

Abstract
Many organelles change their shape in the course of the cell cycle or in response to environmental conditions. Lysosomes undergo drastic changes of shape during microautophagocytosis, which include the invagination of their boundary membrane and the subsequent scission of vesicles into the lumen of the organelle. The mechanism driving these structural changes is enigmatic. We have begun to analyze this process by reconstituting microautophagocytosis in a cell-free system. Isolated yeast vacuoles took up fluorescent dyes or reporter enzymes in a cytosol-, ATP-, and temperature-dependent fashion. During the uptake reaction, vacuolar membrane invaginations, called autophagic tubes, were observed. The reaction resulted in the transient formation of autophagic bodies in the vacuolar lumen, which were degraded upon prolonged incubation. Under starvation conditions, the system reproduced the induction of autophagocytosis and depended on specific gene products, which were identified in screens for mutants deficient in autophagocytosis. Microautophagic uptake depended on the activity of the vacuolar ATPase and was sensitive to GTPγS, indicating a requirement for GTPases and for the vacuolar membrane potential. However, microautophagocytosis was independent of known factors for vacuolar fusion and vesicular trafficking. Therefore, scission of the invaginated membrane must occur via a novel mechanism distinct from the homotypic fusion of vacuolar membranes.
Keywords: budding, autophagocytosis, membrane dynamics, proteolysis, scission
Introduction
Eukaryotic cells have developed multiple pathways for the intracellular degradation of proteins. The proteasome system and direct translocation of proteins through the lysosomal membrane account for only part of the protein turnover (Cuervo and Dice 1998). A major route of degradation is autophagocytosis, a process of controlled self-digestion of cells. Autophagocytosis is constitutive, but it is strongly enhanced under conditions of stress, starvation, or redifferentiation (Seglen and Bohley 1992; Knop et al. 1993). A wide range of eukaryotic cells are capable of autophagocytosis, indicating that the process may be a general characteristic of eukaryotic life.
Current models differentiate two forms of autophagocytosis, macro- and microautophagocytosis. Macroautophagocytosis is characterized by the appearance of a unique type of vesicles. These vesicles (autophagosomes) sequester portions of the cytosol. They can also engulf entire organelles such as mitochondria or peroxisomes. Autophagosomes are surrounded by two or sometimes several membranes that distinguish them from other cellular vesicles. The origin of the autophagosomal membranes is controversial (Dunn 1990; Yamamoto et al. 1990a,Yamamoto et al. 1990b). The outer membrane of autophagosomes can fuse with lysosomes, delivering the inner membrane with its cytosolic contents (called an autophagic body) into the lysosomal lumen for degradation (Seglen and Bohley 1992; Takeshige et al. 1992).
Microautophagocytosis is defined as the uptake of cytosolic components by direct invagination of the lysosomal membrane. It results in vesicles budding into the lumen of the lysosome. The resulting autophagic bodies are finally degraded. Also, the microautophagic pathway is capable of delivering not only soluble proteins, but entire organelles into lysosomes for degradation (Veenhuis et al. 1983; Tuttle et al. 1993; Yokota 1993; Tuttle and Dunn 1995). The mechanisms that drive the profound changes in lysosomal structure during invagination and scission of the nascent microautophagic vesicle are unknown.
Different approaches have been taken to analyze autophagocytosis in various systems. Pharmacological studies have defined agents inducing or inhibiting autophagocytosis in cell culture systems, perfused organs, or entire animals (Seglen and Bohley 1992; Mortimore and Kadowaki 1994). Considerable efforts were also made to isolate autophagosomes (Stromhaug et al. 1998), a prerequisite for analyzing their protein composition, identifying marker proteins, and analyzing their origin.
Pioneering genetic screens in Saccharomyces cerevisiae have revealed at least 16 complementation groups of mutants defective in the induction of autophagocytosis, providing a solid basis for the molecular analysis of the process (Tsukada and Ohsumi 1993; Thumm et al. 1994). These complementation groups overlap strongly with a set of mutants defective in the cytoplasm to vacuole targeting (Cvt) pathway (Harding et al. 1996; Scott et al. 1996). The Cvt pathway leads to the selective and rapid import of aminopeptidase I into vacuoles via vesicular intermediates structurally related to autophagosomes (Baba et al. 1997). Most of the genes found in these screens have now been identified by exploiting the fact that autophagocytosis in yeast is essential for surviving starvation and sporulation. They encode presumptive regulatory components, such as a protein kinase (autophagocytosis [Apg]1p/autophagocytosis [Aut]3p) (Matsuura et al. 1997; Straub et al. 1997). A ubiquitin-like protein conjugation system was found comprising an activation enzyme (Apg7p), a ligase (Apg10p), and two proteins (Apg12p and Apg5p), which become covalently attached to each other by these enzymes (Mizushima et al. 1998; Kim et al. 1999; Shintani et al. 1999). A ubiquitin-activating enzyme is also involved in the autophagic pathway of mammalian cells (Lenk et al. 1992). Homooligomers of Apg16p bind to the Apg5p–Apg12p conjugate (Mizushima et al. 1999). Aut2p and Aut7p/Apg8p were proposed to bind to microtubules and mediate the delivery of autophagosomes to the vacuole (Lang et al. 1998). However, this interpretation was challenged by the recent finding that disassembling microtubules by nocodazole treatment does not block autophagocytosis in yeast (Kirisako et al. 1999). Aut7p/Apg8p is a peripheral component on the membrane of autophagosome precursors and is the first marker protein for autophagosomes (Kirisako et al. 1999). Genetic screens in Pichia pastoris and Hansenula polymorpha have identified another set of mutations affecting peroxisome degradation (Titorenko et al. 1995; Sakai et al. 1998; Yuan et al. 1999). One of the affected genes was Apg7, which provides genetic evidence that autophagocytosis of soluble proteins and peroxisomes may employ a common machinery (Kim et al. 1999; Yuan et al. 1999).
The macroautophagic pathway in S. cerevisiae has been extensively characterized by light and EM (Takeshige et al. 1992; Baba et al. 1994, Baba et al. 1995). The pathway depends on Vam7p and Vam3p, two SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor) proteins that are presumably involved in fusing the outer membrane of autophagosomes with the vacuole (Darsow et al. 1997; Sato et al. 1998). Therefore, macroautophagocytosis seems to involve the conserved fusion machinery that also mediates membrane fusion at many other steps of vesicular trafficking (Rothman 1994). We have recently discovered a microautophagic pathway in S. cerevisiae (Müller et al. 2000, this issue), in which a specialized structure, the autophagic tube, mediates invagination of the vacuolar membrane and scission of vesicles into the lumen. This tube displays a striking lateral heterogeneity of membrane structure. The base of the tube has a normal content of transmembrane proteins, whereas the zone of vesicle formation at the tip of the tube is devoid of visible transmembrane particles. The smooth appearance of this zone matched that of the membrane of autophagic bodies. We also found small areas with very low content of transmembrane particles on the vacuolar surface. These smooth areas often invaginate and appear to be precursors of autophagic tubes.
To study the biochemical basis of the mechanisms behind these profound changes of membrane structure and organellar morphology, we have reconstituted the microautophagic membrane invagination in vitro. We have developed a system based on isolated yeast vacuoles and devised fast assays, which allow light microscopic tracing of the reaction, as well as convenient quantitation. We have used this system to characterize the requirements of microautophagic protein uptake into vacuoles.
Materials and Methods
Yeast Strains and Genetic Manipulations
The strains used were: DBY5734 MATa ade2 lys2 his3 trp1 leu2 ura3 cmd1-Δ1::TRP ade3::HIS3::CMD1 (David Botstein, Stanford University, Stanford, CA); DBY5734-16 MATa pep4Δ::LEU2 ade2 lys2 his3 trp1 leu2 ura3 cmd1-Δ1::TRP ade3::HIS3::CMD1; and DBY5734-19 MATa pho8Δ::URA3 ade2 lys2 his3 trp1 leu2 ura3 cmd1-Δ1::TRP ade3::HIS3::CMD1. The following strains are derivatives of DBY5734 obtained by replacement of the indicated genes with a kanamycin (KAN) resistance cassette: YTS1 (aut1Δ), YTS2 (aut2Δ), YTS3 (aut7Δ), YTS4 (apg1Δ), YTS5 (apg5Δ), and YTS6 (apg13Δ). The strains used for cytosol preparation were derived from K91-1A (MATa pho8::pAL134 pho13::pPH13 lys1; from Y. Kaneko, Osaka University, Osaka, Japan) by replacing the indicated gene with a KAN resistance cassette: YTS11 (aut1Δ), YTS12 (aut2Δ), YTS13 (aut7Δ), YTS14 (apg1Δ), YTS15 (apg5Δ), and YTS16 (apg13Δ). RSY271 (sec18-1) and the corresponding wild-type RSY 249 were from Randy Schekman (University of California, Berkeley, CA).
Pho8 and Pep4 were deleted by one-step gene replacement using plasmid pAR2 (J. Shaw, University of Utah, Salt Lake City, UT) and pTS17 (T. Stevens, University of Oregon, Eugene, OR), respectively.
Deletions of Aut and Apg genes were obtained as follows: using the oligonucleotides listed below, DNA fragments for the chromosomal replacement of the corresponding Aut or Apg genes with a LoxP-KANR-LoxP cassette were created by PCR on plasmid pUG6 (Güldener et al. 1996). The strains YTS1–YTS6 and YTS11–YTS16 were made by transforming the amplified single LoxP-KANR-LoxP cassettes into DBY5734 and K91-1A. The oligonucleotides used were: aut1ΔKAN1, 5′-CGT ATA TCA AGC TAG CTA GAA GTT AGG AAC AAA GAA GTA CCA GCT GAA AGC TTC GTA CGC-3′; aut1ΔKAN2, 5′-GTT TAT CCT GTT TTT TGA CCA CCT GGC TTG CAG CTA ATA GGC ATA GGC CAC TAG TGG ATC TG-3′; aut2ΔKAN1, 5′-ATG GAC GAC TTC TTA TCA CGT ATA GGA GTG ATA TAC ATG CAG AGG CAG CTG AAG CTT CGT ACG C-3′; aut2ΔKAN2, 5′-GCA TTT TTC ATC AAT AGG ACT GTG AAT ACC TAC CGT TTC CTT CTC GCA TAG GCC ACT AGT GGA TCT G-3′; aut7ΔKAN1, 5′-CCT GCC AAA TGT ATT TTC TCC TGA GTA AGT GAC ATA CAA AAA CCC CAG CTG AAG CTT CGT ACG C-3′; aut7ΔKAN2, 5′-ATG AAG TCT ACA TTT AAG TCT GAA TAT CCA TTT GAA AAA AGG AAG GCA TAG GCC ACT AGT GGA TCT G-3′; apg1ΔKAN1, 5′-CAT ATT TTC AAA TCT CTT TTA CAA CAC CAG ACG AGA AAT TCA GCT GAA AGC TTC GTA CGC-3′; apg1ΔKAN2, 5′-GGT CAT TTG TAC TTA ATA AGA AAA CCA TAT TAT GCA TCA CGC ATA GGC CAC TAG TGG ATC TG-3′; apg5ΔKAN1, 5′-GGT TCT AGA AGA ACG GAG ATA GGA AAC CTA TGA TGT AAG TCA GCT GAA GCT TCG TAC GC-3′; apg5ΔKAN2, 5′-GAT ATT TGA ATG ACA CTT TTA AAT GCG TAT ATA ACA GCT CGC ATA GGC CAC TAG TGG ATC TG-3′; apg13ΔKAN1, 5′-AAG AAA GCA GAA CAT ACA GCC CGG TTG AAT AGC ATG AGT CCA GCT GAA AGC TTC GTA CGC-3′; and apg13ΔKAN2, 5′-TAT TTT TCT TTA GTT GTG CCC TTT AAA ATA AAA CTT TAC CGC ATA GGC CAC TAG TGG ATC TG-3′.
Growth of Cells
Yeast was precultured in YPD (1% yeast extract, 2% Bacto peptone, 2% glucose) for 6–8 h at 30°C and then diluted for logarithmic growth overnight (14–16 h, 30°C, 225 rpm) in 2-liter Erlenmeyer flasks with 1 liter of YPD medium. For starving cells, overnight cultures were harvested at an optical density at 600 nm (OD600) of 2, centrifuged (4 min, 4°C, 3,800 g, JLA 10.500 rotor; Beckman), washed with sterile water, resuspended in 1 liter of SD(−N) (0.67% Difco yeast nitrogen base without amino acids and without ammonium sulfate, 2% glucose), and incubated (up to 3 h, 30°C, 225 rpm).
Cytosol Preparation
Cytosol was prepared from strain K91-1A or its derivatives. Overnight cultures were harvested at OD600 = 4.5, centrifuged (4 min, 3,800 g, 4°C, JLA 10.500 rotor), washed with sterile water, harvested again, resuspended in 1 liter of SD(−N), and incubated for 3 h at 30°C and 225 rpm. For preparation of cytosol from nonstarved cells, SD(−N) was replaced by YPD in this incubation. Cells were harvested and washed as described above, first with water, and then with one pellet volume of chilled lysis buffer (40 mM Pipes-KOH, pH 6.8, 0.5 mM MgCl2, 150 mM KCl, 200 mM sorbitol, 1 mM DTT, 0.2 mM PMSF, 0.1 mM pefabloc SC, 0.5 μg/ml pepstatin A, 0.1 μg/ml leupeptin, 50 μM O-phenantroline). After centrifugation (5 min, 3,800 g, 4°C, JLA 10.500 rotor), the pellet was resuspended in a small volume of lysis buffer so that a thick slurry resulted. The suspension was frozen as little nuggets in liquid nitrogen and blended (six to eight times for 30 s) in a Waring blender filled with liquid nitrogen. Cells were thawed and the lysate was centrifuged (10 min, 12,000 g, 4°C, JA 25.50 rotor; Beckman). The supernatant was centrifuged (20 min, 125,000 g, 2°C, TLA45 rotor; Beckman), the fatty top fraction was discarded, and the clarified cytosol was recovered. The protein concentration of this cytosolic fraction was adjusted to 25–30 mg/ml with lysis buffer. Aliquots were frozen in liquid nitrogen and stored at −80°C.
Vacuole Preparation
Cells were grown in YPD overnight to OD600 = 2.5–3.0, harvested (4 min, 3,800 g, 4°C, JLA 10.500 rotor), and resuspended in 50 ml of 30 mM Tris-HCl, pH 8.9, with 10 mM DTT. Cells were incubated (5 min, 30°C), centrifuged as above, resuspended in 15 ml of spheroplasting buffer (50 mM potassium phosphate, pH 7.5, 600 mM sorbitol in YPD with 0.2% glucose and oxalyticase [3,600 U/ml for DBY5734-16, 2,400 U/ml for DBY5734-19, and 3,000 U/ml for DBY5734]) and transferred into 30-ml Corex tubes. Cells were incubated (23 min, 30°C), reisolated (4°C, 1 min at 750 g and 1 min at 1,500 g; JA 25.50 rotor), and resuspended in 2.5 ml 15% Ficoll 400 in PS buffer (10 mM Pipes-KOH, pH 6.8, 200 mM sorbitol) by gentle stirring with a glass rod. DEAE-dextran (150 μl for DBY5734-16, 120 μl for DBY5734-19, and 150 μl for DBY5734) was added from a 0.4-mg/ml stock in 15% Ficoll 400 in PS. The spheroplasts were incubated (2 min at 0°C, then 80 s at 30°C), chilled again, transferred to an SW41 tube, and overlaid with 3 ml 8% Ficoll 400, 3 ml 4% Ficoll 400, and 1.5–2 ml 0% Ficoll 400 in PS buffer. After centrifugation (90 min, 154,000 g, 2°C, SW41 rotor), vacuoles were harvested from the 0–4% interphase.
In Vitro Autophagocytosis Assay
A standard reaction had a volume of 180 μl and was composed of: vacuoles (0.2 mg/ml, from strain DBY5734), 105 mM KCl, 7 mM MgCl2, 2 mM ATP, 100 μM DTT, 80 mM disodium creatine phosphate, 140 U/ml creatine kinase, 1× proteinase inhibitor cocktail (PIC: 75 μM pefabloc SC, 75 ng/ml leupeptin, 37.5 μM O-phenanthroline, 375 ng/ml pepstatin A), 17 μg/ml firefly luciferase, 3 mg/ml cytosol from starved cells, 200 mM sorbitol, and 10 mM Pipes-KOH, pH 6.8. This mixture was incubated for 1 h at 27°C. A 70-μl sample was chilled on ice, diluted with 300 μl 150 mM KCl in PS buffer, centrifuged (6,800 g, 4 min, 2°C, fixed angle–table top centrifuge), washed twice with 300 μl 150 mM KCl in PS buffer, and resuspended in 60 μl 150 mM KCl in PS buffer. 10 μl was used to check for complete vacuole recovery by determining the protein concentration or, alternatively, the activity of endogenous Pho8p, as described previously (Haas et al. 1994). 40 μl of the suspension was treated with proteinase K (15 min, 0°C, 275 μg/ml, added from a 5-mg/ml stock in PS) to digest surface-bound luciferase. Digestion was terminated by adding an equal volume of freshly prepared PMSF solution (0.5 mM PMSF and 150 mM KCl in PS buffer). Luciferase activity was determined from a 25-μl aliquot in a luminometer using a luciferase kit (Berthold Detection Systems), according to the instructions of the supplier.
In Vitro Fusion
When both fusion and autophagocytosis were to be determined, the standard assay was performed as above, but a 1:1 mixture of vacuoles from a Δpep4 and a Δpho8 strain were used. The former carries proalkaline phosphatase (pro-Pho8p), and the latter only the appropriate maturation enzyme (Pep4p). Alkaline phosphatase activity is only generated upon mixing the contents of both vacuole types, resulting in fusion (Haas et al. 1994). Fusion was quantitated by taking a 30-μl aliquot out of the standard assay (after incubation for 1 h at 27°C) and determining alkaline phosphatase activity, as described previously (Haas et al. 1994).
Purification of Fluorescein Dextran 500,000
10 mg fluorescein dextran 500,000 (Molecular Probes) was dissolved in 1 ml of PS buffer, concentrated, and rediluted at least five times by ultrafiltration in Centricon 30 devices. The retentate was used for the experiments if low molecular weight fluorescein was no longer detectable in the filtrate by fluorescence spectroscopy in a PerkinElmer LS 50-B spectrometer.
Microscopic Assay of Autophagocytosis In Vitro
The standard assay was modified by using 1 μM purified fluorescein dextran 500,000 (20-μM stock), instead of luciferase. Samples were taken, and the vacuoles were reisolated and washed, as described for the chemical assay. The vacuoles were resuspended in PS buffer with 50 μM FM4-64 to a protein concentration of ∼1 mg/ml. The suspension was mixed with an equal volume of 0.4% Seaplaque agarose in PS (kept liquid at 35°C). 12 μl was transferred to a slide, chilled at 4°C for 5 min to immobilize the vacuoles, and analyzed by confocal fluorescence microscopy on a Leica TCS system, or by conventional fluorescence microscopy.
Results
Reconstitution of Vacuolar Invagination and Solute Uptake In Vitro
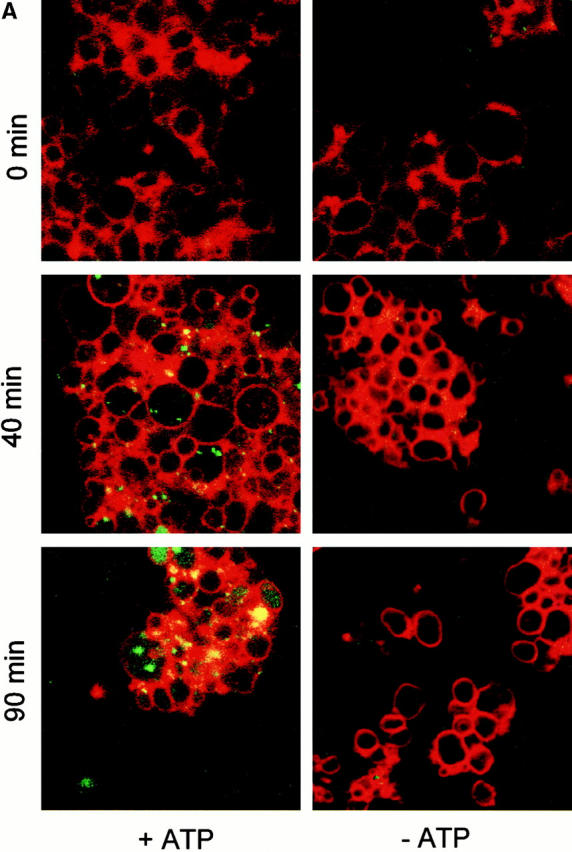
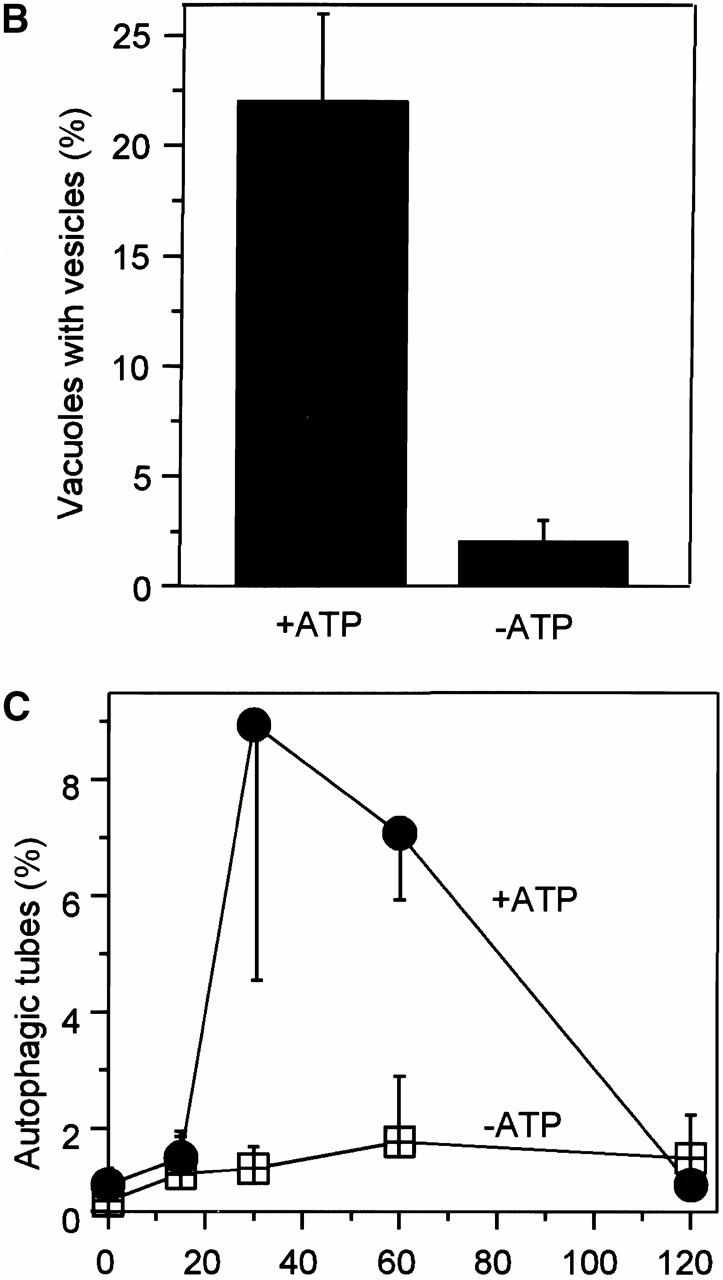
Microautophagocytosis occurs by direct invagination of lysosomal membranes, which is an “inverted” budding process into the lumen of the organelle. Soluble material is entrapped inside the invaginating vesicle and, upon scission of the membrane, becomes inaccessible to the surrounding medium. We have reconstituted this process in vitro using purified vacuoles from yeast. These organelles can be isolated in good purity and yield (Wiemken 1975) and can be readily observed by light microscopy. We incubated isolated vacuoles in the presence of a cytosolic extract, an ATP-regenerating system, and appropriate salt and buffer conditions. To monitor vesicular uptake of soluble material from the medium, the incubation was performed in the presence of high molecular weight dextran coupled to FITC. Dye that had not been taken up was removed by reisolating the vacuoles. Uptake was analyzed by fluorescence microscopy. After 60 min of incubation almost a quarter of the vacuoles contained one or two punctate structures (Fig. 1a and Fig. B) that were mobile in the lumen. Formation of the FITC-dextran–containing vesicles was ATP dependent. Upon longer incubation, these vesicles disappeared. The vacuolar lumen became homogeneously stained (Fig. 1 A), indicating that the vesicles had been lysed inside the vacuoles and their contents dispersed in the vacuolar lumen. Vacuoles were also stained with the membrane dye FM4-64 to assay for the presence of autophagic tubes, the tubular membrane invaginations from which vesicles bud into the lumen of vacuoles in vivo (Müller et al. 2000, this issue). At the beginning of the incubation, <1% of the vacuoles had autophagic tubes (Fig. 1 C). After a lag phase of 15 min, the frequency of autophagic tubes increased to almost 10%. Beyond 60 min of incubation, the tubes disappeared again. The formation of autophagic tubes was ATP dependent, indicating that it was an active process. We conclude that the in vitro reaction reconstitutes the formation of autophagic tubes and the vesicular uptake of soluble material into vacuoles.
Figure 1.
Uptake of dextran-coupled FITC by isolated vacuoles. (A) Isolated vacuoles were incubated in the absence or presence of apyrase (20 U/ml) with 1 μM purified FITC-dextran (molecular mass ∼500 kD) under standard reaction conditions. After the indicated periods of time at 27°C, the vacuoles were reisolated, washed, stained with FM4-64, and analyzed by confocal fluorescence microscopy, as described in the Materials and Methods section. The FITC and rhodamine channels of the microscope were superimposed. (B) Microscopic quantitation of in vitro uptake. A standard uptake reaction with FITC-dextran was performed in the presence or absence of ATP, as in A. After 60 min, the vacuoles were reisolated, washed, and analyzed by fluorescence microscopy. The graph presents the proportion of vacuoles bearing at least one fluorescent inclusion. (C) Frequency of autophagic tubes during the reaction. An uptake reaction was performed in the presence or absence of ATP, as in A. At the indicated times, the vacuoles were reisolated, washed, and analyzed by fluorescence microscopy. The frequency of vacuoles bearing a membrane invagination was determined. For B and C, five independent experiments were averaged. At least 200 vacuoles from 10 randomly selected fields were analyzed for each data point. Error bars indicate SD.
Using these microscopical assays, the concentrations of all components were optimized to yield maximal uptake activity (data not shown). The results of these optimizations were identical to those obtained with the chemical uptake assay described below. We developed this assay, which is based on the uptake of firefly luciferase, to provide a means for more convenient and sensitive quantitation of vesicle formation. Isolated vacuoles were incubated with cytosol and ATP in the presence of purified firefly luciferase. The organelles were reisolated, washed, and treated with increasing concentrations of proteinase K to digest residual luciferase potentially adhering to the surface of the vacuoles. Only luciferase that had been taken up into the vacuoles would resist this treatment. A strong signal of proteinase K–protected luciferase was recovered from samples incubated in the presence of ATP (Fig. 2 A). In the absence of ATP, no protease-protected luciferase cofractionated with the vacuoles. The protease-protected luciferase became accessible to proteinase K after lysis of the vacuoles by detergent (Fig. 2 A), or after rupturing the membranes by freeze–thawing (Fig. 2 B). This demonstrates that protease protection was due to sequestration in the vacuolar lumen. Therefore, the behavior of luciferase matched that of FITC-dextran and could be used as a measure for membrane invagination and solute uptake from the medium.
Figure 2.
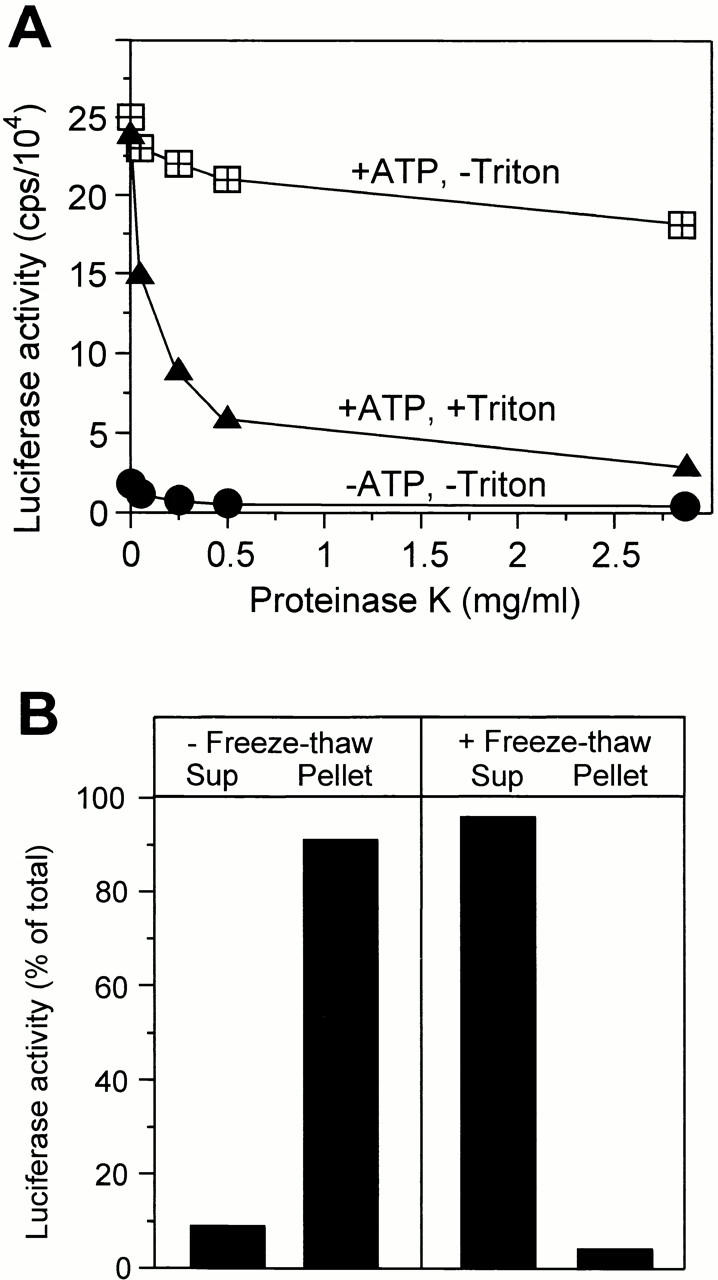
Luciferase sequestration as an assay for in vitro uptake. (A) Luciferase cofractionating with vacuoles is proteinase K protected. In vitro uptake reactions of three times the standard volume were performed (60 min, 27°C) in the presence or absence of the ATP-regenerating system. The vacuoles were reisolated, washed, resuspended in 150 mM KCl in PS buffer to a concentration of 0.23 mg/ml, and split into aliquots. After addition of 1% Triton X-100 or buffer, the mixture was digested with the indicated concentrations of proteinase K (15 min, 0°C). Proteinase treatment was stopped by adding one volume of 0.5 mM PMSF in 150 mM KCl in PS buffer. Then, luciferase activity was assayed. (B) Luciferase can be released from the vacuolar lumen by freeze–thaw treatment. An uptake reaction was performed (60 min, 27°C). The vacuoles were reisolated and washed as described in the Materials and Methods section. The final pellet was resuspended in 90 μl PS buffer with 150 mM KCl and split into two aliquots. One was frozen at −80°C and thawed again slowly, whereas the other remained on ice. The samples were centrifuged (15 min, 145,000 g, 4°C). The supernatants (Sup)were recovered and the pellets were resuspended in an equal volume of PS with 150 mM KCl. Luciferase activity in both fractions was determined. The reaction tubes for this experiment had been coated with BSA (0.5 mg/ml, 15 min at room temperature) to reduce unspecific binding of luciferase to the surface.
The uptake reaction proceeded for ∼60 min, with a lag in the first 30 min, and then entered a plateau at 90 min (Fig. 3 A). This correlates with the time course of formation and disappearance of autophagic tubes (see Fig. 1 C). Longer incubation in the absence of new uptake led to a significant reduction of the luciferase signal inside the vacuoles (Fig. 3 B). Luciferase incubated under the same conditions in reaction buffer remained stable. This is consistent with the fact that the invaginated vesicles became lysed inside the vacuoles (compare with Fig. 1 A). Luciferase released from the vesicles into the vacuolar lumen could then be degraded by vacuolar proteases. Luciferase uptake was temperature dependent, with a narrow optimum at 27°C (Fig. 4 A). The reaction had a strong requirement for cytosol, with the optima ranging from 2 to 4 mg/ml, depending on the individual batch of cytosol (Fig. 4 B). The reaction also required ATP and Mg2+, with optima at 2 and 7 mM, respectively (Fig. 4C and Fig. D). The optima matched those found independently by the visual assay for vesicular uptake of FITC-dextran (data not shown), confirming that both assays measure the same process.
Figure 3.
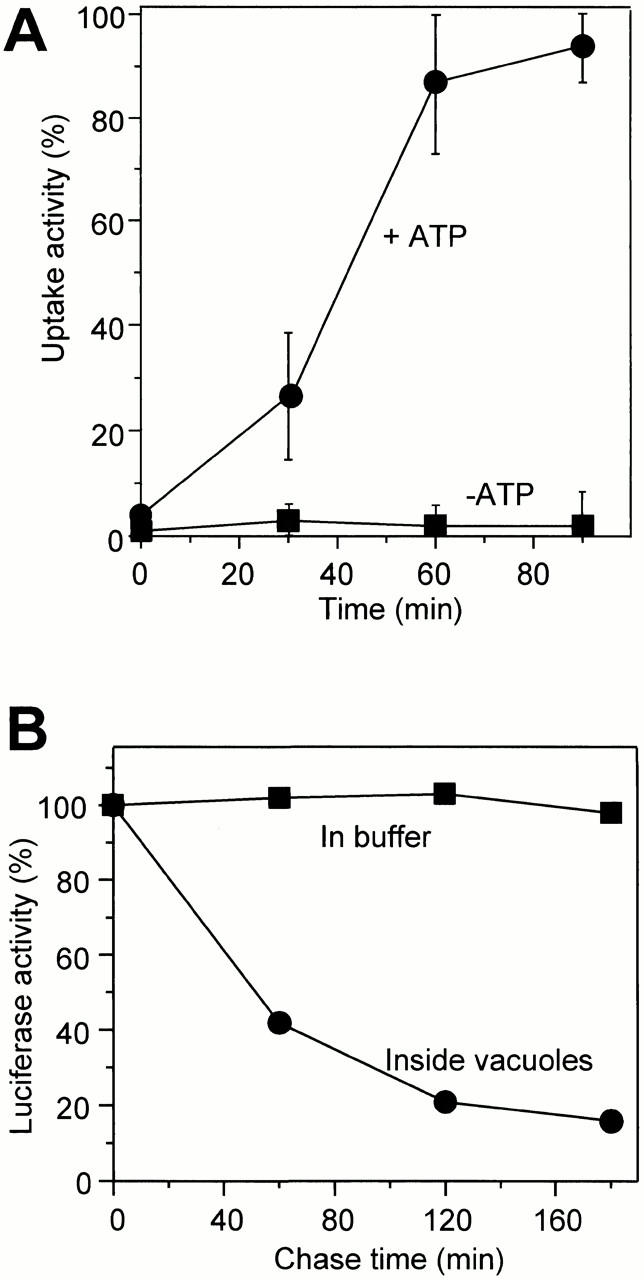
Time course and temperature dependence of uptake. (A) Standard uptake reactions were performed in the presence or absence of the ATP-regenerating system. After the indicated times at 27°C, the vacuoles were reisolated, washed, treated with protease, and luciferase uptake activity was assayed. (B) A standard uptake reaction was performed. After 70 min of incubation, the vacuoles were reisolated, washed, and treated with protease. The vacuoles were reisolated again, resuspended in reaction buffer without luciferase, and incubated for the indicated times at 27°C. Aliquots were withdrawn to assay the total luciferase remaining. As a control, luciferase was incubated in reaction buffer for the same periods of time and assayed for activity.
Figure 4.

Requirements for in vitro uptake. Standard uptake reactions were performed (60 min) (A) at different temperatures, or in the presence of different concentrations of (B) cytosol, (C) ATP, or (D) MgCl2. Luciferase uptake was determined as described in the legend to Fig. 3 A. In B, titration curves for four independent cytosol preparations are shown. The maximal uptake signal of each titration curve was set at 100%. In D, the sample drawn as 0 mM MgCl2 contained 10 mM EDTA to chelate free magnesium in the reaction.
Vesicle Formation Depends on Starvation and on Gene Products Required for Autophagocytosis
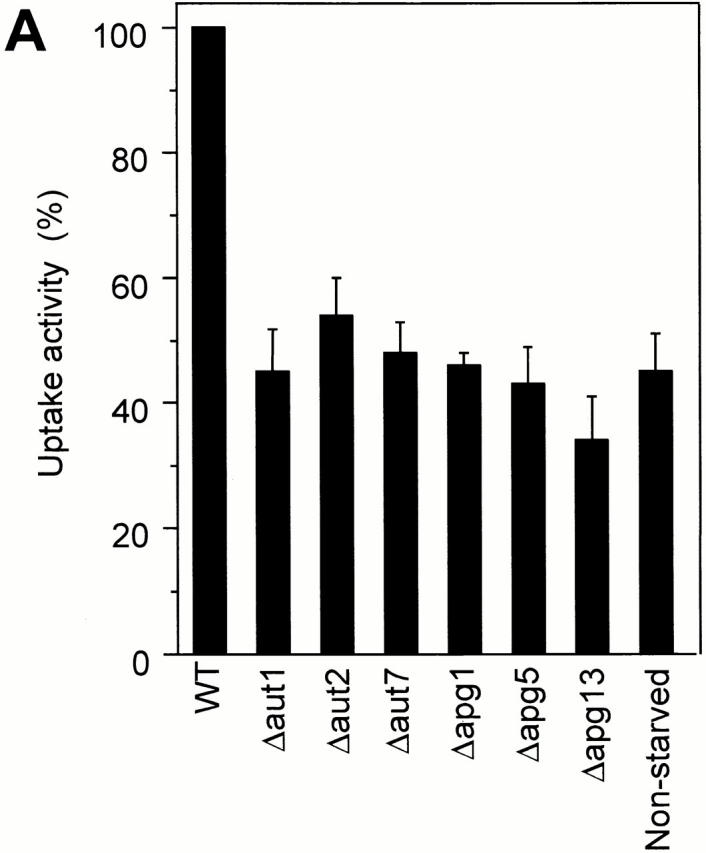
Autophagocytosis in S. cerevisiae is a constitutive process that is induced under conditions of stress or starvation (Takeshige et al. 1992; Noda et al. 1995). Many mutations influencing this process have been isolated (Tsukada and Ohsumi 1993; Thumm et al. 1994; Harding et al. 1995). These mutants affect macroautophagocytosis (Tsukada and Ohsumi 1993; Thumm et al. 1994; Harding et al. 1995) and the formation of autophagic tubes (Müller et al. 2000, this issue) in vivo. Since most of the genes identified so far are predicted not to be integral to the membrane, we tested the activity of cytosols from mutant strains in in vitro reactions. Deletion mutants for Aut1, Aut2, Aut7/Apg8, Apg1, Apg5, and Apg13 and the wild-type control were starved for nitrogen on SD(−N) medium. The cytosol prepared from any of the mutant strains had a significantly lower activity in the in vitro assay than the wild-type cytosol (Fig. 5 A). Also, the activity of the cytosol from wild-type cells grown on a nitrogen-rich source, that is, under conditions not inducing autophagocytosis, was 50–60% lower than that of the cytosol from starved wild-type cells (Fig. 5 A). Performing the assays with both vacuoles and cytosol isolated from Apg/Aut mutants did not increase the difference compared with the control (Fig. 5 B). A combination of Apg/Aut vacuoles with wild-type cytosol showed almost the maximal signal, indicating that a lack of Apg/Aut components on the vacuolar membranes can be complemented by wild-type cytosol. Therefore, the Apg/Aut pathway does not obviously produce persistent changes on the vacuolar membrane. Its influences can be reset by the cytosol during the incubation. In summary, isolated vacuoles perform the uptake of macromolecular solutes into vesicular structures inside the organelle. The in vitro system reproduces the induction of vacuole invagination by starvation and the influence of the Apg/Aut proteins on this process, which was seen in vivo (Müller et al. 2000, this issue). Therefore, the in vitro reaction reconstitutes microautophagocytosis.
Figure 5.
(A) Influence of cytosols from mutants defective in autophagocytosis. Standard uptake assays were performed (60 min, 27°C) with 3 mg/ml of cytosols derived from wild-type (K91-1A) cells, or from isogenic strains with deletions of the indicated genes, both starved on SD(−N) medium to induce autophagocytosis. To assay uptake activity under conditions that do not induce autophagocytosis, one reaction was performed with wild-type cytosol prepared from cells grown on rich medium (YPD). Four independent experiments were averaged, using the signal obtained with starved wild-type cytosol as the 100% reference. Error bars indicate SD. (B) The experiment was performed as in A, but cytosols and vacuoles from wild-type (wt) or mutant cells (Δapg1, Δapg5, and Δaut7) were mixed in the assay in the indicated combinations. Control reactions without ATP or with wild-type cytosol from nonstarved cells are included for comparison.

Formation of a Vesicle In Vitro Is Independent of Proteins Mediating Homotypic Vacuolar Fusion, Biosynthetic Transport to the Vacuole, and Macroautophagocytosis
Scission of the invaginating membrane to form a vesicle is topologically a homotypic fusion reaction of the vacuolar membrane in the autophagic tube. Homotypic vacuolar fusion is part of the normal physiology of the vacuolar membrane. During transmission into the growing daughter cell, the vacuole undergoes a regulated cycle of fragmentation into smaller vesicles, transport of these structures into the bud, and homotypic fusion to reconstitute the new vacuole (Warren and Wickner 1996). This homotypic fusion event has been characterized in vitro and in vivo, and many components involved have been identified. They include, among others, the SNAREs Vam3p, Vam7p, and Nyv1p, Sec17p (α-SNAP), Sec18p (NSF), and Ypt7p (a Rab protein) (Haas et al. 1995, 1996; Nichols et al. 1997; Ungermann and Wickner 1998). Vam3p and Vam7p are also involved in biosynthetic transport to the vacuole and in macroautophagocytosis (Wada et al. 1992; Darsow et al. 1997; Sato et al. 1998; Srivastava and Jones 1998). We asked whether these components would be required for the homotypic fusion process necessary to pinch off the invaginated membrane and release vesicles into the vacuolar lumen. An in vitro uptake reaction was performed in the presence of affinity-purified antibodies to Sec17p, Sec18p, Vam3p, Vam7p, or Nyv1p, or in the presence of Gdi1p, which extracts Ypt7p from the membrane. One aliquot of the reaction was used to quantify the uptake of luciferase into the vacuoles. A second aliquot of the same sample was used to monitor homotypic vacuolar fusion and control for the effect of the inhibitors. Vacuole fusion occurs under the conditions of the uptake reaction, albeit at only 20–30% of the efficiency under optimized conditions (Mayer et al. 1996). None of the inhibitors had a strong influence on the uptake of luciferase into the vacuolar lumen (Fig. 6 A) although they thoroughly blocked the homotypic fusion of vacuoles.
Figure 6.
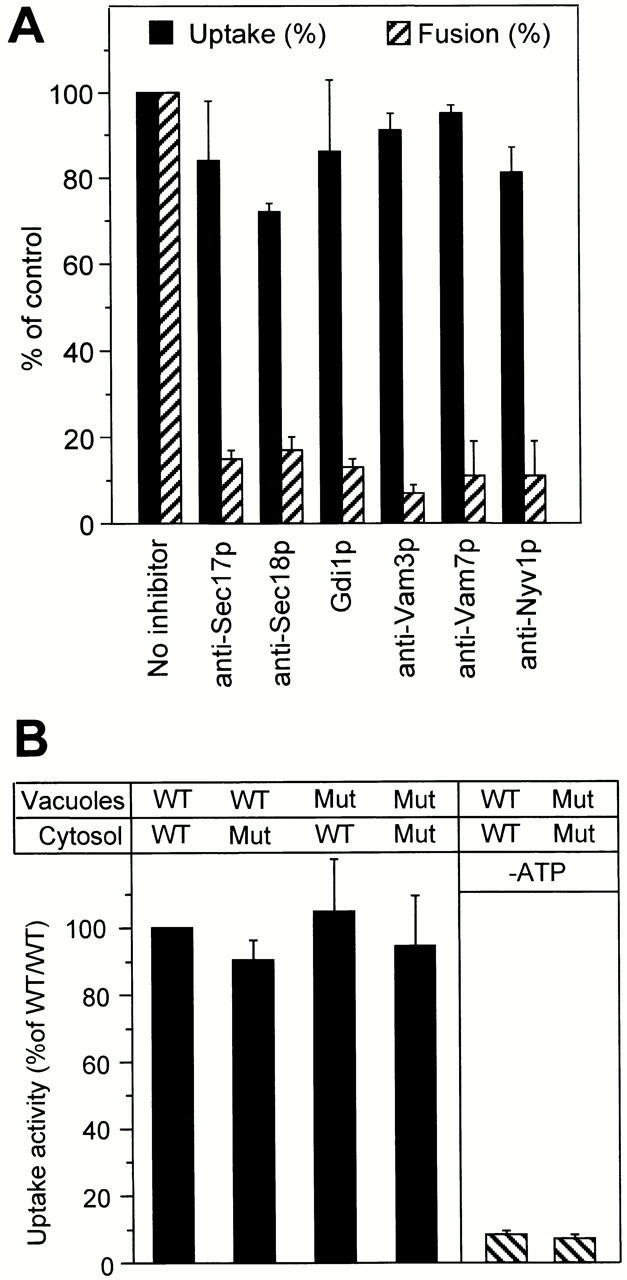
In vitro uptake is independent of components involved in homotypic vacuolar fusion. (A) Standard uptake reactions were performed (60 min, 27°C) in the presence of the indicated reagents using a mixture of vacuoles from strains DBY5734-16 (Δpep4) and DBY5734-19 (Δpho8). Therefore, homotypic vacuolar fusion and luciferase uptake could be assayed simultaneously from the same sample. Four independent experiments were averaged. Error bars indicate SD. For averaging, uptake and fusion activities of the control sample without inhibitor were set at 100%. Inhibitors were used at the following concentrations: anti-Sec17p, 1 μM; anti-Sec18p, 1 μM; Gdi1p, 2.5 μM; anti-Vam3p, 1 μM; anti-Vam7p, 1 μM; and anti-Nyv1p, 1 μM. (B) Vacuoles and cytosols were prepared from sec18-1 and from corresponding wild-type (Wt) cells grown and spheroplasted at 23°C. The isolated vacuoles and the cytosols were exposed to heat treatment (10 min, 37°C). Standard uptake reactions were performed with vacuoles and cytosols in the indicated combinations. Control reactions were performed in the absence of ATP. Five independent experiments were averaged; the sample with vacuoles and cytosol from wild-type cells was used as the 100% reference. Error bars indicate SD. Mut, mutant.
This finding was confirmed by results with a mutant carrying the temperature-sensitive sec18-1 allele. Vacuoles and cytosols from sec18-1 cells, which had been exposed to the restrictive temperature after their isolation from the cells, showed the same uptake activity as those from wild-type cells (Fig. 6 B). Successful inactivation of Sec18-1p was controlled by performing a visual fusion assay. Samples with sec18-1 vacuoles and cytosols had no fusion activity (not shown), whereas those with wild-type components showed many large fusion products (Haas et al. 1994). Like vacuole fusion (Haas et al. 1994), luciferase uptake also depended on guanine nucleotides. Vesicle formation was prevented by the GTP analogue GTPγS, which is poorly hydrolyzed, with an IC50 of 100 μM (Fig. 7 A), indicating that GTP hydrolysis is required. It was also prevented by the addition of the proton uncoupler carbonylcyanid-4-trifluomethoxyphenylhydrazon (FCCP) or the V-ATPase inhibitor concanamycin A (Fig. 7 B). Therefore, the activity of the vacuolar ATPase and the proton gradient across the membrane are prerequisites for the uptake reaction.
Figure 7.
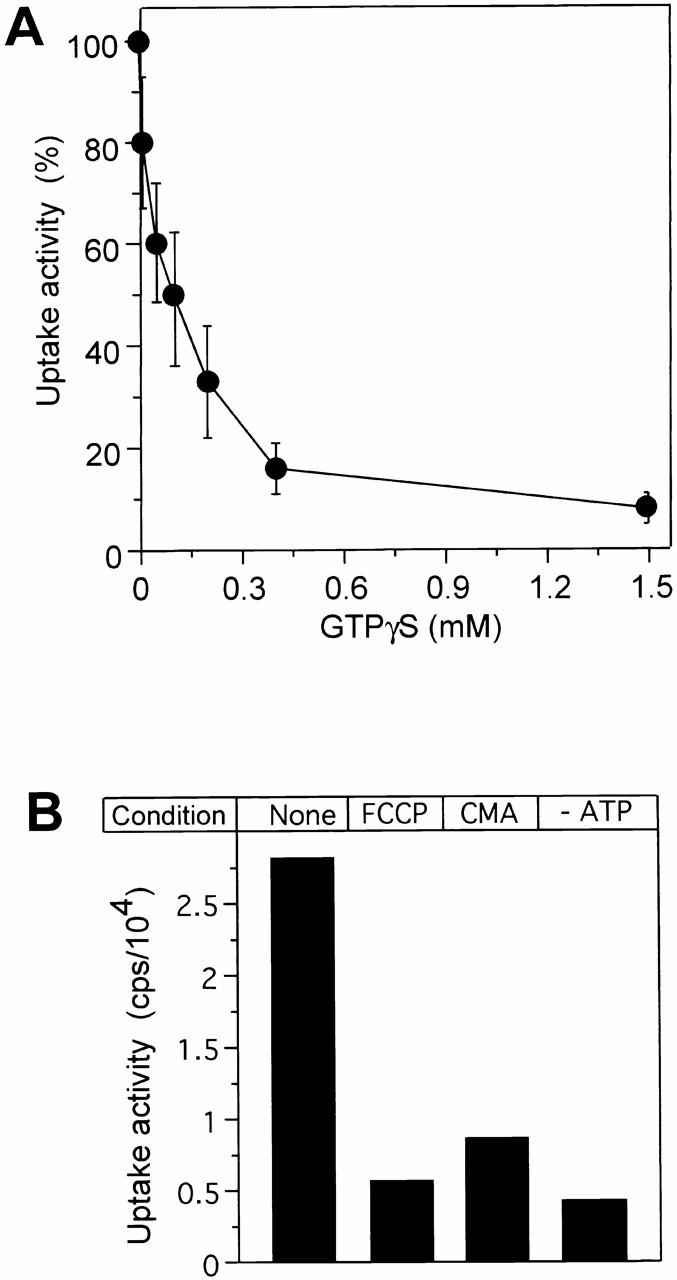
In vitro uptake depends on GTP hydrolysis and on the V-ATPase. (A) Standard uptake reactions were performed in the presence of different concentrations of GTPγS. Luciferase uptake was assayed as in the legend to Fig. 3 A. Four independent experiments were averaged, using the sample without inhibitor as the 100% reference. Error bars indicate SD. GTPγS did not influence luciferase activity by itself (not shown). (B) Standard uptake reactions were performed in the presence of FCCP (CMA, 20 μM) or concanamycin A (20 μM), or in the absence of ATP.
We conclude that the elementary reaction of invagination and scission of a vesicle into the vacuolar lumen involves the action of a GTPase and requires a membrane potential. It occurs independent of homotypic vacuolar fusion, of biosynthetic transport to the vacuole, and of macroautophagocytosis, but it is subjected to control via starvation and components of the Apg/Aut pathway.
Discussion
Transport of proteins into yeast vacuoles can occur via multiple pathways: vesicular transport from endosomes along the alkaline phosphatase, carboxypeptidase Y, or multivesicular body pathways (Wendland et al. 1998); the Cvt pathway, defined by aminopeptidase I (Scott and Klionsky 1998); the Vid pathway, leading to degradation of fructose-1,6-bisphosphatase by inclusion into a novel type of vesicle (Huang and Chiang 1997); direct Hsp70p-dependent translocation across the vacuolar membrane (Horst et al. 1999); and macroautophagocytosis (Takeshige et al. 1992). We have recently identified a microautophagic pathway in S. cerevisiae that operates via a specialized structure, the autophagic tube (Müller et al. 2000, this issue). The autophagic tube is a narrow and long invagination that is filled with cytosol and pinches off vesicles into the lumen of the vacuole. The tip of the tube, where vesicles are formed, is free of transmembrane particles. The smooth ultrastructural appearance of this zone of vesiculation matches that of autophagic bodies, which are also devoid of transmembrane particles (Müller et al. 2000, this issue; Baba et al. 1995).
In vivo analysis indicated that autophagic tubes are only prominent when macroautophagocytosis is induced, that is, under conditions of massive membrane influx towards the vacuolar membrane (Müller et al. 2000, this issue). Also, autophagic bodies, which can be formed both by micro and macroautophagocytosis, can only accumulate inside the vacuoles of protease-deficient cells if the macroautophagic pathway is active (Darsow et al. 1997). Therefore, it appears that the microautophagic pathway depends on the induction of macroautophagy. These observations suggest that a major function of autophagic tubes could be the maintenance of membrane homeostasis, that is, to invaginate excess membrane that is transferred into the vacuolar boundary membrane by its continuous fusion with the outer membrane of newly formed macroautophagosomes.
Microautophagic vacuole invagination in vitro was independent of the t-SNAREs, Vam3p and Vam7p, which are required for macroautophagocytosis, as well as the Cvt, alkaline phosphatase, carboxypeptidase Y, and multivesicular body pathways to the vacuole (Darsow et al. 1997; Sato et al. 1998; Srivastava and Jones 1998; Abeliovich et al. 1999). Furthermore, microautophagocytosis was independent of Sec18p/NSF, Sec17p/α-SNAP, of the vacuolar Rab-like GTPase Ypt7p, and of the vacuolar v-SNARE Nyv1p. This indicates that the core process of microautophagic membrane invagination and vesicle scission may be independent of conventional fusion machinery altogether, although it is topologically a homotypic fusion event between opposing membranes. It should be noted that in the in vitro system, an individual vacuole does not go through many rounds of invagination and vesicle formation. Most of the vacuoles bud only one or two vesicles into their lumen, which can be readily scored (see Fig. 1; our unpublished observations). Therefore, the source membrane for the invagination process is probably not limiting in the in vitro system. In contrast, the in vivo situation involves repetitive budding into the lumen of the organelle. A permanent supply of new vesicles to the vacuolar boundary membrane will be necessary to maintain microautophagic membrane invagination. If this supply is cut, such as by repressing macroautophagocytosis in rich media, or by apg/aut mutations, vacuolar invagination will be impaired by the limitation of source membrane.
Shortage of source membrane is sufficient to explain the effects of the Apg/Aut mutations on microautophagocytosis of soluble components and of peroxisomes in vivo (Müller et al. 2000, this issue). However, it does not readily explain why cytosols from Apg/Aut mutants support in vitro invagination of purified vacuoles significantly less than wild-type cytosols. This observation is consistent with a direct effect of the Apg/Aut components on the formation of autophagic tubes, on the formation of the vesicles at their tips, or on vesicle scission. Two different mechanisms are conceivable in which the same set of components could regulate macro- and microautophagocytosis of soluble and membrane-bound substrates at the same time. First, the Apg/Aut components tested could be part of a regulatory pathway that controls the induction of autophagy as a whole. It could regulate distinct machineries that are directly involved in the structural changes necessary for micro- and macroautophagy of soluble components or organelles. Second, the Apg/Aut components could themselves be part of an apparatus designed to bend the precursor membranes for autophagosomes and autophagic tubes and/or to sort their constituents. For both micro- and macroautophagocytosis, similar membrane curvature must be induced in the future autophagic body membrane, that is, both reactions require topologically equivalent changes. In macroautophagy, large cisternae must be bent to encircle portions of cytosol or an organelle. In microautophagy, invagination of the vacuole requires the induction of an equivalent curvature in part of its boundary membrane. This could explain how similar components and mechanisms may be responsible for both reactions. Detailed morphological and biochemical analysis will be necessary to distinguish between these possibilities.
Ultrastructural analysis revealed that nascent microautophagic vesicles have a strikingly low density of intramembranous particles (Müller et al. 2000, this issue), that is, a very low content of large transmembrane proteins. This unusual structure correlates with the unexpected biochemical properties of vesicle formation, its independence of homotypic fusion factors. An attractive speculation is that scission may be lipid driven. Scission of synaptic vesicles undergoing endocytosis requires the concerted action of the GTPase dynamin and endophilin, a lysophosphatidic acid acyl transferase (Schmidt et al. 1999). The action of this enzyme converts an inverted cone–shaped lipid (lysophosphatidic acid) into a cone-shaped lipid (phosphatidic acid) in the cytoplasmic leaflet of the bilayer, a process that could support formation, as well as scission, of the nascent vesicle. It is unlikely that the same mechanism also acts on microautophagic vesicles, because microautophagic scission is topologically inverse. In endocytosis, enzymes involved in vesicle formation and pinching bind to the outer side of the vesicle and to its neck and induce a negative curvature of the cyoplasmic leaflet. However, in microautophagocytosis they would have to bind to the inside of the nascent structure and locally increase the curvature of the cytoplasmic leaflet. Topologically, microautophagic vesicle formation is more related to the formation of multivesicular bodies, that is, the formation of internal endosomal vesicles. Those vesicles are strongly enriched in a specialized lipid, lysobisphosphatidic acid (Kobayashi et al. 1998). The smooth appearance of the fractured membrane of both vacuolar autophagic bodies and the zone of vesicle formation on autophagic tubes suggests that both membranes are poor in transmembrane proteins and, therefore, rich in lipids. Since vesicle formation is also independent of the enzymes catalyzing homotypic vacuolar membrane fusion, lipids may play an important role in the budding of vesicles into the vacuolar lumen. The in vitro system described here will be an important tool to address this question.
Acknowledgments
We want to thank Scott Emr, Randy Schekman, and David Botstein for supplying yeast strains.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 446) and the Boehringer Ingelheim Foundation to A. Mayer.
Footnotes
Abbreviations used in this paper: Apg and Aut, autophagocytosis; Cvt, cytoplasm to vacuole targeting; KAN, kanamycin; SNARE, soluble N-ethylmaleimide–sensitive factor attachment protein receptor.
References
- Abeliovich H., Darsow T., Emr S.D. Cytoplasm to vacuole trafficking of aminopeptidase I requires a t-SNARE-Sec1p complex composed of Tlg2p and Vps45. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:6005–6016. doi: 10.1093/emboj/18.21.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M., Takeshige K., Baba N., Ohsumi Y. Ultrastructural analysis of the autophagic process in yeastdetection of autophagosomes and their characterization. J. Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M., Osumi M., Ohsumi Y. Analysis of the membrane structures involved in autophagy in yeast by freeze-replica method. Cell Struct. Funct. 1995;20:465–471. doi: 10.1247/csf.20.465. [DOI] [PubMed] [Google Scholar]
- Baba M., Osumi M., Scott S.V., Klionsky D.J., Ohsumi Y. Two distinct pathways for targeting proteins from the cytoplasm to the vacuole/lysosome. J. Cell Biol. 1997;139:1687–1695. doi: 10.1083/jcb.139.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo A.M., Dice J.F. Lysosomes, a meeting point of proteins, chaperones, and proteases. J. Mol. Med. 1998;76:6–12. doi: 10.1007/s001090050185. [DOI] [PubMed] [Google Scholar]
- Darsow T., Rieder S.K., Emr S.D. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W.A., Jr. Studies on the mechanisms of autophagyformation of the autophagic vacuole. J. Cell Biol. 1990;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güldener U., Heck S., Fiedler T., Beinhauer J., Hegemann J.H. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A., Conradt B., Wickner W. G-protein ligands inhibit in vitro reactions of vacuole inheritance. J. Cell Biol. 1994;126:87–97. doi: 10.1083/jcb.126.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A., Schleglmann D., Lazar T., Gallwitz D., Wickner W. The GTPase Ypt7 of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding T.M., Morano K.A., Scott S.V., Klionsky D.J. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding T.M., Hefner-Gravink A., Thumm M., Klionsky D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol. Chem. 1996;271:17621–17624. doi: 10.1074/jbc.271.30.17621. [DOI] [PubMed] [Google Scholar]
- Horst M., Knecht E.C., Schu P.V. Import into and degradation of cytosolic proteins by isolated yeast vacuoles. Mol. Biol. Cell. 1999;10:2879–2889. doi: 10.1091/mbc.10.9.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P.H., Chiang H.L. Identification of novel vesicles in the cytosol to vacuole protein degradation pathway. J. Cell Biol. 1997;136:803–810. doi: 10.1083/jcb.136.4.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Dalton V., Eggerton V., Scott S.V., Klionsky D.J. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell. 1999;10:1337–1351. doi: 10.1091/mbc.10.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T., Baba M., Ishihara N., Kouichi M., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Schiffer H.H., Rupp S., Wolf D.H. Vacuolar/lysosomal proteolysisproteases, substrates, mechanisms. Curr. Opin. Cell Biol. 1993;5:990–996. doi: 10.1016/0955-0674(93)90082-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi T., Stang E., Fang K.S., Demoerloose P., Parton R.G., Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392:193–197. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- Lang T., Schaeffeler E., Bernreuther D., Bredschneider M., Wolf D.H., Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3597–3607. doi: 10.1093/emboj/17.13.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk S.E., Dunn W.A., Jr., Trausch J.S., Ciechanover A., Schwartz A.L. Ubiquitin-activating enzyme E1 is associated with maturation of autophagic vacuoles. J. Cell Biol. 1992;118:301–308. doi: 10.1083/jcb.118.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A., Wickner W., Haas A. Sec18p (NSF)-driven release of Sec17p (α-SNAP) precedes docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Matsuura A., Tsukada M., Wada Y., Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae . Gene. 1997;192:245–250. doi: 10.1016/s0378-1119(97)00084-x. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M.D., Klionsky D.J., Ohsumi M., Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Noda T., Ohsumi Y. Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimore G.E., Kadowaki M. Autophagyits mechanism and regulation. In: Ciechanover A.J., Schwartz A.L., editors. Cellular Proteolytic Systems. Wiley & Liss; New York: 1994. pp. 65–87. [Google Scholar]
- Müller O., Sattler T., Flötenmeyer M., Schwarz H., Plattner H., Mayer A. Autophagic tubesvacuolar invaginations involved in lateral membrane sorting and inverse vesicle budding. J. Cell. Biol. 2000;151:519–528. doi: 10.1083/jcb.151.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols B.J., Ungermann C., Pelham H.R.B., Wickner W.T., Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- Noda T., Matsuura A., Wada Y., Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae . Biochem. Biophys. Res. Commun. 1995;210:126–132. doi: 10.1006/bbrc.1995.1636. [DOI] [PubMed] [Google Scholar]
- Rothman J.E. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Sakai Y., Koller A., Rangell L.K., Keller G.A., Subramani S. Peroxisome degradation in Pichia pastorisidentification of specific steps and morphological intermediates. J. Cell Biol. 1998;141:625–636. doi: 10.1083/jcb.141.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.K., Darsow T., Emr S.D. Vam7p, a SNAP-25-like molecule, and Vam3p, a syntaxin homolog, function together in yeast vacuolar protein trafficking. Mol. Cell. Biol. 1998;18:5308–5319. doi: 10.1128/mcb.18.9.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A., Wolde M., Thiele C., Fest W., Kratzin H., Podtelejnikov A.V., Witke W., Huttner W.B., Söhling H.D. Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature. 1999;401:133–141. doi: 10.1038/43613. [DOI] [PubMed] [Google Scholar]
- Scott S.V., Klionsky D.J. Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr. Opin. Cell Biol. 1998;10:523–529. doi: 10.1016/s0955-0674(98)80068-9. [DOI] [PubMed] [Google Scholar]
- Scott S.V., Hefner-Gravink A., Morano K., Noda T., Ohsumi Y., Klionsky D.J. Cytoplasm to vacuole targeting and autophagy employ the same machinery to deliver proteins to the yeast vacuole. Proc. Natl. Acad. Sci. USA. 1996;93:12304–12308. doi: 10.1073/pnas.93.22.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen P.O., Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–172. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- Shintani T., Mizushima N., Ogawa Y., Matsuura A., Noda T., Ohsumi Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:5234–5241. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A., Jones E.W. Pth1/Vam3p is the syntaxin homolog at the vacuolar membrane of Saccharomyces cerevisiae required for the delivery of vacuolar hydrolases. Genetics. 1998;148:85–98. doi: 10.1093/genetics/148.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub M., Bredschneider M., Thumm M. AUT3, as serine/threonine kinase gene, is essential for autophagocytosis in Saccharomyces cerevisiae . J. Bacteriol. 1997;179:3875–3883. doi: 10.1128/jb.179.12.3875-3883.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromhaug P.E., Berg T.O., Fengsrud M., Seglen P.O. Purification and characterization of autophagosomes from rat hepatocytes. Biochem. J. 1998;335:217–224. doi: 10.1042/bj3350217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshige K., Baba M., Tsuboi S., Noda T., Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumm M., Egner R., Koch B., Schlumpberger M., Straub M., Veenhuis M., Wolf D.H. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae . FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- Titorenko V.I., Keizer I., Harder W., Veenhuis M. Isolation and characterization of mutants impaired in the selective degradation of peroxisomes in the yeast Hansenula polymorpha . J. Bacteriol. 1995;177:357–363. doi: 10.1128/jb.177.2.357-363.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M., Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae . FEBS Lett. 1993;333:168–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Tuttle D.L., Dunn W.A., Jr. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris . J. Cell Sci. 1995;108:25–35. doi: 10.1242/jcs.108.1.25. [DOI] [PubMed] [Google Scholar]
- Tuttle D.L., Lewin A.S., Dunn W.A., Jr. Selective autophagy of peroxisomes in methylotrophic yeasts. Eur. J. Biochem. 1993;60:283–290. [PubMed] [Google Scholar]
- Ungermann C., Wickner W. Vam7p, a vacuolar SNAP-25 homolog, is required for SNARE complex integrity and vacuole docking and fusion. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3269–3276. doi: 10.1093/emboj/17.12.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenhuis M., Douma A., Harder W., Osumi M. Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch. Microbiol. 1983;134:193–203. doi: 10.1007/BF00407757. [DOI] [PubMed] [Google Scholar]
- Wada Y., Ohsumi Y., Anraku Y. Genes for directing vacuolar morphogenesis in Saccharomyces cerevisiae . J. Biol. Chem. 1992;267:18665–18670. [PubMed] [Google Scholar]
- Warren G., Wickner W. Organelle inheritance. Cell. 1996;84:395–400. doi: 10.1016/s0092-8674(00)81284-2. [DOI] [PubMed] [Google Scholar]
- Wendland B., Emr S.D., Riezman H. Protein traffic in the yeast endocytic and vacuolar protein sorting pathways. Curr. Opin. Cell Biol. 1998;10:513–522. doi: 10.1016/s0955-0674(98)80067-7. [DOI] [PubMed] [Google Scholar]
- Wiemken A. Isolation of vacuoles from yeasts. Methods Cell Biol. 1975;12:99–109. doi: 10.1016/s0091-679x(08)60954-1. [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Masaki R., Tashiro Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry J. Histochem. Cytochem. 38 1990. 573 580a [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Masaki R., Tashiro Y. Absence of cytochrome P-450 and presence of autolysosomal membrane antigens on the isolation membranes and autophagosomal membranes in rat hepatocytes J. Histochem. Cytochem. 38 1990. 1571 1581b [DOI] [PubMed] [Google Scholar]
- Yokota S. Formation of autophagosomes during degradation of excess peroxisomes induced by administration of dioctyl phthalate. Eur. J. Cell Biol. 1993;61:67–80. [PubMed] [Google Scholar]
- Yuan W., Stromhaug P.E., Dunn W.A. Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol. Biol. Cell. 1999;10:1353–1366. doi: 10.1091/mbc.10.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]