

Abstract
Interaction of integrins with the extracellular matrix leads to transmission of signals, cytoskeletal reorganizations, and changes in cell behavior. While many signaling molecules are known to be activated within Rac-induced focal complexes or Rho-induced focal adhesions, the way in which integrin-mediated adhesion leads to activation of Rac and Rho is not known. In the present study, we identified clusters of integrin that formed upstream of Rac activation. These clusters contained a Rac-binding protein(s) and appeared to be involved in Rac activation. The integrin clusters contained calpain and calpain-cleaved β3 integrin, while the focal complexes and focal adhesions that formed once Rac and Rho were activated did not. Moreover, the integrin clusters were dependent on calpain for their formation. In contrast, while Rac- and Rho-GTPases were dependent on calpain for their activation, formation of focal complexes and focal adhesions by constitutively active Rac or Rho, respectively, occurred even when calpain inhibitors were present. Taken together, these data are consistent with a model in which integrin-induced Rac activation requires the formation of integrin clusters. The clusters form in a calpain-dependent manner, contain calpain, calpain-cleaved integrin, and a Rac binding protein(s). Once Rac is activated, other integrin signaling complexes are formed by a calpain-independent mechanism(s).
Keywords: cell spreading, β3 integrin, calpain, integrin clusters, Rac activation
Introduction
An important part of the response of cells to growth factors or agonists that act on G protein–coupled receptors is the activation of members of the Rho family of low molecular weight GTPases (Ridley and Hall 1992; Ridley et al. 1992; Hartwig et al. 1995). These GTPases induce cytoskeletal reorganizations. If cells are adherent, the GTPases induce ligand-occupied integrin to become incorporated into complexes with cytoskeletal proteins (Hotchin and Hall 1995). The complexes induced by Cdc42 and Rac1 are smaller than those induced by RhoA and are generally referred to as focal complexes; those induced by RhoA are typically larger arrow-shaped complexes known as focal adhesions. These integrin complexes recruit and activate numerous signaling molecules involved in inducing additional cytoskeletal reorganizations and changes in gene transcription (Burridge and Chrzanowska-Wodnicka 1996). Thus, signals transmitted into a cell that is adherent are different from those transmitted into a cell that remains in suspension. The network of signaling pathways activated by signaling across the various receptors leads to changes in cell behavior appropriate to the specific environmental conditions.
Mechanisms involved in the activation of Rho GTPases by growth-factor receptors and G protein–coupled receptors involves activation of exchange factors. These exchange factors are recruited to the appropriate submembranous location by association with membrane phospholipids or adapter proteins. In both cases, tyrosine phosphorylation has been implicated in the regulation of recruitment (Bollag and McCormick 1991; Boguski and McCormick 1993; Bokoch and Der 1993; Michiels et al. 1997; Ma et al. 1998). Recent evidence has shown that members of the Rho family of GTPases are also activated after signaling across integrins and that they are involved in integrin-induced cell spreading (Clark et al. 1998; Price et al. 1998). Although considerable information is now available on the way in which growth-factor receptors and G protein–coupled receptors activate Rho GTPases, little is known about the way in which integrins activate these proteins.
In the present study, we have shown that clusters of integrin form upstream of integrin-induced Rac activation. We provide evidence that these clusters contain a Rac-binding protein(s) and are involved in Rac activation. These complexes have a different protein composition than Rac-induced focal complexes and Rho-induced focal adhesions. Unlike the Rac-induced focal complexes and Rho-induced focal adhesions, these clusters contain calpain and calpain-cleaved β3-integrin subunit. The presence of calpain inhibitors prevented the formation of any kind of integrin complex. However, the use of constitutively active Rac and Rho showed that Rac- and Rho-induced integrin complexes could form when the activity of calpain was inhibited; in contrast, formation of the early integrin clusters was dependent on calpain.
We suggest that Rac can be activated by more than one pathway. While soluble agonists may activate Rac through calpain-independent mechanisms, integrin engagement leads to Rac activation by a mechanism that involves the activation of calpain, the calpain-dependent formation of clusters of integrin and calpain-cleaved proteins, and association of Rac with these clusters. We refer to these clusters as calpain-induced integrin clusters to distinguish them from the integrin-containing focal complexes and focal adhesions that form by calpain-independent mechanisms after activation of Rac and RhoA.
Materials and Methods
Reagents
Human fibronectin was obtained from Sigma-Aldrich. Human vitronectin was obtained from GIBCO BRL. Calpeptin and MDL (the membrane-permeable inhibitors of calpain) were obtained from Calbiochem. Inhibitors were solubilized in dimethyl sulfoxide. mAb against human vinculin (clone hVIN-1) was obtained from Sigma-Aldrich, mAb against phosphotyrosine (PY20) from Santa Cruz Biotechnology, Inc., mAb against αvβ3 integrin from Chemicon, mAb against μ-calpain from Alexis, mAb against hemagglutinin (HA) epitope (clone 12CA5) from Boehringer, mAb against myc epitope (clone 9E10) from Calbiochem, polyclonal antibodies against β3 integrin from Chemicon, polyclonal antibodies against HA epitope were from Santa Cruz Biotechnology, Inc. Polyclonal antibodies against total calpain were a gift from Dr. Darrel Goll (University of Arizona). TRITC-conjugated phalloidin was purchased from Sigma-Aldrich.
Cell Culture
Bovine aortic endothelial (BAE) cells, provided by Dr. Paul DiCorleto (Cleveland Clinic Foundation), were maintained in DME/F12 (DME and Ham's F12, 1:1; Biowhittaker) medium with 10% FBS (GIBCO BRL), containing penicillin-streptomycin (GIBCO BRL) and glutamine (GIBCO BRL). Cells were used between passages 10 and 20. Clone ST4, overexpressing μ-calpain was described previously (Kulkarni et al. 1999).
Plasmids and Transfection
The plasmid DNA encoding HA-tagged constitutively active Q61L Rac1 was provided by Dr. Charles Abrams (University of Pennsylvania Medical School, Philadelphia, PA). The plasmid DNA encoding myc-tagged dominant-negative Rac1 (N17Rac) was provided by Dr. Allan Hall (University College, London, UK) and plasmid DNA encoding constitutively active RhoA (Q63LRhoA) was provided by Dr. Martin Schwartz (The Scripps Research Institute, La Jolla, CA). The generation of plasmid encoding catalytically inactive μ-calpain has been described previously (Kulkarni et al. 1999).
Transient transfections were carried out using lipofectamine Plus reagent (GIBCO BRL) as described by manufacturer. In brief, BAE cells were plated in 100-mm dishes, grown to ∼80% confluency and washed with serum-free medium once before transfection. Transfections were carried out at 37°C in a total volume of 6.8 ml of serum-free medium containing 40 μl of lipofectamine, 20 μl Plus reagent, and 15 μg DNA per 100-mm dish. After 5 h, transfection medium was replaced with medium containing 10% serum. After 24 h, cells were detached using EDTA (GIBCO BRL), washed with PBS, resuspended in serum-containing medium, and replated on fibronectin-coated coverslips. Cells transfected with constitutively active Rac or constitutively active RhoA were replated on fibronectin-coated coverslips in the presence of calpeptin (100 μg/ml), MDL (250 μM), or a corresponding volume of DMSO alone. After 60 min, cells were fixed and processed for immunofluorescence.
Immunofluorescence
Glass coverslips were coated with human fibronectin (25 μg/ml) overnight at room temperature. Cells were allowed to spread on coverslips for different times, washed with TBS (50 mM Tris, 0.15 mM NaCl, 0.1% NaN3, pH 7.4), fixed with 4% paraformaldehyde in TBS for 10 min, permeabilized with 0.1% Triton X-100 in TBS for 5 min, then blocked with TBS containing 4% horse serum (GIBCO BRL). Coverslips were incubated with appropriate dilutions of primary antibodies (mAb against human vinculin 1:50, mAb against phosphotyrosine 1:500, mAb against αvβ3 integrin 1:200, mAb against calpain 1:50, mAb against HA epitope 1 μg/ml, mAb against myc epitope 3 μg/ml, polyclonal antibodies against β3 integrin 1:200, polyclonal antibodies against autolytic fragment of calpain 1:50, polyclonal antibodies against total calpain 1:200, polyclonal antibodies against calpain-cleaved β3 integrin 1:50, polyclonal antibodies against HA epitope 1 μg/ml) in TBS containing 4% horse serum. Unbound antibodies were removed by extensive washing with TBS containing 0.1% Triton X-100 and coverslips were incubated with biotinylated secondary antibodies (Molecular Probes) diluted 1:500 in TBS containing 4% horse serum, followed by streptavidin conjugated to Alexa 350, Alexa 488, or Alexa 594 diluted 1:500 (Molecular Probes) in TBS containing 4% horse serum or by secondary antibodies conjugated to Alexa 350 (diluted 1:200), Alexa 488, or Alexa 594 (diluted 1:500) (Molecular Probes) in the same buffer. Actin was detected by staining with TRITC-conjugated phalloidin (1 μg/ml). Images were collected using either an immunofluorescence microscope (Carl Zeiss, Inc.) or a TCS-NT confocal laser-scanning microscope (Leica). Laser intensities were adjusted so that the excitation of the fluorochromes did not allow any cross talk between the channels.
Results
β3 Integrin Engagement Leads to the Formation of Integrin Clusters that Contain a Rac-binding Protein and Form Upstream of Rac Activation
As a first step in identifying mechanisms involved in Rac activation by integrins, we plated BAE cells on fibronectin, a substrate for β3-containing integrin (Gerber et al. 1996; Wu et al. 1996), and examined the distribution of β3-containing integrin at increasing times after plating. It is well known that integrins are incorporated into focal adhesions as they spread on an integrin substrate. Focal adhesions were detected with antibodies against β3 integrin as arrowhead structures, but they required several hours to form (Fig. 1 A, arrows). At earlier times of spreading (Fig. 1 A, middle), there were no focal adhesions, integrin at these times was present in tiny complexes all over the cell. When we examined cells that were at an even earlier stage of spreading (Fig. 1 A, left), we detected integrin in yet another type of organization; at this stage, integrin was present in a few clusters at the periphery of the cell. The formation of these early integrin-containing clusters also occurred if cells were plated on vitronectin, another integrin substrate (e.g., one cell in Fig. 1 B, left). However, it did not occur if cells were plated on polylysine, a substrate that allows spreading without integrin engagement (data not shown). The integrin clusters also contained vinculin (e.g., Fig. 1 B, right) and phosphotyrosine (data not shown). The clusters appeared to be transient. Even in the 30-min samples, there were often only a few cells in each field that contained integrin clusters; the rest (e.g., Fig. 1 B) had progressed to the stage of containing numerous tiny focal complexes.
Figure 1.
Distribution of β3 integrin and vinculin in spreading BAE cells. BAE cells were allowed to spread on fibronectin-coated coverslips for 30 min, 1 h, or 8 h (A) or on vitronectin-coated coverslips for 30 min (B). Cells were fixed and permeabilized. Cells were stained with antibodies against β3 integrin or vinculin. Arrows in A indicate focal adhesions. Bars, 20 μm.


To determine which of the various integrin-containing complexes formed upstream of Rac activation, cells were transiently transfected with dominant-negative Rac, detached, and replated on fibronectin (Fig. 2A and Fig. B) or vitronectin (C) for 1 h. During this time, cells spread to the point where most of them contained numerous small integrin complexes. However, cells in which Rac activity was inhibited were arrested at the earlier stage in which the large clusters of integrin were present. The use of antibodies against the myc-tagged Rac revealed that dominant-negative Rac protein colocalized with the integrin clusters. Since inhibition of spreading by dominant-negative Rac is assumed to result from its competition with endogenous Rac, this finding suggests that Rac associates with a molecule present in the integrin clusters and that this is the site of its activation.
Figure 2.

β3 integrin-containing clusters form upstream of Rac activation. BAE cells were grown to 80% confluency and transiently transfected with plasmid encoding myc-tagged dominant-negative N17Rac. Cells were detached and replated on fibronectin-coated coverslips for 1 h (A and B) or on vitronectin-coated coverslips for 1 h (C), fixed, and permeabilized. Cells were stained with antibodies against β3 integrin. Transfected cells were detected with anti–myc antibody and actin filaments were detected with TRITC-labeled phalloidin. Bar, 20 μm.
The Composition and Mechanism of Formation of the Early Calpain-induced Integrin Clusters Are Different from Those of Rac- or Rho-induced Integrin Complexes
The formation of integrin complexes is typically thought to result from activation of Rac and Rho (Nobes and Hall 1995). It is now known that Rac and Rho can be activated by signaling across integrins, but the mechanisms are unknown. The GTPases must presumably be activated by signals transmitted across integrins before the formation of focal complexes and focal adhesions. The finding that the clusters of integrin detected in this study form upstream of Rac activation suggests that they might represent specialized structures used by integrins to induce the activation of Rac. In such a model, these integrin clusters would have a different protein composition and form by a different mechanism than the subsequent Rac-induced focal complexes or Rho-induced focal adhesions.
Based on our previous finding that calpain was required for integrin-induced spreading (Kulkarni et al. 1999), we wondered whether calpain was involved in the formation of the integrin clusters. To gain insight into this possibility, we used an immunofluorescence approach to determine whether calpain was present in the integrin clusters. As shown in Fig. 3, calpain was present in these clusters. Moreover, it appeared to be absent from the focal complexes (Fig. 3 A, middle) and focal adhesions (right). Fig. 3 B shows two additional examples of calpain being present in clusters in cells just beginning to spread. The finding that calpain was absent from focal adhesions was somewhat surprising given the general acceptance of a previous report that calpain is present in these structures (Beckerle et al. 1987). To ensure that our inability to detect calpain in the focal adhesions was not simply the result of lower levels of calpain in these structures than in the early integrin-containing clusters, we investigated the distribution of calpain in BAE cells overexpressing μ-calpain (Kulkarni et al. 1999). Previously, we have shown that these cells express approximately fivefold more μ-calpain than nontransfected BAE cells (Kulkarni et al. 1997). Transfected cells were allowed to spread on fibronectin for times that allowed focal adhesion formation; phosphotyrosine antibodies were used to detect focal adhesions. As in nontransfected cells, we could detect calpain in integrin-containing clusters at the very early stage of the cell spreading (data not shown). However, as shown in Fig. 4, calpain was not present in the focal adhesions.
Figure 3.
Calpain is present in the integrin-containing clusters. BAE cells were allowed to spread on fibronectin-coated coverslips for 30 min, 1 h, or 8 h, fixed, and permeabilized. (A) Cells were stained with antibody against β3 integrin and antibody against calpain. (Right) Arrows indicate focal adhesions. (B) Two additional fields were stained only for calpain. Bars, 20 μm.


Figure 4.

Calpain is absent from the focal adhesions of BAE cells overexpressing μ-calpain. BAE cells overexpressing μ-calpain were allowed to spread on fibronectin-coated coverslips for 24 h, fixed, and permeabilized. Cells were stained with antibodies against phosphotyrosine and calpain. Bar, 20 μm.
The selective association of calpain with the early integrin clusters suggests that this may be a site of calpain activity. When calpain is active, it autolyses. To determine whether calpain in the clusters was active before Rac activation, cells were transiently transfected with dominant-negative Rac. The use of an antibody that only detects the autolysed form of the enzyme demonstrated that calpain fragment was present in the clusters with which dominant-negative Rac associated (Fig. 5).
Figure 5.
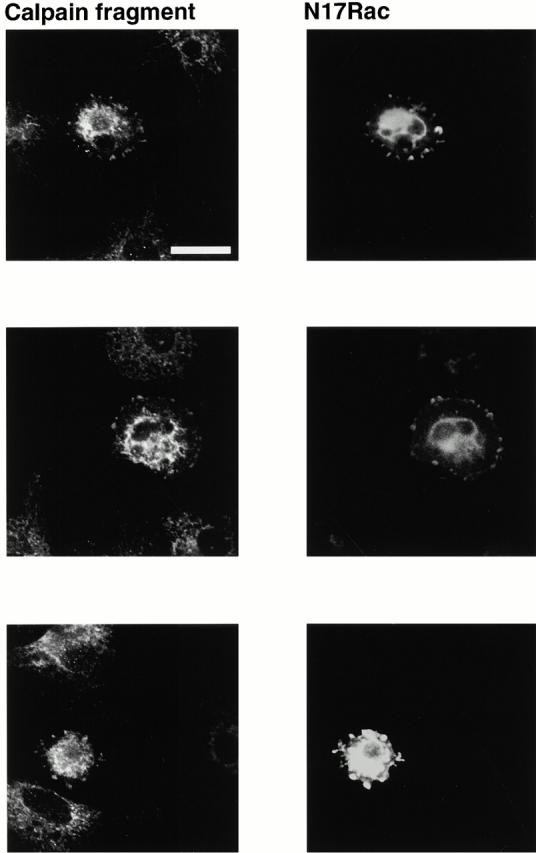
Autolytic fragment of calpain is present in the integrin-containing clusters, which form upstream of Rac activation. BAE cells were transiently transfected with cDNA encoding myc-tagged dominant-negative N17Rac. Cells were detached and replated on fibronectin-coated coverslips for 1 h, fixed, and permeabilized. Transfected cells were detected with anti–myc antibody and autolysed calpain with an antibody that only recognizes the autolysed fragment. Three different transfected cells are shown. Bar, 20 μm.
One of the known substrates of calpain in platelets is the cytoplasmic domain of the β3 integrin subunit (Du et al. 1995). Although the function of the calpain-induced cleavage of β3 integrin is not known, we reasoned that if calpain was present in integrin clusters but not in Rac- or Rho-induced integrin complexes, then β3 may be selectively cleaved in the clusters. We used an antibody that only recognizes the calpain-cleaved form of the β3 subunit to assess this possibility. Although intact β3 integrin was present in early integrin clusters (Fig. 6, top left), focal complexes (top left, arrows), and focal adhesions (bottom left, arrows), calpain-cleaved integrin was only present in the early integrin clusters (Fig. 6, top right). It was not present in focal complexes or in focal adhesions (Fig. 6, right).
Figure 6.

Calpain-cleaved β3 integrin is present in the integrin-containing clusters. BAE cells were allowed to spread on fibronectin-coated coverslips for 45 min or 8 h, fixed, and permeabilized. Cells were stained with antibody against αvβ3 integrin and antibodies against calpain-cleaved β3 integrin. Small arrowheads indicate focal complexes and large arrowheads indicate focal adhesions. Bar, 20 μm.
The observation that calpain and its cleaved substrates were present in the early integrin clusters but not in the Rac- or Rho-induced focal complexes and focal adhesions supports the idea that the integrin clusters might form by a different mechanism than the Rac- and Rho-induced integrin complexes. To assess the possibility that calpain may be involved in formation of the integrin clusters, cells were plated in the presence of membrane-permeable calpain inhibitors, calpeptin and MDL. Previously, we have shown that these inhibitors prevent the formation of Rac- and Rho-induced integrin complexes (Kulkarni et al. 1999). As shown in Fig. 7 A, the inhibitors also prevented the formation of the early integrin clusters. Moreover, cells that were transiently transfected with a catalytically inactive form of μ-calpain (Kulkarni et al. 1999) also failed to form integrin clusters or to spread (Fig. 7 B).
Figure 7.
Calpain inhibitors and dominant-negative μ-calpain prevent formation of integrin-containing clusters. (A) BAE cells were detached and replated on fibronectin-coated coverslips in the presence of calpeptin (100 μg/ml) or MDL (250 μM) for 1 h, fixed, and permeabilized. Cells were stained with antibodies against β3 integrin, actin filaments were detected with TRITC-labeled phalloidin. (B) BAE cells were grown to 80% confluency and transiently transfected with plasmid encoding HA-tagged dominant-negative μ-calpain. Cells were detached and replated on fibronectin-coated coverslips for 1–3 h, fixed, and permeabilized. Cells were stained with antibodies against vinculin. Transfected cells were detected with HA-myc antibody and actin filaments were detected with TRITC-labeled phalloidin. Two independent fields are shown. Bar, 20 μm.


To determine whether Rac-induced focal complexes and Rho-induced focal adhesions form by a mechanism that does not require calpain, cells were transiently transfected with constitutively active Rac (Fig. 8) or RhoA (Fig. 9) and replated on fibronectin in the presence of calpain inhibitors. The spreading of calpain-inhibited cells was restored in cells expressing active Rac (Fig. 8) or RhoA (Fig. 9). The cells transfected with constitutively active Rac were filled with tiny focal complexes (Fig. 8); these complexes were much smaller than the clusters of integrin forming at early times in cells in which calpain had not been inhibited (Fig. 1 Fig. 2 Fig. 3). They were, however, similar to those that formed at intermediate times of spreading (e.g., Fig. 1, middle). Similarly, constitutively active Rho could induce the formation of focal adhesions in cells in which calpain was inhibited. These focal adhesions contained β3 integrin and vinculin (Fig. 9), but, just like the focal adhesions that formed in the absence of calpain inhibitors (Fig. 3 and Fig. 4), the focal adhesions did not contain calpain (data not shown).
Figure 8.

Rac-induced focal complexes contain β3 integrin and vinculin and form by a mechanism that does not involve calpain. BAE cells were grown to 80% confluency and transiently transfected with plasmid encoding HA-tagged constitutively active Q61LRac. Cells were detached and replated on fibronectin-coated coverslips in the presence of calpeptin (100 μg/ml), MDL (250 μM), or corresponding volume of DMSO for 1 h, fixed, and permeabilized. Cells were stained with antibodies against β3 integrin. Transfected cells were detected with anti–HA epitope antibody, actin filaments were detected with TRITC-labeled phalloidin. Bar, 20 μm.
Figure 9.

RhoA-induced focal adhesions contain β3 integrin and vinculin and form by a mechanism that does not require calpain. BAE cells were grown to 80% confluency and transiently transfected with plasmid encoding constitutively active Q63LRhoA. Cells were detached and replated on fibronectin-coated coverslips in the presence of MDL (250 μM) or DMSO for 1 h, fixed, and permeabilized. Cells were stained with antibodies against β3 integrin or vinculin. Transfected cells were detected with anti–HA epitope antibody, actin filaments were detected with TRITC-labeled phalloidin. Bar, 20 μm.
Discussion
Integrin-induced adhesion induces signaling pathways that lead to cytoskeletal reorganizations and altered gene expression (Burridge and Chrzanowska-Wodnicka 1996; Burridge et al. 1997). It has been assumed that the signaling molecules that induce these changes are present in focal adhesions, complexes of integrin and signaling molecules that assemble as a consequence of Rho activation after integrin–ligand interactions (Burridge and Chrzanowska-Wodnicka 1996; Burridge et al. 1997). More recently, it has become apparent that Rac is also activated after integrin–ligand interactions and that this is involved in the formation of smaller clusters of integrin, known as focal complexes (Clark et al. 1998; Price et al. 1998). Although the importance of Rac and Rho in inducing the formation of integrin signaling complexes has been well established, little is known about the way in which these GTPases are themselves activated after integrin–ligand interactions. In the present study, we have identified a new type of integrin complex that forms upstream of Rac activation. We refer to these complexes as integrin clusters to distinguish them from Rac-induced focal complexes and Rho-induced focal adhesions and suggest that they may be involved in Rac activation. We propose a model, shown in Fig. 10, in which the integrin clusters represent transient structures involved in integrin-induced Rac activation. We suggest that the integrin clusters form by a calpain-dependent mechanism. The subsequent Rac-induced focal complexes and Rho-induced focal adhesions form by calpain-independent mechanisms and have a different protein content to the initial integrin clusters.
Figure 10.

Schematic representation of the formation of integrin-containing clusters, focal complexes, and focal adhesions. In this model, integrin engagement leads to calpain activation and formation of transient integrin-containing clusters. The clusters contain calpain-cleaved β3 integrin, active calpain, vinculin, and phosphotyrosine. Recruitment of Rac to the clusters leads to its activation and subsequent formation of Rac-induced focal complexes. These complexes do not require calpain activation for their formation, and they appear to have a different composition than the initial integrin clusters. Rac activation is followed by RhoA activation and formation of arrowhead-shaped focal adhesions, which also form by a mechanism that is independent of calpain activation.
Several lines of evidence suggest that the integrin clusters may be the site of Rac activation. Thus, the integrin clusters accumulated in the presence of dominant-negative Rac. Moreover, dominant-negative Rac accumulated in the integrin clusters, indicating the presence of a Rac binding protein(s) in these clusters. Further, calpain inhibitors prevented the formation of the integrin clusters and activation of Rac. Activation of Rho family GTPases after activation of growth-factor receptors or G protein–coupled receptors is thought to occur by activation of exchange factors that are recruited to submembranous locations by association with exchange factors or adapter proteins to phosphotyrosine residues or lipid metabolites in the membrane (Bollag and McCormick 1991; Boguski and McCormick 1993; Bokoch and Der 1993; Michiels et al. 1997; Ma et al. 1998). Future work will be needed to determine whether activation of Rac in integrin clusters after integrin-induced signaling is initiated through the same adapters and exchange factors or whether novel mechanisms, such as the calpain-induced cleavage of proteins within the integrin clusters are involved in integrin-induced Rac activation.
Several years ago, we showed that calpain was one of the signaling molecules that was activated after integrin engagement (Fox et al. 1983, Fox et al. 1993). We have recently used a variety of approaches to show that calpain is activated very early after integrin engagement in BAE cells, upstream of Rac and Rho activation (Kulkarni et al. 1999). The findings in the present study are consistent with the idea that calpain may be required for Rac and Rho activation because it is required for the formation of integrin clusters in which Rac activation is initiated. Thus, the formation of the integrin clusters was inhibited by two different membrane-permeable calpain inhibitors. Although a recent study has suggested that one of these inhibitors (calpeptin) may also inhibit tyrosine phosphatase (Schoenwaelder and Burridge 1999) at the concentrations used in our studies, we have never observed any evidence for such an action of calpeptin (Fox et al. 1990, Fox et al. 1991) and do not believe that this was a complicating factor in the present study. Thus, inhibition of integrin cluster formation was also observed with MDL, a calpain inhibitor that has not been found to inhibit tyrosine phosphatase (Schoenwaelder and Burridge 1999). Moreover, a dominant-negative form of μ-calpain also prevented cell spreading and focal cluster formation. The idea that the integrin clusters were formed by a mechanism that required calpain activity was supported by the finding that calpain was present in the clusters but not in the Rac- and Rho-induced complexes. Moreover, calpain was active in the clusters (as assessed by the presence of the autolysed form of calpain) and proteins in the clusters had been cleaved by calpain (as assessed by the presence of calpain-cleaved β3-integrin subunit).
We can only speculate on the way in which an action of calpain induces the formation of integrin clusters. In the present study, the availability of antibodies that selectively recognize the calpain-cleaved β3-integrin subunit (Du et al. 1995) allowed us to use this protein as a marker to show that a calpain-cleaved protein was present in the clusters. However, the presence of cleaved integrin does not comment on whether or not cleavage of this protein is involved in formation of the clusters. Many other cytoskeletal proteins and signaling molecules present in integrin complexes are known to be cleaved by calpain. The cell in which the calpain-induced cleavage of proteins involved in integrin signaling has been studied in the most detail is the platelet (Fox et al. 1983, Fox et al. 1985, Fox et al. 1993; Fox and Saido 1999). In this cell, proteins that are cleaved include spectrin, actin-binding protein, dystrophin-related protein, protein kinase C, cortactin, tyrosine phosphatase PTP-1B, or the β3 integrin subunit (Fox et al. 1985, Fox et al. 1987; Tapley and Murray 1985; Frangioni et al. 1993; Du et al. 1995; Earnest et al. 1995; Huang et al. 1997). While we have shown that several of these are cleaved in endothelial cells spreading on an integrin substrate (Kulkarni et al. 1999), it is not possible to conclude which protein must be cleaved for spreading to occur. Likewise, it is not possible at this point to determine which substrate must be cleaved for the integrin clusters identified in this study to form.
The availability of the antibody that selectively recognizes β3 integrin that has been cleaved by calpain allowed us to show that integrin clusters contained cleaved integrin while the β3 integrin present in the Rac- and RhoA-induced complexes that formed at later stages of spreading appeared to be intact. This observation suggests a specific function for the calpain-induced cleavage of β3 integrin in the clusters. Such a function could conceivably be in the formation of the clusters, but it could also be in regulating recruitment or activation of other proteins in the clusters. In platelets, cleavage of β3 integrin by calpain results in the removal of two NXXY motifs (Du et al. 1995). Mutations in this part of the cytoplasmic tail have been reported to prevent the incorporation of αIIbβ3 into focal adhesions and to limit cell spreading and migration (Schaller et al. 1995; Ylanne et al. 1995; Eigenthaler et al. 1997). These findings are consistent with our observation that the calpain-cleaved β3 integrin was not present in focal complexes and focal adhesions and support the idea that the integrin clusters are not simply early versions of focal complexes or focal adhesions, but that they are transient structures with the focal complexes and focal adhesions being formed de novo once Rac and Rho were activated.
The mechanism by which Rac-induced focal complexes and RhoA-induced focal adhesions form has not been elucidated. The finding in the present study that calpain is not required for the formation of the complexes and adhesions is consistent with our observation that neither calpain nor calpain-cleaved integrin were present in these structures. Based on a report several years ago that m-calpain was present in focal adhesions (Beckerle et al. 1987), it has generally been accepted that calpain is a component of RhoA-induced focal adhesions. The reason that calpain was detected in focal adhesions in the early studies, whereas we have been unable to detect it in these structures, is not known. In our studies, we have antibodies that selectively recognize μ- and m-forms of calpain and have been unable to detect either form in focal adhesions (Kulkarni, S., and J.E. Fox, unpublished observations). Moreover, we have used cells that are expressing five times as much μ-calpain as normal (Kulkarni et al. 1999) and have still been unable to detect calpain in focal adhesions. One possibility is that the sites of colocalization of integrin and calpain in the previously reported work were really early integrin-containing clusters, not focal adhesions. Another is that calpain is present in focal adhesions under specific conditions. For example, calpain has been implicated in the breakdown of focal adhesions in migratory cells (Huttenlocher et al. 1997).
The present study indicates a role for calpain in initiating integrin-induced Rac activation and suggests that it does this, at least in part, by inducing the formation of clusters containing calpain-cleaved molecules. It will be interesting to investigate the possibility that calpain is involved in activation of Rac by receptors other than integrins. In platelets, activation of calpain was originally shown to occur after engagement of integrin (Fox et al. 1983, Fox et al. 1993); responses induced by thrombin (an agonist that acts on a G protein–coupled receptor) were retarded by a variety of calpain inhibitors but not completely inhibited (Fox et al. 1990). While this suggested that calpain may play a role in thrombin-induced signaling, it has not been possible to demonstrate thrombin-induced calpain activation as assayed by autolysis of calpain or by cleavage of a platelet protein; all the known substrates for calpain (e.g., spectrin, actin-binding protein, talin, β3 integrin, cortactin, dystrophin-related protein, FAK, PTP-1B, PTPMEG, phospholipase C, and protein kinase C) (Fox et al. 1983, Fox et al. 1985, Fox et al. 1987; Frangioni et al. 1993; Banno et al. 1995; Earnest et al. 1995; Cooray et al. 1996; Gu and Majerus 1996; Huang et al. 1997; Norris et al. 1997) were cleaved only in platelets in which integrin engagement had occurred. Thus, it has generally been assumed that activation of signaling molecules, including Rac, by signaling across the thrombin receptor does not involve calpain activation (Fox and Phillips 1983; Hartwig et al. 1995). However, this conclusion needs to be re-examined based on the recent report that the introduction of calpastat (calpain-inhibiting consensus sequence derived from calpastatin) into platelets prevents thrombin-induced platelet secretion, aggregation, and spreading (Croce et al. 1999). It is of note, however, that in the latter studies, calpastat was introduced into the platelets by a method that could have induced platelet damage. It is certainly possible that activation of calpain after thrombin-induced signaling does not result in cleavage of any of the known substrates, but results in cleavage of an as yet unidentified protein. Future studies will be needed to investigate this possibility. The possibility that calpain is involved in the activation of Rac by growth-factor receptors has not been given much attention, but it has not been excluded either. It is of interest that in the present studies, calpain inhibitors prevented spreading even when serum was present, but did not prevent spreading if constitutively active Rac or Rho were available. Thus, it is conceivable that even growth-factor receptors may need active calpain for the initial activation of Rac and RhoA.
In summary, our studies identify the presence of integrin clusters that form at early stages after integrin-induced adhesion. We suggest that these clusters form by a calpain-dependent mechanism and provide a means of recruiting Rac to sites of occupied integrin. We suggest that recruitment of Rac to these clusters may be an essential step in initiating activation of this Rho family member and that once it is active Rac can initiate the calpain-independent formation of integrin complexes involved in adhesion and cell spreading.
Acknowledgments
This work was supported by research grants HL30657 and HL56264 (J.E.B. Fox) from the National Institutes of Health.
Footnotes
Abbreviations used in this paper: BAE, bovine aortic endothelial; HA, hemagglutinin.
References
- Banno Y., Nakashima S., Hachiya T., Nozawa Y. Endogenous cleavage of phospholipase C-b3 by agonist-induced activation of calpain in human platelets. J. Biol. Chem. 1995;270:4318–4324. doi: 10.1074/jbc.270.9.4318. [DOI] [PubMed] [Google Scholar]
- Beckerle M.C., Burridge K., DeMartino G.N., Croall D.E. Colocalization of calcium-dependent protease II and one of its substrates at sites of cell adhesion. Cell. 1987;51:569–577. doi: 10.1016/0092-8674(87)90126-7. [DOI] [PubMed] [Google Scholar]
- Boguski M.S., McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Bokoch G.M., Der C.J. Emerging concepts in the Ras superfamily of GTP-binding proteins. FASEB J. 1993;7:750–759. doi: 10.1096/fasebj.7.9.8330683. [DOI] [PubMed] [Google Scholar]
- Bollag G., McCormick F. Regulators and effectors of ras proteins. Annu. Rev. Cell Biol. 1991;7:601–632. doi: 10.1146/annurev.cb.07.110191.003125. [DOI] [PubMed] [Google Scholar]
- Burridge K., Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell. Dev. Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Burridge K., Chrzanowska-Wodnicka M., Zhong C. Focal adhesion assembly. Trends Cell Biol. 1997;7:342–347. doi: 10.1016/S0962-8924(97)01127-6. [DOI] [PubMed] [Google Scholar]
- Clark E.A., King W.G., Brugge J.S., Symons M., Hynes R.O. Integrin-mediated signals regulated by members of the Rho family of GTPases. J. Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooray P., Yuan Y., Schoenwaelder S.M., Mitchell C.A., Salem H.H., Jackson S.P. Focal adhesion kinase (pp125FAK) cleavage and regulation by calpain. Biochem. J. 1996;318:41–47. doi: 10.1042/bj3180041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce K., Flaumenhaft R., River M., Furie B., Furie B.C., Herman I.M., Potter D. Inhibition of calpain blocks platelet secretion, aggregation, and spreading. J. Biol. Chem. 1999;274:36321–36327. doi: 10.1074/jbc.274.51.36321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., Saido T.C., Tsubuki S., Indig F.E., Williams M.J., Ginsberg M.H. Calpain cleavage of the cytoplasmic domain of the integrin b3 subunit. J. Biol. Chem. 1995;270:26146–26151. doi: 10.1074/jbc.270.44.26146. [DOI] [PubMed] [Google Scholar]
- Earnest J.P., Santos G.F., Zuerbig S., Fox J.E.B. Dystrophin-related protein in the platelet membrane skeleton. Integrin-induced change in detergent-insolubility and cleavage by aggregating platelets. J. Biol. Chem. 1995;270:27259–27265. doi: 10.1074/jbc.270.45.27259. [DOI] [PubMed] [Google Scholar]
- Eigenthaler M., Hofferer L., Shattil S.J., Ginsberg M.H. A conserved sequence motif in the integrin b3 cytoplasmic domain is required for its specific interaction with b3-endonexin. J. Biol. Chem. 1997;272:7693–7698. doi: 10.1074/jbc.272.12.7693. [DOI] [PubMed] [Google Scholar]
- Fox J.E.B., Austin C.D., Reynolds C.C., Steffen P.K. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J. Biol. Chem. 1991;266:13289–13295. [PubMed] [Google Scholar]
- Fox J.E.B., Goll D.E., Reynolds C.C., Phillips D.R. Identification of two proteins (actin-binding protein and P235) that are hydrolyzed by endogenous Ca2+-dependent protease during platelet aggregation. J. Biol. Chem. 1985;260:1060–1066. [PubMed] [Google Scholar]
- Fox J.E.B., Phillips D.R. Stimulus-induced activation of the calcium-dependent protease within platelets. Cell Motil. 1983;3:579–588. doi: 10.1002/cm.970030524. [DOI] [PubMed] [Google Scholar]
- Fox J.E.B., Reynolds C.C., Austin C.D. The role of calpain in stimulus–response couplingevidence that calpain mediates agonist-induced expression of procoagulant activity in platelets. Blood. 1990;76:2510–2519. [PubMed] [Google Scholar]
- Fox J.E.B., Reynolds C.C., Morrow J.S., Phillips D.R. Spectrin is associated with membrane-bound actin filaments in platelets and is hydrolyzed by the Ca2+-dependent protease during platelet activation. Blood. 1987;69:537–545. [PubMed] [Google Scholar]
- Fox J.E.B., Reynolds C.C., Phillips D.R. Calcium-dependent proteolysis occurs during platelet aggregation. J. Biol. Chem. 1983;258:9973–9981. [PubMed] [Google Scholar]
- Fox J.E.B., Saido T.C. Calpain in signal transduction. In: Wang K.K.W., Yuen P.-W., editors. The Pharmacology and Toxicology of Calpain. Taylor and Francis; Washington, D.C.: 1999. pp. 103–126. [Google Scholar]
- Fox J.E.B., Taylor R.G., Taffarel M., Boyles J.K., Goll D.E. Evidence that activation of platelet calpain is induced as a consequence of binding of adhesive ligand to the integrin, glycoprotein IIb-IIIa. J. Cell Biol. 1993;120:1501–1507. doi: 10.1083/jcb.120.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangioni J.V., Oda A., Smith M., Salzman E.W., Neel B.G. Calpain catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:4843–4856. doi: 10.1002/j.1460-2075.1993.tb06174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber D.J., Pereira P., Huang S.Y., Pelletier C., Tonegawa S. Expression of alpha v beta 3 integrin chains on murine lymphocytes. Proc. Natl. Acad. Sci. USA. 1996;93:14698–14703. doi: 10.1073/pnas.93.25.14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M., Majerus P. The properties of the protein tyrosine phosphatase PTPMEG. J. Biol. Chem. 1996;271:27751–27759. doi: 10.1074/jbc.271.44.27751. [DOI] [PubMed] [Google Scholar]
- Hartwig J.H., Bokoch G.M., Carpenter C.L., Janmey P.A., Taylor L.A., Toker A., Stossel T.P. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Hotchin N.A., Hall A. The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J. Cell Biol. 1995;131:1857–1865. doi: 10.1083/jcb.131.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Tandon N.N., Greco N.J., Ni Y., Wang T., Zhan X. Proteolysis of platelet cortactin by calpain. J. Biol. Chem. 1997;272:19248–19252. doi: 10.1074/jbc.272.31.19248. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A., Palecek S.P., Lu Q., Zhang W., Mellgren R.L., Lauffenburger D.A., Ginsberg M.H., Horwitz A.F. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 1997;272:32719–32722. doi: 10.1074/jbc.272.52.32719. [DOI] [PubMed] [Google Scholar]
- Kulkarni S., Goll D.E., Saido T.C., Suzuki K., Fox J.E.B. Regulation of focal contact formation by calpain. Mol. Biol. Cell. 1997;8:282a. [Google Scholar]
- Kulkarni S., Saido T., Suzuki K., Fox J. Calpain mediates integrin-induced signaling at a point upstream of Rho family members. J. Biol. Chem. 1999;274:21265–21275. doi: 10.1074/jbc.274.30.21265. [DOI] [PubMed] [Google Scholar]
- Ma A.D., Metjian A., Bagrodia S., Taylor S., Abrams C.S. Cytoskeletal reorganization by G protein–coupled receptors is dependent on phosphoinositide 3-kinase g, a Rac guanosine exchange factor, and Rac. Mol. Cell. Biol. 1998;18:4744–4751. doi: 10.1128/mcb.18.8.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels F., Stam J.C., Hordijk P.L., van der Kammen R.A., Ruuls-Van Stalle L., Feltkamp C.A., Collard J.G. Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation. J. Cell Biol. 1997;137:387–398. doi: 10.1083/jcb.137.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C.D., Hall A. Rho, Rac, and Cdc42 GTPase regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Norris F.A., Atkins R.C., Majerus P.W. Inositol polyphosphate 4-phosphatase is inactivated by calpain-mediated proteolysis in stimulated human platelets. J. Biol. Chem. 1997;272:10987–10989. doi: 10.1074/jbc.272.17.10987. [DOI] [PubMed] [Google Scholar]
- Price L.S., Leng J., Schwartz M.A., Bokoch G.M. Activation of Rac and Cdc42 by integrin mediates cell spreading. Mol. Biol. Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J., Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Schaller M.D., Otey C.A., Hildebrand J.D., Parsons J.T. Focal adhesion kinase and paxillin bind to peptides mimicking b integrin cytoplasmic domains. J. Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwaelder S.M., Burridge K. Evidence for a calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase Rho. A novel role for the calpain inhibitor calpeptin in the inhibition of protein-tyrosine phosphatases. J. Biol. Chem. 1999;274:14359–14367. doi: 10.1074/jbc.274.20.14359. [DOI] [PubMed] [Google Scholar]
- Tapley P.M., Murray A.W. Evidence that treatment of platelets with phorbol ester causes proteolytic activation of Ca2+-activated, phospholipid-dependent protein kinase. Eur. J. Biochem. 1985;151:419–423. doi: 10.1111/j.1432-1033.1985.tb09118.x. [DOI] [PubMed] [Google Scholar]
- Wu C., Hughes P.E., Ginsberg M.H., McDonald J.A. Identification of a new biological function for the integrin alpha v beta 3initiation of fibronectin matrix assembly. Cell Adhes. Commun. 1996;4:149–158. doi: 10.3109/15419069609014219. [DOI] [PubMed] [Google Scholar]
- Ylanne J., Huuskonen J., O'Toole T.E., Ginsberg M.H., Virtanen I., Gahmberg C.G. Mutation of the cytoplasmic domain of the integrin beta 3 subunit. Differential effects on cell spreading, recruitment to adhesion plaques, endocytosis, and phagocytosis. J. Biol. Chem. 1995;270:9550–9557. doi: 10.1074/jbc.270.16.9550. [DOI] [PubMed] [Google Scholar]