor leads to recruitment of FADD and subsequent caspase-8 activation (Nagata 1997). Activated caspase-8 cleaves BID, a BH3 domain-only proapoptotic member of the BCL-2 family. The cleaved p15 BID fragment translocates to mitochondria, where it causes release of cytochrome c, probably by changing the conformation of Bax (Li et al. 1998; Luo et al. 1998; Desagher et al. 1999). Bid-deficient hepatocytes do not release cytochrome c in response to agonistic anti–Fas antibody, which confirms the importance for cleaved Bid to induce cytochrome c release in this model (Yin et al. 1999). In our study, C2-ceramide–induced cytochrome c release appears to be caspase independent since a broad caspase inhibitor, zVAD-fmk, inhibits caspase activation but not cytochrome c translocation. The mechanism for caspase-independent release of cytochrome c remains unclear. A recent study (Kennedy et al. 1999) indicates that Akt inhibits the release of cytochrome c from mitochondria after UV irradiation, which may involve activation of cell-surface Fas receptor (Rehemtulla et al. 1997; Aragane et al. 1998). In contrast, our data indicate that Akt fails to inhibit cytochrome c release induced by ceramide via a caspase-independent mechanism. Therefore, the function for Akt to regulate cytochrome c release may vary in response to different apoptotic stimuli or within a different cellular context.
In mammalian cells, the release of cytochrome c from mitochondria has been proposed as the critical event for cells to initiate the apoptotic cascade. In the cytosol, cytochrome c binds to Apaf-1 and triggers Apaf-1–mediated caspase-9 activation. Activated caspase-9 propagates the death signal by activating caspase-3 and other caspases (Slee et al. 1999; Zou et al. 1999). In cell-free assays of apoptosis, exogenously added cytochrome c initiates rapid apoptotic events, including caspase-3 activation and DNA fragmentation (Liu et al. 1996; Li et al. 1997a; Slee et al. 1999). Data from our cell fractionation study and cell-free assay are consistent with Bcl-2 acting to inhibit apoptosis mainly at the mitochondrial level to suppress cytochrome c release, since Bcl-2 inhibits the release of cytochrome c but fails to inhibit caspase activation induced by added cytochrome c in the cell-free assay. Akt phosphorylates and inactivates BAD, which may maintain Bcl-2 and Bcl-xL function, and it has been suggested that Akt inhibits cell death by preventing cytochrome c release from mitochondria (Kennedy et al. 1999). In contrast, our data indicate that Akt protects neural cells from apoptosis at a postmitochondrial stage to inhibit caspase activation. First, active Akt protects cells from apoptosis without blocking cytochrome c release into the cytosol. Second, addition of cytochrome c failed to induce caspase-9 activation, caspase-3 processing, or PARP cleavage in extracts from cells expressing active Akt. Third, addition of active Akt to lysates from control cells blocked cytochrome c–induced caspase activation. Fourth, cells expressing active Akt are resistant to apoptosis induced by microinjection of cytochrome c.
Microinjection of purified cytochrome c into the cytoplasm induces apoptosis in a variety of cell types, whereas microinjection of anti–cytochrome c antibody into sympathetic neurons inhibits NGF withdrawal-induced apoptosis, indicating that cytochrome c is both sufficient and necessary for apoptosis to progress in vivo (Li et al. 1997b; Duckett et al. 1998; Neame et al. 1998; Zhivotovsky et al. 1998). However, recent studies show that certain cell types, including neurons, have postmitochondrial mechanisms to inhibit the cell death machinery after cytochrome c release, suggesting a more complicated mechanism for regulating apoptosis than previously thought (for review, see Newmeyer and Green 1998; Reed and Paternostro 1999). For example, cytochrome c release induced by NGF withdrawal in sympathetic neurons is a reversible event, indicating that redistribution of cytochrome c into the cytosol does not always result in irreversible cell death in neurons (Martinou et al. 1999). Also, microinjection of cytochrome c into sympathetic neurons is not lethal in the presence of NGF. Therefore, NGF signaling appears to block cytochrome c–induced apoptosis in sympathetic neurons. Sympathetic neurons only commit to apoptosis in response to cytochrome c after NGF is removed for a prolonged period. It is proposed that neurons must undergo an unidentified event to develop “competence to die” in addition to the presence of cytochrome c (Deshmukh and Johnson 1998). Our data demonstrate that expressing active Akt inhibits cytochrome c–induced caspase activation in a cell-free assay and also suppresses apoptosis after cytoplasmic injection of cytochrome c, indicating that Akt blocks apoptosis in the presence of cytochrome c. In this regard, loss of Akt signaling may be related to developing neuronal “competence-to-die” in response to cytochrome c since prolonged NGF withdrawal may progressively decrease PI 3-kinase/ Akt activity and lower the threshold for cytochrome c to initiate caspase activation.
Recently, cytochrome c was implicated in the pathogenesis of Parkinson's disease since purified cytochrome c stimulates α-synuclein aggregation in vitro and cytochrome c colocalizes with α-synuclein in Lewy bodies of patients with Parkinson's disease (Hashimoto et al. 1999). Our findings here indicate that Akt protects neural cells from acute apoptosis caused by cytochrome c release and may allow cytosolic cytochrome c to participate in other cellular process such as α-synuclein aggregation. Interestingly, expressing active Akt or treatment with zVAD-fmk inhibited chromatin condensation after cytochrome c injection, yet many rescued cells still shrank, suggesting that cytochrome c may cause cell shrinkage independent of caspase activation. It should be possible to examine additional roles of cytochrome c independent of its effects on apopsome complex formation in Apaf-1 or caspase-9–deficient neurons.
A growing number of downstream targets of Akt have been identified, including glycogen synthase kinase-3, BAD, human caspase-9, and transcription factors such as CREB, Forkhead, and NFκB (Pap and Cooper 1998; Khwaja 1999). While each of these has been implicated as an important target for Akt to inhibit apoptosis, in the present case, caspase-9 appears to be the most likely candidate since Akt phosphorylates human caspase-9 and inhibits its activity (Cardone et al. 1998), which would provide a potential mechanism for Akt to inhibit caspase activation at a postmitochondrial level. However, [32P]orthophosphate labeling of cells expressing active Akt indicated that mouse caspase-9 is not phosphorylated by Akt (data not shown). This may be explained by recent reports that the consensus Akt phosphorylation sites on human caspase-9 are not conserved in caspase-9 from other species such as mouse caspase-9 in HMN1 cells (Fujita et al. 1999; Rodriguez et al. 2000). On the other hand, our data also suggest that there exists an additional, more general mechanism by which Akt can suppress activation of caspases by cytochrome c at a postmitochondrial stage. When added into vector-cell extracts, immunoisolated active Akt is sufficient to inhibit caspase activation induced by cytochrome c, suggesting that active Akt has a direct effect on inhibiting cytochrome c–induced caspase activation. One such direct target for Akt may be Apaf-1, which forms a holoenzyme with caspase-9 and regulates caspase-9 activity (Rodriguez and Lazebnik 1999). Akt phosphorylates Apaf-1 in vitro (our unpublished observations); however, further studies are needed to determine whether cellular Apaf-1 is a direct target of Akt, and how the phosphorylation status of Apaf-1 may regulate caspase-9 activation.
In conclusion, our data based on subcellular fractionation, cell-free assays of apoptosis, and microinjection studies indicate that Akt inhibits apoptosis downstream of cytochrome c release and before activation of caspase-9 (Fig. 10). The ability of Akt to regulate apoptosis at a postmitochondrial stage provides a potential molecular mechanism allowing postmitotic cells like neurons to tolerate leakage of cytochrome c into the cytoplasm when growth factors are present. A key to understanding postmitochondrial regulation of apoptosis by Akt will be future studies to identify the cellular target(s) of Akt.
Figure 10.

Schematic illustration showing that Akt can regulate caspase activation at a premitochondrial level (e.g., Kennedy et al. 1999), as well as at a postmitochondrial level downstream of cytochrome c release and before activation of caspase-9 (present study).
Acknowledgments
We thank Ms. Svetlana Savina for microinjection, Dr. Robert Siman for the caspase-3 antibody, and Dr. Morris Birnbaum (University of Pennsylvania) for the Akt constructs. We also thank members of the Pittman lab for support and helpful suggestions throughout the period of this work.
This work was supported by NS32465 from the National Institutes of Health to R.N. Pittman.
Footnotes
Abbreviations used in this paper: CEB, cell extract buffer; HA, hemagglutinin; HMN1, hybrid motor neuron 1; MAP, mitogen-activated protein; PARP, poly (ADP-ribose) polymerase; PI 3-kinase, phosphoinositide 3 kinase.
References
- Adams J.M., Cory S. The Bcl-2 protein familyarbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Ahmed N.N., Grimes H.L., Bellacosa A., Chan T.O., Tsichlis P.N. Transduction of interleukin-2 antiapoptotic and proliferative signals via Akt protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:3627–3632. doi: 10.1073/pnas.94.8.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragane Y., Kulms D., Metze D., Wilkes G., Poppelmann B., Luger T.A., Schwarz T. Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J. Cell Biol. 1998;140:171–182. doi: 10.1083/jcb.140.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihardjo I., Oliver H., Lutter M., Luo X., Wang X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Cass L.A., Summers S.A., Prendergast G.V., Backer J.M., Birnbaum M.J., Meinkoth J.L. Protein kinase A–dependent and –independent signaling pathways contribute to cyclic AMP-stimulated proliferation. Mol. Cell. Biol. 1999;19:5882–5891. doi: 10.1128/mcb.19.9.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F., Alvarez-Bolado G., Meyer B.I., Roth K.A., Gruss P. APAF1 (CED-4 homologue) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S., Lauper S., Maundrell K., Antonsson B., Martinou J.C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M., Johnson E.M. Evidence of a novel event during neuronal deathdevelopment of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Duckett C.S., Li F., Wang Y., Tomaselli K.J., Thompson C.B., Armstrong R.C. Human IAP-like protein regulates programmed cell death downstream of Bcl-XL and cytochrome c. Mol. Cell. Biol. 1998;18:608–615. doi: 10.1128/mcb.18.1.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt P., Schremser E.J., Cooper G.M. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol. Cell. Biol. 1999;19:5308–5315. doi: 10.1128/mcb.19.8.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eves E.M., Xiong W., Bellacosa A., Kennedy S.G., Tsichlis P.N., Rosner M.R., Hay N. Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neuronal cell line. Mol. Cell. Biol. 1998;18:2143–2152. doi: 10.1128/mcb.18.4.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke T.F., Kaplan D.R., Cantley L.C. PI3Kdownstream AKT ion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Fujita E., Jinbo A., Matuzaki H., Konishi H., Kikkawa U., Momoi T. Akt phosphorylation site found in human caspase-9 is absent in mouse caspase-9. Biochem. Biophys. Res. Commun. 1999;264:550–555. doi: 10.1006/bbrc.1999.1387. [DOI] [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondrial in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Hakem R., Hakem A., Duncan G.S., Henderson J.T., Woo M., Soengas M.S., Elia A., de la Pompa J.L., Kagi D., Khoo W. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Hannun Y.A. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- Hashimoto M., Takeda A., Hsu L.J., Takenouchi T., Masliah E. Role of cytochrome c as a stimulator of α-synuclein aggregation in Lewy body disease. J. Biol. Chem. 1999;274:28849–28852. doi: 10.1074/jbc.274.41.28849. [DOI] [PubMed] [Google Scholar]
- Hengartner M.O., Horvitz H.R. Programmed cell death in Caenorhabditis elegans . Curr. Opin. Genet. Dev. 1994;4:581–586. doi: 10.1016/0959-437x(94)90076-f. [DOI] [PubMed] [Google Scholar]
- Jacobson M.D., Weil M., Raff M.C. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- Kennedy S.G., Kandel E.S., Cross T.K., Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell. Biol. 1999;19:5800–5810. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khwaja A. Akt is more than just a Bad kinase. Nature. 1999;401:33–34. doi: 10.1038/43354. [DOI] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kohn A.D., Takeuchi F., Roth R.A. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J. Biol. Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- Kuida K., Haydar T.F., Kuan C.Y., Gu Y., Taya C., Karasuyama H., Su M.S., Rakic P., Flavell R.A. Reduced apoptosis and cytochrome c–mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Li F., Srinivasan A., Wang Y., Armstrong R.C., Tomaselli K.J., Fritz L.C. Cell-specific induction of apoptosis by microinjection of cytochrome c J. Biol. Chem 272 1997. 30299 30305b [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li P., Nijhawan D., Budihardjo I., Srinivasula S.M., Ahmad M., Alnemri E.S., Wang X. Cytochrome c and dATP-dependent formation of APAF-1/caspase-9 complex initiates an apoptotic protease cascade Cell 91 1997. 479 489a [DOI] [PubMed] [Google Scholar]
- Liu X., Kim C.N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extractsrequirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Los M., Wesselborg S., Schulze-Osthoff K. The role of caspases in development, immunity, and apoptotic signal transductionlessons from knockout mice. Immunity. 1999;10:629–639. doi: 10.1016/s1074-7613(00)80062-x. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Martinou I., Desagher S., Eskes R., Antonsson B., Andre E., Fakan S., Martinou J. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell. Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias S., Pena L.A., Kolesnick R.N. Signal transduction of stress via ceramide. Biochem. J. 1998;335:465–480. doi: 10.1042/bj3350465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- Neame S.J., Rubin L.L., Philpott K.L. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J. Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmeyer D.D., Green D.R. Surviving the cytochrome seas. Neuron. 1998;21:653–655. doi: 10.1016/s0896-6273(00)80580-2. [DOI] [PubMed] [Google Scholar]
- Oppenheim R.W. Cell death during development of the nervous system. Annu. Rev. Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pap M., Cooper G.M. Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 1998;273:19929–19932. doi: 10.1074/jbc.273.32.19929. [DOI] [PubMed] [Google Scholar]
- Pettmann B., Henderson C.E. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- Reed J.C., Paternostro G. Post-mitochondrial regulation of apoptosis during heart failure. Proc. Natl. Acad. Sci. USA. 1999;96:7614–7616. doi: 10.1073/pnas.96.14.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehemtulla A., Hamilton C.A., Chinnaiyan A.M., Dixit V.M. Ultraviolet radiation-induced apoptosis is mediated by activation of CD-95 (Fas/APO-1) J. Biol. Chem. 1997;272:25783–25786. doi: 10.1074/jbc.272.41.25783. [DOI] [PubMed] [Google Scholar]
- Rodriguez J., Lazebnik Y. Caspase-9 and Apaf-1 form an active holoenzyme. Genes Dev. 1999;13:3179–3184. doi: 10.1101/gad.13.24.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J., Chen H., Lin S., Lazebnik Y. Caspase phosphorylation, cell death, and species variability. Science. 2000;282:1318. [Google Scholar]
- Sabbatini P., McCormick F. Phosphoinositide 3-kinase (PI3K) and PKB/Akt delay the onset of p53-mediated, transcriptionally dependent apoptosis. J. Biol. Chem. 1999;274:24263–24269. doi: 10.1074/jbc.274.34.24263. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S., Dixit V.M. Caspasesintracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Slee E.A., Harte M.T., Kluck R.M., Wolf B.B., Casiano C.A., Newmeyer D.D., Wang H., Reed J.C., Nicholson D.W., Alnemri E.S. Ordering the cytochrome c–initiated caspase cascadehierarchical activation of caspase-2,-3, -6, -7, -8, and -10 in a caspase-9–dependent manner. J. Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider W.D. Functions of the neurotrophins during the nervous system developmentwhat the knockout mice are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M., Ahmad M., Fernandes-Alnemri T., Alnemri E.S. Autoactivation of procaspase-9 by APAF-1–mediated oligomerization. Mol. Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Thompson C.B. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A., Lazebnik Y. Caspasesenemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Vaux D.L., Korsmeyer S.J. Cell death in development. Cell. 1999;96:245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J., Peng T., Jones D.P., Wang X. Prevention of apoptosis by BCL-2release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Yin X., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K.A., Korsmeyer S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Kong Y.Y., Yoshida R., Elia A.J., Hakem A., Hakem R., Penninger J.M., Mak T.W. APAF1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Zha J., Harada H., Yang E., Jockel J., Korsmeyer S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhivotovsky B., Orrenius S., Brustugun O.T., Døskeland S.O. Injected cytochrome c induces apoptosis. Nature. 1998;391:449–450. doi: 10.1038/35060. [DOI] [PubMed] [Google Scholar]
- Zhou H., Summers S.A., Birnbaum M.J., Pittman R.N. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J. Biol. Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- Zou H., Henzel W.J., Liu X., Lutschg A., Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c–dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Zou H., Zou Y., Liu X., Wang X. An APAF-1 cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]